

Abstract
Liver fibrosis is mediated by hepatic stellate cells (HSCs), which respond to a variety of cytokine and growth factors to moderate the response to injury and create extracellular matrix at the site of injury. G-protein coupled receptor (GPCR)-mediated signaling, via endothelin-1 (ET-1) and angiotensin II (AngII), increases HSC contraction, migration and fibrogenesis. Regulator of G-protein signaling-5 (RGS5), an inhibitor of vasoactive GPCR agonists, functions to control GPCR-mediated contraction and hypertrophy in pericytes and smooth muscle cells (SMCs). Therefore we hypothesized that RGS5 controls GPCR signaling in activated HSCs in the context of liver injury. In this study, we localize RGS5 to the HSCs and demonstrate that Rgs5 expression is regulated during carbon tetrachloride (CCl4)-induced acute and chronic liver injury in Rgs5LacZ/LacZ reporter mice. Furthermore, CCl4 treated RGS5-null mice develop increased hepatocyte damage and fibrosis in response to CCl4 and have increased expression of markers of HSC activation. Knockdown of Rgs5 enhances ET-1-mediated signaling in HSCs in vitro. Taken together, we demonstrate that RGS5 is a critical regulator of GPCR signaling in HSCs and regulates HSC activation and fibrogenesis in liver injury.
Introduction
Liver fibrosis and its sequelea of cirrhosis and hepatocellular carcinoma (HCC) are responsible for 29,000 deaths a year in the United States, making it the 12th leading cause of death [1]. Injury to the liver results in a wave of cytokine mobilization [2], many of which are secreted by Kupffer cells, the liver's resident macrophages. These cytokines (e.g. tumor necrosis factor α (TNF- α) [3] and transforming growth factor-β1 (TGF- β) [4]) then activate HSCs, which deposit extracellular matrix (e.g. collagen) as part of the wound repair response. With chronic injury, ongoing inflammation and HSC activation results in accumulation of scar tissue and eventual decreased liver function [5].
In the uninjured liver, quiescent HSCs behave like pericytes [6], surrounding the endothelium of the sinusoids. However, upon injury-induced activation, HSCs become the primary collagen-producing cell in the fibrotic liver [7], [8]. HSC activation, in response to platelet derived growth factor BB (PDGF-BB) [9]–[11] and TGFβ [4], [12], is well characterized; however, G-protein coupled receptor (GPCR) signaling pathways also influence their behavior during fibrosis. AngII [13]–[15], ET-1 [16]–[20], and norepinephrine (NE) [21]–[24] have been implicated in promoting HSC activation and thus fibrosis. Therefore, modulating signaling downstream of GPCRs may represent a novel therapeutic target in liver fibrosis, potentially preventing HCC.
RGS5 is a small GTPase activating protein that inhibits Gαq and Gαi-mediated signaling downstream of GPCRs [25]. RGS5 is primarily expressed in vascular smooth muscle cells (SMCs) and pericytes [26]–[29], and inhibits AngII- and ET-1-mediated signaling [30], [31] to regulate blood pressure [26], [32]–[34] and vascular remodeling [35], [36]. Moreover, RGS5 expression correlates with both cardiac [37] and skin [38] fibrosis, and expression is increased in multiple cancers (e.g., breast, ovarian, acute myeloid leukemia, and liver) [39]–[44].
We hypothesized that RGS5 controls liver injury via its ability to modulate GPCR-mediated signaling in activated HSCs. In this study, we localize expression of RGS5 to HSCs in the liver, and demonstrate that Rgs5 expression is regulated in both acute and chronic liver injury. Furthermore, mice lacking RGS5 expression develop increased hepatocyte damage and fibrosis in response to carbon tetrachloride (CCl4). Rgs5 expression is regulated in cultured HSCs in response to fibrogenic agonists, and ET-1-mediated signaling is potentiated in the absence of Rgs5 expression. Taken together, we demonstrate that RGS5 is a critical regulator of GPCR signaling in HSCs, and controls HSC activation and fibrogenesis in response to liver injury.
Materials and Methods
Animals
Rgs5 LacZ mice, which have a nuclear localized β-galactosidase reporter gene knocked into exon 2 of the Rgs5 locus and mice have been backcrossed to a C57BL/6 background, were purchased (Deltagen [45]). To induce acute liver injury, mice were injected (i.p.) with 10 µl/g body weight CCl4 (Sigma-Aldrich) diluted 10% (v/v) in olive oil once. To induce chronic liver injury, mice were injected twice weekly for four weeks. Olive oil-injected animals served as controls for CCl4-injected mice. At the indicated time-points, mice were sacrificed using CO2 inhalation. The Institutional Animal Care and Use Committee of the University of Washington, which is certified by the Association for Assessment and Accreditation of Laboratory Animal Care International, approved all experiments.
X-gal labeling
To preserve β-galactosidase activity, liver tissue was fixed in PLP (4% paraformaldehyde (PFA), 75 mM l-lysine, 10 mM sodium periodate) for 2 hours at 4°C, cryopreserved in 18% sucrose, frozen in optimum cutting temperature compound, and 5-µm cryosections were prepared for X-gal labeling or immunofluorescence (IF). LacZ activity was measured using a standard 5-bromo-4-chloro-3-indolyl-β-d-galactoside (X-gal) staining protocol [46] for 16 hours at 37°C. After washing, sections were post-fixed in 1% PFA for 5 minutes and immunolabeled for additional histologic analysis (see below).
Measurement of relative hepatocyte cytoplasmic clearing
Approximately twenty 10× fields of H&E stained 5 µm paraffin embedded sections liver were imaged, each representing a 1.19x.89 mm area. Using the ImageJ software package, the relative cleared hepatocyte cytoplasmic area was measured. Images were automatically adjusted for contrast, converted to grey scale, and sharpened to enhance borders. The threshold tool isolated the tissue from slide background. Particle analysis included sizes 49 µm2 to 324 µm2 and circularity value between 0.3 and 1.0, yielding total particle area measured as a percentage of slide area. Data was processed by macro to remove potential bias.
Quantification of collagen deposition
To label collagen deposition (indicative of fibrosis), 5 µm paraffin embedded tissue sections were stained with picrosirius red (365548 and P6744, Sigma) and 0.1% Fast Green (F7258, Sigma) at room temperature for 30 minutes. The relative quantification of fibrosis was measured using ImageJ. Ten 4× fields were imaged for each liver, representing a 2.95×2.21 mm area. Using the color threshold tool, red staining was isolated. The image is then converted to grey scale, and threshold is used to subtract remaining background. Measurement of the remaining area yielded the area of the slide with red staining. This was normalized to the area of the tissue (vessel lumen subtracted) for each slide to yield the percent of tissue with picrosirius red staining. Data was processed by macro to remove potential bias.
Immunofluorescence (IF)
IF was performed using standard techniques, with liver sections incubated overnight with the following primary antibodies: rabbit anti-GFAP 1∶1000 (Z0334, Dako), chicken anti-β-gal 1∶1000 (ab9361, Abcam), rabbit anti-SMA1∶200 (ab32575 Abcam), rabbit anti-CRBP1 1∶200 (sc30106, Santa Cruz), rabbit anti-VWF 1∶200 (A0082, Dako), Rat anti-CD-31 1∶100 (553370, BD Pharmingen), and Rat anti-F4/80 1∶200 (122603, Biolegend). Immune complexes were detected with the following secondary antibodies: Alexa 488 conjugated goat anti-Rabbit IgG (A11034, Life Technologies), Alexa 488 conjugated goat anti-rat IgG (A21208, Life Technologies), Alexa 647 conjugated donkey anti-rat IgG (A21247, Life Technologies), Alexa 594 conjugated donkey anti-chicken IgG (703-516-155, Jackson Immunoresearch), antibodies. Sections were mounted with VectaShield (H-1000, Vector Labs) and imaged with a Zeiss Axiovert 200 microscope. Confocal images generated using an Olympus FV1000 confocal microscope.
Cell Culture
The LX-2 HSC cell line was generously provided by Scott L. Friedman [47]. LX-2 cells were passaged in DMEM high glucose (Gibco/Life Technologies) supplemented with 2% FBS and penicillin-streptomycin and used between passages 10 and 20.
HSC isolation
Mouse primary HSCs were prepared by perfusion with collagenase/pronase and density centrifugation using Optiprep (Sigma) as reported previously [48].
siRNA Knockdown of Rgs5 Expression
Rgs5 was knocked down in LX-2 cells using a specific small interfering RNA (siRNA) from Life Technologies (5′-AGGAGAUUAAGAUCAAGUUTT-3′). LX-2 HSCs were transfected with Lipofectamine RNAiMAX Transfection reagent (Life Technologies) following the manufacturer's specifications. Briefly, 5×105 cells were transfected with either Rgs5-specific siRNA (12.5 nM) or siRNA negative control (12.5 nM; Life technologies) and plated at a final density of 105 cells/60 mm dish (for protein isolation) or 4×104 cells/6-well dish (for RNA isolation) and grown in 2% FBS growth media. After 24 hr, the media were changed to serum-free media and cells were starved for 24 hr. Where indicated, cells were stimulated with endothelin-1 (ET-1; 100 nM; Sigma), TGFβ (5 ng/ml; R&D systems), TNFα (5 ng/ml; R&D systems), or PDGF-BB (10 ng/ml; R&D systems).
RNA isolation and quantitative RT-PCR (qPCR)
RNA was isolated from LX-2 cells and mouse livers using the E.Z.N.A. Total RNA Kit I (Omega Bio Tek). For tissue lysates, 27 mm3 were homogenized in TRK lysis buffer using a stator-rotor homogenizer, as per kit instructions. cDNA was prepared by reverse transcription using the High-Capacity cDNA Reverse Transcription Kit; (Applied Biosystems). 20 ng cDNA was used in each qPCR reaction, using PerfeCTa SYBR Green FastMix (Quanta biosciences). Gene expression was calculated by the ΔΔCt method: Fold expression = 2−ΔΔCt. Gene expression was normalized to Gapdh expression within each sample then normalized to individual control treatment conditions within each dataset.
Immunoblot
Lysates were prepared from LX-2 cells 0, 10, and 20, min after ET-1 treatment by resuspending scraped cell pellets in lysis buffer [50 mM Tris·HCl (pH 8.0), 120 mM NaCl, 0.5% Igepal, 1 mM EDTA, with protease inhibitors (Calbiochem)]. After protein quantitation, 10 µg of each protein extract was separated on 10% bis-Tris gels. Proteins were transferred to PVDF and blocked with 5% nonfat dry milk (NFDM) in TBS-T (0.1% Tween). Membranes were incubated with the following primary antibodies diluted in 5% NFDM in TBS-T overnight at 4°C: 1∶1,000 phospho-p42/44 (Thr202/Tyr204; pERK) (Cell Signaling); 1∶5,000 total p42/44 (ERK) [49]. After 3× washes with TBS-T, membranes were incubated with the following secondary antibodies diluted in 5% NFDM in TBS-T at room temperature for 1 h: 1∶8,000 goat α-rabbit IgG HRP conjugate (Bio-Rad); 1∶8,000 goat α-mouse IgG HRP (Bio-Rad). After 4× washes with TBS-T, blots were incubated in ECL reagent (Super Signal West Pico, Pierce) and exposed to autoradiographic film.
Statistics
Quantitative data were analyzed by unpaired t-test in excel. A p-value of less than 0.05 was considered significant. Where indicated, significance was analyzed using a non-parametric Mann-Whitney U test.
Ethics statement
Mice were housed in a specific pathogen-free environment overseen by the Department of Comparative Medicine at the University of Washington with IACUC approval under protocol 4253-01.
Results
RGS5 is expressed in HSCs
Using Rgs5LacZ reporter mice, we localized the expression of RGS5 in the liver by X-gal staining (Fig. 1A). β-gal+ cells, and therefore RGS5+ cells, are observed adjacent to liver sinusoids. As expected, a subset of vascular SMCs of large vessels (arrows, Fig. 1A) express both RGS5 and smooth muscle α-actin (SMA) (arrows, Fig. 1B) by IF, consistent with published findings [35]. Glial fibrillary acidic protein (GFAP) is expressed in HSCs [50], [51], and co-labeling with the GFAP and the β-gal antibody demonstrates co-localization of the RGS5 reporter and GFAP in HSCs adjacent to sinusoids (arrows, Fig. 1C). Another protein expressed in HSCs, cellular retinol-binding protein-1 (CRBP1 [52]), also co-localizes with β-gal, confirming that these cells are HSCs (arrows, Fig. 1D). Von Willebrand Factor (VWF) is expressed in endothelial cells, including those in the liver sinusoids (LSECs) [53]. IF for VWF and β-gal demonstrates that LSECs are distinct from β-gal+ cells (Fig. 1E); LSECs are VWF+, whereas the β-gal+ cells are sparse and evenly distributed, and do not overlap with VWF+ cells. Similarly, F4/80+ Kupffer cells and β-gal+ cells are also distinct, with no co-localization observed (Fig. 1F). Confocal imaging for GFAP and β-gal confirms that the nuclei of GFAP+ HSC are β-gal+ (arrows, Fig. 1G). No co-localization is observed in confocal imaging for CD31+, another endothelial cell marker, and β-gal+ cells (Fig. 1H). Finally, to verify that RGS5 expression is restricted to HSCs, we measured Rgs5 expression in primary HSCs isolated from WT mice [48]. We found that primary HSCs express high levels of Rgs5 relative to other non-parenchymal cell markers (Fig. S1A, B). In summary, our co-localization analyses determined that RGS5 is specifically expressed in HSCs, but not in hepatocytes, LSECs, or Kupffer cells.
Figure 1. Hepatic Stellate Cells express RGS5.

A. Rgs5LacZ/LacZ mouse liver with X-gal labeling. RGS5+ peri-sinusoidal cells are distributed throughout the liver. RGS5 is also expressed in a subset of SMC of the portal vein (yellow arrows). B–G. Immunofluorescence (IF) for cell-specific markers in the Rgs5LacZ/LacZ liver. B. SMA and α-β-gal show RGS5 expression in vascular SMCs, as expected (arrows). C. GFAP and α-β-gal IF in Rgs5LacZ/LacZ mouse liver. β-gal+ nuclei are visible within GFAP+ astrocyte-like HSC cells, localizing RGS5 expression to HSCs (arrows). D. CRBP1 and α-β-gal IF are co-localized in HSC of Rgs5LacZ/LacZ liver (arrows). E. VWF (a marker of endothelial cells) and α-β-gal are not co-localized. VWF extends through all sinusoids, while β-gal+ cells are sparsely distributed. F. F4/80 (a maker of macrophage/Kupffer cells) and β-gal+ cells represent distinct cell populations. G. Confocal image of α-GFAP IF showing co-localization of the nuclear α-β-gal (arrows). H. Confocal image of CD31 (a marker of endothelial cells) and α-β-gal. CD31+ cells have β-gal− nuclei, while β-gal+ nuclei are not associated with CD31+ endothelial cells. All scale bars are 100 µm.
Increased RGS5 expression is associated with liver tumor and liver fibrosis
Multiple studies have demonstrated RGS5 expression in liver tumors [41], [42]. Because these tumors are associated with HSC activation, we examined RGS5 expression in two mouse models of HCC, mice with hepatocyte-specific deletion of either tuberous sclerosis complex 1 (Tsc1) or phosphatase and tensin homolog (Pten). The loss of either of these genes results in disruption of the (PI3K)/AKT/mTORC1 pathway, leading to HCC or cholangiocarcinoma [54]. First we performed trichrome staining on Tsc1fl/fl;AlbCre and Ptenfl/fl;AlbCre liver sections spanning HCCs and adjacent non-tumor liver. Tsc1fl/fl;AlbCre mice had collagen deposition in tumors (Fig. 2C), but not in adjacent non-tumor liver (Fig. 2A), while Ptenfl/fl;AlbCre mice had significant trichrome staining in both tumor and non-tumor liver (Fig. 2B, D). When we macro-dissected tumors from these mice and performed qPCR for Rgs5 expression (Fig. 2E, F), we found that Rgs5 expression mirrored collagen deposition: 1) Rgs5 is elevated in fibrotic Tsc1fl/fl;AlbCre tumors but not adjacent non-fibrotic liver; 2) Rgs5 is elevated in both tumor and surrounding liver (both fibrotic) in Ptenfl/fl;AlbCre mice. The up-regulation of Rgs5 expression in fibrotic liver tissue suggests that Rgs5 expression may be associated with HSC activation, which occurs both in liver injury and in HCC [55].
Figure 2. RGS5 expression is up-regulated in HCC and liver fibrosis.

Sections of liver from Tsc1fl/fl;AlbCre (A,C) and Ptenfl/fl;AlbCre (B,D) mice were stained with Masson's trichrome. A. Tsc1fl/fl;AlbCre non-tumor liver is histologically normal. B. Ptenfl/fl;AlbCre non-tumor liver tissue is steatotic and shows collagen deposition in sinusoids (blue). C. Tsc1fl/fl;AlbCre tumor tissue shows disorganized architecture and high levels of collagen deposition. D. Ptenfl/fl;AlbCre tumor tissue is glandular in appearance, with robust collagen deposition. Scale bars are 100 µm. E–F. RNA was isolated from wild-type normal tissue and matched tumor and non-tumor tissues of Tsc1fl/fl;AlbCre and Ptenfl/fl;AlbCre E. Tsc1fl/fl;AlbCre mice have normal RGS5, SMA, and Collagen expression in non-tumor parenchyma, and elevated expression in tumor tissue. F. Ptenfl/fl;AlbCre mice have elevated expression of RGS5 and collagen in non-tumor parenchyma and in tumors. Data is normalized to expression in wild-type liver tissue. n = 5, error bars = SEM, * = p<0.05.
RGS5 expression is up-regulated in acute CCl4 injury
Acute injection of CCl4 leads to hepatocyte death and a subsequent injury response [56]–[58]. We found that acute CCl4-induced injury induces a 4-fold up-regulation of Rgs5 mRNA expression 48 hours after injection (Fig. 3A). Elevated Rgs5 expression correlates with the increased expression of additional markers of HSC activation and fibrosis in the murine liver, including Sma (Fig. 3B), Desmin (Fig. 3C), and Pdgfrβ (Fig. 3D) [15], [59], which also peak at 48 hours post injury. Pdgfrα (Fig 3E) and Col1a (Fig 3F) are up-regulated at 48 hours, but peak at 72 hours post injury. Taken together, these data support the hypothesis that RGS5 is associated with fibrosis and HSC activation in both tumor and non-tumor associated fibrosis, and that the observed up-regulation of RGS5 expression in fibrotic liver tumors [41], [42] may be due to tumor associated activated HSCs.
Figure 3. RGS5 expression is up-regulated in acute liver injury.

C57BL/6 mice were injected i.p. with 10 µl/g body weight 10% CCl4 diluted in olive oil 10% (v/v). Livers were collected at 24, 48, 72 hr post injection. RNA was isolated and expression of HSC activation markers was determined by qPCR. A. RGS5 expression is elevated from 24 to 72 hr post injury, peaking at 48 hr. Expression of B. SMA, C. Desmin, and D. PDGFRβ is up-regulated at 24 and 48 hours post injury. E. PDGFRα and F. Col 1a are up-regulated 48 hours post injury and remain high at 72 hr. Data is normalized to expression in untreated (ut) samples. n = 3–5, error bars = SEM, * = p<0.05.
RGS5 expression is regulated in response to inflammatory and profibrotic stimuli in vitro
HSCs are activated in response to the growth factor and cytokine milieu released during the response to hepatocyte injury, including TNFα [60], TGFβ [61], PDGF-BB [62], and ET-1 [63]. To determine whether RGS5 expression is affected by any of these factors, we stimulated LX-2 cells and assayed the expression of Rgs5 by qPCR. Rgs5 and endothelin receptor B (ETB) expression were up-regulated by TNFα stimulation but inhibited by TGFβ stimulation (Fig. 4) ETB is a marker of HSC activation [64], and activated receptors are rapidly internalized, serving as a sink for ET-1 agonists [65]. The simultaneous co-regulation of RGS5 and ETB expression makes sense, as ETB sequestering ET-1 agonist and RGS5 inhibiting Gαq-mediated signaling both result in the blockade of ET-1-mediated signal transduction. Regulation of RGS5 in response to cytokines released after hepatocyte injury could enable tunable control of ET-1-mediated signaling in HSCs.
Figure 4. RGS5 expression is regulated by profibrotic cytokines in concert with ETB.
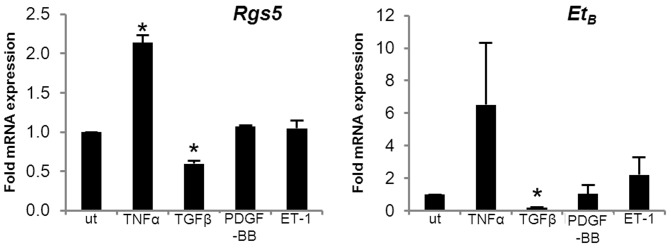
LX2 HSCs were treated with TNFα (5 ng/ml), TGFβ (5 ng/ml), PDGF-BB (10 µM), and ET-1 (100 nm) for 24 hr. RNA was collected for qPCR analysis of RGS5 and ETB expression. Both RGS5 and ETB are up-regulated by TNFα stimulation and down-regulated by TGFβ stimulation. RGS5 and ETB expression are correlated, responding similarly to the same stimuli. n = 3, error bars = SEM, * = p<0.05.
RGS5+ HSCs participate in the response to hepatic injury
As stated above, RGS5 expression is up-regulated in both genetically-induced HCC (Fig. 2) and acutely in response to CCl4-induced liver injury (Fig. 3). To investigate the role of RGS5 in mediating HSC activation and liver fibrosis, we induced liver injury (CCl4 model, as above) in Rgs5LacZ/LacZ mice to assess the changes in RGS5 cellular expression over time during injury. To identify RGS5+ HSCs during the injury response, we co-localized expression of GFAP and β-gal by IF. In uninjured liver tissue, HSCs are sparsely distributed throughout the liver of both Rgs5+/+ and Rgs5LacZ/LacZ mice (Fig. 5A,D). However, 48 hr post CCl4 injury, HSCs are present in the necrotic foci (Fig. 5B). β-gal+ HSCs are clustered in the necrotic foci, and are scarce in the uninjured parenchyma (Fig. 5E). At 96 hours, HSCs are tightly clustered at the foci of injury and are rare in the parenchyma (Fig. 5C, F). Co-localization analysis (Fig. S2) shows 75% of β-gal+ cells are GFAP+, and that this ratio does not significantly change over the course of injury. The expression of RGS5 remains localized to GFAP+ HSCs before and during injury (Fig. 5G–I), and its up-regulation correlates with expression of HSC activation markers (desmin and SMA) [15], [59], peaking at 48 hr post injury (Fig. 3). Since RGS5 functions to inhibit GPCR signaling [25], the up-regulation of RGS5 mRNA in HSCs during the liver injury response suggests a role in controlling GPCR mediated HSC activation.
Figure 5. RGS5+ HSCs participate in the response to acute hepatic injury.

Anti-GFAP and anti-β-gal immunofluorescence were used to localize HSCs in acutely injured liver tissue. A–F. Low magnification images. G–I. High magnification of Rgs5LacZ/LacZ A,D Uninjured liver tissue from Rgs5+/+ and Rgs5LacZ/LacZ mice show sparse HSCs distributed throughout the liver. High magnification in uninjured Rgs5LacZ/LacZ shows HSCs are GFAP+ and β-gal+. B,E. At 48 hours post injury, HSCs are concentrated in the necrotic foci surrounding the central veins. β-gal+ cells (E,H) are associated with GFAP+ cells. C,F At 96 hours post injury, HSCs are tightly clustered at the foci of injury. I. β-gal+ cells are GFAP+. All scale bars are 100 µm.
RGS5 deficient mice have increased hepatocyte damage in response to acute liver injury
Given the induction of RGS5 expression after CCl4 injection, we next used Rgs5LacZ/LacZ mice to assess the functional consequences of loss of RGS5 expression. Uninjured liver appears histologically normal in Rgs5LacZ/LacZ mice (Fig. 6A,D). At 48 hr post injury, foci of necrosis are visible around the central veins (Fig. 6B,E). At 96 hr post injury, necrotic hepatocytes undergo clearance and infiltrating cells remain at the site of injury (denoted by arrow in Fig. 6C&F). In Rgs5LacZ/LacZ mice, necrotic foci are similar to wild-type mice. At 96 hr post injury, the foci of necrosis are diminished; however, hepatocytes throughout the liver have cleared cytoplasm and have a ballooned appearance (Fig. 6F). To determine if Rgs5LacZ/LacZ mice are more susceptible to CCl4-induced liver damage than wild-type mice, we performed careful morphological analysis of H&E stained sections of liver tissue 96 hr after injury. Rgs5+/+ mice have comparatively normal hepatocytes (Fig. 7A, B). In contrast, Rgs5LacZ/LacZ mice demonstrated widespread hepatocyte ballooning (Fig. 7C, D). Furthermore, in Rgs5LacZ/LacZ mice, the hepatocytes are characterized by cleared cytoplasm and central nuclei (Fig. 7D). Quantification of cleared hepatocyte area, using ImageJ, confirms a significant increase in injured hepatocyte area in Rgs5LacZ/LacZ mice (Fig. 7E). Further, by analyzing the liver/body weight ratio in Rgs5+/+ compared to Rgs5LacZ/LacZ mice, we observe injured Rgs5LacZ/LacZ livers are larger than uninjured livers, while Rgs5+/+ liver/body weight ratios do not show an injury-induced increase in size (Fig. S3). Oil red-O staining shows that the cleared hepatocytes do not contain lipids (Fig. S4), nor accumulated glycogen (Fig. S5). TUNEL staining shows no difference in apoptosis between the genotypes, and no association with cleared hepatocytes (Fig. S6).
Figure 6. Rgs5LacZ/LacZ mice have disrupted hepatocyte morphology after injury.

Acute CCl4-induced injury in Rgs5+/+ mice (A–C) and Rgs5LacZ/LacZ (D–F). A,D. Uninjured mice are histologically normal. B,E At 48 hr post CCl4 injection, foci of necrosis are visible central veins in both Rgs5+/+ and Rgs5LacZ/LacZ mice. C,F. At 96 hr post injury, clearance of necrotic hepatocytes is underway and infiltrating cells remain at the site of injury. F. In Rgs5LacZ/LacZ mice, hepatocytes throughout the liver have cleared cytoplasm. Scale bars are 100 µm.
Figure 7. Rgs5LacZ/LacZ mice have increased liver injury and HSC activation following acute injury.

Livers from Rgs5+/+ and Rgs5LacZ/LacZ were collected at 96 hr following a single CCl4 injection. A. and B. H&E stain of Rgs5+/+ mice recover normally from acute injury. Foci of necrosis are centered on the central veins. B. Hepatocytes appear normal. C. and D. Rgs5LacZ/LacZ livers have extensive ballooning of hepatocytes throughout the liver. Foci of necrosis are present. D. Rgs5Lacz/LacZ hepatocytes show cleared cytoplasm and centralized nuclei in hepatocytes distant from necrotic foci. Scale bars are 100 µm. E. Quantification of damaged hepatocyte area reveals a significant increase in ballooning in Rgs5Lacz/LacZ mice compared to Rgs5+/+ mice (n = 8–10; error bars = SEM; * = p<0.05) F. mRNA expression of markers of HSC activation (Desmin, PDGFRβ, and ETB) are elevated in Rgs5Lacz/LacZ mice compared to Rgs5+/+ mice following acute CCl4 injury. n = 4–6, * = p<0.05 by Mann-Whitney U test.
We propose that in the absence of RGS5 expression in HSCs, HSCs are increasingly sensitive to GPCR agonists, such as ET-1 and AngII, which contribute to HSC activation, portal hypertension, and the severity of fibrosis [14]–[19]. Excessive ET-1 or AngII signaling in Rgs5LacZ/LacZ mice may induce inappropriate or excessive activation of HSCs, causing increased stress on hepatocytes during injury. Therefore, we compared the mRNA expression of HSC activation markers in Rgs5+/+ and Rgs5LacZ/LacZ mice in acutely injured liver tissue. Expression of desmin, endothelin-1 receptor B (ETB), and PDGFRβ [15], [59] is increased in Rgs5LacZ/LacZ mouse liver tissue relative to Rgs5+/+ littermates at 48 hr post CCl4 injury (Fig. 7F). The observed increase in expression of HSC activation markers may be attributed to an increase in HSC proliferation or an increase in HSC activation throughout the liver of Rgs5LacZ/LacZ mice. Taken together these data suggest RGS5 controls HSC activation in acute liver injury.
RGS5 suppresses ET-1 signaling in HSCs
As stated above, the increased hepatocyte injury and fibrosis observed in the Rgs5LacZ/LacZ mouse may be due to excessive signaling through GPCRs, since RGS5 inhibits Gαq signal transduction [25]. To elucidate the role of RGS5 in HSCs during activation and fibrosis, we utilized a human HSC cell line (LX-2 HSCs [47]) to investigate RGS5-mediated ET-1 signaling in vitro. LX-2 cells are transfected with RGS5 siRNA or non-specific siRNA, stimulated with ET-1, and the activation of MAPK signaling was determined. As shown in Fig. 8A and 8B, knock-down of RGS5 expression significantly increases ERK1/2 phosphorylation in response to ET-1 in LX-2 HSCs. Therefore, in the context of increased ET-1-mediated signaling during liver injury, the absence of RGS5 expression could further exacerbate fibrogenic signaling.
Figure 8. Knock-down of RGS5 expression enhances endothelin-1-mediated signaling in LX-2 HSCs.
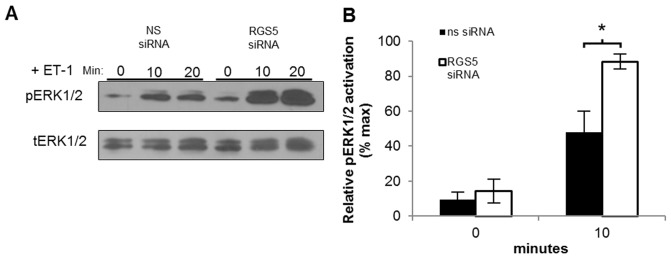
LX2 cells were treated with Rgs5 siRNA or non-specific siRNA for 24 hours, then stimulated with 100 nM ET-1 for the indicated times. Whole cell protein extracts were isolated and analyzed by Western blot. A. A representative immunoblot against pERK1/2 demonstrates increased ET-1-mediated signaling in the absence of RGS5 expression; tERK serves as loading control. B. Quantitation of densitometry of (A) n = 7, error bars = SEM, * = p<0.05.
RGS5 expression is up-regulated in chronic liver injury
While acute injury models provide insight to the HSC response to damage, HSCs produce detectable fibrosis and scarring during chronic liver injury. Chronic exposure to CCl4 in mice recapitulates the fibrosis observed in human chronic liver disease [58]. Rgs5+/+ and Rgs5LacZ/LacZ mice were repeatedly injected with CCl4 over a period of 4 weeks. In damaged Rgs5LacZ/LacZ liver tissue, SMA expression localizes activate HSCs in fibrotic septa (Fig. 9A). HSCs (GFAP+, βGAL+) were associated with fibrotic septa bridging the portal veins, near the sites of collagen deposition (Fig. 9B). Interestingly, the β-gal reporter is limited to these GFAP expressing cells in the chronically injured liver. RGS5 mRNA expression is increased 8-fold in the chronic CCl4 injured liver (Fig. 9C), while HSC activation markers desmin, SMA, PDGFRα, and PDGFRβ are elevated, yet below the levels observed in acute CCl4 injury (Fig. 3). Elevated expression of RGS5 in HSCs during the chronic CCl4 injury indicates an ongoing role during fibrosis, while classic markers of HSC activation have subsided, suggesting RGS5's function may extend beyond controlling HSC activation.
Figure 9. RGS5 expression is up-regulated with HSC activation in chronic liver injury.

Rgs5LacZ/LacZ mice were chronically injected with CCl4 (or oil), twice weekly for 4 weeks. RNA was isolated from whole liver and analyzed by qPCR for expression of Rgs5 and HSC activation markers. A. Chronic CCl4 treated Rgs5LacZ/LacZ dab labeled with anti-SMA shows activated HSCs. B. Rgs5LacZ/LacZ liver immunolabeled with α-β-gal and α-GFAP antibodies. GFAP+ HSCs are visible along fibrotic septa bridging the portal veins. β-gal+ HSCs are visible in Rgs5LacZ/LacZ (B) around the fibrotic septa. Scale bars are 100 µm. C. qPCR of Rgs5+/+ liver RNA shows Rgs5 is up-regulated in chronic CCl4 injury. Collagen 1α expression is elevated and multiple HSC activation markers (PDGFRα, PDGFRβ, and SMA) are increased relative to oil-injected mice. Data is normalized to oil injected mice. n = 6; error bars = SEM; * = p<.05.
RGS5 deficient mice have increased liver fibrosis during chronic injury
In the acute liver injury model, Rgs5LacZ/LacZ mice were characterized by an increase in ballooning of hepatocytes (Fig. 7E) and increased liver/body weight ratio (Fig. S3), as compared to Rgs5+/+ mice. To determine whether this phenotype persists in the chronic liver injury setting, hepatic fibrosis was visualized and quantified using picrosirius red. Fibular collagen is stained red, and the relative degree of fibrosis is quantitated by ImageJ (Fig. 10 A–D). Chronic CCl4-treated Rgs5+/+ mice develop scar tissue bridging between portal veins (Fig. 10A), while oil-treated control mice are histologically normal, with minimal picrosirius red staining in Rgs5+/+ (Fig. 10C) and Rgs5LacZ/LacZ (Fig. 10D). In contrast, chronic CCl4-treated Rgs5LacZ/LacZ mice develop severe fibrosis, as demonstrated by fibrotic septa bridging between portal veins and encircling hepatic lobules (Fig. 10B). Quantification of picrosirius red staining reveals significantly increased fibrosis in Rgs5LacZ/LacZ mice relative to Rgs5+/+ mice (Fig. 10E). Taken together with the finding that RGS5 is up-regulated in chronic injury (Fig. 9A), the increase in fibrosis in Rgs5LacZ/LacZ mice suggests a RGS5-dependent role in controlling HSC-mediated collagen deposition.
Figure 10. Rgs5LacZ/LacZ mice have increased fibrosis during chronic liver injury.

Mice were injected with CCl4 (or oil), twice weekly for 4 weeks. Formalin fixed, paraffin embedded liver sections were stained with picrosirius red to assess fibrosis. A. Rgs5+/+ mice show periportal fibrosis and bridging fibrosis between portal veins. B. Rgs5LacZ/LacZ mice show severe periportal fibrosis bridging between portal veins and surrounding lobules. C. Rgs5+/+ and D. Rgs5LacZ/LacZ oil injected mice have minimal fibrosis. Scale bars are 500 µm. E. ImageJ quantitation of total picrosirius red staining in chronic CCl4 injury shows significantly more fibrosis in Rgs5LacZ/LacZ mice, compared to Rgs5+/+ mice. n = 6, * = p<0.05 by Mann-Whitney U test.
Discussion
In this study, we have identified RGS5 as a marker of HSCs and a regulator of GPCR-mediated signaling in the liver. Up-regulation of Rgs5 expression correlates with HSC activation during liver injury, and Rgs5 is highly expressed in the fibrotic liver. RGS5 deficient mice develop more severe liver injury following acute CCl4 exposure, and increased fibrosis after chronic CCl4 administration. In vitro, profibrotic and pro-inflammatory mediators regulate Rgs5 expression in HSCs, and RGS5 knockdown in HSCs resulted in increased ERK1/2 signaling in response to ET-1, a possible mechanism for its effects in the liver. Taken together, these data suggest that the regulation of RGS5 in HSCs allows for tunable sensitivity to ET-1 signaling following hepatic injury. Loss of RGS5 disrupts this fine level of control, leading to increased HSC activation, hepatic injury, and fibrosis.
The Rgs5LacZ reporter mouse provides a robust and sensitive method to specifically localize RGS5 expression in vivo. Co-localization of β-galactosidase activity and traditional HSC markers (GFAP [50] and CRBP1 [66]) in these mice establishes RGS5 as a marker of HSCs in the liver. RGS proteins have been implicated in the progression of HCC [42], [67], and an earlier study localized hepatic Rgs5 expression to endothelial cells in HCC [41] using in situ hybridization (ISH). However, the nature of ISH makes precise localization and discrimination between adjacent cells difficult. IF labeling of endothelial cells with an antibody to detect VWF confirms that RGS5+ HSCs are distinct from endothelial cells. A recent publication correlates RGS5 expression with increased vascular invasion, tumor recurrence, and decreased survival in patients with HCC [42]. However, HSCs are the major stromal cell in the tumor microenvironment, and promote HCC proliferation and invasion [68]–[70]. The correlation of increased RGS5 expression with decreased survival may reflect the level of HSC activation within the tumor, therefore predicting tumor metastasis and proliferation. High RGS5 expression has also been found in select patient-derived HCC cell lines, suggesting expression by transformed hepatocytes. These data should be interpreted with caution, however, as high passage number in culture conditions containing serum and associated growth factors may select for a phenotype that does not reflect in vivo expression. We therefore propose that the specific co-localization of β-gal expression (and therefore RGS5 expression) with markers of HSCs, and not with markers of other non-parenchymal cells, establishes RGS5 as a marker of HSCs.
We observed that RGS5 deficient mice had widespread hepatocyte clearing after a single CCl4 injection, while this phenotype was not observed in Rgs5+/+ mice (Fig. 7). The hepatocyte cytoplasmic clearing observed after acute CCl4 treated Rgs5LacZ/LacZ mice is similar to ballooning observed in human cases of non-alcoholic fatty liver disease (NAFLD). Conflicting evidence suggests RGS5 plays a role in maintaining body weight and steatosis, with one group reporting that Rgs5−/− mice exhibit spontaneous hepatic steatosis and obesity [71] while another study demonstrates RGS5−/− mice have low body weight [34]. Although RGS proteins have been implicated in the control of hepatic fatty acid oxidation [72] and homeostasis [73], we did not observe any differences in baseline mouse body weight or oil red-o staining (Fig. S3, S4) after CCl4 injection, indicating that the ballooning is not due to lipid accumulation. The differences in our findings may be due to differences in mouse strains or housing conditions used in the different institutions, and the overall effects of RGS5 expression on body weight are unclear. Glycogen storage can also induce ballooning in hepatocytes, but periodic acid-schiff staining showed no difference between Rgs5LacZ/LacZ mice and wild type littermates (Fig. S5). Hepatocyte swelling reflects reversible cell injury [74] in Rgs5LacZ/LacZ mice, and it has been suggested that ballooning is protective in hepatotoxicity [59], [75]. To explore this possibility, TUNEL staining was conducted to determine whether ballooned hepatocytes were undergoing apoptosis. However, there was minimal staining in hepatocytes 96 hrs after CCl4 injection, and no difference was observed between Rgs5LacZ/LacZ mice and wild type littermates (Fig. S6). We similarly did not appreciate a difference in liver necrosis between the genotypes, indicating that the centrilobular zonal expression of cytochrome p450, and thus CCl4 metabolism, is unchanged in Rgs5LacZ/LacZmice. Further, Rgs5LacZ/LacZ mice survive repeated doses of CCl4 in the chronic liver injury model, suggesting that the hepatocyte swelling is reversible.
Feathery hepatocyte degeneration is commonly observed in cholestasis [76] and is characterized by enlarged periportal hepatocytes with a flocculent cytoplasm. Intrahepatic cholestasis due to endotoxemia or drug toxicity can induce feathery degeneration; however, we did not observe pigmented hepatocytes in Rgs5LacZ/LacZ mice, and the hepatocyte swelling was equally distributed throughout the liver lobule. Whether hepatocyte swelling enhances hepatocyte survival or is simply a surrogate for more severe injury, Rgs5LacZ/LacZ mice show extensive hepatocyte swelling after a single injection of CCl4, while Rgs5+/+ mice have normal histology.
Potential mechanisms for the increased injury observed in Rgs5LacZ/LacZ mice include portal hypertension or increased activation of HSCs. ET-1 and AngII-mediated sinusoidal constriction by HSCs induces portal hypertension [19], [77], [78]; loss of RGS5 expression enhances GPCR signaling [31] in response to these agonists, and may thus result in sinusoidal constriction. AngII infusion exacerbates fibrosis in rodent models of fibrosis [14] and ET-1 infusion increases HSC contraction and portal hypertension [79]. These effects are similar to what is observed in the Rgs5LacZ/LacZ mice during CCl4 injury. Loss of VEGF induces sinusoidal capillarization, portal hypertension and HSC activation [80] in mice. However no parenchymal injury was observed, invalidating portal hypertension as a potential mechanism.
The increased hepatocyte injury in Rgs5LacZ/LacZ mice may be due to enhanced HSC activation via GPCR signaling. ET-1-mediated control of HSC activation has been demonstrated in previous studies, in which pharmaceutical inhibition of AngII and ET-1 reduces HSC activation in vitro [13], [17] and in rat models of cirrhosis [15], [19]. Our findings of increased expression of HSC markers (desmin, ETB, and PDGFRβ) in Rgs5LacZ/LacZ mice are consistent with this hypothesis, as removal of ET-1 inhibition would enhance expression of markers of HSC activation. ET-1 induced TGFβ secretion [81], [82] would be expected to increase in the absence of RGS5 expression, further activating HSCs and contributing to enhanced fibrosis after chronic CCl4 administration. This hypothesis could be further confirmed by rescue of the RGS5-null phenotype through ET-1 antagonism. For instance, the ETA antagonist BQ-123 and the ETB antagonist BQ-788 could be administered to Rgs5LacZ/LacZ mice during CCl4 injury, and would be expected to mitigate the hepatocyte swelling and increased fibrosis that we found in these mice.
RGS5-deficient mice exhibit enhanced arterial hypertrophy and perivascular fibrosis in a hypertension-induced vascular injury model [83]. This pathogenic remodeling was attributed to enhanced MEK/ERK and Rho kinase (ROCK) signaling via increased AngII-induced Gαq signaling in RGS5-null mice. Enhanced ERK activity due to RGS5 knock-down has been previously reported in aortic SMCs [31], and we observed enhanced ERK signaling in LX-2 cells following RGS5 siRNA treatment (Fig. 8A,B). ET-1 induced Rho activation in HSCs has been shown to enhance migration in vitro [84], and inhibition of ROCK improves fibrosis in choline deficient diet fed rats [85]. Loss of RGS5-mediated inhibition of ROCK could be one mechanism behind the enhanced HSC activation and fibrosis observed in Rgs5LacZ/LacZ mice.
RGS5 inhibition of ET-1 signaling has potential in the treatment of liver fibrosis and cirrhosis. While animal models of cirrhosis benefit from ET-1 antagonism, patients risk liver failure [86] due the hepatotoxic effects these drugs. RGS5 inhibition of ET-1 signaling specifically in HSCs provides a novel avenue for future therapies. Enhancing the expression of RGS5 in HSCs could reduce their activation and subsequent development of fibrosis and cirrhosis. The anti-fibrotic effects of RGS5 overexpression could be validated with studies of chronic CCl4 administration to mice in which RGS5 expression is conditionally overexpressed in HSCs. High RGS5 expression would be expected to block ET-1 mediated signaling in HSCs, potentially reducing HSC activation, contraction, migration, and ultimately fibrosis.
Conclusions
RGS5 is a marker of HSCs that is up-regulated as HSCs respond to injury. RGS5 controls HSC activation via inhibition of ET-1 signaling and reduction of ERK activation. The critical role of RGS5 in liver injury is demonstrated by enhanced HSC activation, hepatocyte ballooning, and fibrosis in CCl4 treated RGS5-deficient mice. RGS5-mediated control of GPCR signaling in the liver is a novel mechanism by which HSC activation can be controlled, and a potential target of therapeutic intervention for liver fibrosis.
Supporting Information
RGS5 is expressed in freshly isolated primary HSCs. 1° HSC have astrocyte-like morphology. A. Isolated 1° HSCs have an astrocyte-like phenotype and store vitamin A in lipid droplets. B. Isolated 1° HSCs are characterized by high RGS5 expression and low Tie1 and CD68 expression, indicating there are few contaminating cells. Low PDGFRβ expression demonstrates the 1° HSCs are in a quiescent state.
(TIF)
Co-localization of β-gal+ nuclei and GFAP staining does not change during injury. Frozen sections of CCl4 injected mouse liver were immunofluorescently labeled with antibodies for β-gal and GFAP. Co-localization analysis using ImagePro showed that the fraction of β-gal+ cells that are β-gal+ and GFAP+ does not significantly change during the course of injury. n = 3 mice per group.
(TIF)
Liver weight to body weight ratio is moderately elevated in Rgs5lacZ/lacZ mice at 96hr post CCl4 injury. Body and liver weights of CCl4 injected mice were measured at time of sacrifice. Rgs5LacZ/LacZ mice had elevated liver to body weight ratio 96 h post injury. Rgs5+/+ liver to body weight ratio was not significantly different from untreated mice. * p = .001, ‡ p = .06, n = 4–6.
(TIF)
Hepatocyte clearing is not associated with lipid accumulation. Oil red O staining of mouse liver 96 hr post CCl4 injection was used to assess lipid accumulation. A. Lipid droplets are visible around the site of injury in both Rgs5+/+ A,C and Rgs5LacZ/LacZ B,D mice. Cleared hepatocytes are visible in B, distant from the site of injury and droplets of lipid. C,D. Oil red O staining is present, but there is no accumulation of lipid in hepatocytes. Scale bar is 100 µm.
(TIF)
Hepatocyte clearing is not associated with glycogen accumulation. Periodic acid schiff stain of frozen mouse liver 96 hr post CCl4 injection was used to assess glycogen accumulation. A. Hepatocytes appear normal in Rgs5+/+ liver. Intense magenta staining labels glycogen in the hepatocytes. B. Cleared hepatocytes are visible in Rgs5LacZ/LacZ and glycogen staining is present in hepatocytes. High magnification of C Rgs5+/+ and D Rgs5LacZ/LacZ liver show RGS5 that cleared hepatocytes do not contain accumulated glycogen. Scale bar is 100 µm.
(TIF)
Cleared hepatocytes are not apoptotic. TUNEL staining of mouse liver 96 hr post CCl4 injection was used to assess apoptosis. A. Rgs5+/+ liver shows TUNEL minimal staining 96 hr post injection. B. Cleared hepatocytes are visible in Rgs5LacZ/LacZ liver 96 hr post CCl4. No difference in TUNEL staining is visible in cleared hepatocytes. C. DNAse I treated positive control shows intense staining. D. Uninjured Rgs5LacZ/LacZ has minimal TUNEL staining. Scale bar is 100 µm.
(TIF)
Acknowledgments
The authors would like to thank Dr. Morayma Reyes, Nick Ieronimakis, and Kelly Hudkins (University of Washington, Department of Pathology) for assistance and reagents for immunohistochemistry, Ken Lindsey and Ron Seifert for assistance in confocal imaging, Dr. Scott L. Friedman (Mount Sinai School of Medicine) for generously providing the human hepatic stellate cell line (LX2 HSCs), and Joseph Beavo for thoughtful scientific advice.
Data Availability
The authors confirm that all data underlying the findings are fully available without restriction. All relevant data are within the paper and its Supporting Information files.
Funding Statement
Research reported in this publication was supported by the Heart, Blood and Lung Institute of the National Institutes of Health (NIH), under award HL087513 and HL094374 and University of Washington, Department of Pathology start-up funds (WMM). In addition, AJB and BJH were supported by NIH T32HL007312 and the University of Washington-Howard Hughes Medical Institute program in Molecular Medicine. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health or the Howard Hughes Medical Institute. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Heron M (2011) Deaths: leading causes for 2007. Natl Vital Stat Rep 59: 1–95. [PubMed] [Google Scholar]
- 2. Hernandez-Gea V, Friedman SL (2011) Pathogenesis of liver fibrosis. Annu Rev Pathol 6: 425–456 doi:10.1146/annurev-pathol-011110-130246 [DOI] [PubMed] [Google Scholar]
- 3. Tarrats N, Moles A, Morales A, García-Ruiz C, Fernández-Checa JC, et al. (2011) Critical role of TNF-receptor 1 but not 2 in hepatic stellate cell proliferation, extracellular matrix remodeling and liver fibrogenesis. Hepatology. doi:10.1002/hep.24388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Breitkopf K, Godoy P, Ciuclan L, Singer MV, Dooley S (2006) TGF-beta/Smad signaling in the injured liver. Z Gastroenterol 44: 57–66 doi:10.1055/s-2005-858989 [DOI] [PubMed] [Google Scholar]
- 5. Bataller R, Brenner DA (2005) Liver fibrosis. J Clin Invest 115: 209–218 doi:10.1172/JCI24282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Hellerbrand C (2013) Hepatic stellate cells—the pericytes in the liver. Pflugers Arch 465: 775–778 doi:10.1007/s00424-012-1209-5 [DOI] [PubMed] [Google Scholar]
- 7. Sato M, Suzuki S, Senoo H (2003) Hepatic stellate cells: unique characteristics in cell biology and phenotype. Cell Struct Funct 28: 105–112. [DOI] [PubMed] [Google Scholar]
- 8. Ankoma-Sey V, Wang Y, Dai Z (2000) Hypoxic stimulation of vascular endothelial growth factor expression in activated rat hepatic stellate cells. Hepatology 31: 141–148 doi:10.1002/hep.510310122 [DOI] [PubMed] [Google Scholar]
- 9. Reichenbach V, Fernández-Varo G, Casals G, Oró D, Ros J, et al. (2012) Adenoviral dominant-negative soluble PDGFRβ improves hepatic collagen, systemic hemodynamics, and portal pressure in fibrotic rats. J Hepatol 57: 967–973 doi:S0168-8278(12)00570-3 [pii] 10.1016/j.jhep.2012.07.012 [DOI] [PubMed] [Google Scholar]
- 10. Friedman SL (2008) Mechanisms of hepatic fibrogenesis. Gastroenterology 134: 1655–1669 doi:S0016-5085(08)00429-0 [pii] 10.1053/j.gastro.2008.03.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Breitkopf K, Roeyen C, Sawitza I, Wickert L, Floege J, et al. (2005) Expression patterns of PDGF-A, -B, -C and -D and the PDGF-receptors alpha and beta in activated rat hepatic stellate cells (HSC). Cytokine 31: 349–357 doi:S1043-4666(05)00193-6 [pii] 10.1016/j.cyto.2005.06.005 [DOI] [PubMed] [Google Scholar]
- 12. Inagaki Y, Okazaki I (2007) Emerging insights into Transforming growth factor beta Smad signal in hepatic fibrogenesis. Gut 56: 284–292 doi:56/2/284 [pii] []10.1136/gut.2005.088690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Bataller R, Sancho-Bru P, Ginès P, Lora JM, Al-Garawi A, et al. (2003) Activated human hepatic stellate cells express the renin-angiotensin system and synthesize angiotensin II. Gastroenterology 125: 117–125 doi:S0016508503006954 [pii] [DOI] [PubMed] [Google Scholar]
- 14. Bataller R, Gäbele E, Parsons CJ, Morris T, Yang L, et al. (2005) Systemic infusion of angiotensin II exacerbates liver fibrosis in bile duct-ligated rats. Hepatology 41: 1046–1055 doi:10.1002/hep.20665 [DOI] [PubMed] [Google Scholar]
- 15. Bahde R, Kebschull L, Stöppeler S, Zibert A, Siaj R, et al. (2011) Role of angiotensin-1 receptor blockade in cirrhotic liver resection. Liver Int 31: 642–655 doi:10.1111/j.1478-3231.2011.02493.x [DOI] [PubMed] [Google Scholar]
- 16. Rockey DC, Fouassier L, Chung JJ, Carayon A, Vallee P, et al. (1998) Cellular localization of endothelin-1 and increased production in liver injury in the rat: potential for autocrine and paracrine effects on stellate cells. Hepatology 27: 472–480 doi:S027091399800069X [pii] 10.1002/hep.510270222 [DOI] [PubMed] [Google Scholar]
- 17. Rockey DC, Chung JJ (1996) Endothelin antagonism in experimental hepatic fibrosis. Implications for endothelin in the pathogenesis of wound healing. J Clin Invest 98: 1381–1388 doi:10.1172/JCI118925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Cho JJ, Hocher B, Herbst H, Jia JD, Ruehl M, et al. (2000) An oral endothelin-A receptor antagonist blocks collagen synthesis and deposition in advanced rat liver fibrosis. Gastroenterology 118: 1169–1178 doi:S0016508500421190 [pii] [DOI] [PubMed] [Google Scholar]
- 19. Anselmi K, Subbotin VM, Nemoto E, Gandhi CR (2002) Accelerated reversal of carbon tetrachloride-induced cirrhosis in rats by the endothelin receptor antagonist TAK-044. J Gastroenterol Hepatol 17: 589–597 doi:2705 [pii] [DOI] [PubMed] [Google Scholar]
- 20. Li T, Shi Z, Rockey DC (2012) Preproendothelin-1 expression is negatively regulated by IFNγ during hepatic stellate cell activation. Am J Physiol Gastrointest Liver Physiol 302: G948–57 doi:ajpgi.00359.2011 [pii] 10.1152/ajpgi.00359.2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Oben JA, Roskams T, Yang S, Lin H, Sinelli N, et al. (2003) Norepinephrine induces hepatic fibrogenesis in leptin deficient ob/ob mice. Biochem Biophys Res Commun 308: 284–292. [DOI] [PubMed] [Google Scholar]
- 22. Oben JA, Yang S, Lin H, Ono M, Diehl AM (2003) Norepinephrine and neuropeptide Y promote proliferation and collagen gene expression of hepatic myofibroblastic stellate cells. Biochem Biophys Res Commun 302: 685–690. [DOI] [PubMed] [Google Scholar]
- 23. Oben JA, Yang S, Lin H, Ono M, Diehl AM (2003) Acetylcholine promotes the proliferation and collagen gene expression of myofibroblastic hepatic stellate cells. Biochem Biophys Res Commun 300: 172–177. [DOI] [PubMed] [Google Scholar]
- 24. Hsu CT (1992) The role of the sympathetic nervous system in promoting liver cirrhosis induced by carbon tetrachloride, using the essential hypertensive animal (SHR). J Auton Nerv Syst 37: 163–173. [DOI] [PubMed] [Google Scholar]
- 25. Zhou J, Moroi K, Nishiyama M, Usui H, Seki N, et al. (2001) Characterization of RGS5 in regulation of G protein-coupled receptor signaling. Life Sci 68: 1457–1469 doi:S0024320501009390 [pii] [DOI] [PubMed] [Google Scholar]
- 26. Nisancioglu MH, Mahoney WM, Kimmel DD, Schwartz SM, Betsholtz C, et al. (2008) Generation and characterization of rgs5 mutant mice. Mol Cell Biol 28: 2324–2331 doi:MCB.01252-07 [pii] 10.1128/MCB.01252-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Mitchell TS, Bradley J, Robinson GS, Shima DT, Ng YS (2008) RGS5 expression is a quantitative measure of pericyte coverage of blood vessels. Angiogenesis 11: 141–151 doi:10.1007/s10456-007-9085-x [DOI] [PubMed] [Google Scholar]
- 28. Bondjers C, Kalén M, Hellström M, Scheidl SJ, Abramsson A, et al. (2003) Transcription profiling of platelet-derived growth factor-B-deficient mouse embryos identifies RGS5 as a novel marker for pericytes and vascular smooth muscle cells. Am J Pathol 162: 721–729 doi:10.1016/S0002-9440(10)63868-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Cho H, Kozasa T, Bondjers C, Betsholtz C, Kehrl JH (2003) Pericyte-specific expression of Rgs5: implications for PDGF and EDG receptor signaling during vascular maturation. FASEB J 17: 440–442 doi:10.1096/fj.02-0340fje [DOI] [PubMed] [Google Scholar]
- 30. Anger T, Klintworth N, Stumpf C, Daniel WG, Mende U, et al. (2007) RGS protein specificity towards Gq- and Gi/o-mediated ERK 1/2 and Akt activation, in vitro. J Biochem Mol Biol 40: 899–910. [DOI] [PubMed] [Google Scholar]
- 31. Gunaje JJ, Bahrami AJ, Schwartz SM, Daum G, Mahoney WM (2011) PDGF-dependent regulation of regulator of G protein signaling-5 expression and vascular smooth muscle cell functionality. Am J Physiol Cell Physiol 301: C478–89 doi:ajpcell.00348.2010 [pii] 10.1152/ajpcell.00348.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Faruque MU, Chen G, Doumatey A, Huang H, Zhou J, et al. (2011) Association of ATP1B1, RGS5 and SELE polymorphisms with hypertension and blood pressure in African-Americans. J Hypertens 29: 1906–1912 doi:10.1097/HJH.0b013e32834b000d [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Chang YP, Liu X, Kim JD, Ikeda MA, Layton MR, et al. (2007) Multiple genes for essential-hypertension susceptibility on chromosome 1q. Am J Hum Genet 80: 253–264 doi:S0002-9297(07)62683-4 [pii] 10.1086/510918 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Cho H, Park C, Hwang IY, Han SB, Schimel D, et al. (2008) Rgs5 targeting leads to chronic low blood pressure and a lean body habitus. Mol Cell Biol 28: 2590–2597 doi:MCB.01889-07 [pii] 10.1128/MCB.01889-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Li J, Adams LD, Wang X, Pabon L, Schwartz SM, et al. (2004) Regulator of G protein signaling 5 marks peripheral arterial smooth muscle cells and is downregulated in atherosclerotic plaque. J Vasc Surg 40: 519–528 doi:10.1016/j.jvs.2004.06.021 [DOI] [PubMed] [Google Scholar]
- 36. Wang X, Adams LD, Pabón LM, Mahoney WM, Beaudry D, et al. (2008) RGS5, RGS4, and RGS2 expression and aortic contractibility are dynamically co-regulated during aortic banding-induced hypertrophy. J Mol Cell Cardiol 44: 539–550 doi:S0022-2828(07)01323-5 [pii] 10.1016/j.yjmcc.2007.11.019 [DOI] [PubMed] [Google Scholar]
- 37. Li H, He C, Feng J, Zhang Y, Tang Q, et al. (2010) Regulator of G protein signaling 5 protects against cardiac hypertrophy and fibrosis during biomechanical stress of pressure overload. Proc Natl Acad Sci U S A 107: 13818–13823 doi:1008397107 [pii] 10.1073/pnas.1008397107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Fleming JN, Nash RA, McLeod DO, Fiorentino DF, Shulman HM, et al. (2008) Capillary regeneration in scleroderma: stem cell therapy reverses phenotype? PLoS One 3: e1452 doi:10.1371/journal.pone.0001452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Boss CN, Grünebach F, Brauer K, Häntschel M, Mirakaj V, et al. (2007) Identification and characterization of T-cell epitopes deduced from RGS5, a novel broadly expressed tumor antigen. Clin Cancer Res 13: 3347–3355 doi:10.1158/1078-0432.CCR-06-2156 [DOI] [PubMed] [Google Scholar]
- 40. Hurst JH, Mendpara N, Hooks SB (2009) Regulator of G-protein signalling expression and function in ovarian cancer cell lines. Cell Mol Biol Lett 14: 153–174 doi:10.2478/s11658-008-0040-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Chen X, Higgins J, Cheung S-T, Li R, Mason V, et al. (2004) Novel endothelial cell markers in hepatocellular carcinoma. Mod Pathol 17: 1198–1210 doi:10.1038/modpathol.3800167 [DOI] [PubMed] [Google Scholar]
- 42. Hu M, Chen X, Zhang J, Wang D, Fang X, et al. (2013) Over-expression of regulator of G protein signaling 5 promotes tumor metastasis by inducing epithelial-mesenchymal transition in hepatocellular carcinoma cells. J Surg Oncol 108: 192–196 doi:10.1002/jso.23367 [DOI] [PubMed] [Google Scholar]
- 43. Chen X, Cheung ST, So S, Fan ST, Barry C, et al. (2002) Gene expression patterns in human liver cancers. Mol Biol Cell 13: 1929–1939 doi:10.1091/mbc.02-02-0023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Bilger A, Bennett LM, Carabeo RA, Chiaverotti TA, Dvorak C, et al. (2004) A potent modifier of liver cancer risk on distal mouse chromosome 1: linkage analysis and characterization of congenic lines. Genetics 167: 859–866 doi:10.1534/genetics.103.024521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Eppig JT, Bult CJ, Kadin JA, Richardson JE, Blake JA, et al. (2005) The Mouse Genome Database (MGD): from genes to mice—a community resource for mouse biology. Nucleic Acids Res 33: D471–5 doi:10.1093/nar/gki113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Humphreys BD, Lin S-L, Kobayashi A, Hudson TE, Nowlin BT, et al. (2010) Fate tracing reveals the pericyte and not epithelial origin of myofibroblasts in kidney fibrosis. Am J Pathol 176: 85–97 doi:10.2353/ajpath.2010.090517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Xu L, Hui AY, Albanis E, Arthur MJ, O'Byrne SM, et al. (2005) Human hepatic stellate cell lines, LX-1 and LX-2: new tools for analysis of hepatic fibrosis. Gut 54: 142–151 doi:54/1/142 [pii] 10.1136/gut.2004.042127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Maschmeyer P, Flach M, Winau F (2011) Seven steps to stellate cells. J Vis Exp pii: 2710. doi:10.3791/2710. [DOI] [PMC free article] [PubMed]
- 49. Seger R, Seger D, Reszka AA, Munar ES, Eldar-Finkelman H, et al. (1994) Overexpression of mitogen-activated protein kinase kinase (MAPKK) and its mutants in NIH 3T3 cells. Evidence that MAPKK involvement in cellular proliferation is regulated by phosphorylation of serine residues in its kinase subdomains VII and VIII. J Biol Chem 269: 25699–25709. [PubMed] [Google Scholar]
- 50. Friedman SL (2008) Hepatic stellate cells: protean, multifunctional, and enigmatic cells of the liver. Physiol Rev 88: 125–172 doi:88/1/125 [pii] 10.1152/physrev.00013.2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Baratta JL, Ngo A, Lopez B, Kasabwalla N, Longmuir KJ, et al. (2009) Cellular organization of normal mouse liver: a histological, quantitative immunocytochemical, and fine structural analysis. Histochem Cell Biol 131: 713–726 doi:10.1007/s00418-009-0577-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Lepreux S, Bioulac-Sage P, Gabbiani G, Sapin V, Housset C, et al. (2004) Cellular retinol-binding protein-1 expression in normal and fibrotic/cirrhotic human liver: different patterns of expression in hepatic stellate cells and (myo)fibroblast subpopulations. J Hepatol 40: 774–780 doi:10.1016/j.jhep.2004.01.008 [DOI] [PubMed] [Google Scholar]
- 53. Ning H, Lin G, Lue TF, Lin C-S (2011) Mesenchymal stem cell marker Stro-1 is a 75 kd endothelial antigen. Biochem Biophys Res Commun 413: 353–357 doi:10.1016/j.bbrc.2011.08.104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Kenerson HL, Yeh MM, Kazami M, Jiang X, Riehle KJ, et al. (2013) Akt and mTORC1 have different roles during liver tumorigenesis in mice. Gastroenterology 144: 1055–1065 doi:10.1053/j.gastro.2013.01.053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Faouzi S, Lepreux S, Bedin C, Dubuisson L, Balabaud C, et al. (1999) Activation of cultured rat hepatic stellate cells by tumoral hepatocytes. Lab Invest 79: 485–493. [PubMed] [Google Scholar]
- 56. Starkel P, Leclercq IA (2011) Animal models for the study of hepatic fibrosis. Best Pract Res Clin Gastroenterol 25: 319–333 doi:10.1016/j.bpg.2011.02.004 [DOI] [PubMed] [Google Scholar]
- 57. Liu Y, Meyer C, Xu C, Weng H, Hellerbrand C, et al. (2013) Animal models of chronic liver diseases. Am J Physiol Gastrointest Liver Physiol 304: G449–68 doi:10.1152/ajpgi.00199.2012 [DOI] [PubMed] [Google Scholar]
- 58. Weiler-Normann C, Herkel J, Lohse AW (2007) Mouse models of liver fibrosis. Z Gastroenterol 45: 43–50 doi:10.1055/s-2006-927387 [DOI] [PubMed] [Google Scholar]
- 59.Puche JE, Lee YA, Jiao J, Aloman C, Fiel MI, et al. (2012) A novel murine model to deplete hepatic stellate cells uncovers their role in amplifying liver damage. Hepatology. doi:10.1002/hep.26053. [DOI] [PMC free article] [PubMed]
- 60. Zimmermann HW, Trautwein C, Tacke F (2012) Functional role of monocytes and macrophages for the inflammatory response in acute liver injury. Front Physiol 3: 56 doi:10.3389/fphys.2012.00056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Dooley S, ten Dijke P (2012) TGF-β in progression of liver disease. Cell Tissue Res 347: 245–256 doi:10.1007/s00441-011-1246-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Mallat A, Gallois C, Tao J, Habib A, Maclouf J, et al. (1998) Platelet-derived growth factor-BB and thrombin generate positive and negative signals for human hepatic stellate cell proliferation. Role of a prostaglandin/cyclic AMP pathway and cross-talk with endothelin receptors. J Biol Chem 273: 27300–27305. [DOI] [PubMed] [Google Scholar]
- 63.Khimji AK, Rockey DC (2011) Endothelin and hepatic wound healing. Pharmacol Res. doi:S1043–6618(11)00069-7 [pii] 10.1016/j.phrs.2011.03.005. [DOI] [PMC free article] [PubMed]
- 64. Pinzani M, Milani S, De Franco R, Grappone C, Caligiuri A, et al. (1996) Endothelin 1 is overexpressed in human cirrhotic liver and exerts multiple effects on activated hepatic stellate cells. Gastroenterology 110: 534–548. [DOI] [PubMed] [Google Scholar]
- 65. Boyd R, Rätsep MT, Ding LL, Wang HD (2011) ETA and ETB receptors are expressed in vascular adventitial fibroblasts. Am J Physiol Hear Circ Physiol 301: H2271–8 doi:ajpheart.00869.2010 [pii] 10.1152/ajpheart.00869.2010 [DOI] [PubMed] [Google Scholar]
- 66. Van Rossen E, Vander Borght S, van Grunsven LA, Reynaert H, Bruggeman V, et al. (2009) Vinculin and cellular retinol-binding protein-1 are markers for quiescent and activated hepatic stellate cells in formalin-fixed paraffin embedded human liver. Histochem Cell Biol 131: 313–325 doi:10.1007/s00418-008-0544-2 [DOI] [PubMed] [Google Scholar]
- 67. Sokolov E, Iannitti DA, Schrum LW, McKillop IH (2011) Altered expression and function of regulator of G-protein signaling-17 (RGS17) in hepatocellular carcinoma. Cell Signal 23: 1603–1610 doi:10.1016/j.cellsig.2011.05.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Yang JD, Nakamura I, Roberts LR (2011) The tumor microenvironment in hepatocellular carcinoma: current status and therapeutic targets. Semin Cancer Biol 21: 35–43 doi:10.1016/j.semcancer.2010.10.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Han S, Han L, Yao Y, Sun H, Zan X, et al. (2014) Activated hepatic stellate cells promote hepatocellular carcinoma cell migration and invasion via the activation of FAK-MMP9 signaling. Oncol Rep 31: 641–648 doi:10.3892/or.2013.2872 [DOI] [PubMed] [Google Scholar]
- 70. Amann T, Bataille F, Spruss T, Mühlbauer M, Gäbele E, et al. (2009) Activated hepatic stellate cells promote tumorigenicity of hepatocellular carcinoma. Cancer Sci 100: 646–653 doi:10.1111/j.1349-7006.2009.01087.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Deng W, Wang X, Xiao J, Chen K, Zhou H, et al. (2012) Loss of regulator of G protein signaling 5 exacerbates obesity, hepatic steatosis, inflammation and insulin resistance. PLoS One 7: e30256 doi:PONE-D-11-21348 [pii] 10.1371/journal.pone.0030256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Pashkov V, Huang J, Parameswara VK, Kedzierski W, Kurrasch DM, et al. (2011) Regulator of G protein signaling (RGS16) inhibits hepatic fatty acid oxidation in a carbohydrate response element-binding protein (ChREBP)-dependent manner. J Biol Chem 286: 15116–15125 doi:M110.216234 [pii] 10.1074/jbc.M110.216234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Iankova I, Chavey C, Clapé C, Colomer C, Guérineau NC, et al. (2008) Regulator of G protein signaling-4 controls fatty acid and glucose homeostasis. Endocrinology 149: 5706–5712 doi:10.1210/en.2008-0717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Abbas AK, Fausto N, Robbins SL, Cotran RS, Kumar SUPPLS disc Health Books QZ 4 R6354 2005 text AVAILABLE Health Books QZ 4 R6354 2005 disc DUE 10-02-12 Health Books QZ 4 R6354 2005 disc AVAILABLE VCN-HBQZ 4 R 2005 text (2005) Robbins and Cotran pathologic basis of disease. 7th ed. Philadelphia: Elsevier Saunders.
- 75. Bergasa NV, Borque MJ, Wahl LM, Rabin L, Jones EA (1992) Modulation of thioacetamide-induced hepatocellular necrosis by prostaglandins is associated with novel histologic changes. Liver 12: 168–174. [DOI] [PubMed] [Google Scholar]
- 76. Li MK, Crawford JM (2004) The pathology of cholestasis. Semin Liver Dis 24: 21–42 doi:10.1055/s-2004-823099 [DOI] [PubMed] [Google Scholar]
- 77. Cavasin MA, Semus H, Pitts K, Peng Y, Sandoval J, et al. (2010) Acute effects of endothelin receptor antagonists on hepatic hemodynamics of cirrhotic and noncirrhotic rats. Can J Physiol Pharmacol 88: 636–643 doi:y10-038 [pii] 10.1139/Y10-038 [DOI] [PubMed] [Google Scholar]
- 78. Watanabe N, Takashimizu S, Nishizaki Y, Kojima S, Kagawa T, et al. (2007) An endothelin A receptor antagonist induces dilatation of sinusoidal endothelial fenestrae: implications for endothelin-1 in hepatic microcirculation. J Gastroenterol 42: 775–782 doi:10.1007/s00535-007-2093-1 [DOI] [PubMed] [Google Scholar]
- 79. Rockey DC, Weisiger RA (1996) Endothelin induced contractility of stellate cells from normal and cirrhotic rat liver: implications for regulation of portal pressure and resistance. Hepatology 24: 233–240 doi:10.1002/hep.510240137 [DOI] [PubMed] [Google Scholar]
- 80. May D, Djonov V, Zamir G, Bala M, Safadi R, et al. (2011) A transgenic model for conditional induction and rescue of portal hypertension reveals a role of VEGF-mediated regulation of sinusoidal fenestrations. PLoS One 6: e21478 doi:10.1371/journal.pone.0021478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Gandhi CR, Kuddus RH, Uemura T, Rao AS (2000) Endothelin stimulates transforming growth factor-beta1 and collagen synthesis in stellate cells from control but not cirrhotic rat liver. Eur J Pharmacol 406: 311–318 doi:S001429990000683X [pii] [DOI] [PubMed] [Google Scholar]
- 82. Koda M, Bauer M, Krebs A, Hahn EG, Schuppan D, et al. (2006) Endothelin-1 enhances fibrogenic gene expression, but does not promote DNA synthesis or apoptosis in hepatic stellate cells. Comp Hepatol 5: 5 doi:1476-5926-5-5 [pii] 10.1186/1476-5926-5-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Holobotovskyy V, Manzur M, Tare M, Burchell J, Bolitho E, et al. (2013) Regulator of G-protein signaling 5 controls blood pressure homeostasis and vessel wall remodeling. Circ Res 112: 781–791 doi:10.1161/CIRCRESAHA.111.300142 [DOI] [PubMed] [Google Scholar]
- 84. Shafiei MS, Rockey DC (2012) The function of integrin-linked kinase in normal and activated stellate cells: implications for fibrogenesis in wound healing. Lab Invest 92: 305–316 doi:[]labinvest2011155 [pii] 10.1038/labinvest.2011.155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Kitamura K, Tada S, Nakamoto N, Toda K, Horikawa H, et al. (2007) Rho/Rho kinase is a key enzyme system involved in the angiotensin II signaling pathway of liver fibrosis and steatosis. J Gastroenterol Hepatol 22: 2022–2033 doi:[]JGH4735 [pii] 10.1111/j.1440-1746.2006.04735.x [DOI] [PubMed] [Google Scholar]
- 86. Lavelle A, Sugrue R, Lawler G, Mulligan N, Kelleher B, et al. (2009) Sitaxentan-induced hepatic failure in two patients with pulmonary arterial hypertension. Eur Respir J 34: 770–771 doi:34/3/770 [pii] 10.1183/09031936.00058409 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
RGS5 is expressed in freshly isolated primary HSCs. 1° HSC have astrocyte-like morphology. A. Isolated 1° HSCs have an astrocyte-like phenotype and store vitamin A in lipid droplets. B. Isolated 1° HSCs are characterized by high RGS5 expression and low Tie1 and CD68 expression, indicating there are few contaminating cells. Low PDGFRβ expression demonstrates the 1° HSCs are in a quiescent state.
(TIF)
Co-localization of β-gal+ nuclei and GFAP staining does not change during injury. Frozen sections of CCl4 injected mouse liver were immunofluorescently labeled with antibodies for β-gal and GFAP. Co-localization analysis using ImagePro showed that the fraction of β-gal+ cells that are β-gal+ and GFAP+ does not significantly change during the course of injury. n = 3 mice per group.
(TIF)
Liver weight to body weight ratio is moderately elevated in Rgs5lacZ/lacZ mice at 96hr post CCl4 injury. Body and liver weights of CCl4 injected mice were measured at time of sacrifice. Rgs5LacZ/LacZ mice had elevated liver to body weight ratio 96 h post injury. Rgs5+/+ liver to body weight ratio was not significantly different from untreated mice. * p = .001, ‡ p = .06, n = 4–6.
(TIF)
Hepatocyte clearing is not associated with lipid accumulation. Oil red O staining of mouse liver 96 hr post CCl4 injection was used to assess lipid accumulation. A. Lipid droplets are visible around the site of injury in both Rgs5+/+ A,C and Rgs5LacZ/LacZ B,D mice. Cleared hepatocytes are visible in B, distant from the site of injury and droplets of lipid. C,D. Oil red O staining is present, but there is no accumulation of lipid in hepatocytes. Scale bar is 100 µm.
(TIF)
Hepatocyte clearing is not associated with glycogen accumulation. Periodic acid schiff stain of frozen mouse liver 96 hr post CCl4 injection was used to assess glycogen accumulation. A. Hepatocytes appear normal in Rgs5+/+ liver. Intense magenta staining labels glycogen in the hepatocytes. B. Cleared hepatocytes are visible in Rgs5LacZ/LacZ and glycogen staining is present in hepatocytes. High magnification of C Rgs5+/+ and D Rgs5LacZ/LacZ liver show RGS5 that cleared hepatocytes do not contain accumulated glycogen. Scale bar is 100 µm.
(TIF)
Cleared hepatocytes are not apoptotic. TUNEL staining of mouse liver 96 hr post CCl4 injection was used to assess apoptosis. A. Rgs5+/+ liver shows TUNEL minimal staining 96 hr post injection. B. Cleared hepatocytes are visible in Rgs5LacZ/LacZ liver 96 hr post CCl4. No difference in TUNEL staining is visible in cleared hepatocytes. C. DNAse I treated positive control shows intense staining. D. Uninjured Rgs5LacZ/LacZ has minimal TUNEL staining. Scale bar is 100 µm.
(TIF)
Data Availability Statement
The authors confirm that all data underlying the findings are fully available without restriction. All relevant data are within the paper and its Supporting Information files.