

Abstract
Calcium signaling has a versatile role in many important cellular functions. Despite its importance, regulation of calcium signaling in bone marrow stromal cells (BMSCs, also known as bone marrow-derived mesenchymal stem cells) has not been explored extensively. Our previous study revealed that cyclic adenosine monophosphate (cAMP) enabled BMSCs to generate calcium signal upon stimulation by dopamine, KCl and glutamate. Concurrently, cAMP transiently activated the transcription factor cAMP response element binding protein (CREB) in BMSCs. Activity of CREB can be modulated by the calcium/calmodulin-dependent kinase signaling pathway, however, whether the calcium signaling observed in cAMP-induced BMSCs requires CREB has not been investigated. In an effort to uncover the role of CREB in the generation of calcium signaling in response to modulators such as dopamine and KCl, we knocked down CREB activity in BMSCs. Our study indicated that BMSCs, but not its close relative fibroblasts, are responsive to dopamine and KCl after cAMP treatment. Calcium signal elicited by dopamine depends, in part, on calcium influx whereas that elicited by KCl depends completely on calcium influx. Knock-down of CREB activity significantly reduced or abolished the cAMP-induced calcium response, and reintroducing a constitutively active CREB partially restored the calcium response.
Keywords: CREB, bone marrow stromal cells, calcium signal, morphology
1. Introduction
Bone marrow stromal cells (BMSCs), also known as bone marrow-derived mesenchymal stem cells, have the potential to undergo osteogenic, chondrogenic and adipogenic differentiation [1]. Calcium is one of the many important players that regulates differentiation [2], such as osteogenic [3, 4], chondrogenic [5, 6] and adipogenic differentiation [7-9]. Regulation of calcium signaling in BMSCs has been explored to some extent. Human BMSCs has exhibited spontaneous calcium oscillations without agonist stimulation [10-12]. Inhibition of calcium influx through L-type voltage-gated calcium channel reduced osteogenic differentiation of BMSCs [4], while stimulating cytosolic calcium oscillation facilitated osteogenic differentiation in human BMSC [3]. Tropel et al. demonstrated that mouse BMSCs induced by basic fibroblast growth factor (bFGF) are able to generate calcium signal upon stimulation by dopamine, glutamate and veratridine [13]. While these studies demonstrated the importance of calcium signaling in BMSCs, studies regarding calcium signaling in BMSCs is still at an early stage.
Rise in cytosolic calcium concentration mainly results from calcium release from internal stores or calcium influx from external medium [2, 14]. Endoplasmic reticulum (ER) is generally recognized as the intracellular calcium stores. Release of calcium from ER is mediated by second messengers, such as inositol-1,4,5-trisphosphate (Ins(1,4,5)P3) and calcium itself [14, 15]. It has been shown that spontaneous calcium oscillations observed in BMSCs mainly occurred through calcium release from internal stores, mediated by the inositol-1,4,5-trisphosphate receptor (InsP3R) [10]. External medium is another source of cytosolic calcium. Calcium influx through ion channels on the cell membrane, including voltage-gated channels and receptor-gated channels, contribute to the dynamical changes in cytosolic calcium concentrations [2, 14]. Voltage-gated ion channels or receptor-gated channels are predominantly expressed in excitable cells [15]. However, a previous study showed that many ion channels, such as L-type voltage-gated Ca2+ channels, voltage-gated potassium channels, Ca2+-activated potassium channels and voltage-gated sodium channels, are expressed in undifferentiated rat BMSCs [16]. In addition to voltage-gated channels, influx of calcium can also be mediated by receptor-gated channels, i.e., G-protein coupled dopamine receptors. Dopamine induces calcium rise mainly through binding to G-protein coupled dopamine receptors, the D1-like (including D1 and D5) and the D2-like (including D2, D3 and D4) dopamine receptors [17, 18].
Cyclic AMP is another important intracellular second messenger that can crosstalk with calcium signaling and regulates cell function. cAMP activates the downstream transcription factor cAMP response element binding protein (CREB), which serves as a hub for many cellular processes including metabolism, survival, immune response as well as learning and memory [19, 20]. Phosphorylation of CREB at the key serine 133 site by kinases such as protein kinase A (PKA), Ca2+/calmodulin-dependent kinases (CaMKs), and mitogen-activated protein kinases (MAPKs) is required for its transcriptional activity [21, 22]. Previously, we observed that after cAMP treatment, BMSCs exhibited robust calcium response upon stimulation by dopamine and KCl [23]. Dopamine not only plays an important role in the central nervous system, but also functions in the peripheral system. Many conditions, including stress, hypovolemia and exercise, can increase the dopamine level in the plasma [24]. In mouse bone marrow, evidence of dopamine uptake was observed, suggestive of the presence of dopamine receptors in the bone marrow [25]. In further support, dopamine receptor D1 protein was detected in uninduced human BMSCs which further increased with brain-derived neurotrophic factor (BDNF) treatment [26].
Although it is well known that the cAMP downstream signaling component CREB can be modulated by the Ca2+/calmodulin dependent kinases (CaMKs) [21], little is known regarding the role of the cAMP-CREB pathway on calcium signaling. In particular, the impact of cAMP-CREB pathway on calcium signaling in BMSCs has not been explored. cAMP is increased in many pathophysiological conditions, such as fibrous dysplasia of the bone [27, 28], hyperparathyroidism [29, 30] and myocardial infarction [31, 32]. In other cases, elevation of cAMP has been beneficial for treating a number of ailments, such as spinal cord injury [33, 34] and autoimmune diseases [35, 36]. Reagents that are able to boost cAMP levels are also used widely in modulating osteogenic- and adipogenic-differentiation of BMSCs [37-40]. BMSCs are excellent sources for cell-based therapies, due to their differentiation potential, secretion of a plethora of trophic factors, and immunomodulatory capability [41]. Currently, clinical trials using BMSCs are being studied for a variety of diseases, such as Graft-versus-Host disease, myocardial infarction, spinal cord injury, stroke, and amyotrophic lateral sclerosis (ALS) [41, 42]. Accordingly, it is important to understand how BMSCs would respond to calcium modulators in an environment with elevated cAMP levels.
Previous studies showed that certain dopamine receptors and voltage-gated calcium channels are expressed on BMSCs [16, 26], therefore, we set out to determine how the cAMP-CREB pathway might modulate calcium signaling through receptor-gated channels stimulated by dopamine and through voltage-gated channels stimulated by KCl. In this study, we examined intracellular calcium signaling in BMSCs through fluorescence imaging. CREB activity was modulated by siRNA silencing and the generation of a dominant negative CREB cell line. Our results suggested that the calcium response to dopamine and KCl in cAMP-induced BMSCs requires calcium influx from an external source. Long term knock-down of CREB activity significantly reduced the calcium response to dopamine and KCl, along with dramatic morphological changes and increased apoptosis in cAMP-induced BMSCs.
2. Materials and Methods
2.1 Cell culture and materials
All procedures in the cell isolation were approved by the Institutional Animal Care and Use Committee at Michigan State University. Bone marrow stromal cells (BMSCs) were isolated from 6-8 week old Sprague-Dawley female rat as previously described [43]. In brief, femurs and tibias were taken from the hind legs of 6-8 week old rat. The marrow was flushed out with Dulbecco's Modified Eagle Medium (DMEM) and filtered through a 65 um nylon mesh to remove bone debris and blood aggregates. Cells were cultured in DMEM (Invitrogen, Carlsbad, CA, USA) supplemented with 10% fetal bovine serum (Invitrogen), 100 ug/mL streptomycin (Invitrogen) and 100 U/mL penicillin (Invitrogen) and placed in an incubator with a humidified atmosphere containing 5% CO2 at 37 °C. Non-adherent cells were removed on the second day after plating. After culturing for two passages, magnetic cell sorting using markers CD54 and CD90 was applied to enrich BMSCs. Cell sorting results were verified by flow cytometry as shown in [43].
NIH3T3 fibroblasts were purchased from American Type Culture Collection (ATCC). Cells were cultured in DMEM (high glucose (4.5 g/L) (Invitrogen) supplemented with 10% FBS, 100 μg/mL streptomycin, and 100 U/mL penicillin and placed in an incubator with a humidified atmosphere containing 10% CO2 at 37 °C.
Forskolin (Sigma, St. Louis, MO, USA) and isobutylmethylxanthine (IBMX) (Sigma) were used to increase intracellular cAMP levels at concentrations of 10 μM and 100 μM, respectively.
2.2 Cell proliferation assay
Cell proliferation was measured by the CyQUANT NF cell proliferation assay kit from Invitrogen. This assay is based on the binding of a fluorescent dye to DNA and therefore enabled the quantification of cellular DNA. Briefly, cells were cultured in 96-well culture plate and assayed at the desired time. After washing with Hank's balanced salt solution (HBSS), the cells were incubated in the dye-binding solution for 45 min at 37 °C. Fluorescence was measured by Spectra MAX GEMINI EM plate reader at excitation of 485 nm and emission of 530 nm.
2.3 Western blot
Whole cell extracts lysed with CelLytic (Sigma) were assayed for protein concentrations by Bradford assay (Bio-Rad). 15-30 μg protein samples were separated by 10% Tris-HCl gel and transferred to nitrocellulose membrane. Membranes were then blocked with 5% milk in 0.05% Tween 20-Tris buffered saline (T-TBS) (USB corporation, Cleveland, Ohio, USA) for one hour and incubated with primary antibodies, glyceraldehyde 3-phosphate dehydrogenase (GAPDH) (Cell signaling, Danvers, MA, USA), CREB (Cell signaling), dopamine receptor D1 (Novus Biologicals), c-fos (Cell Signaling), TATA-binding protein (TBP) (Sigma) and inducible cAMP early repressor (ICER) (a kind gift from Dr. Carlos Molina) overnight at 4 °C. After removing excessive primary antibodies, anti-mouse or anti-rabbit horseradish peroxidase (HRP)-conjugated secondary antibodies (Thermo Scientific) were added and the blots were incubated for one hour at room temperature. The blots were then washed three times with 0.05% T-TBS and visualized by SuperSignal West Femto maximum sensitivity substrate (Thermo Scientific).
2.4 Immunocytochemistry
Triple staining for actin filaments, microtubules and nucleus was performed as previously described [44]. In brief, actin filaments were stained with Texas Red-X phalloidin (Invitrogen), microtubules were stained with α-tubulin (Invitrogen) primary antibody followed by Alexa Fluor 488-conjugated anti-mouse IgG secondary antibody (Invitrogen), and the nucleus was stained with DAPI (4′, 6-diamidino-2-phenylindole) (Invitrogen). Stained glass coverslips were mounted in ProLong Gold (Invitrogen). Fluorescence images were taken by confocal microscope Olympus FluoView 1000.
2.5 Annexin V and propidium iodide (PI) staining
Apoptosis and necrosis were measured by the annexin V and propidium iodide (PI) staining kit (Invitrogen), respectively, according to the manufacturer's instructions. In brief, cells were stained with Alexa Fluor 488 conjugated annexin V and PI in 1X annexin binding buffer for 15 minutes at room temperature and then subjected to flow cytometry analysis by BD FACSVantage. Early apoptotic cells were identified as those stained by Alexa Fluor 488 but not PI, late apoptotic cells were those stained by both Alexa Fluor 488 and PI, and necrotic cells were those stained by PI but not Alexa Fluor 488.
2.6 Quantitative real time polymerase chain reaction (RT-PCR)
Cells were treated as prescribed and the mRNA was extracted by the RNA extraction kit from Qiagen according to the manufacture's instruction. mRNAs were then reverse transcribed into cDNA using the cDNA synthesis kit from Bio-Rad. The following primer sets (Eurofins MWG Operon) were used for PCR: actin (5′-CTCTTCCAGCCTTCCTTCCT-3′, 5′-AATGCCTGGGTACATGGTG-3′), dopamine receptor 1 (D1) (5′-GCCATAGAGACGGTGAGCAT-3′, 5′-ATTCCACCAGCCTCTTCCTT-3′), dopamine receptor 2 (D2) (5′-TATGGCTTGAAGAGCCGTGCCA-3′, 5′-TACAGCCATGCACACCAGCACA-3′), dopamine receptor 3 (D3) (5′-CAGCCGCATTTGCTGTGACGTT-3′, 5′-AGCAAAAGCCAGCACCCACACA-3′), dopamine receptor 4 (D4) (5′-TGCTGCTCATCGGCATGGTGT-3′, 5′-AGCCACAAACCTGTCCACGCT-3′), dopamine receptor 5 (D5) (5′-TGGAGCCTATGAACCTGACC-3′, 5′-GAAGAAAGGCAACCAGCAAC-3′). Amplification of the cDNA templates were detected by SYBR Green supermix (Bio-Rad) using Real-Time PCR Detection System (Bio-Rad). The cycle threshold (CT) values for each condition were determined by the MyIQ software.
2.7 Calcium imaging
Calcium imaging was performed according to the protocol described in [13]. Cells were cultured in 4-well chambered cover-glass (Thermo Fisher Scientific). After the desired treatment, the cells were loaded with 4 μM non-ratiometric dye Fluo-4 (Invitrogen) in ACSF-HEPES (artificial cerebral spinal fluid with HEPES: 119 mM NaCl, 2.5 mM KCl, 1.3 mM MgCl2, 2.5 mM CaCl2, 1 mM NaH2PO4, 26.2 mM NaHCO3, 11 mM dextrose, 10 mM HEPES, pH=7.4) for 30 min at 37 °C• • Excess dye was removed by washing the cells twice with PBS and placing them into a 37 °C chamber on the stage of Olympus FluoView 1000. 0.5 ml ACSF-HEPES was added to the well to begin imaging. Images were captured every 1.137 seconds and fluorescence intensity is represented by a spectral table (warmer colors represent higher intensity whereas cooler colors represent lower intensity). After 15~20 images, 0.5 ml ACSF-HEPES buffer containing the following compounds were added: 200 μM glutamate (final concentration 100 μM), 200 μM dopamine (final concentration 100 μM), 100 mM KCl (final concentration 50 mM), 100 μM veratridine (final concentration 50 μM) or 200 μM ATP (final concentration 100 μM). A total of 100~200 images were recorded and the data was analyzed by the FluoView 1000 software. Changes in the fluorescence intensity of the Ca2+ signal are represented as Ft/F0, where Ft represents the fluorescence intensity at time t after stimulation and F0 represents the fluorescence intensity before stimulation. The percent of responsive cells is calculated as the number of cells with a Ft/F0 signal greater than 20% divided by the total number of cells. A total of around 150 cells are counted for each experiment, and every experiment is repeated for 3 or more times.
2.8 Transfection
For siRNA silencing, siRNAs targeting CREB mRNA (CREB siRNA #1 and #2) as well as a scramble siRNA (negative control) were purchased from Ambion. In brief, the transfection reagent Lipofectamine RNAiMAX (Invitrogen) was diluted in Opti-MEM (Invitrogen) reduced media The siRNAs were also diluted in Opti-MEM and then mixed with RNAiMAX to allow the formation of siRNA-RNAiMAX complex at room temperature for 20 minutes. Cell culture medium was replaced with media without antibiotics. The siRNA-RNAiMAX complexes were then added to the corresponding wells to reach a final concentration of 10 nM siRNA. Medium was replaced after 4~6 hours incubation in a 5% CO2 incubator at 37 °C• • Treatments were performed 24 hours from the start of silencing.
The constitutively active form CREB (VP16-CREB) and the dominant negative form CREB (serine 133 mutated to alanine, named M1-CREB) and the empty vector pCMV are kind gifts from Dr. David Ginty. In brief, cells were transfected with 1.5 μg pCMV, VP16-CREB or M1-CREB using Lipofectamine 2000 (Invitrogen) according to the manufacture's instruction. Medium was replaced after 4~6 hours and cells were incubated in fresh culture medium for up to 24 hours until treatment was carried out. For establish a stable cell line expressing M1-CREB, BMSCs that are transiently transfected with M1-CREB for 24 hours were trypsinized and replated at low density in media containing 500 μg/ml geneticin (Invitrogen) for selection. For control, BMSCs were also transfected with an empty pCMV vector containing neomycin resistance to establish a control cell line. The geneticin-containing media was replaced every 3 days for two weeks. Colonies formed from surviving cells were isolated by cloning cylinders (Sigma) and maintained in culture media containing geneticin. Clone 2 from Fig. 3A was used for experiments in this study.
Fig. 3.
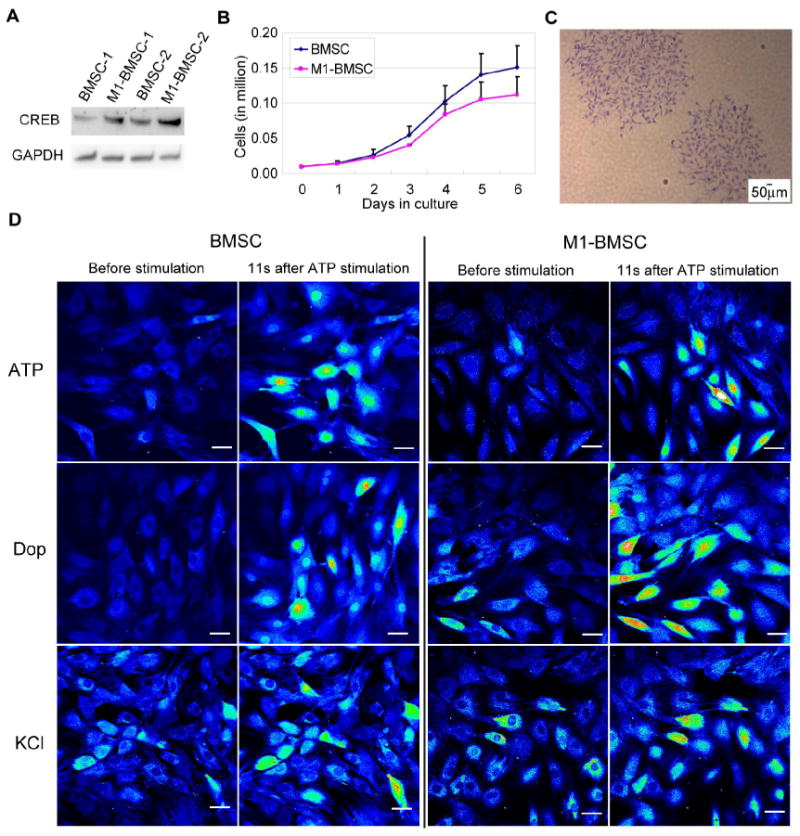
Evaluation of BMSCs expressing the dominant negative CREB (M1-CREB). (A) Expression of CREB in BMSCs expressing the control vector and BMSCs expressing the dominant negative M1-CREB (M1-BMSCs). Clone 2 in (A) was used for the following experiments. (B) Proliferation curves for BMSCs and M1-BMSCs during culture. (C) Colony formation of M1-BMSCs (expressing M1-CREB). (D) Calcium signaling upon stimulation by ATP, dopamine and KCl in BMSCs and M1-BMSCs. Cells were loaded with Fluo-4 and images were taken under time-lapse mode upon stimulation with 100 μM ATP, 100 μM dopamine or 50 mM KCl for a total of around 100 seconds. Fluorescence images represented by a spectral table; warmer colors indicate higher fluorescence intensities and cooler colors indicate lower fluorescence intensities. Gain and offset were held constant in each experiment (before stimulation until the end of stimulation). Scale bar: 50 μm.
2.9 Statistical analysis
All experiments were performed at least three times and results were shown as mean ± standard deviation. Statistical analysis were carried out by an unpaired, two tail Student's t-test. * indicates p<0.05, ** indicates p<0.01 and *** indicates p<0.001.
3. Results
3.1 BMSCs but not fibroblasts are responsive to dopamine and KCl
In our previous study, we reported that BMSCs exhibited enhanced calcium response upon stimulation by dopamine and KCl when the cells were treated with 10 μM forskolin and 100 μM IBMX (reagents used to increase cAMP levels, abbreviated as FI) [23]. Fibroblasts, which share many mesenchymal phenotypes with BMSCs [45], showed similar morphological changes upon FI treatment and these morphological changes remain at one day post treatment (Supplementary Fig. S1) [23]. Since FI-induced morphological changes are not unique to BMSCs and also occurred in fibroblasts, we therefore set out to determine if the ability of BMSCs to generate enhanced calcium signal after FI induction is a universal effect due to the FI treatment. Although some control BMSCs were responsive to dopamine and KCl stimulation (Supplementary Fig. S2), FI treatment further increased the number of cells responsive to dopamine and KCl (Fig. 1). Unlike the BMSCs, neither control fibroblasts nor FI-treated fibroblasts were responsive to dopamine or KCl stimulation (Supplementary Fig. S2 and Fig. 1). These results suggest that although FI induced morphological changes in both BMSCs and fibroblasts, it resulted in dopamine and KCl responses only in BMSCs. As the morphologies of the FI-treated fibroblasts were altered dramatically, we evaluated whether the decrease or loss of response to dopamine and KCl was through general damage of membrane bound channels and intracellular calcium stores such as ER. We applied a positive control, ATP, which can cause calcium influx through the P2X inotropic receptors, and cause calcium release from ER through stimulating Ins(1,4,5)P3 production by binding to the P2Y metabotropic receptors [46, 47]. The FI-treated fibroblasts were able to respond to ATP similarly as the FI-treated BMSCs (Fig. 1), suggesting that the lack of calcium rise in response to dopamine and KCl is not likely due to general damage to the membrane bound channels or ER. These results indicate that the ability of BMSCs to respond to dopamine and KCl is not a universal effect of cAMP treatment.
Fig. 1.
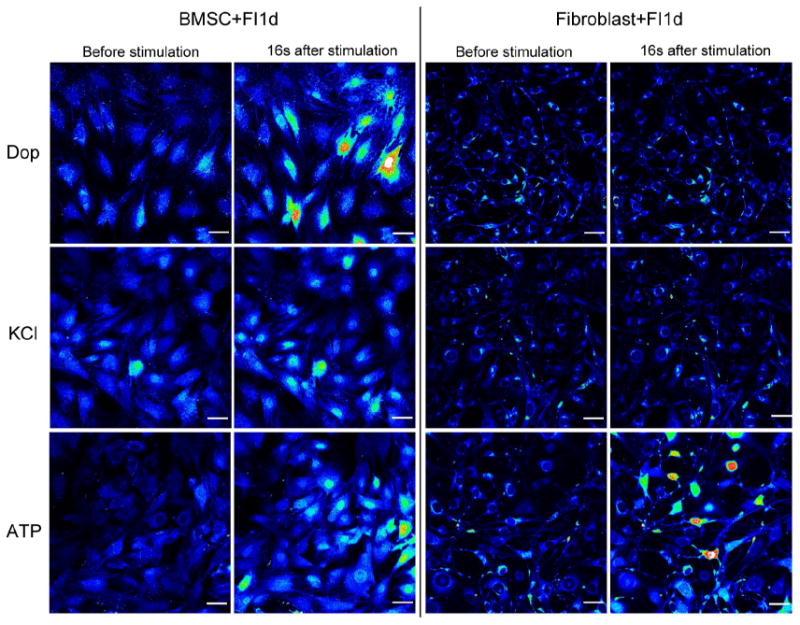
BMSCs and fibroblasts in response to dopamine, KCl and ATP stimulation. BMSCs and fibroblasts were treated with 10 μM forskolin and 100 μM IBMX (FI) for 1 day. After FI treatment, cells were loaded with Fluo-4 and images were taken under time-lapse mode upon stimulation with 100 μM dopamine, 50 mM KCl or 100 μM ATP. Representative images before stimulation and 16 seconds after stimulation are shown. Fluorescence images represented by a spectral table; warmer colors indicate higher fluorescence intensities (yellow to red) and cooler colors indicate lower fluorescence intensities (blue to green). The basal fluorescence intensity is blue or very light green, which is around 500 relative fluorescence units (RFU). When the cells were stimulated, the highest RFU that was obtained was around 3000 (colored red) and the majority of the responsive cells had RFU between 1000~2000 (colored yellow to red). Gain and offset were held constant in each experiment (before stimulation until the end of stimulation). Scale bar: 50 μm.
3.2 Silencing of CREB reduces the number of dopamine and KCl responsive cells in cAMP induced BMSCs
CREB was transiently activated by cAMP in BMSCs as indicated by our previous study [23]. To determine if CREB plays a role in modulating calcium signal, we silenced CREB in BMSCs and assessed the dopamine and KCl response upon cAMP induction (Fig. 2). Upon silencing with either a scramble siRNA (control) or CREB siRNA #1 (Fig. 2B) or CREB siRNA #2 (Supplementary Fig. S3), the BMSCs in control medium or cAMP induction medium were stimulated with dopamine or KCl. CREB siRNA silenced BMSCs in control medium (no FI) responded similarly to the scramble siRNA silenced BMSCs. However, the number of dopamine and KCl responsive cells were reduced in the CREB silenced BMSCs when treated with FI for one day (Fig. 2C and 2D). Average fold change at peak intensity for the responsive cells appears to be only slightly lower when CREB is knocked down (Fig. 2E and 2F). When the dopamine responsive cells were divided into four groups based on the fold change (1.2-2.0, 2.0-3.0, 3.0-4.0 and >4.0 fold change), the FI treatment led to more cells with stronger dopamine response, in particular, those with greater than 4-fold change (Supplementary Fig. S4). Notably, no statistical difference was observed between cells transfected with the scramble siRNA and CREB siRNA (Supplementary Fig. S4), suggesting that CREB silencing reduced the number of dopamine and KCl responsive cells but not the intensity of the calcium signal.
Fig. 2.
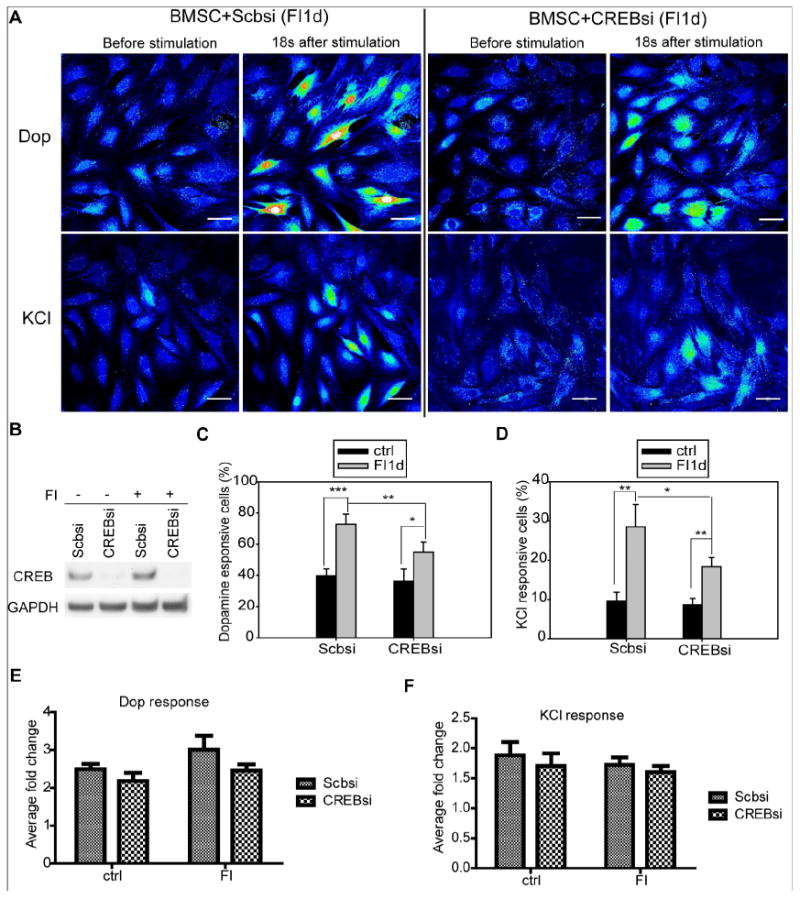
Calcium response to dopamine and KCl stimulation after silencing of CREB. (A) Calcium imaging in response to dopamine and KCl. BMSCs were transfected with scramble siRNA (Scbsi) or CREB siRNA (CREBsi) and induced with 10 μM forskolin and 100 μM IBMX (FI) for 1 day. Representative images before dopamine stimulation and 18 seconds after dopamine stimulation were presented. Fluorescence images were represented by a spectral table; warmer colors indicate higher fluorescence intensities (yellow to red) and cooler colors indicate lower fluorescence intensities (blue to green). Gain and offset are held constant in each experiment (before stimulation until the end of stimulation). Scale bar: 50 μm. (B) Protein levels of CREB for cells transfected with scramble or CREB siRNA and incubated in the absence (−) or presence (+) of FI. (C-D) Quantification of the percentage of cells responsive to dopamine (C) and KCl (D) (n=4). (E-F) Quantification of average fold change at peak intensity for cells responsive to dopamine (E) and KCl (F) (n=3). *: p <0.05, **: p <0.01, ***: p <0.001.
3.3 Knock-down of CREB activity minimally affect calcium signaling in BMSCs without cAMP treatment
To assess further the long term effect of CREB on calcium signaling, we engineered BMSCs to stably express the dominant negative form of CREB (M1-CREB, whose serine 133 site is mutated to alanine) and compared their response to BMSCs stably transfected with a control vector that contains neomycin-resistance. BMSCs stably expressing M1-CREB, denoted as M1-BMSCs, have higher total CREB expression than the control BMSCs (Fig. 3A). The M1-BMSCs grew slightly slower as compared with the BMSCs expressing the control vector (Fig. 3B) and are able still to form colonies (Fig. 3C). The intracellular calcium stores, such as the Ins(1,4,5)P3 regulated calcium stores, also appear to be functioning properly in the M1-BMSC when stimulated with the positive control ATP (Fig. 3D). The response of the M1-BMSCs to dopamine and KCl is similar to BMSCs prior to FI treatment (Fig. 3D). To assess whether knock-down of CREB activity affects the intensity of calcium signal, fold change at the peak intensity for the individual cells were taken and averaged. BMSCs and M1-BMSCs showed very similar average fold change for both dopamine and KCl responses (Fig. 4A). When the cells were divided into different groups based on the fold change at the peak intensity, no difference was observed between BMSCs and M1-BMSCs (Fig. 4B and 4C), suggesting that knock-down of CREB activity did not influence calcium signaling in the control condition.
Fig. 4.
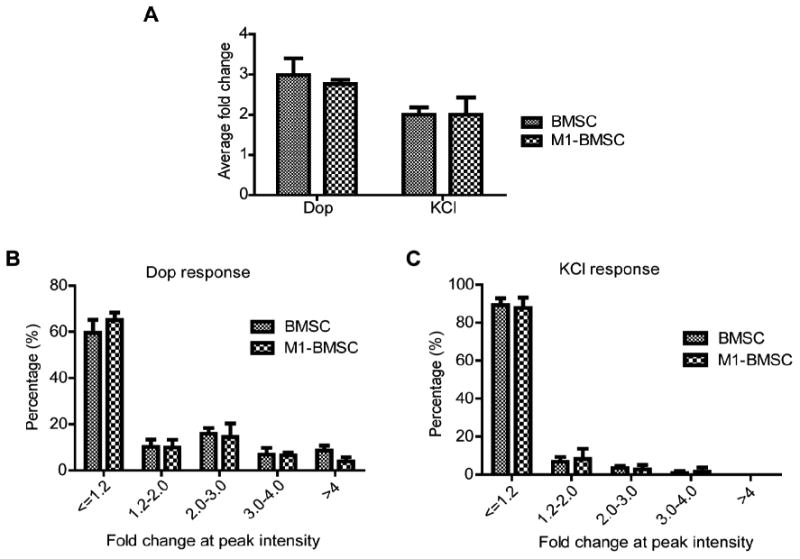
Quantification of fold changes of dopamine and KCl responsive cells. (A) Quantification of average fold change at peak intensity for cells responsive to dopamine and KCl in BMSCs and M1-BMSCs (n=3). (B) Distribution of dopamine responsive BMSCs and M1-BMSCs into different categories (<=1.2 (non-responsive), 1.2-2.0, 2.0-3.0, 3.0-4.0 and >4.0) based on the fold change at peak intensity (n=3). (C) Distribution of KCl responsive BMSCs and M1-BMSCs into different categories (<=1.2 (non-responsive), 1.2-2.0, 2.0-3.0, 3.0-4.0 and >4.0) based on the fold change at peak intensity (n=3).
Although individual cells respond differently to dopamine, the overall trend of their response to dopamine and KCl over time is similar for the BMSCs and M1-BMSCs (Fig. 5). However, the pattern of dopamine response and KCl response are quite different. While dopamine elicited a stronger calcium signal which decayed rapidly (Fig. 5A), KCl stimulated calcium signal is weaker and remained stable over time (Fig. 5B). In addition to the different pattern, the dependence on the external calcium source also appears to differ for dopamine and KCl. When calcium is removed from the external medium, the dopamine responsive cells reduced to ~40% and almost no cells responded to KCl (Fig. 6A and 6B). Average peak intensity of the dopamine response changed ~2.92-fold in the presence of calcium, and ~2.56-fold in the absence of calcium. The difference between the two conditions is not statistically significant, with a p-value of 0.059. However, more cells generated stronger dopamine response in the presence of calcium (Fig. 6C). When the dopamine responsive cells were separated into different categories based on their fold change in peak intensity, significantly higher number of cells showed a greater than 3-fold change in peak intensity (Fig. 6C), indicating the presence of external calcium not only enabled more cells to respond to dopamine but also more cells to generate stronger dopamine response. Peak intensity of KCl response changed 1.75-fold in the presence of calcium, and almost zero in the absence of calcium. The loss of KCl responsive cells in the absence of calcium indicates that calcium influx through membrane bound channels is required for generating a robust KCl-induced calcium signal in the FI-treated BMSCs. Unlike KCl, calcium signal elicited by dopamine is partially dependent on external calcium influx. The reduction but not complete loss of dopamine responsive cells upon external calcium removal suggests the dopamine responsive cells are likely generating calcium signals from internal calcium stores. As such, the stronger signal generated by dopamine stimulation may due to the cumulative effect of both calcium influx and calcium release from internal stores.
Fig. 5.
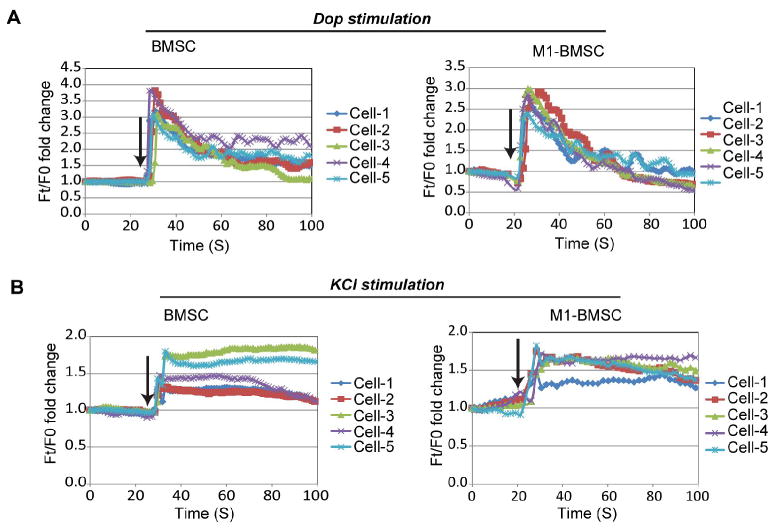
Fluorescence intensity profiles of uninduced BMSCs and M1-BMSCs stimulated by dopamine and KCl. (A) Fluorescence profile for five representative cells stimulated with 100 μM dopamine. Fluorescence intensity at time t (Ft) is normalized to fluorescence intensity at the starting point (F0). Arrows indicate the time of dopamine addition. (A) Fluorescence profile for five representative cells stimulated with 50 mM KCl. Fluorescence intensity at time t (Ft) is normalized to fluorescence intensity at the starting point (F0). Arrows indicate the time of KCl addition
Fig. 6.
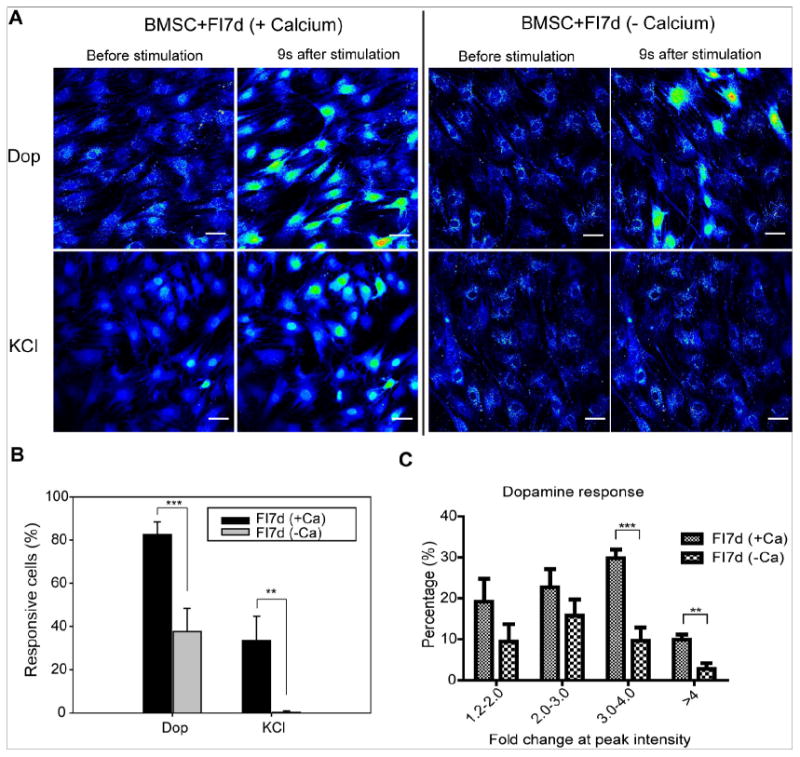
Increase in calcium signal upon agonist stimulation requires calcium influx. (A) Calcium imaging in response to dopamine and KCl stimulation. BMSCs were treated with 10 μM forskolin and 100 μM IBMX (FI) for 1 week before stimulation. After FI treatment, cells were loaded with Fluo-4 in ACSF-HEPES buffer or calcium-free ACSF-HEPES buffer. To check whether calcium influx is the mechanism for signal increase, either ACSF-HEPES buffer or calcium-free ACSF-HEPES buffer was used during imaging. Images were taken under time-lapse mode upon stimulation with 100 μM dopamine or 50 mM KCl. Representative images before stimulation and 9 seconds after stimulation are shown. Fluorescence images represented by a spectral table; warmer colors indicate higher fluorescence intensities (yellow to red) and cooler colors indicate lower fluorescence intensities (blue to green). Gain and offset are held constant in each experiment (before stimulation until the end of stimulation). Scale bar: 50 μm. (B) Quantification of dopamine and KCl responsive cells in the presence (+Ca) or absence (−Ca) of calcium (n=4). (C) Distribution of dopamine responsive cells into different categories (1.2-2.0, 2.0-3.0, 3.0-4.0 and >4.0) based on the fold change at peak intensity (n=3). **: p <0.01, ***: p <0.001.
3.4 Knock-down of CREB activity greatly reduces calcium signaling in cAMP-induced BMSCs
After FI induction for one week, however, M1-BMSCs lost their ability to respond to dopamine as compared to BMSCs (Fig. 7A). Quantification of the results suggests that the uninduced M1-BMSCs and uninduced BMSCs respond similarly to dopamine and KCl (Fig. 7B). However, the response of M1-BMSCs to dopamine and KCl reduced dramatically after one week of FI induction (Fig. 7B). When a constitutively active form of CREB (VP16-CREB) was transiently introduced into the M1-BMSCs, the number of dopamine responsive cells was significantly increased, while the KCl responsive cells increased only slightly and non-significantly (Fig. 7B), suggesting a role of CREB in the calcium rise in response to these two modulators. Since the responses of the M1-BMSCs to dopamine and KCl were compromised (Fig. 7B), we evaluated whether the cell integrity was affected after one week of FI treatment. Apoptosis and necrosis staining indicate that ~93% of the attached BMSCs and ~90% of the attached M1-BMSCs are still healthy (Supplementary Fig. S5), indicating the reduction or loss of calcium response to dopamine and KCl stimulation is not likely from loss of cellular integrity. Second, we used a positive control, ATP, to evaluate whether M1-BMSCs are still able to generate calcium signal when stimulated. The M1-BMSCs induced with FI for a week remained responsive to ATP (Fig. 7C), indicating that calcium signaling is still functioning properly in cAMP induced M1-BMSCs, although these cells are significantly less responsive to dopamine and KCl.
Fig. 7.
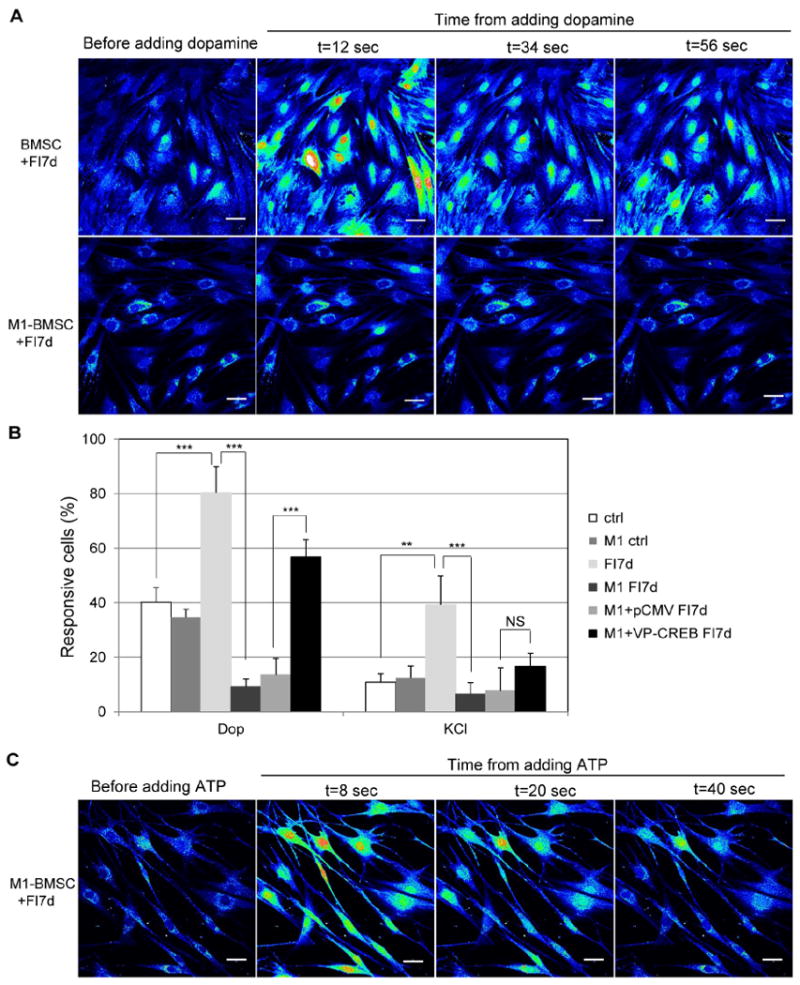
Calcium response upon stimulation by dopamine and KCl in cAMP-induced cells. (A) BMSCs (expressing control vector) and M1-BMSCs (expressing M1-CREB) before dopamine stimulation and 12, 34 and 56 seconds after 100 μM dopamine stimulation. Cells were induced with 10 μM forskolin and 100 μM IBMX (FI) for 7 days before dopamine stimulation. Scale bar: 50 μm. (B) Quantification of the percentage of cells that respond to dopamine and KCl (n=4). Ctrl: control cells; M1: M1-BMSCs; FI7d: control cells treated with FI for 7 days; M1 FI7d: M1-BMSCs treated with FI for 7 days; M1+pCMV FI7d: M1-BMSCs transfected with control pCMV vector and treated with FI for 7 days; M1+VP-CREB FI7d: M1-BMSCs transfected with the constitutively active VP16-CREB and treated with FI for 7 days. *: p <0.05; **: p<0.01, ***: p <0.001; NS: not significant. (C) Calcium imaging in response to the positive control ATP for M1-BMSCs induced with FI for 7 days. Cells were loaded with Fluo-4 and stimulated with 100 μM ATP. Representative images before ATP stimulation and 8, 20 and 40 seconds after ATP stimulation were shown. Scale bar: 50 μm.
3.5 CREB alters expression of certain dopamine receptors
As the dopamine response is stronger and more robust as compared to the KCl response, we focused further analysis on the dopamine response. To investigate how CREB modulates the calcium signaling upon stimulation by dopamine, we assessed whether CREB could modulate the transcription of dopamine receptors. To our surprise, knock down of CREB activity increased rather than decreased the mRNA levels of the dopamine receptor D1 (Fig. 8A). The mRNA levels of the dopamine receptor D2 was undetectable while the dopamine receptors D3, D4 and D5 were not affected by knocking-down CREB activity in the BMSCs (Fig. 8A). When the BMSCs were treated with FI, D1 mRNA increased ~3.2 fold as compared to control BMSCs (Fig. 8A). After FI treatment, D1 mRNA level was significantly higher in M1-BMSCs than in BMSCs (Fig. 8A). Since the D1 mRNA level exhibited greater differential change with both CREB knock-down and FI treatment, we further assessed the regulation of D1 by overexpression and silencing studies. Overexpression is achieved by introducing the constitutively active form of VP16-CREB and silencing through CREB siRNA #1. Overexpression of CREB led to a ~20% reduction in D1 mRNA level, silencing resulted in approximately 60% increase in D1 mRNA level (Fig. 8B), These results support our observation that knocking down CREB activity in the M1-BMSCs caused an increase in D1 mRNA levels after FI treatment for 7 days.
Fig. 8.
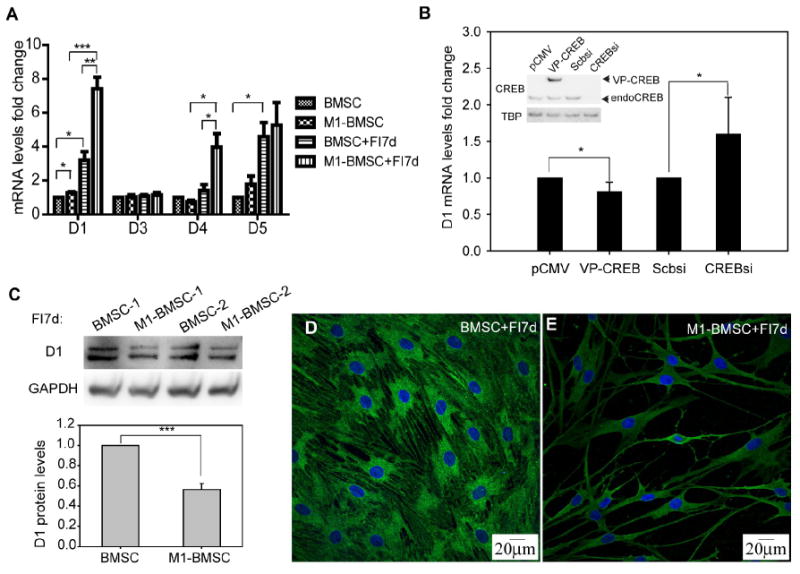
Expression of dopamine receptors. (A) mRNA levels of dopamine receptors D1, D3, D4 and D5 for control cells and cells treated with 10 μM forskolin and 100 μM IBMX (FI) for 7 days (n=3). BMSCs: expressing the control vector; M1-BMSCs: expressing M1-CREB. (B) D1 mRNA levels for BMSCs with CREB overexpression or silencing. CREB overexpression was achieved by transfection of the constitutively active VP16-CREB (denoted as VP-CREB) and silencing was achieved by CREB siRNA #1. pCMV and scramble siRNA were used as controls (n=5). The western blot insert in figure (B) indicates the overexpression and silencing results of CREB. Endogenous CREB is denoted as endoCREB. (C) Protein levels of dopamine receptor 1 (D1) for BMSCs and M1-BMSCs treated with FI for 7 days (n=3). (D) Dopamine receptor D1 staining for BMSCs. (E) Dopamine receptor D1 staining for M1-BMSCs. Green: D1 staining; blue: nucleus staining. *: p<0.05. **: p<0.01, ***: p<0.001.
Promoter analysis revealed that the D1 promoter contains a half CRE site that could be regulated by the CREB/ATF family transcription factors (Supplementary Fig. S6A). CREB is a transcriptional activator, and thus knock-down of CREB is expected to increase rather than decrease D1 gene expression. A potential reason for the upregulation of D1 mRNA after CREB knock-down is: while CREB is an activator of the cAMP-responsive genes, it could induce the expression of a transcriptional inhibitor, inducible cAMP early repressor (ICER), which could compete with CREB for binding to the CRE sites to thereby repress the transcription of cAMP-responsive genes [19]. We observed that cAMP treatment resulted in a constant upregulation of ICER (Supplementary Fig. S6B). It is possible that ICER could bind to the CRE site and inhibit D1 transcription. Therefore, when CREB activity is knocked down, expression of ICER would be inhibited, thereby relieving the transcriptional inhibition of D1. Alternatively, CREB may modulate D1 transcription indirectly through the regulation of other transcription factors. We have observed that c-fos was transiently upregulated by cAMP (Supplementary Fig.S6C). c-fos has been shown to upregulate Sp3 [48], a transcription factor that inhibit D1 expression [49]. Therefore, it could be possible that CREB indirectly inhibits D1 transcription by upregulating c-fos expression, which increases Sp3 expression and in turn represses D1 transcription.
While D1 mRNA level is higher in the M1-BMSCs as compared to the BMSCs upon FI treatment, the change in protein level was reversed. Immunoblotting results suggest that the M1-BMSCs treated with FI for 7 days express less D1 than the BMSCs treated with FI for 7 days (Fig. 8C). The immunostaining results further confirmed the immunoblotting results, i.e. down-regulating CREB activity decreased the dopamine receptor D1 protein level. Fluorescence intensity of individual M1-BMSCs (expressing M1-CREB) immunostained against the D1 antibody also appeared to be lower than that of the control BMSCs (Fig. 8D and 8E). The reduced protein levels of dopamine receptor D1 could contribute, in part, to the reduced dopamine response in M1-BMSCs.
4. Discussion
In this study, we have shown that CREB plays an important role in regulating the calcium signal induced by dopamine and KCl in cAMP-induced BMSCs. Both silencing CREB and a dominant negative CREB (M1-CREB) reduced the calcium response (Fig. 2 and 7), and reintroducing a constitutively active CREB into the M1-BMSCs partially restored the calcium response (Fig. 7B), implicating a role of CREB in the regulation of calcium signaling. One possible reason for the partial, i.e. incomplete, restoration after reintroducing the constitutively active CREB (VP-CREB) is the transient transfection in which not all of the cells present are transfected. However, the transfection efficiency is sufficiently high (>60%) for assessing the effect of VP-CREB on calcium imaging in the M1-BMSCs.
How CREB regulates calcium signaling remains a mystery. Potential mechanisms could involve the down-regulation of the membrane receptors that respond to the modulators. We investigated the impact of CREB on the expression of the dopamine receptors, since dopamine elicited the most robust response. Knock-down of CREB activity in the M1-BMSCs reduced the protein level of D1 (Fig. 8C, 8D and 8E). A reduction of the dopamine receptors could contribute, in part, to the observed reduction in the dopamine response. However, this does not preclude other mechanisms that might contribute to the CREB-mediated calcium response elicited by dopamine. One of the mechanisms that the D1 receptor modulate intracellular calcium signaling is through stimulating phospholipase C (PLC) activity, which leads to hydrolysis of phosphatidylinositol (PI) and subsequent production of inositol 1,4,5-trisphosphate (Ins(1,4,5)P3) that triggers calcium release from internal stores [17]. In addition, signaling initiated by the D1 receptor may result in calcium channel phosphorylation and affect its activity. It has been shown that calcium currents in rat striatal neurons is blocked by PKA inhibitors [50]. Recent identification of the dopamine-receptor interacting proteins (DRIPs) adds another level of complexity to dopamine stimulated calcium signaling. The DRIP proteins includes a cohort of receptors and channels, cytoskeletal proteins, signaling proteins, and adaptors that act in concert to transduce dopamine stimulated calcium signal [51]. Moreover, dopamine can also generate calcium through potentiation of the N-methyl-D-aspartate (NMDA) receptor. A previous study demonstrated that application of a D1 agonist increased the activity of NMDA receptors [52]. Another study indicated that NMDA receptors are expressed on rat BMSCs [53]. Therefore, it is likely that CREB could have both direct and indirect effects on the calcium signal elicited by dopamine. Taken together, calcium signal generated by dopamine stimulation can be regulated by a number of mechanisms and is quite complicated. In contrast, calcium signal generated by KCl stimulation is relatively simpler and mainly occurs through activation of voltage-gated calcium channels (VGCCs) by depolarizing the membrane. The mechanistic difference upon dopamine and KCl stimulation could explain in part the differential calcium signaling patterns (Fig. 5). VGCCs are composed by a complex of proteins, including a pore forming α1 subunit (10 known isoforms), a transmembrane α2 and δ subunit (4 known isoforms), an intracellular β subunit (4 known isoforms), and sometimes a transmembrane γ subunit [54]. Li and co-workers reported that the L-type calcium channel α2 subunit (CCHL2a) is expressed in rat BMSCs [16]. It is postulated that KCl induced calcium signal in cAMP-induced BMSCs could occur through activation of certain types of VGCCs, however, further experiments are required to characterize the specific channels. Since the regulation of the calcium response by the modulators used in this study involves a variety of signaling components [17, 54-56], unraveling the complete mechanism by which CREB regulates calcium signaling remains an important task for the future.
It is unclear why the D1 mRNA level increased while its protein level decreased. In fact, many times mRNA and protein levels poorly correlate with each other due to several reasons: complex posttranscriptional regulation, varied protein half life, and noise in data processing [57]. As an example, D1 expression can be posttranscriptionally regulated by microRNAs (miRNAs), which bind to the 3′-untranslated (3′-UTR) regions of the mRNA and suppress translation [58]. A recent study suggested that the expression of D1 can be modulated by miRNA miR-504 [59], although it is unknown how miR-504 expression is regulated. Analysis of the miR-504 promoter region reveals that it contains a half CRE site (TGACG), indicating that it could be regulated by CREB [60, 61]. Thus a possible scenario is that knock-down of CREB activity results in an upregulation of the miR-504 levels through a yet unknown mechanism, to downregulate the D1 protein level.
In addition to the impact on calcium signaling, knock-down of CREB activity also appeared to affect the cell morphology. The M1-BMSCs initially appeared similar to the BMSCs, but continuous remodeling of the cytoskeleton during FI induction resulted in long projections in the M1-BMSCs (Supplementary Fig. S7). Potential regulators of the morphological changes are the Rho GTPase family proteins, Rho, Rac and Cdc42, which are key players in regulating cytoskeletal reorganization [62]. Activity of Rho GTPases is regulated by the association with GTP (active state) or GDP (inactive state) [63]. Guanine nucleotide exchange factors (GEFs) are Rho activators which switch Rho to a GTP-bound state, whereas GTPase-activating proteins (GAPs) are inhibitors which switch Rho to a GDP-bound state [63, 64]. It has been shown that CREB can transcriptionally regulate a RhoA GTPase named p190RhoGAP [65] and two p190RhoGAP inhibitors, Par6C and Rnd3 [66]. In addition, a search of a CREB target gene database suggests that the Rho signaling downstream component, p21-activated kinase 1 (PAK1), can also be transcriptionally regulated by CREB [61]. Therefore, it is possible that knock-down of CREB activity influences the Rho signaling pathway and in turn modulates cytoskeletal changes in the cAMP-induced BMSCs.
In conclusion, we examined the role of CREB in mediating calcium signaling elicited by modulators in cAMP-induced BMSCs. The absence of CREB activity greatly reduced the calcium response to dopamine and KCl in cAMP-induced BMSCs. The lack of CREB activity also appears to result in dramatic morphological changes and render the cells more prone to apoptosis.
Supplementary Material
Highlights.
BMSCs but not fibroblasts are responsive to dopamine and KCl.
Silencing of CREB reduces the number of dopamine and KCl responsive cells in cAMP induced BMSCs.
Knock-down of CREB activity minimally affect calcium signaling in BMSCs without cAMP treatment.
Knock-down of CREB activity greatly reduces calcium signaling in cAMP-induced BMSCs.
CREB alters expression of certain dopamine receptors.
Acknowledgments
We thank Dr. David Ginty at Johns Hopkins University for kindly providing the pCMV control vector, VP16-CREB and M1-CREB plasmids, Dr. Carlos A. Molina from Montclair State University for kindly providing the ICER antibody, Dr. William Atchinson for his advice and insightful discussion on calcium imaging, Dr. Melinda Frame from the Center for Advanced Microscopy at Michigan State University (MSU) for help with confocal microscopy imaging and Dr. Louis King from Research Technology Support Facility at MSU for help with flow cytometry. This study was supported in part by the National Science Foundation (CBET 0941055 and CBET 1049127), and the National Institute of Health (R01GM079688, R01GM089866, R21RR024439 and R21CA176854).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Chamberlain G, Fox J, Ashton B, Middleton J. Concise review: mesenchymal stem cells: their phenotype, differentiation capacity, immunological features, and potential for homing. Stem Cells. 2007;25:2739–49. doi: 10.1634/stemcells.2007-0197. [DOI] [PubMed] [Google Scholar]
- 2.Berridge MJ, Lipp P, Bootman MD. The versatility and universality of calcium signalling. Nat Rev Mol Cell Biol. 2000;1:11–21. doi: 10.1038/35036035. [DOI] [PubMed] [Google Scholar]
- 3.Sun S, Liu Y, Lipsky S, Cho M. Physical manipulation of calcium oscillations facilitates osteodifferentiation of human mesenchymal stem cells. FASEB J. 2007;21:1472–80. doi: 10.1096/fj.06-7153com. [DOI] [PubMed] [Google Scholar]
- 4.Wen L, Wang Y, Wang H, Kong L, Zhang L, Chen X, Ding Y. L-type calcium channels play a crucial role in the proliferation and osteogenic differentiation of bone marrow mesenchymal stem cells. Biochem Biophys Res Commun. 2012;424:439–45. doi: 10.1016/j.bbrc.2012.06.128. [DOI] [PubMed] [Google Scholar]
- 5.Matta C, Fodor J, Szijgyarto Z, Juhasz T, Gergely P, Csernoch L, Zakany R. Cytosolic free Ca2+ concentration exhibits a characteristic temporal pattern during in vitro cartilage differentiation: a possible regulatory role of calcineurin in Ca-signalling of chondrogenic cells. Cell Calcium. 2008;44:310–23. doi: 10.1016/j.ceca.2007.12.010. [DOI] [PubMed] [Google Scholar]
- 6.Oca P, Zaka R, Dion AS, Freeman TA, Williams CJ. Phosphate and calcium are required for TGFbeta-mediated stimulation of ANK expression and function during chondrogenesis. J Cell Physiol. 2010;224:540–8. doi: 10.1002/jcp.22155. [DOI] [PubMed] [Google Scholar]
- 7.Ntambi JM, Takova T. Role of Ca2+ in the early stages of murine adipocyte differentiation as evidenced by calcium mobilizing agents. Differentiation. 1996;60:151–8. doi: 10.1046/j.1432-0436.1996.6030151.x. [DOI] [PubMed] [Google Scholar]
- 8.Shi H, Halvorsen YD, Ellis PN, Wilkison WO, Zemel MB. Role of intracellular calcium in human adipocyte differentiation. Physiol Genomics. 2000;3:75–82. doi: 10.1152/physiolgenomics.2000.3.2.75. [DOI] [PubMed] [Google Scholar]
- 9.Szabo E, Qiu Y, Baksh S, Michalak M, Opas M. Calreticulin inhibits commitment to adipocyte differentiation. J Cell Biol. 2008;182:103–16. doi: 10.1083/jcb.200712078. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 10.Kawano S, Shoji S, Ichinose S, Yamagata K, Tagami M, Hiraoka M. Characterization of Ca(2+) signaling pathways in human mesenchymal stem cells. Cell Calcium. 2002;32:165–74. doi: 10.1016/s0143416002001240. [DOI] [PubMed] [Google Scholar]
- 11.Kawano S, Otsu K, Shoji S, Yamagata K, Hiraoka M. Ca(2+) oscillations regulated by Na(+)-Ca(2+) exchanger and plasma membrane Ca(2+) pump induce fluctuations of membrane currents and potentials in human mesenchymal stem cells. Cell Calcium. 2003;34:145–56. doi: 10.1016/s0143-4160(03)00069-1. [DOI] [PubMed] [Google Scholar]
- 12.Kawano S, Otsu K, Kuruma A, Shoji S, Yanagida E, Muto Y, Yoshikawa F, Hirayama Y, Mikoshiba K, Furuichi T. ATP autocrine/paracrine signaling induces calcium oscillations and NFAT activation in human mesenchymal stem cells. Cell Calcium. 2006;39:313–24. doi: 10.1016/j.ceca.2005.11.008. [DOI] [PubMed] [Google Scholar]
- 13.Tropel P, Platet N, Platel JC, Noel D, Albrieux M, Benabid AL, Berger F. Functional neuronal differentiation of bone marrow-derived mesenchymal stem cells. Stem Cells. 2006;24:2868–76. doi: 10.1634/stemcells.2005-0636. [DOI] [PubMed] [Google Scholar]
- 14.Berridge MJ, Bootman MD, Roderick HL. Calcium signalling: dynamics, homeostasis and remodelling. Nat Rev Mol Cell Biol. 2003;4:517–29. doi: 10.1038/nrm1155. [DOI] [PubMed] [Google Scholar]
- 15.Parekh AB, Putney JW., Jr Store-operated calcium channels. Physiol Rev. 2005;85:757–810. doi: 10.1152/physrev.00057.2003. [DOI] [PubMed] [Google Scholar]
- 16.Li GR, Deng XL, Sun H, Chung SS, Tse HF, Lau CP. Ion channels in mesenchymal stem cells from rat bone marrow. Stem Cells. 2006;24:1519–28. doi: 10.1634/stemcells.2005-0307. [DOI] [PubMed] [Google Scholar]
- 17.Missale C, Nash SR, Robinson SW, Jaber M, Caron MG. Dopamine receptors: from structure to function. Physiol Rev. 1998;78:189–225. doi: 10.1152/physrev.1998.78.1.189. [DOI] [PubMed] [Google Scholar]
- 18.Nicola SM, Surmeier J, Malenka RC. Dopaminergic modulation of neuronal excitability in the striatum and nucleus accumbens. Annu Rev Neurosci. 2000;23:185–215. doi: 10.1146/annurev.neuro.23.1.185. [DOI] [PubMed] [Google Scholar]
- 19.Mayr B, Montminy M. Transcriptional regulation by the phosphorylation-dependent factor CREB. Nat Rev Mol Cell Biol. 2001;2:599–609. doi: 10.1038/35085068. [DOI] [PubMed] [Google Scholar]
- 20.Haus-Seuffert P, Meisterernst M. Mechanisms of transcriptional activation of cAMP-responsive element-binding protein CREB. Mol Cell Biochem. 2000;212:5–9. [PubMed] [Google Scholar]
- 21.Johannessen M, Delghandi MP, Moens U. What turns CREB on? Cell Signal. 2004;16:1211–27. doi: 10.1016/j.cellsig.2004.05.001. [DOI] [PubMed] [Google Scholar]
- 22.Shaywitz AJ, Greenberg ME. CREB: a stimulus-induced transcription factor activated by a diverse array of extracellular signals. Annu Rev Biochem. 1999;68:821–61. doi: 10.1146/annurev.biochem.68.1.821. [DOI] [PubMed] [Google Scholar]
- 23.Zhang L, Seitz LC, Abramczyk AM, Liu L, Chan C. cAMP initiates early phase neuron-like morphology changes and late phase neural differentiation in mesenchymal stem cells. Cell Mol Life Sci. 2010;68:863–76. doi: 10.1007/s00018-010-0497-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rubi B, Maechler P. Minireview: new roles for peripheral dopamine on metabolic control and tumor growth: let's seek the balance. Endocrinology. 2010;151:5570–81. doi: 10.1210/en.2010-0745. [DOI] [PubMed] [Google Scholar]
- 25.Basu S, Dasgupta PS, Lahiri T, Chowdhury JR. Uptake and biodistribution of dopamine in bone marrow, spleen and lymph nodes of normal and tumor bearing mice. Life Sci. 1993;53:415–24. doi: 10.1016/0024-3205(93)90645-j. [DOI] [PubMed] [Google Scholar]
- 26.Trzaska KA, King CC, Li KY, Kuzhikandathil EV, Nowycky MC, Ye JH, Rameshwar P. Brain-derived neurotrophic factor facilitates maturation of mesenchymal stem cell-derived dopamine progenitors to functional neurons. J Neurochem. 2009;110:1058–69. doi: 10.1111/j.1471-4159.2009.06201.x. [DOI] [PubMed] [Google Scholar]
- 27.Riminucci M, Fisher LW, Shenker A, Spiegel AM, Bianco P, Gehron Robey P. Fibrous dysplasia of bone in the McCune-Albright syndrome: abnormalities in bone formation. Am J Pathol. 1997;151:1587–600. [PMC free article] [PubMed] [Google Scholar]
- 28.Marie PJ, de Pollak C, Chanson P, Lomri A. Increased proliferation of osteoblastic cells expressing the activating Gs alpha mutation in monostotic and polyostotic fibrous dysplasia. Am J Pathol. 1997;150:1059–69. [PMC free article] [PubMed] [Google Scholar]
- 29.Hanson AS, Linas SL. Parathyroid hormone/adenylate cyclase coupling in vascular smooth muscle cells. Hypertension. 1994;23:468–75. doi: 10.1161/01.hyp.23.4.468. [DOI] [PubMed] [Google Scholar]
- 30.Nickols GA, Metz MA, Cline WH., Jr Endothelium-independent linkage of parathyroid hormone receptors of rat vascular tissue with increased adenosine 3′,5′-monophosphate and relaxation of vascular smooth muscle. Endocrinology. 1986;119:349–56. doi: 10.1210/endo-119-1-349. [DOI] [PubMed] [Google Scholar]
- 31.Schulz R, Rose J, Martin C, Brodde OE, Heusch G. Development of short-term myocardial hibernation. Its limitation by the severity of ischemia and inotropic stimulation. Circulation. 1993;88:684–95. doi: 10.1161/01.cir.88.2.684. [DOI] [PubMed] [Google Scholar]
- 32.Lubbe WF, Podzuweit T, Opie LH. Potential arrhythmogenic role of cyclic adenosine monophosphate (AMP) and cytosolic calcium overload: implications for prophylactic effects of beta-blockers in myocardial infarction and proarrhythmic effects of phosphodiesterase inhibitors. J Am Coll Cardiol. 1992;19:1622–33. doi: 10.1016/0735-1097(92)90629-2. [DOI] [PubMed] [Google Scholar]
- 33.Pearse DD, Pereira FC, Marcillo AE, Bates ML, Berrocal YA, Filbin MT, Bunge MB. cAMP and Schwann cells promote axonal growth and functional recovery after spinal cord injury. Nat Med. 2004;10:610–6. doi: 10.1038/nm1056. [DOI] [PubMed] [Google Scholar]
- 34.Cai D, Qiu J, Cao Z, McAtee M, Bregman BS, Filbin MT. Neuronal cyclic AMP controls the developmental loss in ability of axons to regenerate. J Neurosci. 2001;21:4731–9. doi: 10.1523/JNEUROSCI.21-13-04731.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Shenoy P, Agarwal V. Phosphodiesterase inhibitors in the management of autoimmune disease. Autoimmun Rev. 9:511–5. doi: 10.1016/j.autrev.2010.02.012. [DOI] [PubMed] [Google Scholar]
- 36.Kumar N, Goldminz AM, Kim N, Gottlieb AB. Phosphodiesterase 4-targeted treatments for autoimmune diseases. BMC Med. 11:96. doi: 10.1186/1741-7015-11-96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jia B, Madsen L, Petersen RK, Techer N, Kopperud R, Ma T, Doskeland SO, Ailhaud G, Wang J, Amri EZ, Kristiansen K. Activation of protein kinase A and exchange protein directly activated by cAMP promotes adipocyte differentiation of human mesenchymal stem cells. PLoS One. 2012;7:e34114. doi: 10.1371/journal.pone.0034114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kao R, Lu W, Louie A, Nissenson R. Cyclic AMP signaling in bone marrow stromal cells has reciprocal effects on the ability of mesenchymal stem cells to differentiate into mature osteoblasts versus mature adipocytes. Endocrine. 2012;42:622–36. doi: 10.1007/s12020-012-9717-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kim JM, Choi JS, Kim YH, Jin SH, Lim S, Jang HJ, Kim KT, Ryu SH, Suh PG. An activator of the cAMP/PKA/CREB pathway promotes osteogenesis from human mesenchymal stem cells. J Cell Physiol. 2013;228:617–26. doi: 10.1002/jcp.24171. [DOI] [PubMed] [Google Scholar]
- 40.Yang DC, Tsay HJ, Lin SY, Chiou SH, Li MJ, Chang TJ, Hung SC. cAMP/PKA regulates osteogenesis, adipogenesis and ratio of RANKL/OPG mRNA expression in mesenchymal stem cells by suppressing leptin. PLoS One. 2008;3:e1540. doi: 10.1371/journal.pone.0001540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Caplan AI. Adult mesenchymal stem cells for tissue engineering versus regenerative medicine. J Cell Physiol. 2007;213:341–7. doi: 10.1002/jcp.21200. [DOI] [PubMed] [Google Scholar]
- 42.Parekkadan B, Milwid JM. Mesenchymal stem cells as therapeutics. Annu Rev Biomed Eng. 12:87–117. doi: 10.1146/annurev-bioeng-070909-105309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhang L, Chan C. Isolation and enrichment of rat mesenchymal stem cells (MSCs) and separation of single-colony derived MSCs. J Vis Exp. 2010 doi: 10.3791/1852. http://www.jove.com/index/Details.stp?ID=1852. [DOI] [PMC free article] [PubMed]
- 44.Bell PB, Jr, Safiejko-Mroczka B. Improved methods for preserving macromolecular structures and visualizing them by fluorescence and scanning electron microscopy. Scanning Microsc. 1995;9:843–57. discussion 858-60. [PubMed] [Google Scholar]
- 45.Alt E, Yan Y, Gehmert S, Song YH, Altman A, Vykoukal D, Bai X. Fibroblasts share mesenchymal phenotypes with stem cells, but lack their differentiation and colony-forming potential. Biol Cell. 2011;103:197–208. doi: 10.1042/BC20100117. [DOI] [PubMed] [Google Scholar]
- 46.Vassort G. Adenosine 5′-triphosphate: a P2-purinergic agonist in the myocardium. Physiol Rev. 2001;81:767–806. doi: 10.1152/physrev.2001.81.2.767. [DOI] [PubMed] [Google Scholar]
- 47.Dubyak GR, el-Moatassim C. Signal transduction via P2-purinergic receptors for extracellular ATP and other nucleotides. Am J Physiol. 1993;265:C577–606. doi: 10.1152/ajpcell.1993.265.3.C577. [DOI] [PubMed] [Google Scholar]
- 48.Tapias A, Ciudad CJ, Noe V. Transcriptional regulation of the 5′-flanking region of the human transcription factor Sp3 gene by NF-1, c-Myb, B-Myb, AP-1 and E2F. Biochim Biophys Acta. 2008;1779:318–29. doi: 10.1016/j.bbagrm.2008.02.006. [DOI] [PubMed] [Google Scholar]
- 49.Yang Y, Hwang CK, Junn E, Lee G, Mouradian MM. ZIC2 and Sp3 repress Sp1-induced activation of the human D1A dopamine receptor gene. J Biol Chem. 2000;275:38863–9. doi: 10.1074/jbc.M007906200. [DOI] [PubMed] [Google Scholar]
- 50.Surmeier DJ, Bargas J, Hemmings HC, Jr, Nairn AC, Greengard P. Modulation of calcium currents by a D1 dopaminergic protein kinase/phosphatase cascade in rat neostriatal neurons. Neuron. 1995;14:385–97. doi: 10.1016/0896-6273(95)90294-5. [DOI] [PubMed] [Google Scholar]
- 51.Bergson C, Levenson R, Goldman-Rakic PS, Lidow MS. Dopamine receptor-interacting proteins: the Ca(2+) connection in dopamine signaling. Trends Pharmacol Sci. 2003;24:486–92. doi: 10.1016/S0165-6147(03)00232-3. [DOI] [PubMed] [Google Scholar]
- 52.Chen G, Greengard P, Yan Z. Potentiation of NMDA receptor currents by dopamine D1 receptors in prefrontal cortex. Proc Natl Acad Sci U S A. 2004;101:2596–600. doi: 10.1073/pnas.0308618100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Woodbury D, Reynolds K, Black IB. Adult bone marrow stromal stem cells express germline, ectodermal, endodermal, and mesodermal genes prior to neurogenesis. J Neurosci Res. 2002;69:908–17. doi: 10.1002/jnr.10365. [DOI] [PubMed] [Google Scholar]
- 54.Catterall WA. Structure and regulation of voltage-gated Ca2+ channels. Annu Rev Cell Dev Biol. 2000;16:521–55. doi: 10.1146/annurev.cellbio.16.1.521. [DOI] [PubMed] [Google Scholar]
- 55.Dingledine R, Borges K, Bowie D, Traynelis SF. The glutamate receptor ion channels. Pharmacol Rev. 1999;51:7–61. [PubMed] [Google Scholar]
- 56.Conn PJ, Battaglia G, Marino MJ, Nicoletti F. Metabotropic glutamate receptors in the basal ganglia motor circuit. Nat Rev Neurosci. 2005;6:787–98. doi: 10.1038/nrn1763. [DOI] [PubMed] [Google Scholar]
- 57.Greenbaum D, Colangelo C, Williams K, Gerstein M. Comparing protein abundance and mRNA expression levels on a genomic scale. Genome Biol. 2003;4:117. doi: 10.1186/gb-2003-4-9-117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Pillai RS, Bhattacharyya SN, Filipowicz W. Repression of protein synthesis by miRNAs: how many mechanisms? Trends Cell Biol. 2007;17:118–26. doi: 10.1016/j.tcb.2006.12.007. [DOI] [PubMed] [Google Scholar]
- 59.Huang W, Li MD. Differential allelic expression of dopamine D1 receptor gene (DRD1) is modulated by microRNA miR-504. Biol Psychiatry. 2009;65:702–5. doi: 10.1016/j.biopsych.2008.11.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Karolchik D, Baertsch R, Diekhans M, Furey TS, Hinrichs A, Lu YT, Roskin KM, Schwartz M, Sugnet CW, Thomas DJ, Weber RJ, Haussler D, Kent WJ. The UCSC Genome Browser Database. Nucleic Acids Res. 2003;31:51–4. doi: 10.1093/nar/gkg129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zhang X, Odom DT, Koo SH, Conkright MD, Canettieri G, Best J, Chen H, Jenner R, Herbolsheimer E, Jacobsen E, Kadam S, Ecker JR, Emerson B, Hogenesch JB, Unterman T, Young RA, Montminy M. Genome-wide analysis of cAMP-response element binding protein occupancy, phosphorylation, and target gene activation in human tissues. Proc Natl Acad Sci U S A. 2005;102:4459–64. doi: 10.1073/pnas.0501076102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Hall A. Rho GTPases and the actin cytoskeleton. Science. 1998;279:509–14. doi: 10.1126/science.279.5350.509. [DOI] [PubMed] [Google Scholar]
- 63.Etienne-Manneville S, Hall A. Rho GTPases in cell biology. Nature. 2002;420:629–35. doi: 10.1038/nature01148. [DOI] [PubMed] [Google Scholar]
- 64.Jaffe AB, Hall A. Rho GTPases: biochemistry and biology. Annu Rev Cell Dev Biol. 2005;21:247–69. doi: 10.1146/annurev.cellbio.21.020604.150721. [DOI] [PubMed] [Google Scholar]
- 65.Chava KR, Tauseef M, Sharma T, Mehta D. Cyclic AMP response element-binding protein prevents endothelial permeability increase through transcriptional controlling p190RhoGAP expression. Blood. 2012;119:308–19. doi: 10.1182/blood-2011-02-339473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Lesiak A, Pelz C, Ando H, Zhu M, Davare M, Lambert TJ, Hansen KF, Obrietan K, Appleyard SM, Impey S, Wayman GA. A genome-wide screen of CREB occupancy identifies the RhoA inhibitors Par6C and Rnd3 as regulators of BDNF-induced synaptogenesis. PLoS One. 2013;8:e64658. doi: 10.1371/journal.pone.0064658. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.