

Abstract
Vagal activation can reduce inflammation and disease activity in various animal models of intestinal inflammation via the cholinergic anti-inflammatory pathway. In the current model of this pathway, activation of descending vagal efferents is dependent on a signal initiated by stimulation of vagal afferents. However, little is known about how vagal afferents are activated, especially in the context of subclinical or clinical pathogenic bacterial infection. To address this question, we first determined if selective lesions of capsaicin-sensitive vagal afferents altered c-Fos expression in the nucleus of the solitary tract (nTS) after mice were inoculated with either Campylobacter jejuni or Salmonella typhimurium. Our results demonstrate that the activation of nTS neurons by intraluminal pathogenic bacteria is dependent on intact, capsaicin sensitive vagal afferents. We next determined if inflammatory mediators could cause the observed increase in c-Fos expression in the nTS by a direct action on vagal afferents. This was tested by the use of single-cell calcium measurements in cultured vagal afferent neurons. We found that tumor necrosis factor alpha (TNFα) and lipopolysaccharide (LPS) directly activate cultured vagal afferent neurons and that almost all TNFα and LPS responsive neurons were sensitive to capsaicin. We conclude that activation of the afferent arm of the parasympathetic neuroimmune reflex by pathogenic bacteria in the gut is dependent on capsaicin sensitive vagal afferent neurons and that the release of inflammatory mediators into intestinal tissue can be directly sensed by these neurons.
Keywords: Vagus, Cholinergic anti-inflammatory pathway, Campylobacter, Salmonella, TNFα, LPS
1. Introduction
Gastrointestinal inflammation can arise from different factors including genetics, diet, and gastrointestinal infection, and is a major financial burden in industrialized countries (Danese et al., 2004; Hulisz, 2004). While the initiating causes may be diverse, they all eventually involve a dysregulation of the balance between pro- and anti-inflammatory cytokines that are essential for maintaining the immunological function of the gut (Maloy and Powrie, 2011). This dysregulation facilitates the initiation and propagation of pro-inflammatory pathophysiology that ultimately results in disease symptoms (Artis, 2008; Hooper and Macpherson, 2010).
The gastrointestinal tract and its mucosal lining provide an essential barrier from nearly continuous exposure to foreign pathogens. To maintain this protection, multiple systems have evolved to coordinate the release of pro- and anti-inflammatory cytokines which mediate the initiation and control of immune responses. Recently the significance of the parasympathetic nervous system, and specifically the vagus nerve, in regulating inflammatory responses in the intestinal tract has been demonstrated (Borovikova et al., 2000; Tracey, 2007; van der Zanden et al., 2009); leading to the concept of a vagally dependent cholinergic anti-inflammatory pathway (Tracey, 2002). This cholinergic anti-inflammatory pathway has been proposed to be part of a vago-vagal immune reflex in which signals arising from the gut activate vagal afferents, which in turn activate the descending cholinergic anti-inflammatory pathway (Tracey, 2009). The majority of work studying this reflex has concentrated on the cholinergic efferent branch. Much less is known regarding the afferent branch, and more specifically the mechanisms through which vagal afferents are activated in response to a localized immune challenge. The primary goal of the experiments presented in this communication is to delineate the role(s) of vagal afferent neurons in detecting gastrointestinal infection.
Numerous studies have used direct recordings from vagal afferent neurons or hindbrain c-Fos expression to demonstrate that vagal afferent neurons can be activated by direct peripheral injections of lipopolysaccharides (LPS) (Liu et al., 2007), the pro-inflammatory cytokines interleukin-1β (IL-1β) (Niijima, 1996; Ek et al., 1998) or tumor necrosis factor-α (TNFα) (Rogers et al., 2006). While the use of injections of purified pro-inflammatory agents may produce a reliable response, there are limits to this approach that may lead to effects not observed in the normal sequela of events that follow intestinal colonization by pathogenic bacteria. For example, the purified component may have access to sites other than those accessible in a normal host/pathogen response, the concentration of the exogenously applied component may exceed that in a normal host/pathogen response, and the normal host/pathogen response might include factors other than the purified components. Prior work by Goehler and colleagues employed inoculation with Campylobacter jejuni (Gaykema et al., 2004; Goehler et al., 2005) to initiate a gastrointestinal immune challenge. These studies demonstrated that oral inoculation with C. jejuni induced c-Fos in the nTS within hours of inoculation, consistent with a neural (vagally) mediated detection of pro-inflammatory agents. They further demonstrated neuronal activation in more rostral brain regions, such as the hypothalamic paraventricular nucleus and central amygdala, without any detectable increase in circulating cytokines (IL-1β, IL-6, TNFα). This suggests that vagal afferent detection is sufficient to activate higher-level brain structures that are involved in generating systemic immune responses (Konsman et al., 1999). However, in these studies the neuronal activation in the nTS is assumed to be due to direct activation of vagal afferents, but this was not definitively tested through lesions of the vagus nerve.
In a similar study Wang et al. (2002) inoculated rats with Salmonella typhimurium. Oral inoculation of rats with S. typhimurium resulted in increased hypothalamic c-Fos expression, which was attenuated by subdiaphragmatic vagotomy (Wang et al., 2002). This study indicates that immune information from the gut to the brain is partially transmitted by an intact vagus nerve. While this study strongly supports the role of vagal afferents in immune monitoring of the gut, key characterization of the neural circuitry was not described. Specifically, no results showing markers of neuronal activation in the hindbrain or in the nTS were presented. Thus, whether the attenuation of S. typhimurium-induced hypothalamic activation following sub-diaphragmatic vagotomy was a result of decreased vagal signaling to the nTS or from decreased circulating cytokine levels was not clear.
In this study we used systemic injections of capsaicin to destroy afferent neurons that express the transient receptor potential type V1 ion channel (TRPV1). Capsaicin is an agent that causes activation of TRPV1 (Caterina et al., 1997) and high concentrations of capsaicin can induce neuronal death due to excessive stimulation of ion influx. Expression of TRPV1 is commonly interpreted to be a marker for C-type sensory neurons that have unmyelinated axons (Holzer, 1991). Approximately 70% of the afferent vagal fibers express TRPV1 (Li and Schild, 2007). Capsaicin lesions have previously been shown to cause nearly complete elimination of intraganglionic laminar endings in the upper and lower small intestines, cecum, and colon, while leaving a significant proportion (60–90%) of the intraganglionic laminar endings as well as intramuscular arrays in the stomach and esophagus intact (Berthoud et al., 1997). Since the primary site for C. jejuni colonization is the cecum (Jesudason et al., 1989) and the primary site for S. typhimurium is the large intestine proximal to the cecum (Nevola et al., 1985), we hypothesized that neuronal activation in the nTS caused by intestinal colonization by these pathogenic bacteria would be blocked by capsaicin lesions of vagal afferent neurons. We found that capsaicin lesions did indeed prevent activation of neurons in the nTS following inoculations with C. jejuni and S. typhimurium, and further found that almost all vagal afferent neurons that are acutely responsive to TNFα or LPS are also capsaicin sensitive. These results demonstrate that capsaicin-sensitive vagal afferent neurons are a critical component in signaling the presence of pathogenic bacteria in the intestines to the brain.
2. Materials and methods
2.1. Animals
Male BALB/c and CF-1 mice (4–8 weeks old) were purchased from Harlan Laboratories (Indianapolis, IN) and used as subjects for the bacterial inoculation experiments. All animals were housed individually in Association for Assessment and Accreditation of Laboratory Animal Care (AALAC)-accredited quarters under a 12:12 hour light: dark cycle with lights on at 7:00 am. Animals had ad libitum access to pelleted chow (Purina #5001) and water except when indicated.
Adult male Sprague-Dawley rats (200–240 g) were purchased from Simonsen Laboratories and used as a tissue source for nodose ganglia. All animals were housed in AALAC-accredited quarters under a 12:12-h light-dark cycle and had ad libitum access to pelleted chow and water. The Washington State University Institutional Animal Care and Use Committee approved all procedures performed (IACUC protocol numbers ASAF 4086 and ASAF 3914).
2.2. Capsaicin treatment
Capsaicin (E-Capsaicin, Tocris #0462) was dissolved with 5% ethanol and 13% Tween-80 in sterile saline (0.9%) and was injected into the intraperitoneal cavity. Control injections were identical minus capsaicin. Animals received three capsaicin/vehicle injections over a 24-hour period. Prior to the first capsaicin injections (25 mg/kg), animals were fasted ~10 h. Animals were fasted ~4 h for the second injection (50 mg/kg) and had ad libitum access to food prior to the third injection (50 mg/kg). Ten minutes prior to each capsaicin/vehicle injection, animals received atropine (0.27 mg/kg, i.p.). For each capsaicin/vehicle injection, animals were deeply anesthetized with isoflurane (3%) and monitored under anesthesia 20–40 min after injection. Animals were allowed to recover from the capsaicin treatment for 12–14 days before inoculation with bacteria. To test for lesion completeness, 6 days prior to the inoculation we used an eye-wipe test (lesion of capsaicin-sensitive afferent neurons was considered successful if the animal failed to respond with vigorous eye-wiping following a brief exposure to the mild irritant 1% ammonium hydroxide).
2.3. Bacteria
C. jejuni was grown on Mueller–Hinton (MH) agar plates or in MH broth with shaking at 37 °C in a 10% CO2 environment. The C. jejuni strains used were: 11168 (human isolate, mouse adapted, streptomycin resistant), H34 (human isolate, streptomycin resistant), F38011 (human isolate, mouse adapted, streptomycin resistant), H41 (human isolate, streptomycin resistant), 81–176 (mouse adapted, streptomycin resistant). S. typhimurium strain SL1344 (wild-type) was grown on Luria-Bertani (LB) agar plates or in LB broth, at 37° C. Prior to inoculation, C. jejuni was introduced into MH broth supplemented with 0.01% deoxycholate and incubated for 18 h with shaking to keep the bacteria in suspension. S. typhimurium was introduced into LB broth and incubated without shaking for 18 h. Both bacterial cultures were centrifuged to pellet the bacteria, and re-suspended in phosphate-buffered saline (PBS) to 3.0 O.D.540 using a Genesys 10 spectrophotometer (Thermo Scientific) for the final inoculum.
2.4. Inoculation and tissue collection
Forty-eight hours prior to inoculation, streptomycin (20 μg/mL) was added to the drinking water of all animals. Each mouse was inoculated by oral gavage with a volume of 0.15 mL bacteria in PBS. Successful gavages were determined by the absence of sinus discharge of inoculum as well as subsequent documented gut colonization. The number of C. jejuni or S. typhimurium within the inoculum and gastrointestinal tract were enumerated by serial dilution in PBS followed by plating on Campy-Cefex agar (a selective growth medium for cephalothin-resistant Campylobacter species such as C. jejuni) or xylene-lysine-deoxycholate agar (a selective growth medium for Salmonella species), respectively. Mice were orally gavaged with ~1.3–6.3×1010 C. jejuni (F38011) or 1.0×1010 S. typhimurium suspended in 0.1 M PBS. Mice were inoculated at 15 min intervals and sacrificed at the same interval 24 h post inoculation. During the 24 h following inoculation cardboard igloos were placed in each cage to provide enrichment and minimize anxiety. All food was removed 4 h prior to sacrifice. At time of sacrifice, animals were deeply anesthetized with isoflurane. Blood was collected by cardiac puncture and animals were perfused transcardially with 0.1 M PBS. Intestines and cecum were collected and placed in MH broth. For C. jejuni inoculations, the area of the gut collected encompassed tissue 1 cm proximal to the cecum to 3 cm distal to the cecum. For S. typhimurium inoculations, the gut area collected was approximately a 3-inch section of ilium proximal to the ileocecal valve. Animals were then perfused with 4% paraformaldehyde and brains were removed and post fixed overnight. Once post-fixed, brains were placed in 0.1 M PBS+25% sucrose for ~24 h and then frozen at −80 °C. Samples remained frozen until sectioned.
2.5. Immunohistochemistry
Frozen brains were cut on a Leica CM1900 cryostat (−22 °C) in 30 μm sections. Sections were divided and placed in either 0.1 M PBS or cryoprotectant (50% 0.05 M PBS pH 7.3, 30% ethylene glycol, 20% glycerol). Sections placed in 0.1 M PBS were stored at 4 °C until reacted with an immunoperoxidase method using the avidin–biotin labeling procedure and nickel-intensified staining with 3′ 3-diaminobenzidine (Ni-DAB) (Vectastain ABC kit from Vector Laboratories, procedure as per manufacturer’s protocol). For the 1° antibody we used anti-c-Fos, Ab-5 rabbit, from Calbiochem (Lot# D00100487; 1:20,000 dilution) and for the 2° antibody we used Biotin-SP-conjugated Pure Donkey Anti-rabbit IgG from Jackson ImmunoResearch (1:500 dilution). Following the Ni-DAB reaction sections were rinsed in TPBS and stored at 4 °C until mounted on Colorfrost Plus microscope slides. Once mounted on microscope slides, sections were allowed to dry, dehydrated with EtOH (95%, 100%) and 2-propanol, washed in Histoclear and cover slipped with Di-N-butyl phthalate in xylene (DPX).
2.6. Microscopic analysis
The c-Fos immunoreactivity in the nTS was evaluated using a Nikon Eclipse 80i microscope. For nuclear stained c-Fos quantification in the nTS, a series of three bright field images were taken using a Plan Apo 20×/0.75 objective with a Nikon DS-Qi1Mc digital camera and stitched together. Images were processed and analyzed using NIS Elements Advanced Research v3.2. All images were captured at similar exposure times and illumination levels, and were normalized by subtracting the background adaptively. Regions of interest (ROI) outlining the nTS in each serial section were equal between brains. Nuclear stained c-Fos positive neurons were identified using a cell specific representative ROI and measuring the signal to background ratio. Neurons were considered positively stained if the following criteria were met: 1) signal to background ratio value ≥5; 2) c-Fos staining of the nucleus was darker than the surrounding tissue; 3) staining was circular in shape with smooth edges; and 4) it was within the nTS ROI. Cell counting was performed by an observer blind to the nature of the treatment of the individual animal. The nTS counts were performed bilaterally at six levels: −7.76 mm, −7.64 mm, −7.56 mm, −7.48 mm, −7.32 mm and −7.20 mm from bregma. Area postrema (AP) counts were performed bilaterally at three levels: −7.76 mm, −7.56 mm, and −7.32 mm from bregma. The counts of c-Fos positive nuclei were summated to yield the total count for each animal.
2.7. Plasma cytokine assay
TNFα levels at time of sacrifice were determined by ELISA. Blood was collected as described above and stored at −80 °C until analyzed. The ELISA kit (Mouse TNFα ELISA Ready-SET-Go!) was purchased from eBioscience and used according to manufacturer’s instructions. The sensitivity of the kit was rated at 8 pg/mL.
2.8. Dissociation and culture of vagal afferent neurons
Isolated nodose ganglion preparations were performed as previously described (Simasko et al., 2002). Briefly, rats were anesthetized and both left and right nodose ganglia were collected and placed in ice-cold Ca2+ and Mg2+-free Hank’s balanced salt solution (HBSS). After desheathing, the remaining tissue was minced into ~1-mm3 blocks and digested for 90 min at 37 °C (1 mg/ml dispase II and 1 mg/ml collagenase type Ia in HBSS). After the digestion the tissue fragments were triturated, washed by gentle centrifugation, and the liberated cells placed onto poly-L-lysine coated coverslips (100 μg/ml for 30 min). The dissociated neurons were then maintained in Hepes-buffered Dulbecco’s modified Eagle’s medium supplemented with 10% fetal calf serum at 37 °C. All measurements with cultured cells were performed within 36 h of isolation. Media components were obtained from Gibco/Life Technologies Corp., collagenase was purchased from Sigma (St. Louis, MO), and dispase from Boehringer Mannheim (Indianapolis, IN).
2.9. Intracellular calcium measurements
All experiments were performed at room temperature (22 °C). Calcium was monitored by use of the fluorescent calcium indicator fura-2 AM (Molecular Probes, Eugene, OR). MetaFluor Software (Universal Imaging, West Chester, PA) was used to collect and analyze results. Cells were loaded with 1 μM fura-2 AM in physiological bath (in mm: 140 NaCl, 5 KCl, 2 CaCl2, 1 MgCl2, 6 glucose, and 10 Hepes, pH adjusted to 7.4 with Tris-base) for 45 min followed by a 15 min wash. After the fura-2 AM incubation/wash, coverslips were mounted into an open chamber and constantly perfused with physiological bath. Neurons were challenged with agents of interest by changing solutions applied through a common manifold upstream of the perfusion chamber (agents reached cells ~10 s after the switch). Ratios of fluorescence intensity were converted to calcium concentrations using a standard curve obtained in a bath containing 10 μM fura-2, 130 mm KCl, 10 mm MOPS buffered to pH 7.4 with KOH, 10 mm EGTA, and various concentrations of CaCl2 (0–10 mm). Image pairs (340 and 380 nM excitation, 510 emissions) were collected every 6 s. Buffer components were obtained from Sigma (St. Louis, MO).
Nodose neurons were easily identified and selected based on their large, round cell bodies. In most cases basal calcium levels were below 100 nM. Any neuron with an initial basal level above 250 nM was not included in the analysis. Further, in some neurons the calcium level did not return to basal levels after challenge with the agent of interest. In the summarized analyses we included data from only those neurons in which calcium increases returned to baseline levels after each drug challenge. A positive response to a particular challenge needed to be at least 20 nM in amplitude; a value chosen that easily exceeded the typical noise in the basal level of calcium (generally <5 nM). Finally, at the end of each run, a high potassium (Hi-K) solution (in mm: 90 NaCl, 55 KCl, otherwise same as physiological saline) was briefly applied to depolarize the cells and determine whether or not neurons were viable. Only those neurons that responded to the Hi-K challenge with a rapid and reversible calcium response of at least 50 nM in amplitude were included in the analysis.
2.10. Drugs
Agents used in the calcium measurements were: (E)-capsaicin (Cap), from Tocris (Minneapolis, MN); lipopolysaccharide from Escherichia coli 0111:B4 (LPS), from Sigma (St. Louis, MO); tumor necrosis factor alpha (TNFα), from R&D Systems (Minneapolis, MN). Initial stock solutions were made in distilled water except for TNFα (made in PBS with 0.1% bovine serum albumin), and Cap (made in 95% ethanol).
2.11. Statistical analysis
Positive oral inoculation was assumed if bacteria were recovered in intestinal samples after sacrifice. Only bacterial inoculated animals with positive colonization were used in the final analysis. No C. jejuni or S. typhimurium were found in the intestines from control animals. For testing of statistical significance, we used a Student’s t-test when only two groups were compared. For multiple comparisons, we used one-way or two-way analysis of variances (ANOVA) followed by pairwise multiple comparisons using the Holm-Sidak method. We used the non-parametric Mann-Whitney test when colonization levels were compared because values varied by orders of magnitude between animals. In the calcium imaging experiments we used Chi-squared analysis to determine the significance of the overlap between responsiveness to TNFα and LPS vs capsaicin. Results with p<0.05 were considered statistically significant. Statistical analyses were conducted using SigmaStat software.
3. Results
3.1. Optimization of intestinal colonization by C. jejuni
We found in initial experiments that 14-week-old BALB/c mice treated with C. jejuni strain 11168 did not produce a reliable c-Fos signal in the nTS (data not shown). Thus we investigated various parameters to optimize the inoculations. We began to treat our mice with streptomycin in their drinking water prior to the inoculation to reduce competition from endogenous flora. Further, we also investigated the effectiveness of various strains of C. jejuni and different strains and ages of mice since these are known factors that affect virulence (Vuckovic et al., 1998; Bell et al., 2009). We found that strain F38011 colonized mice better than other strains of C. jejuni (Supplementary Fig. 1A). We also found that 8-week-old BALB/c mice inoculated with F38011 had a higher cecal colonization compared to 8-week-old CF-1 mice and 14-week-old BALB/c mice (Supplementary Fig. 1B). Thus all subsequent experiments were performed with C. jejuni strain F38011 in 8-week-old BALB/c mice treated with streptomycin prior to the inoculation.
3.2. Capsaicin lesions attenuate C. jejuni induced c-Fos-ir in the nTS of BALB/c mice
To address the question of which type of vagal afferent is responsible for conveying the presence of pathogenic bacteria in the intestine to the brain, we eliminated TRPV1 expressing neurons by treatment with the neurotoxin capsaicin. We found that systemic treatment with capsaicin ~12 days prior to inoculation prevented the increase in c-Fos-ir within the nTS produced by intestinal inoculation with C. jejuni (Fig. 1).
Fig. 1.

Capsaicin lesions attenuate C. jejuni induced c-Fos-ir in BALB/c mice. (A) Photomicrographs of c-Fos staining in the nTS after C. jejuni inoculation. Sections shown are approximately −7.56 mm from bregma. Left hand photomicrographs: inoculated with the phosphate buffered saline vehicle (PBS). Right hand photomicrographs: inoculated with ~6.3×1010 C. jejuni of the F38011 strain. Top set of photomicrographs: treated with vehicle prior to inoculation (Intact). Bottom set of photomicrographs: treated with capsaicin prior to inoculation (see Materials and methods section for details). AP — area postrema; NTS — nucleus of the solitary tract; CC — central canal. Bar in each photomicrograph indicates 100 μm. (B) Quantification of c-Fos-ir in nTS. Bar graph shows the sum of the c-Fos-ir neurons over 3 serial sections (−7.64 to −7.48 mm from bregma). Region for counting indicated on photomicrographs by dashed line. Number of animals treated: Intact: PBS (n=6); Intact: C. jejuni (n=13); Cap: PBS (n=8); Cap: C. jejuni (n=14). Two-way ANOVA p=0.033; *p=0.01 and **p=0.003 by post-hoc Holm–Sidak test.
Although all animals were inoculated with equivalent amounts of bacteria, the resulting degree of colonization was highly variable (~5×102 to 2×105 CFU; see x-axis distribution in Fig. 2B). We found a positive relationship between the amount c-Fos-ir in the nTS and the degree of C. jejuni colonization in the intact animals, but not in capsaicin lesioned animals (Figs. 2A and 2B). This variability in colonization created a de facto dose response of bacterial loads. If only results from animals with low levels of colonization (<1×104 CFU) are considered, there was no difference in the average c-Fos expression in the nTS between intact and capsaicin lesioned animals (Intact: n=6; 57±10 vs. Cap: n=3; 40±20; p=0.44, t-test). However, when the colonization levels were >1×104 CFU, the difference in c-Fos-ir labeled neurons between intact and capsaicin animals was highly significant (Fig. 2B; results from all animals with CFUs >1×104: Intact: n=7; 164±33 vs. Cap: n=11; 52±10; p=0.001, t-test). Although on average the intestinal colonization was slightly higher in capsaicin treated animals, there was no statistically significant difference between groups (Intact: n=13; 1.9×104±0.6×104 CFU; Cap: n=14; 3.5×104±1.0×104 CFU; p=0.17, Mann–Whitney test).
Fig. 2.

Increased c-Fos-ir from oral C. jejuni inoculation is dependent on cecal colonization in intact BALB/c mice. (A) Representative examples of c-Fos-ir expression in hindbrains of mice versus degree of cecal colonization determined at the time of sacrifice (approximate cecal CFU levels indicated above photomicrographs). Representative examples from animals with low CFUs in left hand photomicrographs and from animals with high CFUs in right hand photomicrographs. Top set of photomicrographs are from intact animals (vehicle) and the lower set of photomicrographs are from capsaicin treated animals. Sections shown are approximately −7.56 mm from bregma. Abbreviations and scale bar are as in Fig. 1. (B) Quantification of the number of c-Fos-ir neurons versus number of cecal CFUs in individual animals. As in Fig. 1, c-Fos-ir cells were summed over 3 serial sections. Open circles are from intact animals (n=13), filled circles are from capsaicin treated animals (n=14). These results are from the same animals as reported in Fig. 1. Solid line is fit to data from intact animals excluding the black filled circle (an intact animal), R2=0.68. The dashed line indicates the average value obtained from intact PBS-treated animals.
3.3. Capsaicin lesions attenuate S. typhimurium induced c-Fos-ir in the nTS of BALB/c mice in a colonization independent manner
In contrast to C. jejuni, which do not establish a systemic infection in BALB/c mice, S. typhimurium will eventually produce a colonized systemic infection. Oral inoculation with S. typhimurium is known to increase c-Fos-ir in the hypothalamus via a mechanism that is in part dependent on an intact vagus nerve (Wang et al., 2002). Here we used S. typhimurium to determine whether lesions of capsaicin sensitive afferents were capable of preventing activation of neurons in the nTS even when a highly virulent strain of bacteria is responsible for the intestinal irritation. We found that oral inoculation with S. typhimurium caused a dramatic increase in c-Fos-ir in the nTS of intact BALB/c mice, and this increase was completely prevented in animals that received the capsaicin lesions (Fig. 3). Further, the maximal degree of c-Fos induction in the nTS was significantly higher with S. typhimurium versus C. jejuni (291±21 versus 114±23, respectively, Figs. 1B and 3B; p=0.001, t-test). However, unlike C. jejuni in which the magnitude of the c-Fos induction was dependent on the degree of colonization, we found no relationship between intestinal colonization (CFU) and number of c-Fos-ir cells in the nTS (data not shown). Like C. jejuni, there was a trend for capsaicin lesioned animals to have slightly higher colonization, but this difference was not statistically significant (Intact: n=4; 3.0×104±1.6×104 CFU; Cap: n=5; 1.6×105±0.9×105 CFU; p=0.29, Mann–Whitney test).
Fig. 3.
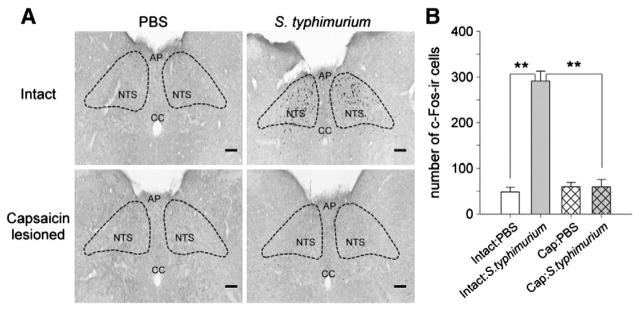
Capsaicin lesions attenuate S. typhimurium induced c-Fos-ir in the nTS of BALB/c mice. (A) Photomicrographs of c-Fos staining in the nTS after oral inoculation with S. typhimurium (see experimental details in Materials and Methods). Sections shown are approximately −7.56 mm from bregma. Organization of photomicrographs, abbreviations, and scale bar are as in Fig. 1. (B) Quantification of c-Fos-ir in mouse nTS. Bar graph shows the sum of the c-Fos-ir neurons over 3 serial sections (−7.64 to −7.48 mm from bregma). Number of animals treated: Intact: PBS (n=5); Intact: S. typhimurium (n=4); Cap: PBS (n=5); Cap: S. typhimurium (n=5). Two-way ANOVA p <0.001; **p <0.001 by post-hoc Holm-Sidak test.
3.4. Anatomical distribution of c-Fos-ir in the nTS
We also examined if there was an anatomic difference in the rostral-caudal distribution of c-Fos-ir in the nTS with inoculations of C. jejuni versus S. typhimurium, and if the capsaicin lesion might cause a loss of c-Fos at a particular rostral-caudal position. At all rostral-caudal positions examined, the number of c-Fos-ir neurons was greater in both the C. jejuni and S. typhimurium treated animals versus the intact animals (Fig. 4A). Comparing S. typhimurium treated animals to C. jejuni treated animals there were no significant differences in the number of c-Fos-ir neurons at any rostral-caudal levels (Fig. 4A). For both C. jejuni and S. typhimurium treated animals the most extreme caudal position (−7.76 mm from bregma) had significantly less c-Fos-ir than the middle positions (Fig. 4A). For C. jejuni treated animals the number of c-Fos-ir neurons in the most rostral position (−7.20 mm from bregma) was significantly less than the number of c-Fos-ir neurons at −7.48 and −7.32 mm from bregma (Fig. 4A). In the capsaicin treated mice the increase of c-Fos-ir by either C. jejuni or S. typhimurium was prevented at all rostral-caudal positions (Fig. 4B).
Fig. 4.

Quantification of neurons expressing c-Fos-ir at different rostral-caudal levels of the nTS and AP. (A) Graph shows number of c-Fos-ir neurons in the nTS from each of 6 section spanning −7.76 mm to −7.20 mm from bregma from control animals (Intact). For C. jejuni (filled triangles) the number of c-Fos-ir neurons at −7.76 mm from bregma (#) was significantly less than c-Fos-ir neurons at −7.56, −7.48, and −7.32 mm from bregma, p<0.005, Holm–Sidak post-hoc test. Also the number of c-Fos-ir neurons at −7.20 mm from bregma (#) was significantly less than those at −7.48 and −7.32 mm from bregma, p<0.005, Holm–Sidak post-hoc test. For S. typhimurium (filled squares) the number of c-Fos-ir neurons at −7.76 mm (*) was significantly less than c-Fos-ir positive neurons at all other positions, p<0.005, Holm-Sidak test. Number of animals in each group: Intact: C. jejuni (n=4; filled triangles), Intact: S. typhimurium (n=4; filled squares), Intact: PBS (n=8; open circles). (B) Graph shows number of c-Fos-ir neurons in the nTS from each of 6 section spanning −7.76 mm to −7.20 mm from bregma from capsaicin treated animals (Cap). Number of animals in each group: Cap: C. jejuni (n=4; filled triangles), Cap: S. typhimurium (n=4; filled squares), Cap: PBS (n=8; open circles). (C) Graph shows number of c-Fos-ir neurons in the AP from each of 3 section spanning from −7.76 to −7.32 from bregma from control animals: Number of animals in each group: Intact: C. jejuni (n=4; filled triangles), Intact: S. typhimurium (n=4; filled squares), Intact: PBS (n=8; open circles). (D) Graph shows number of c-Fos-ir neurons in the AP from each of 3 section spanning from −7.76 to −7.32 from bregma from capsaicin treated animals: Number of animals in each group: Cap: C. jejuni (n=4; filled triangles), Cap: S. typhimurium (n=4; filled squares), Cap: PBS (n=8; open circles).
3.5. C. jejuni and S. typhimurium inoculations do not increase c-Fos-ir in the area postrema (AP) or increase circulating TNFα
To investigate whether the inoculations were inducing a systemic release of cytokines we examined both c-Fos-ir in the AP and circulating TNFα at the time the animals were sacrificed. Because neurons in the AP are outside the blood-brain barrier, they are more sensitive to systemic release of pro-inflammatory markers. Neither bacterial inoculation produced an increase in c-Fos-ir in the AP (Fig. 4C), nor did the capsaicin treatment alter c-Fos-ir expression in this region (Fig. 4D). There was also no significant increase in circulating TNFα in any of the treatment conditions (Fig. 5). It is interesting to note that in each condition the capsaicin treated animals had slightly increased amounts of TNFα, an effect that just missed statistical significance in the two-way ANOVA (main effect of capsaicin, p=0.054).
Fig. 5.

Systemic TNFα levels did not increase in bacterial challenged animals. Graph illustrates the average TNFα levels in serum collected at time of sacrifice. Intact are the vehicle controls; Cap indicates capsaicin treated. PBS indicates the inoculation control. Dashed line represents the threshold of detection for the ELISA kit. Animal numbers were: Intact: PBS (n=10), Cap: PBS (n=13), Intact: C. jejuni (n=13), Cap: C. jejuni (n=14), Intact: S. typhimurium (n=4), Cap: S. typhimurium (n=5).
3.6. Direct activation of nodose neurons by TNFα and LPS
The means by which the presence of pathogenic bacteria in the intestine causes activation of vagal afferents is not known. Because the above results suggest that TRPV1 expressing neurons are responsible to transmitting the presence of pathogenic bacteria from the gut to the brainstem, we next asked if it is possible that inflammatory mediators such as TNFα and LPS could directly activate vagal afferent neurons and whether this was confined to those neurons that expressed TRPV1. We performed this experiment on cultured nodose neurons because in this preparation any action of the applied agent has to be mediated directly on the neurons under study and thus is not confounded by the possibility that the agent under study releases some other bioactive substance that ultimately activates the vagal neuron. We found that when we applied TNFα to cultured nodose neurons an increase in intracellular calcium concentration occurred (Fig. 6A and B) in 14% of the tested neurons (14 of 98). The response was dose dependent with an EC50 between 0.1 and 1 ng/ml. While the time between challenges to TNFα (~10 min) were usually adequate to allow for full recovery, we frequently observed some desensitization between 1 ng/mL and 10 ng/mL dose. We found that 93% of TNFα responsive neurons (13 of 14) were also responsive to capsaicin, a highly significant degree of overlap (Fig. 6B). These results demonstrate that TNFα responsive cells are primarily TRPV1 expressing C-type vagal afferent neurons, but that not all TRPV expressing neurons are sensitive to TNFα.
Fig. 6.

TNFα and LPS induce calcium responses in a subset of capsaicin sensitive neurons. (A) A representative trace of the intracellular Ca2+ level in a TNFα sensitive neuron in response to different concentrations of TNFα. Bars over the traces represent when the test solution was applied. Bars labeled with TNFα and numbers indicate the concentration of TNFα (in ng/mL). Bar labeled with Cap indicates that application of capsaicin (100 nM). Bar labeled with HiK indicates when neurons were challenged with 55 mM K+. Veh is the highest concentration of the vehicle (bovine serum albumin) used for the TNFα solutions. (B) Distribution of TNFα responsive (TNF+) and TNFα non-responsive (TNF−) neurons relative to capsaicin responsive (Cap+) and capsaicin non-responsive (Cap−) neurons. Numbers of neurons in each group were: Cap+/TNF− 56; Cap+/TNF+13; Cap−/TNF− 28; Cap−/TNF+1. Chi-squared analysis indicated the segregation of TNF responsive neurons with capsaicin responsive neurons was significant: χ2=4.05, p=0.04. (C) A representative trace of the intracellular Ca2+ level in an LPS sensitive neuron in response to different concentrations of LPS. Bars over the trace represent when the test solution was applied. Bars labeled with LPS and numbers indicate the concentration of LPS (in ng/ml). Bars labeled with Cap indicate that application of capsaicin (100 nM). (D) Distribution of LPS responsive (LPS+) and LPS non-responsive (LPS−) neurons relative to capsaicin responsive (Cap+) and non-responsive (LPS−) neurons. Numbers of neurons in each group were: Cap+/LPS− 24; Cap+/LPS+14; Cap−/LPS− 17; Cap−/LPS+0. Chi-squared analysis indicated the segregation of LPS responsive neurons with capsaicin responsive neurons was highly significant: χ2 = 15.1, p <0.001.
We also examined the effects of LPS on cultured vagal afferent neurons and found LPS directly activated 25% of the neurons tested (14 of 55) (Fig. 6C and D). The response was dose dependent with an EC50 just over ~100 ng/ml. Like TNFα, we also found a highly significant overlap between responsiveness to LPS and capsaicin in that all cells responsive to LPS also responded to capsaicin, but not all capsaicin responsive cells responded to LPS (Fig. 6D).
4. Discussion
In this study we found that oral inoculation of mice with either C. jejuni or S. typhimurium causes activation of neurons in the nTS via a capsaicin sensitive pathway. In comparison to a physical vagotomy, the use of capsaicin to induce a lesion not only confines the loss of afferent neurons to a specific subtype of afferent neuron, it also preserves vagal efferent neurons. This avoids complications in the interpretation of the effects of the lesion that might be caused by a loss of efferent activity, such as gastrointestinal stasis. Previously Wang and co-workers (Wang et al., 2002) found that a physical vagotomy partially prevented the induction of c-Fos-ir in hypothalamic nuclei by oral inoculation with S. typhimurium. We now extend their findings by showing that neurons within the nTS are also activated by oral inoculation with S. typhimurium, and that this effect is dependent on vagal afferent neurons that express TRPV1. Similarly, Goehler and colleagues found that oral inoculation with C. jejuni in mice was capable of activating central nervous system effects (Gaykema et al., 2004; Goehler et al., 2005). While previous reports demonstrated peripheral administration of cytokines can induce behavioral effects via a vagal-dependent pathway (Fleshner et al., 1995; Goehler et al., 1995; Watkins et al., 1995; Fleshner et al., 1997; Fleshner et al., 1998; Goehler et al., 1998), whether brainstem c-Fos activation produced by inoculation with C. jejuni is dependent on an intact vagus nerve has not been reported. We now demonstrate that the intestinal presence of C. jejuni is signaled to the brain by a pathway that is sensitive to capsaicin, indicating the neurons that underlie the signal must express TRPV1. Because afferent neurons in both the vagus nerve and spinal afferents express TRPV1, from our experiments we cannot rule out that spinal pathways also participate in this response. On the other hand, physical vagotomies block the behavioral responses to an oral C. jejuni inoculation (Fleshner et al., 1995; Goehler et al., 1995; Watkins et al., 1995; Fleshner et al., 1997; Fleshner et al., 1998; Goehler et al., 1998), thus suggesting that the vagus nerve is the relevant neuronal pathway communicating intestinal challenges to the nTS. Additionally, Mueller et al. (Mueller et al., 2006) have shown that inflammation induced by intestinal manipulations during surgery is also capable of inducing c-Fos-ir in the brainstem via a pathway sensitive to capsaicin treatments. Our results now show that irritation caused by bacterial agents appears to activate a similar population of neurons as physical manipulation. Finally, we found evidence that a subset of isolated vagal neurons in culture could respond directly to TNFα and LPS, and that almost all neurons responsive to these pro-inflammatory mediators were responsive to capsaicin. In total, these observations provide support for the conclusion that TRPV1 expressing afferent fibers of the vagus are responsible for the direct detection of inflammatory mediators in the intestine, and that this is likely to represent the neural population that participates in initiating the afferent arm of the neuroimmune reflex outlined by Tracey and co-workers (Andersson and Tracey, 2012).
4.1. Role of the vagus in detecting cytokines
Peripheral administration of LPS or the pro-inflammatory cytokines IL-1β or TNFα induce many effects associated with sickness that are mediated by the central nervous system such as hyperthermia, social withdrawal, decreased food motivated behaviors, increased sleep, and release of corticosterone (Elmquist et al., 1996; Mascarucci et al., 1998; Konsman et al., 1999; Emch et al., 2001; Hermann et al., 2001). What has remained controversial is the role of the vagus in these effects, with many investigators finding that a physical vagotomy blocks the behavioral or physiological responses (Bret-Dibat et al., 1995; Fleshner et al., 1995; Goehler et al., 1995; Watkins et al., 1995; Bluthe et al., 1996; Fleshner et al., 1997; Hansen and Krueger, 1997; Fleshner et al., 1998), but others finding that under certain circumstances vagotomies do not block similar responses (Bluthe et al., 1996; Hansen and Krueger, 1997; Hansen et al., 2001; Hermann et al., 2001). In several of the studies in which vagotomy failed to block the response, part of the explanation appears to be either the magnitude of the challenge (large magnitude challenges bypass the vagus; Hansen and Krueger, 1997; Hansen et al., 2001) or the route of the challenge (intraperitoneal challenges require the vagus but subcutaneous or intravenous challenges do not; Bluthe et al., 1996). In our study we wanted to restrict our stimuli to a host-pathogen response in which evidence for systemic involvement is minimized, and thus we could be confident in our conclusion that activation of vagal afferents is an event that can occur within intestinal tissue itself.
4.2. C. jejuni as a model
While C. jejuni can colonize the intestine, they do not establish a systemic infection in naive mice (Vuckovic et al., 1998; Young et al., 2000; Dorrell and Wren, 2007). This would confine the stimulation produced by C. jejuni to the gut itself. In fact we had difficulty in our initial experiments finding conditions in which the particular strain of C. jejuni and strain and age of mouse could reliably induce c-Fos-ir in the nTS. This supports the conclusion that this was a very mild stimulant, but under the correct conditions it was capable of causing enough stimulation to activate TRPV1 expressing vagal afferents. The observation that we could detect no increase in circulating TNFα at the time of sacrifice (24 h post inoculation) is similar to the results found by Gaykema et al. (Gaykema et al., 2004) in which they observed no increase in circulating IL-6, IL-1β, or TNFα at 18 or 42 h after inoculation. These findings support the conclusion that the C. jejuni induced response was localized to the gut and therefore likely to induce activation of neurons in the brain primarily by activation of intestinally innervating fibers rather than the release of a circulating factor.
4.3. S. typhimurium as a model
In contrast to C. jejuni, S. typhimurium will establish a systemic infection. While others have observed large increases in circulating TNFα within 24 h of inoculation (~250 pg/mL at 24 h which increased to >4000 pg/mL 72 h post-inoculation) (Jotwani et al., 1995); we did not detect a significant increase in circulating TNFα in blood at the time of sacrifice (24 h post-inoculation). However, we have observed overt disease and death over an 8 day period with our strain of S. typhimurium when we use higher number of bacteria in the inoculation, so we know that our strain of bacteria is pathogenic (data not shown). The failure to observe a significant increase in TNFα at 24 h suggests the systemic infection with our strain of S. typhimurium was delayed in breeching the intestinal barrier. Nevertheless, the lack of a statistically significant increase in circulating TNFα at a time when there was a large increase in c-Fos-ir in the nTS supports the conclusion that activation of neurons in the nTS occurs before a systemic response is manifest, and thus activation via a neural pathway is likely to play a significant role. Our conclusion that we had yet to induce a systemic response is further supported by the observation that we could observe no increase in c-Fos-ir in the AP, a brain area that lies outside the blood-brain barrier and which is known for being able to respond to circulating cytokines with an increase in c-Fos-ir (Sagar et al., 1995; Hermann and Rogers, 2008). Even though we did not have evidence for a systemic response at the time of sacrifice, the induction of c-Fos-ir in the nTS was much more robust with S. typhimurium than C. jejuni, suggesting the pathogenic challenge with S. typhimurium produced a stronger activation of TRPV1 expressing vagal afferents. However, we were unable to detect any significant differences in the pattern of c-Fos-ir between inoculations with S. typhimurium versus C. jejuni, indicating the difference in vagal activation between these two bacteria was one more of degree of activation than activation of different subpopulations of neurons.
4.4. Direct activation of vagal afferents by TNFα and LPS
Although we do not know the specific signaling molecule(s) responsible for activating vagal afferents by the bacterial challenges we used, our studies with isolated nodose neurons suggest that the neurons themselves must be considered as possible targets for pro-inflammatory factors generated by intestinal bacterial challenges. TNFα caused an increase in intracellular calcium in a subpopulation of capsaicin sensitive neurons that required only seconds to become observable. The ability of TNFα to cause such rapid calcium responses in isolated neuronal and neuron-like cells is variable, having been reported to occur in 34% of dorsal root ganglia neurons (Pollock et al., 2002), but not in cultured hippocampal neurons (De et al., 2003). A slow, irreversible increase in calcium is induced by TNFα in astroglial cells (Koller et al., 1996), but since this is irreversible, it is difficult to separate this from a possible effect to compromise the health of the cells. Hermann et al. (Hermann and Rogers, 2008) examined the actions of TNFα on the central terminals of vagal afferents and concluded that TNFα weakly increased the pre-synaptic calcium signal by itself but amplified terminal calcium signals produced by known agonists (such as ATP). However, since this was a slice preparation, it is possible that the effects of TNFα were indirectly mediated on another component within the slice. While our findings suggest vagal afferents can be directly activated by TNFα, it is possible that during physiological responses TNFα may work in concert with other factors to produce a robust activation of vagal afferents.
Prior studies have demonstrated the presences of TLR4, the receptor for LPS, in the nodose (Hosoi et al., 2005). Our observations confirm that TLR4 observed by Hosoi et al. in the nodose ganglia is physiologically active. LPS has also been shown to induce a calcium response in 50% of neurons from the trigeminal ganglia (Diogenes et al., 2011). Similar to this action in trigeminal neurons, we found that 25% of nodose neurons responded to LPS with a robust increase in cytosolic calcium. We did not test TNFα and LPS on the same neuron so we do not know if these two agents activate the same population of neurons, but the actions of both agents were primarily confined to a subpopulation of neurons that express TRPV1.
4.5. Implications for the inflammatory reflex
How the afferent arm of the inflammatory reflex is activated remains poorly understood. A myriad of sites sensitive to pro-inflammatory factors, including vagal paraganglia (Goehler et al., 1997), vagal afferent cell bodies in the nodose ganglia (Hosoi et al., 2005), or a direct action of cytokines within the brain (Bluthe et al., 1996; Hansen and Krueger, 1997; Hansen et al., 2001; Hermann et al., 2001) have been suggested. However, activation from these sites would require that pro-inflammatory factors had been released into the systemic circulation before activation would occur. Notably, this is something that also occurs when LPS or pro-inflammatory cytokines are given systemically. Thus few prior studies have directly assessed if an inflammatory response confined to intestinal tissue itself is capable of activating the reflex. Our study, in which a stimulus confined to the intestinal tract was used, demonstrates that a non-systemic response is adequate to activate what is likely to be the afferent arm of the reflex.
It is interesting to note that the descending pathway in the reflex has both a systemic component mediated by an action within the spleen, and potentially a localized component within the intestine (Andersson and Tracey, 2012). Briefly, the best characterized aspect of the descending cholinergic anti-inflammatory pathway involves the release acetylcholine from vagal efferents in the celiac ganglia onto norepinephrine expressing neurons that project to the spleen (Rosas-Ballina et al., 2008) although this specific component has recently been questioned by others, (see Bratton et al., 2012). The release of norepinephrine from these neurons activates acetylcholine-synthesizing T cells in the spleen (Rosas-Ballina et al., 2011), which in turn release acetylcholine onto neighboring macrophages. Through activation of α7 nicotinic acetylcholine receptors on these resident macrophages (Wang et al., 2003), systemic release of cytokines is inhibited. Because acetylcholine has a short half-life, it is capable of only suppressing activity of macrophages in the near vicinity of its release, and thus this splenic release of acetylcholine would unlikely directly affect macrophage activity in other tissues. Thus this component would be most significant when an insult has reached the point that systemic release of pro-inflammatory cytokines has occurred, and the system would not be selective in targeting suppression to a specific location.
On the other hand, a direct release of acetylcholine by vagal efferents within intestinal tissue has also been hypothesized, both in the original conception of the immune reflex (Tracey, 2002) and in more current views (Andersson and Tracey, 2012). In this situation an activated macrophages within a specific location could be inhibited without inhibiting responses elsewhere, thus targeting the inflammatory control to a precise location. Such a response would have the advantage of maintaining local tissue integrity by control of a local inflammatory reaction, while at the same time not compromising an immune response that is needed to occur elsewhere. Such a system would require that when the immune reflex is activated within tissue, it activates a corresponding efferent pathway back into the same tissue area. However, it is possible that when the response has gone systemic, and the afferent arm has been activated at the level of vagal paraganglia, nodose cell bodies, or directly in the brain, the descending splenic component is recruited to protect the individual from an overzealous systemic response. This suggests that there might be multiple levels of activation of the reflex. A deeper understanding of both the afferent and efferent side of the reflex will be needed to test this idea.
4.6. Conclusions
The results from this study demonstrate that gastrointestinal bacterial colonization induces a central nervous system response conveyed by a subpopulation of TRPV1 expressing vagal afferents independent of increased circulating TNFα levels or increased c-Fos expression in the AP. Although the precise means by which these specific bacterial challenges lead to afferent neuron activation remains to be elucidated, we also found via single cell calcium measurements on isolated cells a direct activation of vagal afferent cell bodies by TNFα and LPS. These actions occur almost exclusively on capsaicin sensitive neurons, consistent with the first set of observations that capsaicin-induced lesions prevented the transmission of the presence of an intestinal insult to the brain. We conclude that vagal innervation of the GI tract is capable of detecting host-pathogen interactions in the intestine prior to the challenge producing a systemic response. These findings deepen our understanding of the specific pathways conveying neuroimmune information from the gut to the brain and suggest that additional exploration of this afferent arm may result in new therapeutic interventions that provide an additional avenue for treatment of inflammatory intestinal diseases.
Supplementary Material
Footnotes
Supplementary data to this article can be found online at http://dx.doi.org/10.1016/j.jneuroim.2013.01.009.
References
- Andersson U, Tracey KJ. Reflex principles of immunological homeostasis. Annu Rev Immunol. 2012;30:313–335. doi: 10.1146/annurev-immunol-020711-075015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Artis D. Epithelial-cell recognition of commensal bacteria and maintenance of immune homeostasis in the gut. Nat Rev Immunol. 2008;8:411–420. doi: 10.1038/nri2316. [DOI] [PubMed] [Google Scholar]
- Bell JA, St Charles JL, Murphy AJ, Rathinam VA, Plovanich-Jones AE, Stanley EL, Wolf JE, Gettings JR, Whittam TS, Manfield LS. Multiple factors interact to produce responses resembling spectrum of human disease in Campylobacter jejuni infected C57BL/6 IL-10−/− mice. BMC Microbiol. 2009;9:57. doi: 10.1186/1471-2180-9-57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berthoud HR, Patterson LM, Willing AE, Mueller K, Neuhuber WL. Capsaicin-resistant vagal afferent fibers in the rat gastrointestinal tract: anatomical identification and functional integrity. Brain Res. 1997;746:195–206. doi: 10.1016/s0006-8993(96)01222-x. [DOI] [PubMed] [Google Scholar]
- Bluthe RM, Michaud B, Kelley KW, Dantzer R. Vagotomy blocks behavioural effects of interleukin-1 injected via the intraperitoneal route but not via other systemic routes. Neuroreport. 1996;7:2823–2827. doi: 10.1097/00001756-199611040-00083. [DOI] [PubMed] [Google Scholar]
- Borovikova LV, Ivanova S, Zhang M, Yang H, Botchkina GI, Watkins LR, Wang H, Abumrad N, Eaton JW, Tracey KJ. Vagus nerve stimulation attenuates the systemic inflammatory response to endotoxin. Nature. 2000;405:458–462. doi: 10.1038/35013070. [DOI] [PubMed] [Google Scholar]
- Bratton BO, Martelli D, McKinley MJ, Trevaks D, Anderson CR, Mcallen RM. Neural regulation of inflammation: no neural connection from vagus to splenic sympathetic neurons. Exp Physiol. 2012;45:1080–1085. doi: 10.1113/expphysiol.2011.061531. [DOI] [PubMed] [Google Scholar]
- Bret-Dibat JL, Bluthe RM, Kent S, Kelley KW, Dantzer R. Lipopolysaccharide and interleukin-1 depress food-motivated behavior in mice by a vagal-mediated mechanism. Brain Behav Immun. 1995;9:242–246. doi: 10.1006/brbi.1995.1023. [DOI] [PubMed] [Google Scholar]
- Caterina MJ, Schumacher MA, Tominaga M, Rosen TA, Levine JD, Julius D. The capsaicin receptor: a heat-activated ion channel in the pain pathway. Nature. 1997;389:816–824. doi: 10.1038/39807. [DOI] [PubMed] [Google Scholar]
- Danese S, Sans M, Fiocchi C. Inflammatory bowel disease: the role of environmental factors. Autoimmun Rev. 2004;3:394–400. doi: 10.1016/j.autrev.2004.03.002. [DOI] [PubMed] [Google Scholar]
- De A, Krueger JM, Simasko SM. Tumor necrosis factor alpha increases cytosolic calcium responses to AMPA and KCl in primary cultures of rat hippocampal neurons. Brain Res. 2003;981:133–142. doi: 10.1016/s0006-8993(03)02997-4. [DOI] [PubMed] [Google Scholar]
- Diogenes A, Ferraz CC, Akopian AN, Henry MA, Hargreaves KM. LPS sensitizes TRPV1 via activation of TLR4 in trigeminal sensory neurons. J Dent Res. 2011;90:759–764. doi: 10.1177/0022034511400225. [DOI] [PubMed] [Google Scholar]
- Dorrell N, Wren BW. The second century of Campylobacter research: recent advances, new opportunities and old problems. Curr Opin Infect Dis. 2007;20:514–518. doi: 10.1097/QCO.0b013e3282a56b15. [DOI] [PubMed] [Google Scholar]
- Ek M, Kurosawa M, Lundeberg T, Ericsson A. Activation of vagal afferents after intravenous injection of interleukin-1beta: role of endogenous prostaglandins. J Neurosci. 1998;18:9471–9479. doi: 10.1523/JNEUROSCI.18-22-09471.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elmquist JK, Scammell TE, Jacobson CD, Saper CB. Distribution of Fos-like immunoreactivity in the rat brain following intravenous lipopolysaccharide administration. J Comp Neurol. 1996;371:85–103. doi: 10.1002/(SICI)1096-9861(19960715)371:1<85::AID-CNE5>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- Emch GS, Hermann GE, Rogers RC. TNF-alpha-induced c-Fos generation in the nucleus of the solitary tract is blocked by NBQX and MK-801. Am J Physiol Regul Integr Comp Physiol. 2001;281:R1394–R1400. doi: 10.1152/ajpregu.2001.281.5.R1394. [DOI] [PubMed] [Google Scholar]
- Fleshner M, Goehler LE, Hermann J, Relton JK, Maier SF, Watkins LR. Interleukin-1 beta induced corticosterone elevation and hypothalamic NE depletion is vagally mediated. Brain Res Bull. 1995;37:605–610. doi: 10.1016/0361-9230(95)00051-f. [DOI] [PubMed] [Google Scholar]
- Fleshner M, Silbert L, Deak T, Goehler LE, Martin D, Watkins LR, Maier SF. TNF-alpha-induced corticosterone elevation but not serum protein or corticosteroid binding globulin reduction is vagally mediated. Brain Res Bull. 1997;44:701–706. doi: 10.1016/s0361-9230(97)00258-x. [DOI] [PubMed] [Google Scholar]
- Fleshner M, Goehler LE, Schwartz BA, Mcgorry M, Martin D, Maier SF, Watkins LR. Thermogenic and corticosterone responses to intravenous cytokines (IL-1beta and TNF-alpha) are attenuated by subdiaphragmatic vagotomy. J Neuroimmunol. 1998;86:134–141. doi: 10.1016/s0165-5728(98)00026-5. [DOI] [PubMed] [Google Scholar]
- Gaykema RP, Goehler LE, Lyte M. Brain response to cecal infection with Campylobacter jejuni: analysis with Fos immunohistochemistry. Brain Behav Immun. 2004;18:238–245. doi: 10.1016/j.bbi.2003.08.002. [DOI] [PubMed] [Google Scholar]
- Goehler LE, Busch CR, Tartaglia N, Relton J, Sisk D, Maier SF, Watkins LR. Blockade of cytokine induced conditioned taste aversion by subdiaphragmatic vagotomy: further evidence for vagal mediation of immune-brain communication. Neurosci Lett. 1995;185:163–166. doi: 10.1016/0304-3940(95)11251-q. [DOI] [PubMed] [Google Scholar]
- Goehler LE, Relton JK, Dripps D, Kiechle R, Tartaglia N, Maier SF, Watkins LR. Vagal paraganglia bind biotinylated interleukin-1 receptor antagonist: a possible mechanism for immune-to-brain communication. Brain Res Bull. 1997;43:357–364. doi: 10.1016/s0361-9230(97)00020-8. [DOI] [PubMed] [Google Scholar]
- Goehler LE, Gaykema RP, Hammack SE, Maier SF, Watkins LR. Interleukin-1 induces c-Fos immunoreactivity in primary afferent neurons of the vagus nerve. Brain Res. 1998;804:306–310. doi: 10.1016/s0006-8993(98)00685-4. [DOI] [PubMed] [Google Scholar]
- Goehler LE, Gaykema RP, Opitz N, Reddaway R, Badr N, Lyte M. Activation in vagal afferents and central autonomic pathways: early responses to intestinal infection with Campylobacter jejuni. Brain Behav Immun. 2005;19:334–344. doi: 10.1016/j.bbi.2004.09.002. [DOI] [PubMed] [Google Scholar]
- Hansen MK, Krueger JM. Subdiaphragmatic vagotomy blocks the sleep- and fever-promoting effects of interleukin-1beta. Am J Physiol. 1997;273:R1246–R1253. doi: 10.1152/ajpregu.1997.273.4.R1246. [DOI] [PubMed] [Google Scholar]
- Hansen MK, O’Connor KA, Goehler LE, Watkins LR, Maier SF. The contribution of the vagus nerve in interleukin-1beta-induced fever is dependent on dose. Am J Physiol Regul Integr Comp Physiol. 2001;280:R929–R934. doi: 10.1152/ajpregu.2001.280.4.R929. [DOI] [PubMed] [Google Scholar]
- Hermann GE, Rogers RC. TNFalpha: a trigger of autonomic dysfunction. Neuroscientist. 2008;14:53–67. doi: 10.1177/1073858407305725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hermann GE, Emch GS, Tovar CA, Rogers RC. c-Fos generation in the dorsal vagal complex after systemic endotoxin is not dependent on the vagus nerve. Am J Physiol Regul Integr Comp Physiol. 2001;280:R289–R299. doi: 10.1152/ajpregu.2001.280.1.R289. [DOI] [PubMed] [Google Scholar]
- Holzer P. Capsaicin as a tool for studying sensory neuron functions. Adv Exp Med Biol. 1991;298:3–16. doi: 10.1007/978-1-4899-0744-8_1. [DOI] [PubMed] [Google Scholar]
- Hooper LV, Macpherson AJ. Immune adaptations that maintain homeostasis with the intestinal microbiota. Nat Rev Immunol. 2010;10:159–169. doi: 10.1038/nri2710. [DOI] [PubMed] [Google Scholar]
- Hosoi T, Okuma Y, Matsuda T, Nomura Y. Novel pathway for LPS-induced afferent vagus nerve activation: possible role of nodose ganglion. Auton Neurosci. 2005;120:104–107. doi: 10.1016/j.autneu.2004.11.012. [DOI] [PubMed] [Google Scholar]
- Hulisz D. The burden of illness of irritable bowel syndrome: current challenges and hope for the future. J Manag Care Pharm. 2004;10:299–309. doi: 10.18553/jmcp.2004.10.4.299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jesudason MV, Hentges DJ, Pongpech P. Colonization of mice by Campylobacter jejuni. Infect Immun. 1989;57:2279–2282. doi: 10.1128/iai.57.8.2279-2282.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jotwani R, Tanaka Y, Watanabe K, Tanaka K, Kato N, Ueno K. Cytokine stimulation during Salmonella typhimurium sepsis in Itys mice. J Med Microbiol. 1995;42:348–352. doi: 10.1099/00222615-42-5-348. [DOI] [PubMed] [Google Scholar]
- Koller H, Thiem K, Siebler M. Tumour necrosis factor-alpha increases intracellular Ca2+ and induces a depolarization in cultured astroglial cells. Brain. 1996;119 (Pt 6):2021–2027. doi: 10.1093/brain/119.6.2021. [DOI] [PubMed] [Google Scholar]
- Konsman JP, Kelley K, Dantzer R. Temporal and spatial relationships between lipopolysaccharide-induced expression of Fos, interleukin-1beta and inducible nitric oxide synthase in rat brain. Neuroscience. 1999;89:535–548. doi: 10.1016/s0306-4522(98)00368-6. [DOI] [PubMed] [Google Scholar]
- Li BY, Schild JH. Electrophysiological and pharmacological validation of vagal afferent fiber type of neurons enzymatically isolated from rat nodose ganglia. J Neurosci Methods. 2007;164:75–85. doi: 10.1016/j.jneumeth.2007.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu CY, Mueller MH, Grundy D, Kreis ME. Vagal modulation of intestinal afferent sensitivity to systemic LPS in the rat. Am J Physiol Gastrointest Liver Physiol. 2007;292:G1213–G1220. doi: 10.1152/ajpgi.00267.2006. [DOI] [PubMed] [Google Scholar]
- Maloy KJ, Powrie F. Intestinal homeostasis and its breakdown in inflammatory bowel disease. Nature. 2011;474:298–306. doi: 10.1038/nature10208. [DOI] [PubMed] [Google Scholar]
- Mascarucci P, Perego C, Terrazzino S, De Simoni MG. Glutamate release in the nucleus tractus solitarius induced by peripheral lipopolysaccharide and interleukin-1 beta. Neuroscience. 1998;86:1285–1290. doi: 10.1016/s0306-4522(98)00105-5. [DOI] [PubMed] [Google Scholar]
- Mueller MH, Kampitoglou D, Glatzle J, Hahn J, Kreis ME. Systemic capsaicin inhibits neuronal activation in the brainstem during postoperative ileus in the mouse. Langenbecks Arch Surg. 2006;391:88–95. doi: 10.1007/s00423-006-0042-8. [DOI] [PubMed] [Google Scholar]
- Nevola JJ, Stocker BA, Laux DC, Cohen PS. Colonization of the mouse intestine by an avirulent Salmonella typhimurium strain and its lipopolysaccharide-defective mutants. Infect Immun. 1985;50:152–159. doi: 10.1128/iai.50.1.152-159.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niijima A. The afferent discharges from sensors for interleukin 1 beta in the hepatoportal system in the anesthetized rat. J Auton Nerv Syst. 1996;61:287–291. doi: 10.1016/s0165-1838(96)00098-7. [DOI] [PubMed] [Google Scholar]
- Pollock J, McFarlane SM, Connell MC, Zehavi U, Vandenabeele P, MacEwan DJ, Scott RH. TNF-alpha receptors simultaneously activate Ca2+ mobilisation and stress kinases in cultured sensory neurones. Neuropharmacology. 2002;42:93–106. doi: 10.1016/s0028-3908(01)00163-0. [DOI] [PubMed] [Google Scholar]
- Rogers RC, Van Meter MJ, Hermann GE. Tumor necrosis factor potentiates central vagal afferent signaling by modulating ryanodine channels. J Neurosci. 2006;26:12642–12646. doi: 10.1523/JNEUROSCI.3530-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosas-Ballina M, Ochani M, Parrish WR, Ochani K, Harris YT, Huston JM, Chavan S, Tracey KJ. Splenic nerve is required for cholinergic antiinflammatory pathway control of TNF in endotoxemia. Proc Natl Acad Sci U S A. 2008;105:11008–11013. doi: 10.1073/pnas.0803237105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosas-Ballina M, Olofsson PS, Ochani M, Valdes-Ferrer SI, Levine YA, Reardon C, Tusche MW, Pavlov VA, Andersson U, Chavan S, Mak TW, Tracey KJ. Acetylcholine-synthesizing T cells relay neural signals in a vagus nerve circuit. Science. 2011;334:98–101. doi: 10.1126/science.1209985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sagar SM, Price KJ, Kasting NW, Sharp FR. Anatomic patterns of Fos immunostaining in rat brain following systemic endotoxin administration. Brain Res Bull. 1995;36:381–392. doi: 10.1016/0361-9230(94)00217-o. [DOI] [PubMed] [Google Scholar]
- Simasko SM, Wiens J, Karpiel A, Covasa M, Ritter RC. Cholecystokinin increases cytosolic calcium in a subpopulation of cultured vagal afferent neurons. Am J Physiol Regul Integr Comp Physiol. 2002;283:R1303–R1313. doi: 10.1152/ajpregu.00050.2002. [DOI] [PubMed] [Google Scholar]
- Tracey KJ. The inflammatory reflex. Nature. 2002;420:853–859. doi: 10.1038/nature01321. [DOI] [PubMed] [Google Scholar]
- Tracey KJ. Physiology and immunology of the cholinergic antiinflammatory pathway. J Clin Invest. 2007;117:289–296. doi: 10.1172/JCI30555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tracey KJ. Reflex control of immunity. Nat Rev Immunol. 2009;9:418–428. doi: 10.1038/nri2566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Der Zanden EP, Snoek SA, Heinsbroek SE, Stanisor OI, Verseijden C, Boeckxstaens GE, Peppelenbosch MP, Greaves DR, Gordon S, De Jonge WJ. Vagus nerve activity augments intestinal macrophage phagocytosis via nicotinic acetylcholine receptor alpha4beta2. Gastroenterology. 2009;137:1029–1039. 1039 e1–4. doi: 10.1053/j.gastro.2009.04.057. [DOI] [PubMed] [Google Scholar]
- Vuckovic D, Abram M, Doric M. Primary Campylobacter jejuni infection in different mice strains. Microb Pathog. 1998;24:263–268. doi: 10.1006/mpat.1997.0194. [DOI] [PubMed] [Google Scholar]
- Wang X, Wang BR, Zhang XJ, Xu Z, Ding YQ, Ju G. Evidences for vagus nerve in maintenance of immune balance and transmission of immune information from gut to brain in STM-infected rats. World J Gastroenterol. 2002;8:540–545. doi: 10.3748/wjg.v8.i3.540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Yu M, Ochani M, Amella CA, Tanovic M, Susarla S, Li JH, Wang H, Yang H, Ulloa L, Al-Abed Y, Czura CJ, Tracey KJ. Nicotinic acetylcholine receptor alpha7 subunit is an essential regulator of inflammation. Nature. 2003;421:384–388. doi: 10.1038/nature01339. [DOI] [PubMed] [Google Scholar]
- Watkins LR, Goehler LE, Relton JK, Tartaglia N, Silbert L, Martin D, Maier SF. Blockade of interleukin-1 induced hyperthermia by subdiaphragmatic vagotomy: evidence for vagal mediation of immune-brain communication. Neurosci Lett. 1995;183:27–31. doi: 10.1016/0304-3940(94)11105-r. [DOI] [PubMed] [Google Scholar]
- Young VB, Schauer DB, Fox JG. Animal models of Campylobacter infection. In: Blaser MJ, editor. Campylobacter. 2. ASM Press; Washington, D.C: 2000. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.