

Abstract
Prospective data on the value of allogeneic hematopoietic stem cell transplantation (alloHSCT) in Philadelphia chromosome–positive (Ph+) acute lymphoblastic leukemia (ALL) are limited. The UKALLXII/ECOG 2993 study evaluated the outcome of assigning alloHSCT with a sibling (sib) or matched unrelated donor (MUD) to patients younger than 55 years of age achieving complete remission (CR). The CR rate of 267 patients, median age 40, was 82%. Twenty-eight percent of patients proceeded to alloHSCT in first CR. Age older than 55 years or a pre-HSCT event were the most common reasons for failure to progress to alloHSCT. At 5 years, overall survival (OS) was 44% after sib alloHSCT, 36% after MUD alloHSCT, and 19% after chemotherapy. After adjustment for sex, age, and white blood count and excluding chemotherapy-treated patients who relapsed or died before the median time to alloHSCT, only relapse-free survival remained significantly superior in the alloHSCT group (odds ratio 0.31, 95% confidence interval 0.16-0.61). An intention-to-treat analysis, using the availability or not of a matched sibling donor, showed 5-year OS to be nonsignificantly better at 34% with a donor versus 25% with no donor. This prospective trial in adult Ph+ ALL indicates a modest but significant benefit to alloHSCT. This trial has been registered with clinicaltrials.gov under identifier NCT00002514 and as ISRCTN77346223.
Introduction
Patients with acute lymphoblastic leukemia (ALL) in whom the Philadelphia chromosome is detected (Ph+)1 constitute the largest molecularly defined subgroup, approximately 25%, of adults with ALL.2 The poor prognostic relevance of the Ph+ ALL is well established.3–8 It has long been concluded that the outcome with standard ALL chemotherapy alone is sufficiently poor to recommend a sibling hematopoietic stem cell transplantation (sib allo HSCT) in first complete remission (CR1). A recent retrospective case series from the City of Hope National Medical Center/Stanford University,9 the largest study of allogeneic HSCT to date in Ph+ ALL, details the outcome of 79 patients (10 of whom were children) treated with alloHSCT in CR1 or CR2 and demonstrates that survival of 54% can be achieved in patients in CR1. Such a study, however, is unlikely to be generally representative of all patients with Ph+ ALL and may overestimate the potential benefits of alloHSCT, due to selection bias.
The international adult ALL trial UKALLXII/ECOG E2993, initiated in 1993, was designed to evaluate the overall hypothesis that alloHSCT was the optimal therapy for adults with ALL. Patients with Ph+ ALL were assigned to a “high-risk” arm in which all eligible patients who achieved CR after standard induction were allocated to sib alloHSCT (if a sibling donor was available) or to matched unrelated donor (MUD) HSCT. Patients without a donor or those unsuitable for alloHSCT were eligible for the autologous HSCT versus chemotherapy randomization. In practice, very few patients were randomized, and the majority of patients not receiving alloHSCT were treated with chemotherapy alone. The present analysis reports on the results of 267 patients in the Ph+ arm of the study diagnosed between 1993 and 2004, before the general implementation of imatinib therapy in the treatment of this disease. For more recent patients on this study, a protocol modification introduced imatinib into intensification and then later into induction, but these patients are not reported here. This report documents the largest prospective study of patients with Ph+ ALL and is also the largest study of alloHSCT in this disease. The dataset defines the limits of application and the outcome of HSCT in a real-world, multicenter patient population with Ph+ ALL in the pre-imatinib era and provides an important baseline comparator against which the addition of newer and more targeted therapies can be evaluated.
Methods
Study eligibility
This trial was jointly conducted by the Medical Research Council (MRC) of the United Kingdom and the Eastern Cooperative Oncology Group (ECOG) of the United States. Eligible patients were aged 15 to 60 years (ECOG) or 55 years (MRC) with newly diagnosed, untreated ALL and no prior malignancy. The Ethics Committee or Institutional Review Board of each participating center approved the study. Informed consent was obtained from all subjects in accordance with the Declaration of Helsinki. There were no exclusion criteria for abnormal renal or hepatic function or poor performance status at diagnosis. All comers with a confirmed diagnosis were eligible for inclusion.
Diagnosis
Diagnosis of ALL was established by documenting more than 25% marrow lymphoblasts. Confirmation of the diagnosis of ALL by morphology or immunophenotyping (ECOG only) central review was required in both the United Kingdom and the United States. The Philadelphia chromosome [t(9;22)(q34;q11.2)] or BCR-ABL fusion was detected by conventional cytogenetics, fluorescence in situ hybridization (FISH), reverse transcription–polymerase chain reaction (RT-PCR), or a combination in all cases and was confirmed centrally in both the United Kingdom and the United States, as previously described.2
Treatment
Induction phases 1 and 2 were administered as described by Goldstone et al.10 Those patients deemed to be in hematologic CR after completion of phases 1 and 2 who were aged 50 years or older, lacked a suitable allogeneic donor, or had contraindications to allogeneic transplantation were eligible for the randomization between autologous HSCT and continuing chemotherapy. Patients who were in remission then received intensification as previously described.10 After intensification, those aged younger than 50 years (United States) or 55 years (United Kingdom) with a sibling donor proceeded immediately to either an HLA-identical sib alloHSCT, or MUD HSCT if no sibling donor was available. Patients awaiting transplantation continued chemotherapy as per protocol. Patients randomized to autograft were also to receive their transplant at this stage. In practice, very few patients received autograft (n = 7). In the early years of the protocol, bone marrow was used as a source of stem cells. Latterly, mobilized peripheral blood stem cells were recommended. Mobilization of stem cells was achieved after administering mitoxantrone 30 mg/m2 on days 1 and 2 of week 15, combined with Ara-C 2 g/m2 on days 1 through 3 and rhuG-CSF daily.
The conditioning regimens for transplantation consisted of fractionated total body irradiation (TBI) 1320 cGy in 6 fractions twice daily on days −6 and −4, along with 400 cGy testicular boost in males and high-dose etoposide 60 mg/kg intravenously on day −3 originally proposed by Blume et al.11 T-cell depletion was not recommended, but the final decision was left to the transplant center. Recommended graft-versus-host disease prophylaxis was standard cyclosporine and short-course methotrexate. Response was measured at day 21 (although in many cases the day 21 result was indeterminate due to hypocellular marrow) and at the end of phases 1 and 2 of induction, at approximately days 28 and 56 after the start of induction therapy. Hematologic remission was defined as less than 5% lymphoblasts. The date of remission is taken as the date on which the patient's doctor recorded remission to the data reporting center.
Statistical analysis
All patients were centrally registered by telephone at the Clinical Trial Service Unit (CTSU) in Oxford, United Kingdom, for MRC patients or at the ECOG operations office for ECOG patients. All outcomes were measured from the start of treatment. The primary outcome measure was overall survival (OS). Other outcomes analyzed were event-free survival (EFS), defined as the time to relapse or death; relapse-free survival (RFS), defined as time to relapse, excluding patients who never entered remission and censoring at death in remission; and death in remission, excluding nonremitters and censoring at relapse. Patients who did not relapse or die within the follow-up period were censored at the earlier of (1) the date of last contact or (2) October 31, 2007.
χ2 tests were used for comparing groups by initial characteristics. The Mann-Whitney U test was used to test for differences between remitters and nonremitters for continuous variables and between treatment groups. Kaplan-Meier curves were used, and univariate comparisons were made by the log-rank method. Odds ratios (ORs) were calculated and are given with their 95% confidence intervals (CIs). Unless otherwise indicated, an OR of less than 1 indicates a worse prognosis in the second group compared with the first group.
An intention-to-treat analysis on related donor transplantation was planned for the entire trial. For this, information on whether tissue typing was being carried out and, if so, whether a sibling donor was found was collected prospectively to obtain an unbiased estimate of the effect of aiming to carry out a sibling donor transplantation on all those with a suitable donor. For the Ph+ group, the number of patients who had an unrelated donor HSCT considerably weakens this comparison, so these analyses were repeated, censoring at unrelated donor transplantation.
Multivariate Cox regression analysis was carried out using stepwise regression, adding variables of age, white blood count (WBC), sex, BCR-ABL breakpoint, and cytogenetics to the model one at a time in order of most to least significant (as long as their P value was < .05). Age and WBC were treated as continuous variables. Due to the high number of missing cases for central nervous system (CNS) disease at entry, this was not included. Analyses were carried out on the subsets of patients with data for age, WBC, and sex (n = 265 for OS and EFS; n = 218 for RFS and death in remission); age, WBC, sex, and CNS disease at entry (n = 185 for OS and EFS; n = 151 for RFS and death in remission); age, WBC, sex, and BCR-ABL breakpoint (n = 140 for OS and EFS; n = 109 for RFS and death in remission); and age, WBC, sex, and cytogenetics (n = 220 for OS and EFS; n = 183 for RFS and death in remission).
Results
Patients
Recruitment ran from January 1993 to May 2004. Only patients registered onto the original protocol before an amendment introduced imatinib to the therapy are included in this analysis. Of a total of 1533 patients registered, 268 were registered as Ph+. One patient was later excluded because of misdiagnosis. Thus, a total of 267 patients are included in this report. The median follow-up in the 51 survivors is 8 years 2 months (range, 3 years 2 months to 14 years 3 months). Five patients were lost to follow-up, 3 after relapse, and 2 cases (lost at 1 day and at 2 months) are known to have later died. Four patients are being followed up for survival only, with no information available on disease status, from 2 months, 3 months (n = 2), and 15 months. Patient characteristics are summarized in Table 1.
Table 1.
Patient characteristics at diagnosis
| Characteristic | Value |
|---|---|
| Male sex, n (%) | 150 (56) |
| Presenting WBC × 109/L (2 unknown) | |
| < 30, n (%) | 140 (52) |
| ≥ 30, n (%) | 125 (47) |
| Median (range) | 26.8 (1.5-438) |
| Immunophenotype (8 unknown), n (%) | |
| B lineage (8 null) | 247 (95) |
| T cell | 1 (< 1) |
| Mature B | 2 (1) |
| Aberrant myeloid markers | 6 (2) |
| Other | 3 (1) |
| Age, y | |
| < 20, n (%) | 12 (4) |
| 20-29, n (%) | 53 (20) |
| 30-39, n (%) | 68 (25) |
| 40-49, n (%) | 88 (33) |
| ≥ 50, n (%) | 46 (17) |
| Median (range) | 40 (15-60) |
| CNS disease (82 unknown), n (%) | 14 (8) |
| Ph/BCR-ABL detection method,* n (%) | |
| Cytogenetics | 209 (78) |
| FISH | 76 (28) |
| RT-PCR | 218 (82) |
| BCR breakpoint (124 unknown), n (%) | |
| Major | 42 (29) |
| Minor | 100 (70) |
| Both | 1 (< 1) |
| Additional cytogenetic abnormalities (46 unknown), n (%) | |
| Extra Ph chromosome [+der(22)] | 49 (22) |
| −7 | 31 (14) |
| +8 | 24 (11) |
| del(9p) | 24 (11) |
| High hyperdiploidy (HeH) | 28 (13) |
| t(4;11), t(1;19), t(8;14), or low hypodiploidy/near tridiploidy | 0 |
N = 267 patients.
One hundred fifteen cases were positive by cytogenetics and RT-PCR, 47 by RT-PCR alone, 45 by all 3 methods, 29 by cytogenetics alone, 17 by cytogenetics and FISH, 12 by RT-PCR and FISH, and 2 by FISH alone.
Response to induction therapy
Of 267 patients, 262 were fully assessable for remission. The 5 in whom the date of remission was not known are assumed to have achieved remission at some point: 2 relapsed, 1 died at 13 months, and the other 2 are still alive at 6 and 7 years. Figure 1 is a flow chart that details the outcomes of all patients in the study. Of262 patients, 176 (67%) achieved remission after protocol induction therapy phases 1 and 2. This is in contrast with 87% for those patients with Ph− disease.12 A further 39 remitted with additional (non-protocol) therapy; 19 stopped induction early and 20 had not remitted by the end of phase 2; remission followed transplantation in 9 cases. Together with the 5 who remitted at an unknown time, this gives an overall remission rate of 82%, compared with 93% for those with Ph− disease.
Figure 1.
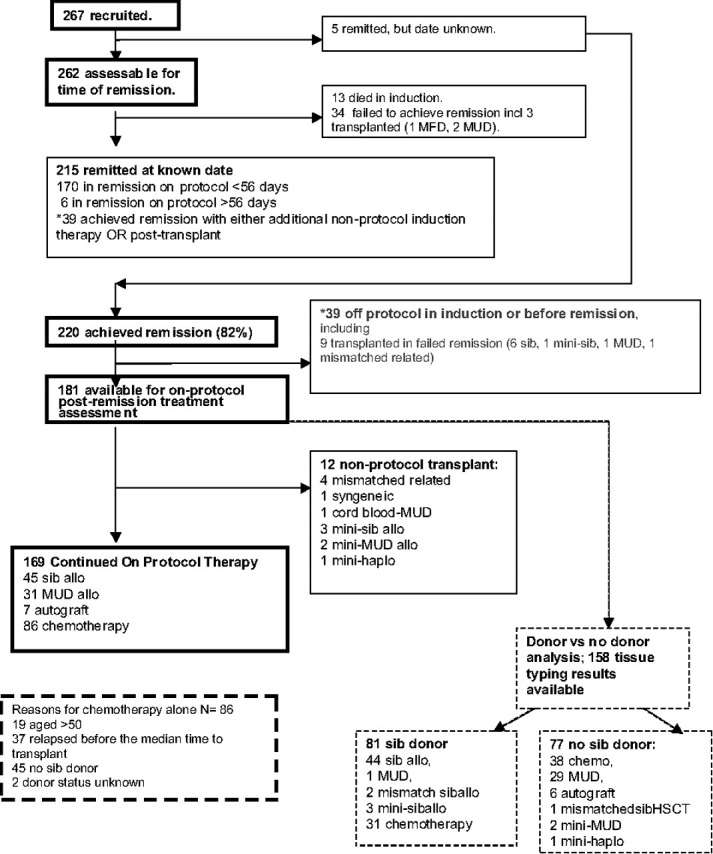
Flow chart showing all patients and treatments received.
There were 13 deaths during induction (5%). Nine patients died of infection alone, 1 of infection plus bleeding, 1 of infection plus necrotizing enterocolitis, and 1 with acute respiratory syndrome plus myocardial infarction. There was one suicide. Of the 34 other patients who did not achieve remission at any point, including 3 patients who received an allogeneic transplant (2 MUD, 1 sibling donor), all died, at a median of 7 months (range, 2-20 months).
Factors predictive for achievement of CR at any point were age (88% CR rate in those aged under 30, 85% in age 30-49, falling to 67% in those aged ≥ 50 years; P = .03) and presenting WBC (89% CR rate in WBC < 30 × 109/L vs 75% CR rate in WBC ≥ 30 × 109/L, P = .005). There were no statistically significant differences in overall CR rate by sex, CNS disease at diagnosis, or additional cytogenetic abnormalities.
Outcome by postremission therapy
Survival.
The median EFS in all 267 Ph+ patients was 9 months, and the median OS was 13 months. Estimated EFS at 5 years and 10 years was 17% (95% CI = 13%-22%) and 15.5% (95% CI = 11%-20%), respectively. Estimated OS at 5 and 10 years was 22% (95% CI = 17%-27%) and 18% (95% CI = 13%-23%), respectively.
Of the 220 patients who achieved remission at some point, 181 remained on protocol and were eligible for analysis of postremission therapy (as shown in flow chart in Figure 1). Twelve patients who received nonprotocol transplants were excluded from the analysis, leaving 76 patients who received a per-protocol myeloablative alloHSCT in first remission (45 with cells from a sibling and 31 with cells from a MUD). Sibling donor alloHSCT was carried out significantly earlier during therapy (median 153 days, range 79-284 days from start of therapy) than unrelated donor alloHSCT (median 191 days, range 113-276 days, P < .001). A total of 86 patients received chemotherapy alone. The reasons for these patients not receiving a transplant are shown in the flow chart in Figure 1. Among those receiving transplant, there was no statistically significant difference between patients receiving MUD and sib alloHSCT in terms of age, sex, WBC, CNS disease at diagnosis, or additional cytogenetic abnormalities. Only 7 patients were treated with autologous transplantation, too few to carry out meaningful analysis.
Comparison of chemotherapy and alloHSCT.
For those who achieved remission on protocol, the outcome of patients who received transplant was compared with the outcome of those who received chemotherapy. Only 82 of the 86 chemotherapy-treated patients who remained in remission at 12 weeks, which was the scheduled time for transplantation, were included in the comparative analysis. Table 2 shows the outcomes, OS, RFS, EFS, and survival free from death in remission by treatment received, and Figure 2 illustrates OS by treatment received. At 5 years, OS was 44% for sib alloHSCT, 36% for MUD alloHSCT, and 19% for chemotherapy. At 10 years, OS was 39% for sib alloHSCT, 31% for MUD alloHSCT, and 13% for chemotherapy.
Table 2.
Outcome at 5 years by treatment received
| Outcome measure | Sib alloHSCT (n = 45) |
MUD alloHSCT (n = 31) |
Chemo (n = 82) |
Auto (n = 7) |
||||
|---|---|---|---|---|---|---|---|---|
| Events, n | Event-free at 5 y, % (95% CI) | Events, n | Event-free at 5 y, % (95% CI) | Events, n | Event-free at 5 y, % (95% CI) | Events, n | Event-free at 5 y, % (95% CI) | |
| Survival | 27 | 44 (29-59) | 21 | 36 (19-52) | 69 | 19 (10-28) | 6 | 29 (0-62) |
| Event-free survival | 27 | 41 (27-56) | 21 | 36 (19-52) | 74 | 9 (3-15) | 6 | 29 (0-62) |
| Relapse-free survival* | 15 | 57 (40-73) | 9 | 66 (48-85) | 68 | 10 (3-18) | 4 | 44 (1-88) |
| Survival free from death in remission* | 12 | 73 (59-87) | 12 | 53 (33-74) | 6 | 83 (33-74) | 2 | 64 (23-100) |
Excludes patients who did not achieve remission on protocol and chemotherapy-treated patients in whom treatment failed within 12 weeks.
Actuarial survivals. For relapse, a relapse is counted as an event and times are censored at death in remission; for death in remission, relapses are censored and death in remission is counted as an event.
Figure 2.
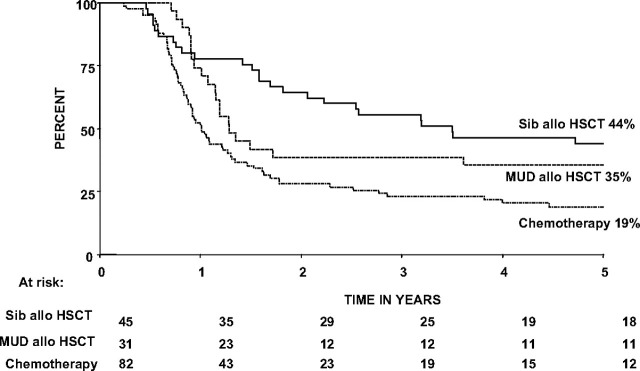
Kaplan-Meier plot of overall survival by treatment received. Patients who failed to achieve remission on protocol and chemotherapy-treated patients who relapsed or died within 12 weeks of the start of treatment (the scheduled time for transplantation) are excluded from the analysis.
The difference in outcome between sib alloHSCT and MUD alloHSCT groups was not statistically significant, but this may be due to the small numbers involved in the analysis. Sib alloHSCT had a nonsignificant 9% higher actuarial relapse risk at 5 years than MUD alloHSCT, but MUD alloHSCT had a nonsignificant 20% higher actuarial risk of death in remission at 5 years. Comparing the outcome after any alloHSCT with the outcome after chemotherapy alone, OS (P = .001), EFS (P < .001), and RFS (P < .001) were all significantly superior for patients receiving any alloHSCT over those receiving chemotherapy alone. There was a marked difference in the cause of death between alloHSCT and chemotherapy recipients. Whereas the leading cause of death in chemotherapy-treated patients was relapse, the leading cause of death after transplantation was treatment related mortality (TRM), which was 27% after sib HSCT and 39% after MUD HSCT.
As expected, the groups receiving alloHSCT and chemotherapy differed significantly from each other in age (Mann-Whitney U test; P = .004) with a preponderance of older patients among the patients treated with chemotherapy. Of patients who received alloHSCT, 95% were younger than 50 years of age (72/76) compared with 77% (63/82) of the chemotherapy patients. This clear selection bias is explained by both the upper age limit for HSCT years and the relative increase in contraindications to HSCT in older patients. There was also a significant difference in presenting WBC between the groups, with those receiving alloHSCT tending to have lower presenting WBC than those receiving chemotherapy (Mann-Whitney U test; P = .007). There is no obvious explanation for this difference, but it might reflect the fact that patients with lower presenting WBC were more likely to enter CR and progress to alloHSCT, or the fact that patients with high presenting WBC were more likely to relapse before HSCT or that an association between WBC and some other, unknown, factor exists. There was no significant difference between the groups by sex, CNS disease at presentation, or additional cytogenetic abnormalities.
Although chemotherapy-treated patients who relapsed or died before the 12-week scheduled transplantation date were excluded from the previous analysis, an additional 32 chemotherapy patients relapsed or died before the median time to alloHSCT (day 160), whereas none relapsed within this time frame in the HSCT arm. To take as many known differences between the groups into account as possible, the data were reanalyzed, adjusting for sex, age, and WBC and excluding chemotherapy-treated patients who relapsed or died before the median time to alloHSCT. RFS remained significantly superior in the HSCT group compared with the chemotherapy group (unadjusted OR 0.17 [0.11-0.26], adjusted OR 0.31 [0.16-0.61]). After adjustment, however, the difference in TRM between those receiving alloHSCT and those receiving chemotherapy alone reached significance (unadjusted OR 1.54 [0.69-3.45], adjusted OR 6.22 [1.98-19.56]). As a consequence, there was no longer a significant difference in OS (unadjusted OR 0.48 [0.33-0.69], adjusted OR 1.31 [0.73-2.35]) or EFS (unadjusted OR 0.27 [0.19-0.40], adjusted OR 0.67 [0.37-1.19]) between the 2 groups.
Because analyses by treatment received are potentially biased, we also carried out an intention-to-treat analysis using the availability (or not) of a matched related sibling donor. Information on sibling donor availability was available for 158 patients (Figure 1 flow chart). Figure 3 shows the Kaplan-Meier survival analysis of sib donor versus no sib donor. OS was nonsignificantly better (OR 0.80 [95% CI, 0.55-1.15], P = .2) for the group with a sibling donor. At 5 years, OS was 34% (95% CI, 24%-45%) with a sibling donor and 25% (95% CI, 15%-34%) without a sibling donor. At 10 years, OS was 30% (95% CI, 19%-40%) and 19.5% (95% CI, 10%-29%) for these groups.
Figure 3.
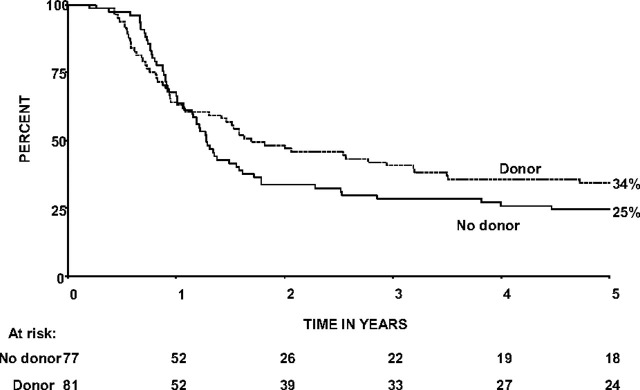
Kaplan-Meier plot of overall survival by availability of sibling donor among those in whom tissue typing was carried out and reported.
As a substantial proportion of patients in the no sibling donor group received a MUD or mismatched alloHSCT (30 full-intensity and 3 nonmyeloablative), this analysis was repeated but censoring at MUD/mismatched and nonmyeloablative alloHSCT. This slightly increased the difference between the groups, with an OR of 0.74 (0.50-1.11), 5-year survival at 36% (95% CI, 25%-46%) for the sibling donor group and 23% (95% CI, 12%-34%) for the no donor group. Although not statistically significant, these results suggest that the apparent superiority of sib alloHSCT over chemotherapy partly truly reflects an inherent, though moderate, superiority of alloHSCT and is not entirely due to selection bias.
Prognostic factors for outcome
Effect of graft-versus-host disease.
To assess the contribution of a graft-versus-leukemia (GVL) effect to the superior EFS and RFS seen after HSCT, we analyzed the relationship between graft-versus-host disease (GVHD) and outcome in patients who achieved remission on protocol. Acute GVHD of any grade was reported in 29 of the 45 sib HSCTs (6 patients had grade 3/4 GVHD) and 15 of the 31 MUD HSCTs (2 patients had grade 3/4 GVHD). The occurrence of GVHD was unknown in 7 sib and 5 MUD HSCTs. We examined relapses and deaths in remission by the presence or absence of GVHD. The data are shown in Table 3. There were significantly more relapses at 5 years from HSCT in the absence of GVHD (65%; 95% CI, 43%-87%), than in the presence of any grade of acute GVHD, (32%; 95% CI 15%-49%; log rank P = .01). There was also an apparent increase in deaths in remission (14%; 95% CI 0%-32%) at 5 years in the absence of GVHD vs 43%; 95% CI 27%-59%) in the presence of GVHD, but this was not statistically significant (P = .2). There was no significant difference in EFS.
Table 3.
Relationship between acute graft-versus-host disease and outcome in patients who achieved remission on protocol and went on to receive an HSCT in first remission
| GVHD |
OR* (95% CI), log rank P | ||
|---|---|---|---|
| Yes | No | ||
| No. of patients | 44 | 20 | |
| Relapses | 10 | 12 | |
| Deaths in remission (TRM) | 17 | 3 | |
| Any event | 27 | 15 | |
| Actuarial risk of relapse at 5 y (95% CI) | 32% (15%-49%) | 65% (43%-87%) | 3.63 (1.44-9.18), .01† |
| Actuarial risk of TRM at 5 y (95% CI) | 43% (27%-59%) | 14% (0%-32%) | 0.51 (0.19-1.34), > 0.1 |
| Actuarial risk of any event at 5 y (95% CI) | 62% (47%-76%) | 70% (50%-90%) | 1.42 (0.73-2.78), > 0.1 |
OR indicates odds ratio; and CI, confidence interval.
OR greater than 1 indicates an increased risk of the observed event in the absence of GVHD.
Statistically significant P values.
Impact of additional cytogenetic abnormalities and BCR breakpoint.
The impact of additional chromosome abnormalities and BCR breakpoint on OS, EFS, RFS, and death in remission were examined by univariate analysis (Table 4). Both an extra Ph chromosome [+der (22)] and high hyperdiploidy (HeH) were associated with a significantly better RFS. It should be noted that 25 of 28 (89%) cases with HeH also had +der(22). The RFS results were broadly consistent with the effects seen on OS and EFS, but these did not reach statistical significance. BCR breakpoint was a marginally significant prognostic factor for RFS and del(9p) had worse RFS, but this was not reflected in OS.
Table 4.
Relationship between additional chromosome abnormalities, BCR breakpoint, and outcome
| Death |
Any event |
Relapse |
Death in CR |
|||||
|---|---|---|---|---|---|---|---|---|
| OR (95% CI) | P | OR (95% CI) | P | OR (95% CI) | P | OR (95% CI) | P | |
| BCR breakpoint, minor vs major | 1.22 (0.82-1.83) | > .1 | 1.31 (0.88-1.96) | > .1 | 1.78 (1.03-3.08) | .05* | 0.77 (0.31-1.94) | > .1 |
| Extra Ph chromosome [+der(22)] | 0.82 (0.58-1.16) | > .1 | 0.77 (0.55-1.07) | > .1 | 0.64 (0.42-0.99) | .04* | 1.38 (0.70-2.72) | > .1 |
| HeH | 0.72 (0.47-1.08) | > .1 | 0.69 (0.46-1.03) | .06 | 0.55 (0.33-0.91) | .01* | 1.29 (0.58-2.85) | > .1 |
| −7 | 1.11 (0.72-1.70) | > .1 | 1.10 (0.72-1.66) | > .1 | 1.06 (0.60-1.89) | > .1 | 1.37 (0.56-3.38) | > .1 |
| +8 | 0.72 (0.46-1.12) | > .1 | 0.72 (0.47-1.11) | > .1 | 0.76 (0.44-1.34) | > .1 | 0.69 (0.29-1.67) | > .1 |
| del(9p) | 0.92 (0.58-1.45) | > .1 | 1.40 (0.84-2.34) | > .1 | 2.45 (1.27-4.72) | .01* | 0.59 (0.21-1.71) | > .1 |
An OR greater than 1 indicates an increased risk of the event observed in relationship to the presence of the particular chromosome abnormality studied. In the case of major BCR breakpoint, the comparator is minor breakpoint.
OR indicates odds ratio; and CI, confidence interval.
Statistically significant P values.
Other factors affecting outcome.
Presenting WBC was significant for all endpoints, with a worse prognosis with increasing WBC (P < .001 for OS, EFS, and RFS; P = .05 for death in remission) in this group of 267 Ph+ patients. Increasing age corresponded to increasingly worse prognosis for OS (P = .005) and death in remission (P = .03) but was only marginally significant for EFS (P = .06) and was not significant for RFS (P > .1). Sex and CNS disease at presentation were not found to be statistically significant. Data about the source of stem cells for transplantation were not routinely collected, but the majority of transplants in the first half of the study would have been bone marrow, whereas peripheral blood was more common in the second half of the trial. Analysis of outcome by era of transplantation (first half vs second half of study) did not show any statistically significant differences.
Multivariate analysis
By multivariate analysis, age and presenting WBC were prognostic factors for several outcome measures as shown in Table 5. As most HeH cases also had +der(22), only the latter was considered in the multivariate model. The existence of +der(22) remained prognostic for better RFS, whereas del(9p) was still significant for a poorer RFS. BCR breakpoint and CNS disease at diagnosis were not statistically significant on multivariate analysis.
Table 5.
Multivariate analysis of prognostic factors
| Variable | Death |
Any event |
Relapse |
Death in CR |
||||
|---|---|---|---|---|---|---|---|---|
| HR (95% CI) | P | HR (95% CI) | P | HR (95% CI) | P | HR (95% CI) | P | |
| WBC at presentation | 1.004 (1.002-1.005) | < .001 | 1.003 (1.001-1.004) | < .001 | 1.004 (1.002-1.006) | .001 | NS | |
| Age | 1.02 (1.007-1.034) | .002 | 1.013 (1.000-1.025) | .04 | NS | 1.032 (1.005-1.061) | .02 | |
| Extra Ph chromosome [+der(22)] | NS | NS | 0.569 (0.342-0.946) | .03 | NS | |||
| del(9p) | NS | NS | 1.742 (1.036-2.928) | .04 | NS | |||
HR greater than 1 (unity) indicates increased risk of the event in relationship to the characteristic studied.
HR indicates hazard ratio; CI, confidence interval; and NS, not significant.
Discussion
This is the largest prospective study of Ph+ ALL. The data confirm the generally poor outcome for patients with Ph+ ALL with an overall CR rate of 82%, considerably less than in Ph negative disease. The need for a third course of induction therapy to achieve remission in 20 patients is consistent with the data reported from the LALA94 study, in which the corresponding figure was 18%.6 The OS was 22% at 5 years. These remission and survival figures are entirely consistent with previously published, smaller studies.4,6,13 The factors predicting achievement of CR in our study were age and presenting WBC count.
The first striking finding is the number of patients (76 of 267, 28%) who actually received the proposed HSCT. The reasons for patients not receiving HSCT were, in the majority of cases, either being beyond the age limit or having an early event that prevented transplantation even when a donor was available. In some cases, patient or physician choice was the reason given. “Lack of donor availability” was also common. Our understanding of this situation is not precise, as in some cases tissue typing was not carried out in cases in which this is given as a reason, which may indicate no sibling was available but does not explain why an unrelated donor was not sought. However, because this was a multicenter international study, the low transplantation rate was unlikely to have been due to individual physician or national bias and likely represents a realistic assessment of the therapeutic milieu in Ph+ ALL. The relevance of such a low transplantation rate in a prospective study of Ph+ ALL is 2-fold. First, it sets the valuable results from the transplantation-only series into a more realistic context by supplying a denominator, which can illuminate the potential magnitude of the selection bias that can occur in such studies. Second, these data provide an international transplantation rate baseline against which studies of novel induction therapies, including tyrosine kinase inhibitors, can be evaluated.
Despite the low transplantation rate overall, sufficient numbers of patients received transplants to enable comparisons between outcomes of those who received HSCT and chemotherapy. Adjusting as far as possible to allow for the selection bias favoring the receipt of transplant by adjusting for sex, age, and presenting WBC as well as excluding chemotherapy-treated patients who relapsed or died before the median time to HSCT (160 days), RFS remained significantly superior in the HSCT group compared with the chemotherapy group (P = .001), but the difference in TRM between the groups became statistically significant, so that the OR for OS changed from 0.48 (95% CI, 0.33-0.69) unadjusted (ie, OS significantly better in the HSCT group) to an adjusted figure of 1.31 (95% CI, 0.73-2.35), that is, OS nonsignificantly worse in the HSCT group. This suggests that the advantage of HSCT over chemotherapy alone in terms of relapse must be carefully weighed against the disadvantage in terms of TRM. This finding is consistent with high TRM in high-risk Ph-negative patients in the UKALLXII/ECOG2993 study. The only factor clearly associated with the high TRM in the high-risk group was older age.10
The adoption of MUD alloHSCT in this study paradoxically provides a barrier to assessing the true role of alloHSCT, as the sibling donor versus no donor analysis is complicated by the fact that those with no sibling donor can also receive alloHSCT, considerably weakening the power of this analysis to demonstrate the true role of alloHSCT. With this in mind, it is not surprising that the sib donor versus no donor analysis of this study did not demonstrate a statistically significant advantage to alloHSCT. Despite the limitations of the analysis in this context, however, the data shown in Figure 3 show a trend toward a survival advantage for the “sib donor” arm, which is entirely consistent with the hypothesis that HSCT provides a survival advantage in Ph+ ALL in adults. Hence, our data support and extend previous “transplantation-only” studies9,14–18 and are consistent with the conclusions of LALA94.6 Peritransplantation care remains an important area to focus on, with a view to reducing posttransplantation TRM. It is possible that reduced-intensity conditioned transplantation19 may benefit the older patients at highest risk of TRM, but the benefit of this procedure for Ph+ ALL remains untested in prospective study.
As confirmation of the therapeutic relevance of the GVL effect in this disease, the occurrence of any acute GVHD was associated with a significant reduction in relapse, although unsurprisingly a concomitant significant increase in death in remission was seen when GVHD was reported. Overall, EFS was better in the presence of GVHD, but this difference was not statistically significant. This contrasts with the findings of Laport et al,9 in whose study acute GVHD was associated with less good OS and EFS. One speculative explanation for this difference may be that the widespread use of alemtuzumab (Campath; Genzyme, Cambridge, MA) in conditioning regimens in the United Kingdom cohort was a possible limiting factor in the severity of acute GVHD; only 8 of 41 (20%) of patients with acute GVHD level recorded had grade 3 or 4 GVHD. We did not collect sufficient data on alemtuzumab administration to formally test this hypothesis. Similarly, chronic GVHD was not sufficiently well documented to assess an effect.
In concordance with our previous analysis, we found that none of the additional chromosomal abnormalities affected EFS or OS.2 This more extensive analysis, however, revealed that patients with +der(22) or del(9p) affected RFS. We found that patients with +der(22) had a lower risk of relapse, which is in contrast to previous studies by Cancer and Leukaemia Group B (CALGB)20 and the Japanese Adult Leukemia Study Group (JALSG).21 Both of these studies, however, were based on fewer patients, and neither result was confirmed by multivariate analysis. In our study, virtually all HeH patients also had +der(22). Therefore, it was not surprising that univariate analysis of HeH also showed a reduced RFS. HeH is known to confer a good prognosis when it occurs alone,2 and it has been hypothesized to improve outcome when coexisting with t(9;22).22 We observed that patients with del(9p) had a increased risk of relapse, which is in agreement with the recent JALSG study.21 Deletions of 9p, which are a surrogate marker for CDKN2A deletions, occur in all cytogenetic subgroups at varying frequencies, and their prognostic relevance has yet to be firmly established.23 It is noteworthy that, in similarity to the present study, other investigators observed differences in RFS only and not on other outcome measures. The therapeutic relevance of these additional chromosome abnormalities is not clear, but because the chromosome abnormalities in the JALSG study retained their poor prognostic significance in an entirely imatinib-treated cohort, it is tempting to speculate that knowledge of these cytogenetic abnormalities and further investigation of their pathologic significance could contribute to a possible therapeutic stratification of adult Ph+ ALL in the future.
In summary, the UKALLXII/ECOG2993 study demonstrates the prognostic relevance of additional chromosome abnormalities in Ph+ ALL. We also show for the first time in an unselected cohort that alloHSCT, using either a sibling or a matched unrelated donor, may be a better treatment for Ph+ ALL than conventional chemotherapy alone. TRM, however, remains a significant problem. The data provide an important baseline against which developments in HSCT technology and promising novel therapies with tyrosine kinase inhibitors24–27 can be evaluated.
Acknowledgments
We thank all participating centers, physicians, and patients.
A.K.F. is supported by the Leukaemia Research Fund (United Kingdom).
Footnotes
The online version of this article contains a data supplement.
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Authorship
Contribution: A.K.F. wrote the paper; all authors contributed to writing the paper, checked the final version, and participated in data collection, study design, and coordination; S.M.R. and G.B. controlled and analyzed data; A.V.M., E.P., G.D., and L.F. supervised cytogenetic or molecular diagnosis or review; and A.H.G. and J.M.R. were study chairs in the United Kingdom and United States, respectively.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
A list of the participating centers of the United Kingdom Medical Research Council Adult Leukemia Working Party and Eastern Cooperative Oncology Group can be found in the Appendix (available on the Blood website; see the Supplemental Materials link at the top of the online article).
Correspondence: Adele K. Fielding, University College London, Royal Free Campus, Department of Haematology, Rowland Hill St, London NW3 2PF, United Kingdom; e-mail: A.Fielding@medsch.ucl.ac.uk.
References
- 1.Gleissner B, Gokbuget N, Bartram CR, et al. Leading prognostic relevance of the BCR-ABL translocation in adult acute B-lineage lymphoblastic leukemia: a prospective study of the German Multicenter Trial Group and confirmed polymerase chain reaction analysis. Blood. 2002;99:1536–1543. doi: 10.1182/blood.v99.5.1536. [DOI] [PubMed] [Google Scholar]
- 2.Moorman AV, Harrison CJ, Buck GA, et al. Karyotype is an independent prognostic factor in adult acute lymphoblastic leukemia (ALL): analysis of cytogenetic data from patients treated on the Medical Research Council (MRC) UKALLXII/Eastern Cooperative Oncology Group (ECOG) 2993 trial. Blood. 2007;109:3189–3197. doi: 10.1182/blood-2006-10-051912. [DOI] [PubMed] [Google Scholar]
- 3.Jones LK, Saha V. Philadelphia positive acute lymphoblastic leukaemia of childhood. Br J Haematol. 2005;130:489–500. doi: 10.1111/j.1365-2141.2005.05611.x. [DOI] [PubMed] [Google Scholar]
- 4.Faderl S, Kantarjian HM, Thomas DA, et al. Outcome of Philadelphia chromosome-positive adult acute lymphoblastic leukemia. Leuk Lymphoma. 2000;36:263–273. doi: 10.3109/10428190009148847. [DOI] [PubMed] [Google Scholar]
- 5.Preti HA, O'Brien S, Giralt S, Beran M, Pierce S, Kantarjian HM. Philadelphia-chromosome-positive adult acute lymphocytic leukemia: characteristics, treatment results, and prognosis in 41 patients. Am J Med. 1994;97:60–65. doi: 10.1016/0002-9343(94)90049-3. [DOI] [PubMed] [Google Scholar]
- 6.Dombret H, Gabert J, Boiron JM, et al. Outcome of treatment in adults with Philadelphia chromosome-positive acute lymphoblastic leukemia–results of the prospective multicenter LALA-94 trial. Blood. 2002;100:2357–2366. doi: 10.1182/blood-2002-03-0704. [DOI] [PubMed] [Google Scholar]
- 7.Larson RA, Dodge RK, Burns CP, et al. A five-drug remission induction regimen with intensive consolidation for adults with acute lymphoblastic leukemia: cancer and leukemia group B study 8811. Blood. 1995;85:2025–2037. [PubMed] [Google Scholar]
- 8.Secker-Walker LM, Craig JM, Hawkins JM, Hoffbrand AV. Philadelphia positive acute lymphoblastic leukemia in adults: age distribution, BCR breakpoint and prognostic significance. Leukemia. 1991;5:196–199. [PubMed] [Google Scholar]
- 9.Laport GG, Alvarnas JC, Palmer JM, et al. Long-term remission of Philadelphia chromosome-positive acute lymphoblastic leukemia after allogeneic hematopoietic cell transplantation from matched sibling donors: a 20-year experience with the fractionated total body irradiation-etoposide regimen. Blood. 2008;112:903–909. doi: 10.1182/blood-2008-03-143115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Goldstone AH, Richards SM, Lazarus HM, et al. In adults with standard-risk acute lymphoblastic leukemia, the greatest benefit is achieved from a matched sibling allogeneic transplantation in first complete remission, and an autologous transplantation is less effective than conventional consolidation/maintenance chemotherapy in all patients: final results of the International ALL Trial (MRC UKALL XII/ECOG E2993). Blood. 2008;111:1827–1833. doi: 10.1182/blood-2007-10-116582. [DOI] [PubMed] [Google Scholar]
- 11.Blume KG, Forman SJ, O'Donnell MR, et al. Total body irradiation and high-dose etoposide: a new preparatory regimen for bone marrow transplantation in patients with advanced hematologic malignancies. Blood. 1987;69:1015–1020. [PubMed] [Google Scholar]
- 12.Rowe JM, Buck G, Burnett AK, et al. Induction therapy for adults with acute lymphoblastic leukemia: results of more than 1500 patients from the international ALL trial: MRC UKALL XII/ECOG E2993. Blood. 2005;106:3760–3767. doi: 10.1182/blood-2005-04-1623. [DOI] [PubMed] [Google Scholar]
- 13.Wrzesien-Kus A, Robak T, Pluta A, et al. Outcome of treatment in adults with Philadelphia chromosome-positive and/or BCR-ABL–positive acute lymphoblastic leukemia-retrospective analysis of Polish Adult Leukemia Group (PALG). Ann Hematol. 2006;85:366–373. doi: 10.1007/s00277-006-0099-z. [DOI] [PubMed] [Google Scholar]
- 14.Stockschlader M, Hegewisch-Becker S, Kruger W, et al. Bone marrow transplantation for Philadelphia-chromosome-positive acute lymphoblastic leukemia. Bone Marrow Transplant. 1995;16:663–667. [PubMed] [Google Scholar]
- 15.Sierra J, Radich J, Hansen JA, et al. Marrow transplants from unrelated donors for treatment of Philadelphia chromosome-positive acute lymphoblastic leukemia. Blood. 1997;90:1410–1414. [PubMed] [Google Scholar]
- 16.Forman SJ, O'Donnell MR, Nademanee AP, et al. Bone marrow transplantation for patients with Philadelphia chromosome-positive acute lymphoblastic leukemia. Blood. 1987;70:587–588. [PubMed] [Google Scholar]
- 17.Dunlop LC, Powles R, Singhal S, et al. Bone marrow transplantation for Philadelphiachromosome-positive acute lymphoblastic leukemia. Bone Marrow Transplant. 1996;17:365–369. [PubMed] [Google Scholar]
- 18.Barrett AJ, Horowitz MM, Ash RC, et al. Bone marrow transplantation for Philadelphia chromosome-positive acute lymphoblastic leukemia. Blood. 1992;79:3067–3070. [PubMed] [Google Scholar]
- 19.Mohty M, Labopin M, Tabrizzi R, et al. Reduced intensity conditioning allogeneic stem cell transplantation for adult patients with acute lymphoblastic leukemia: a retrospective study from the European Group for Blood and Marrow Transplantation. Haematologica. 2008;93:303–306. doi: 10.3324/haematol.11960. [DOI] [PubMed] [Google Scholar]
- 20.Wetzler M, Dodge RK, Mrozek K, et al. Additional cytogenetic abnormalities in adults with Philadelphia chromosome-positive acute lymphoblastic leukaemia: a study of the Cancer and Leukaemia Group B. Br J Haematol. 2004;124:275–288. doi: 10.1046/j.1365-2141.2003.04736.x. [DOI] [PubMed] [Google Scholar]
- 21.Yanada M, Takeuchi J, Sugiura I, et al. Karyotype at diagnosis is the major prognostic factor predicting relapse-free survival for patients with Philadelphia chromosome-positive acute lymphoblastic leukemia treated with imatinib-combined chemotherapy. Haematologica. 2008;93:287–290. doi: 10.3324/haematol.11891. [DOI] [PubMed] [Google Scholar]
- 22.Tauro S, McMullan D, Griffiths M, Craddock C, Mahendra P. High-hyperdiploidy in Philadelphia positive adult acute lymphoblastic leukaemia: case-series and review of literature. Bone Marrow Transplant. 2003;31:763–766. doi: 10.1038/sj.bmt.1703913. [DOI] [PubMed] [Google Scholar]
- 23.Sulong S, Moorman AV, Irving JA, et al. A comprehensive analysis of the CDKN2A gene in childhood acute lymphoblastic leukemia reveals genomic deletion, copy number neutral loss of heterozygosity, and association with specific cytogenetic subgroups. Blood. 2009;113:100–107. doi: 10.1182/blood-2008-07-166801. [DOI] [PubMed] [Google Scholar]
- 24.Wassmann B, Pfeifer H, Stadler M, et al. Early molecular response to posttransplantation imatinib determines outcome in MRD+ Philadelphia-positive acute lymphoblastic leukemia (Ph+ ALL). Blood. 2005;106:458–463. doi: 10.1182/blood-2004-05-1746. [DOI] [PubMed] [Google Scholar]
- 25.Wassmann B, Pfeifer H, Goekbuget N, et al. Alternating versus concurrent schedules of imatinib and chemotherapy as front-line therapy for Philadelphia-positive acute lymphoblastic leukemia (Ph+ ALL). Blood. 2006;108:1469–1477. doi: 10.1182/blood-2005-11-4386. [DOI] [PubMed] [Google Scholar]
- 26.Ottmann OG, Wassmann B, Pfeifer H, et al. Imatinib compared with chemotherapy as front-line treatment of elderly patients with Philadelphia chromosome-positive acute lymphoblastic leukemia (Ph+ALL). Cancer. 2007;109:2068–2076. doi: 10.1002/cncr.22631. [DOI] [PubMed] [Google Scholar]
- 27.Ottmann O, Dombret H, Martinelli G, et al. Dasatinib induces rapid hematologic and cytogenetic responses in adult patients with Philadelphia chromosome positive acute lymphoblastic leukemia with resistance or intolerance to imatinib: interim results of a phase 2 study. Blood. 2007;110:2309–2315. doi: 10.1182/blood-2007-02-073528. [DOI] [PubMed] [Google Scholar]