

Abstract
Mutants of tumor suppressor p53 not only lose the activity in genome stabilizing and in tumor suppression, but also exhibit oncogenic function in cancer cells. Most efforts in restoring p53 biological activity focus on either altering mutant-protein conformation or introducing an exogenous p53 gene into cells to eliminate p53-mutant cancer cells. Being different from these, we report that ceramide can restore the expression of wild-type p53 and induce p53-dependent apoptosis in deletion-mutant cancer cells. We show that endogenous long-carbon chain ceramide species (C16- to C24-ceramides) and exogenous C6-ceramide, rather than other sphingolipids, restore wild-type mRNA (intact exon-5), phosphorylated protein (Ser15 in exon-5) of p53, and p53-responsive proteins, including p21 and Bax, in ovarian cancer cells, which predominantly express a deleted exon-5 of p53 mutant before treatments. Consequently, the restored p53 sensitizes these p53-mutant cancer cells to DNA damage-induced growth arrest and apoptosis. Furthermore, we elucidate that ceramide activates protein phosphatase-1, and then the dephosphorylated serine/arginine-rich splicing-factor 1 (SRSF1) is translocated to the nucleus, thus promoting pre-mRNA splicing preferentially to wild-type p53 expression. These findings disclose an unrecognized mechanism that pre-mRNA splicing dysfunction can result in p53 deletion-mutants. Ceramide through SRSF1 restores wild-type p53 expression versus deletion-mutant and leads cancer cells to apoptosis. This suggests that heterozygous deletion-mutants of p53 can be restored in posttranscriptional level by using epigenetic approaches.
Keywords: Ceramide, Pre-mRNA splicing, p53 mutant, Restoration, SRSF1, Cancer
1. Introduction
The p53 protein, encoded by human gene TP53, is a key tumor suppressor that stabilizes the genome, preventing tumorigenesis and cancer progression [1]. As an essential transcription factor, p53 activates the expression of p21, Bax, Puma, Fas and other p53-responsive genes, consequently promoting cell division-arrest, apoptosis, DNA repair and cell differentiation [2]. Mutants of p53, which are detected more frequently than any other gene, compromise its functions [3,4]. p53 mutants not only lose their activities in suppressing tumor, but they also confer dominant-negative activity and oncogenic function in cancer cells [2,3]. These changes promote tumor progression and result in drug resistance; therefore, p53 mutants have become the most common prognostic indicator both for tumor recurrence and for cancer death [2,5]. Most tumors that exhibit disrupted p53-signaling pathways remain addicted to p53 mutants, and p53 mutants have emerged as perhaps the most important target to improve cancer treatments [2,5]. Current approaches targeting p53 mutants mainly focus on replacing wild-type p53 by introducing an exogenous p53 gene, reactivating p53 mutants by altering mutant-protein conformation and augmenting wild-type p53 by inhibiting MDM2-mediated degradation [6–8]. In cancers, the dominant-negative activity and gain-of-function of p53 mutants potentially compromise the efficacy of these approaches [8,9]. To develop consistently effective approaches targeting p53 mutants, greater promise would seem to rest in the possibility of regulating the expression of wild-type p53 versus mutants in cancer cells, which are mostly heterozygous for the p53 gene.
Ceramide is the central metabolite of sphingolipids, and has myriad effects on cell function, including cell growth arrest, senescence, apoptosis and autophagy [10]. Furthermore, several reports have shown that ceramide is involved in regulating gene expression [11–14]. For example, ceramide upregulates the expression of p21 [11,12], cyclooxygenase-2 [15], and glucosylceramide synthase (GCS) [14]; it down-regulates the expression of c-myc [11] and human telomerase reverse transcriptase (hTERT) [16]. By activation of Sp1 binding to the promoter, ceramide increases the expression of GCS [14]; conversely, ceramide decreases hTERT promoter activity by rapid proteolysis of the ubiquitin-conjugated c-myc [16]. Interestingly, ceramide can modulate alternative pre-mRNA splicing process and allow cells to express apoptotic isoforms of bcl-x and caspase-9 [17,18]. Our previous report shows that suppression of ceramide glycosylation restores the expression of wild-type p53 protein in p53-mutant cells [19]. Current study examines whether or not ceramide modulates pre-mRNA splicing to regulate the expression of wild-type p53 protein in p53-mutant cells.
2. Materials and methods
2.1. Cell culture and treatments
Human NCI/ADR-RES ovary cancer cell line, which presents multi-drug resistance and a 7-amino acid deletion in exon-5 of tumor suppressor p53 [20], was kindly provided by Dr. Kenneth Cowan (UNMC Eppley Cancer Center, Omaha, NE, USA) and Dr. Merrill Goldsmith (National Cancer Institute, Bethesda, MD, USA). Cells were cultured in RPMI-1640 medium containing 10% fetus bovine serum (FBS), 100 units/ml penicillin, 100 μg/ml streptomycin, and 584 mg/l L-glutamine. Cells were maintained in an incubator humidified with 95% air and 5% CO2 at 37 °C.
For treatments, cells (3 × 106/100-mm dish; 4000 cells/well in 96-well plate) were grown in 10% FBS RPMI-1640 medium overnight and then treated with C6-ceramide (C6-Cer, 5 μM), C6-dihydroceramide (C6-diH-Cer, 5 μM), D-threo-1-phenyl-2-decanoylamino-3-morpholino-1-propanol (PDMP, 10 μM), okadaic acid (OA, 10 nM) and calyculin A (Cal A, 5 nM) in Opti-MEM reduced-serum medium for 4 h and then cultured in 5% FBS medium containing 2.5 μM doxorubicin (Dox) for additional 48 h, as described previously [14]. In combination groups, cells were pretreated with OA (10 nM) or Cal A (5 nM) in Opti-MEM reduced-serum medium for 4 h, and then treated with C6-Cer (5 μM) in 5% FBS medium for additional 48 h. C6-ceramide and C6-dihydroceramide were purchased from Biomol (Plymouth Meeting, PA). D-threo-1-phenyl-2-decanoylamino-3-morpholino-1-propanol HCI (PDMP) was purchased from Matreya (Pleasant Gap, PA). Sphingomyelinase (acidic, human placenta, 100 units/mg proteins) and doxorubicin hydrochloride were purchased from Sigma-Aldrich (St. Louis, MO). Calyculin A (Cal A) and okadaic acid (OA) were purchased from Cell Signaling Technology Inc. (Danvers, MA).
For Geimsa staining, cells in 35-mm dishes were fixed with ice-cold methanol, and then stained with Karyomax® Giemsa stain improved R66 solution (Invitrogen, Carlsbad, CA) at room temperature for 2 min. Following wash with deionized water, cells were photomicrographed (×100 magnification) under Nikon Eclipse TS-100 microscope equipped with a digital camera.
2.2. Gene silencing of GCS and SRSF1
To silence GCS expression, mixed-backbone oligonucleotide against human GCS (MBO-asGCS, 0–200 nM) was introduced into cells (3 × 106/100-mm dish; 4000 cells/well in 96-well plate) after overnight growth, facilitating with Lipofectamine 2000 in Opti-MEM reduced-serum medium (Invitrogen) for 4 h. The cells continuously grew in 5% FBS medium containing 2.5 μM Dox for an additional 48 h, as described previously [21]. In combination groups, cells were pretreated with FB1 (100 μM) or Cal A (5 nM) and OA (10 nM) in Opti-MEM medium for 4 h, and then transfected with MBO-asGCS (100 nM), and further grown in 5% FBS medium containing 2.5 μM Dox for an additional 48 h. To silence serine/arginine splicing factor 1 (SRSF1), cells were transfected with siRNA targeting SRSF1 (siSRSF1, 100 nM) or scrambled control (siRNA-SC, 100 nM) in Opti-MEM medium and grew in 5% FBS medium containing 2.5 μM Dox for an additional 48 h. To co-silence GCS and SRSF1, cells were transfected with both MBO-asGCS (100 nM) and siSRSF1 (100 nM) or siRNA-SC simultaneously. Mixed-backbone oligonucleotide against human GCS (MBO-asGCS) and scrambled control (MBO-SC) [21] were purchased from Integrated DNA Technologies (Coralville, IA). siRNA targeting human SRSF1 (a pool of 3 target-specific 20–25 nt siRNA, sc-38319) [22] and its scrambled control (siRNA-SC, sc-37007) were purchased from Santa Cruz Biotechnology (Santa Cruz, CA).
2.3. SRSF1 transfection
pSRSF1-EGFP plasmid (previously named as EGFP-SF2/ASF), which was generated by Tom Misteli [23] inserting human SRSF1 into the HindIII and PstI restriction sites of pEGFP-C1, was generously provided by Addgene (#17990; Cambridge, MA). The pEGFP-C1 was purchased from Clontech Laboratories (Mountain View, CA). Four micrograms of each plasmid was introduced into NCI/ADR-RES cells (3 × 106/100-mm dish) after overnight growth, facilitating with Lipofectamine 2000 in Opti-MEM reduced-serum medium (Invitrogen). After 4 h transfection, cells were cultured in 10% FBS RPMI medium, and geneticin G418 (400 μg/ml) was added into culture after overnight growth. Transfected SRSF1 cells were treated with indicated agents as described above, and the cells transfected with pEGFP-C1 were used as mock control.
2.4. ESI/MS/MS analysis of sphingolipids
Endogenous sphingomyelin molecular species were performed on a Thermo-Fisher TSQ Quantum triple quadrupole mass spectrometer, operating in a Multiple Reaction Monitoring (MRM) positive ionization mode, as described previously [24,25]. Total cells, fortified with internal standards, were extracted with ethyl acetate/iso-propanol/water (60/30/10 v/v), evaporated to dryness and reconstituted in 100 μl of methanol. The reconstituted samples were injected on the Surveyor/TSQ Quantum LC/MS system and gradient eluted from the BDS Hypersil C8 column (150 × 3.2 mm, 3 μm particle size) with 1.0 mM methanolic ammonium formate/2 mM aqueous ammonium formate mobile phase system. The peaks for the target analytes and internal standards were collected and processed using the Xcalibur software. Calibration curves were constructed by plotting peak area ratios of synthetic standards, representing each target analyte, to the corresponding internal standard. The target analyte peak area ratios from the samples were similarly normalized to their respective internal standards and compared with the calibration curves using a linear regression model. The levels of sphingolipids of samples were normalized against cellular protein, and expressed as pmol/μg protein.
2.5. RNA extraction and reverse transcription-polymerase chain reaction
After treatment, total RNA was extracted from NCI/ADR-RES cells using a SV total RNA isolation kit (Promega, Madison, WI). Equal amounts of total RNA (500 ng) were used to synthesize first strand DNA using the SuperSrcriptR III kit (Invitrogen) and 5 μl of the first strand DNA reaction from each sample was amplified by using the Platinum® Blue PCR SuperMix kit (Invitrogen) [14,19]. To detect the deletion in exon-5 of p53, a 400-bp fragment in the region of human TP53 mRNA (ORF 113–512; accession number BC003596.1) was generated by using the upstream primer (5′-TCACTGCCATGGAGGAG-3′) and downstream primer (5′-TTGAGGGCAGGGGAG-3′). This 400-bp product that includes the deleted region of exon-5 (codons 126–133) would be absent in the p53 deletion mutant. Another 393-bp p53 fragment was generated in the region of the TP53 mRNA (223–490) to detect the expression of p53 mRNA using the upstream primer (5′-TTGCCGTCCCAAGCAATG-3′) and downstream primer (5′-AAGTCACAGACTTGGCTGTC CCAGA-3′). This product that does not include the deletion region of exon-5 (codons 126–133) is present in both mutant and wild-type p53 mRNAs. As internal control of RNA loading, a 200-bp product from the glyceraldehyde-3-phosphate dehydrogenase (GAPDH) mRNA was generated using upstream primer 5′-ATGGGGAAGGTGAAGGTCGG-3′; and the downstream primer 5′-TCCACCACCCTGTTGCTG TA-3′. The PCR amplification was performed in 35 cycles by denaturation at 94 °C for 30 s, annealing at 58 °C for 30 s, and extension at 72 °C for 60 s.
2.6. Western blot analysis
After treatment, cells were collected with trypsin–EDTA and lysed in NP40 cell lysis buffer (Biosource, Camarillo, CA, USA) to extract total cellular protein for Western blot. Nuclear protein was extracted using Tween-20 lysis buffer and centrifugation, as described previously [26,27]. Protein was measured by using a bicinchoninic acid (BCA) protein assay kit (Pierce, Rockford, IL, USA). Western blot analysis was conducted as described previously [19,26]. Briefly, equal amounts of protein (50 μg/lane) were resolved by 4–20% gradient SDS-PAGE, and transferred to a nitrocellulose membrane. The blots were blocked in 5% fat-free milk in PBST (0.05% Tween-20, 20 mM phosphate buffered saline, pH 7.4) for 60 min at room temperature, and then incubated with specific primary antibodies against p53 (clone PAB1801, #13-4000, Invitrogen), phosphorylated p53 at Ser15 (pp53; #9286S, Cell Signaling Technology), p63 (D-9, sc-25268), p73 (S-20, sc-9651), p21 (F-5, sc-6246), Bax (2D2, sc-20067), SRSF1 (H-110, sc-28724, Santa Cruz Biotechnology) and active caspase-7 (04-441, EMD Chemicals) (1:500–1:5000 dilution) in 5% fat-free milk PBS, respectively, at 4 °C, overnight. After washing, these blots were incubated with appropriate horseradish peroxide-conjugated secondary antibodies (1:5000 dilution) in 5% fat-free milk PBST for an hour at room temperature, and developed using the SuperSignal® West Pico ECL substrate (Thermo Scientific, Rockford, IL). Glyceraldehydrodes-3-phosphate dehydrogenase (GAPDH, sc-137179, Santa Cruz Biotechnology) and β-tubulin (DM1A, T6199, Sigma-Aldrich) were used as a loading control for cellular protein, and for nuclear protein, respectively.
2.7. Immunocytochemistry
Cells (10,000 cells/chamber) after treatments were grown in 4-chamber slides for 48 h and immunocytochemistry was performed as described previously [19,26]. After fixation with ice cold methanol and PBS wash, cells were blocked with 5% goat serum PBS (block solution) and then incubated with antibodies against ceramide (1:500, clone MID 15B4 from Sigma), SRSF1 (1:500, Santa Cruz Biotechnology) and pp53 (1:500, Cell Signaling Technology) in blocking solution at 4 °C, overnight. Cellular ceramide, SRSF1 and pp53 tagged with antibodies were recognized by corresponding Alexa Fluor 555- or 488-conjugated goat IgG (1:1000), respectively. Cell nuclei were counter-stained with DAPI (4′, 6-diamidino-2-phenylindole) in mounting solution (Vector Laboratories). Images (×200 magnification) were captured using the EVOS FL cell imaging system with color CCD camera (Life Technologies, Grand Island, NY).
2.8. Cell viability assay
Cell viability was determined by quantitation of ATP, an indicator of live cells, using the CellTiter-Glo luminescent cell viability assay (Promega, Madison, WI) kit, as described previously [19,21]. Briefly, cells (4000 cells/well) were grown in 96-well plates with 10% FBS RPMI-1640 medium overnight. Cells were pre-treated with MBO-asGCS (100 nM) for 6 days, and FB1 (100 μM), C6-Cer (5 μM), C6-diH-Cer (5 μM), SMase (0.5 unit/ml), and PDMP (10 μM) for 72 h. Simultaneously, cells were exposed to Dox (2.5 μM) for 72 h. Cell viability was determined by the measurement of luminescent ATP in a Synergy HT microplate reader (BioTek, Winnooski, VT, USA) following incubation with CellTiter-Glo reagent.
2.9. Apoptosis analysis by flow cytometry
Apoptosis was evaluated by using propidium iodide (PI) staining and flow cytometry, as described previously [21,28] with minimal modification. Briefly, treated cells were harvested by trypsinization and centrifugation. Cell pellets were resuspended and exposed to 0.01% PI in staining solution (0.1% sodium citrate, 0.3% Triton X-100, 2 mg/ml ribonuclease A) at 4 °C for 30 min, followed by flow cytometry analysis using FACSCalibur (BD Biosciences, San Jose, CA). For each sample, 10,000 events were counted three times and cell cycle histograms were generated. All analyses were performed using CellQuest Pro program, where subphase G1/G0 was defined as indicative of apoptotic cells.
2.10. Protein phosphatase assay
Protein phosphatase (PP) activity was measured using ProFluor™ Ser/Thr phosphatase assay kit (Promega, Madison, WI), following manufacturer’s instructions [29]. Briefly, after treatments, cells were harvested with trypsin–EDTA and lysed in NP40 cell lysis buffer. Cell lysates (100 μg proteins) from each sample were incubated with R110 substrate, and protease solution in 96-well plates at 25 °C for 10 min. PP activity was determined by measurement of fluorescence with excitation/emission at 485 nm/530 nm in Synergy HT microplate reader.
2.11. Data analysis
All experiments were repeated 2 or 3 times. The data are expressed as the mean ± SD. Two-tailed Student’s t tests were used to compare the continuous variables between groups, using a Prism v5 program (GraphPad software, San Diego, CA). All p < 0.05 was considered statistically significant.
3. Results
3.1. Ceramide restores wild-type p53 expression in deletion-mutant cells
Ceramide is central in sphingolipid metabolism, and ceramide glycosylation catalyzed by GCS converts ceramide to glucosylceramide [30, 31]. Disruption of ceramide glycosylation can alter cellular levels of several sphingolipid molecules, including sphingosine and sphingosine 1 phosphate (S1P), although ceramide alteration might be predominant. Moreover, ceramide is a family of closely related molecules and constitutes more than 50 species possessing various saturated/unsaturated acyl chains [32,33]. Individual ceramide molecular species are regulated by specific biochemical pathways in distinct subcellular compartments and execute distinct functions. To understand how suppression of ceramide glycosylation restores the expression of wild-type p53 [19], we analyzed endogenous sphingolipids in p53 deletion-mutant NCI/ADR-RES cells treated with MBO-asGCS, a mixed-backbone oligonucleotide that specifically silences GCS expression [21]. Among 13 detectable ceramide species, MBO-asGCS treatment significantly increased C16-, C18-, C20-, C22- C24-, C24:1- and C26-Cer species. Cellular C18-Cer was increased by 2-fold (0.073 vs. 0.023 pmol/μg protein), C20-Cer increased by 2-fold, C22-Cer by 1-fold and C24-Cer by 2-fold (1.98 vs. 0.66 pmol/μg protein) respectively, compared to the scrambled oligonucleotide control (MBO-SC) (Fig. 1A). Among the unsaturated species, only C24:1-Cer was increased by 1-fold (0.394 vs. 0.179 pmol/μg protein). In contrast to increasing ceramide levels, MBO-asGCS treatment did not significantly alter other sphingolipids, including C16-dihydroceramide, dihydrosphingosine, sphingosine and S1P in NCI/ADR-RES cells, as compared to cells treated with MBO-SC or vehicle control (Fig. 1B). These data indicate that suppression of ceramide glycosylation with MBO-asGCS mainly increases ceramide level, particularly saturated C18- and C24-Cer species.
Fig. 1.

Endogenous sphingolipids and p53 expression. A) Ceramide species. NCI/ADR-RES cells were treated with MBOs (100 nM, 48 h) and extracted lipids were analyzed by LC/MS. *, p < 0.001 compared to MBO-SC. B) Sphingolipids analyzed by ESI/MS/MS analysis. DHC16-Cer, C16-dihydroceramide; DHSph, dihydrosphingosine; Sph, sphingosine; Sph-1P, sphingosine 1 phosphate. C) Ceramide and p53 expression in NCI/ADR-RES cells. Cells were treated with agents, including MBO-asGCS (100 nM), FB1 (100 μM), C6-Cer (5 μM); C6-diH-Cer (5 μM), SMase (0.5 U/ml), and PDMP (10 μM) for 48 h. RT-PCR analyses were utilized to detect the pan p53 mRNA and deleted region in exon-5. Equal amounts of cellular protein (100 μg/lane) were used for Western analyses. pp53, phosphorylated p53; pp53/p53, the ratios of optical densities of pp53 bands normalized against total p53. *, p < 0.001 compared to cells treated with vehicle and exposed to Dox.
We used several different ways to evaluate ceramide effects on p53 expression. PDMP inhibits GCS activity, and acidic SMase hydrolyzes sphingomyelin, thus increasing endogenous ceramide [34]. C6-Cer, a short carbon-chain ceramide, is cell-permeable and also can increase cellular ceramide [19]. Conversely, fumonisin B1 (FB1) inhibits de novo ceramide synthesis [35], depleting cellular ceramide. C6-dihydroceramide (C6-diH-Cer), which lacks bioactivity, was used as control. NCI/ADR-RE cells have mutated p53 with a 21-bp deletion spanning codons 126–133 in exon-5 of p53 mRNA [20]. The treatments with MBO-asGCS, PDMP, C6-Cer, and SMase, which all increase cellular ceramide levels, induced the expression of wild-type p53 mRNA (intact exon-5) in RT-PCR and phosphorylated p53 (Ser15, 7 aa more than deletion-mutant) in Western blotting. MBO-asGCS, C6-Cer, SMase, and PDMP significantly increased the pp53 levels by approximately 6-, 7-, 8-, and 7-fold, respectively, as compared to vehicle control (Fig. 1C). Consequently, the protein levels of p21 and Bax were substantially increased in cells after these ceramide-increasing treatments (Fig. 1C). On the other hand, agents that do not affect ceramide levels or inhibit ceramide generation had no effect on restoration of wild-type p53 expression. In the same conditions, we further examined p63 and p73 proteins, which both exert p53-like tumor-suppressive activities through their ability to activate a common set of target genes [36,37]. NCI/ADR-RES cells express p63 and p73 at the similar levels as mutant p53, rather than ΔNp63 or ΔNp73 isoforms that can act as dominant-negative inhibitors of p53 [36,38]; ceramide treatments increase both protein levels, however, these changes are not significant, compared to vehicle controls (Fig. 1C). Although the p53 promoter elements for PE21 and Sp1 are essential for p53 transactivation [39], we found that MBO-asGCS treatments do not have any significant effect on activating the wild-type or truncated p53 promoter reporters (Fig. S1). Together, this evidence indicates that ceramide restores p53 expression in deletion-mutant cancer cells.
These findings were further verified by immunostaining of ceramide and pp53, because the induced nuclear pp53 was coincident with cytoplasmic ceramide in most NCI/ADR-RES cells treated with MBO-asGCS, C6-Cer, SMase and PDMP, respectively (Fig. 2). However, neither ceramide nor pp53 was detected in cells treated with vehicle and either C6-diH-Cer or MBO-asGCS combined with FB1 (Fig. 2).
Fig. 2.

Cellular ceramide and wild-type p53. NCI/ADR-RES cells were treated with agents, including MBO-asGCS (100 nM), FB1 (100 μM), C6-Cer (5 μM), C6-diH-Cer (5 μM), SMase (0.5 U/ml), and PDMP (10 μM) for 48 h and exposed to Dox. Red, Alexa Fluor 555-Cer; green, Alexa Fluor 488-pp53; blue, DAPI-nucleus. Fluorescence micrographs (x 200 magnification) were captured by EVOS imaging system.
3.2. Restored wild-type p53 expression sensitizes cells to apoptosis
To determine whether the restored wild-type p53 is able to execute its role as a tumor suppressor, we pretreated NCI/ADR-RE cells with the reagents, which we showed can restore wild-type p53 expression, and then assessed the cell response to doxorubicin treatments. We found that MBO-asGCS treatment substantially decreased cell viability more than 4-fold; however, MBO-asGCS combined with FB1, which inhibited ceramide de novo synthesis and restoration of p53 expression, did not significantly affect cell viability (Fig. 3A). Consistently, C6-Cer decreased cell viability by 4-fold (91% vs. 17% of control), but C6-diH-Cer did not. Treatments with SMase, and PDMP, all of which can induce p53 expression, significantly decreased cell viability by approximately 2-, 4- and 7-fold, respectively (Fig. 3A). To account for the decreased cell viability observed on stimulation of ceramide levels, we examined induction of apoptosis. We found that MBO-asGCS treatment substantially increased apoptotic cells by approximately 4-fold (81% vs. 15% of total cells) (Fig. 3B top), due to increased level of active caspase 7 (Fig. 3B bottom); however, MBO-asGCS combined with FB1 did not significantly affect apoptosis (20% vs. 15%) or the level of active caspase 7. Similarly, C6-Cer dramatically increased apoptotic cells by 4-fold (80% vs. 15% of total cells), but C6-diH-Cer only increased apoptotic cells by 1-fold (30% vs. 15%) and did not significantly increase the active caspase 7 (Fig. 3B bottom). Further, treatment with SMase or PDMP significantly increased apoptotic cells by approximately 3- and 4-fold (Fig. 3B top), respectively, accompanied by increased levels of active caspase 7 (Fig. 3B bottom), as compared to vehicle control.
Fig. 3.
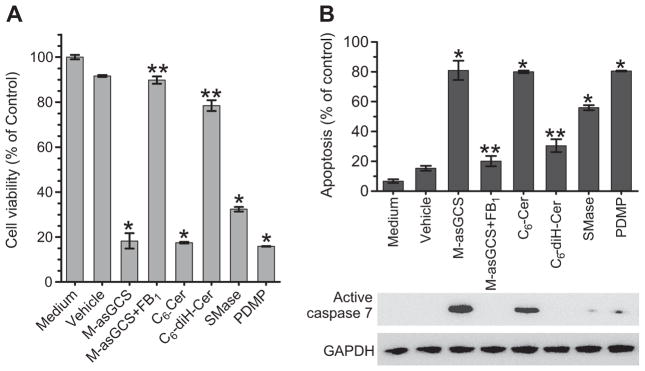
Restored expression of wild-type p53 induces cell death. NCI/ADR-RES cells were pretreated with M-asGCS (MBO-asGCS, 100 nM), C6-Cer (5 μM), C6-diH-Cer (5 μM), FB1 (100 μM), and PDMP (10 μM), and then treated with Dox (2.5 μM). A) Cell viability after 72 h of treatments. B) Apoptotic cells were analyzed by flow cytometry after 48 h of treatments. Active caspase 7 levels were detected by using Western blotting. *, p < 0.001 compared to vehicle (Dox alone); **, p < 0.001 compared to M-asGCS or C6-Cer, respectively.
3.3. Ceramide modulates the activity of protein phosphate 1 and increases nuclear SRSF1
It has been reported that protein phosphatase 1 (PP1) as well as protein phosphatase 2A (PP2A), can be activated by ceramide and play an important role in mediating the pharmacological effects of ceramide [40,41]. PP1 is engaged in ceramide-mediated the dephosphorylation of serine/arginine-rich proteins (SR proteins) [42,43]. To explore whether PP1 or PP2A is involved in ceramide-mediated restoration of p53 expression, we tested phosphatase inhibitors in NCI/ADR-RES cells treated with MBO-asGCS and C6-Cer. Both MBO-asGCS and C6-Cer significantly increased the activity of PP1 to 165% (76,376 vs. 46,155 FLU/100 μg protein, p < 0.001) and 173% (79,412 vs. 46,155 FLU/100 μg protein, p < 0.001), respectively, as compared to vehicle control (Fig. 4A). Calyculin A (Cal A) has been reported to inhibit both PP1 and PP2a [44,45]. Pretreatment with Cal A substantially reduced PP1 activity to 39% (30,071 vs. 76,376 FLU/100 μg protein, p < 0.001) and 47% (37,214 vs. 79,412 FLU/100 μg protein, p < 0.001) in cells treated with MBO-asGCS and C6-Cer, respectively. Okadaic acid (OA), a specific inhibitor of PP2, did not significantly decrease PP activity of cells treated with either MBO-asGCS or C6-Cer (Fig. 4A).
Fig. 4.

PP1 and SRSF1 are associated with p53 restoration. A) PP1 activities. NCI/ADR-RES cells were pretreated with calyculin (Cal A, 5 nM) or okadaic acid (OA 10 nM), and treated with MBO-asGCS (M-asGCS, 100 nM) or C6-Cer (5 μM) in medium containing Dox. FLU, fluorescence units. *, p < 0.001 compared to vehicle; **, p < 0.001 compared to M-asGCS or C6-Cer. B) Western blotting for Nuclear SRSF1. NCI/ADR-RES cells were treated with MBO-asGCS (0–200 nM) for 6 days and then exposed to Dox in the last 48 h. Equal amounts of nuclear protein were used for immunoblotting. Nuclear SRSF1 levels are represented by SRSF1/tubulin, ratios of SRSF1 optical densities normalized against β-tubulin. C) RT-PCR analysis of wild-type p53 exon-5. D) Immunostaining of SRSF1 and pp53. SRSF1 and pp53 tagged with antibodies were further recognized by corresponding Alexa Fluor 488 or 555-conjugated goat IgG (×200, magnification).
We next assessed the effect of MBO-asGCS treatment on spliceosome proteins in NCI/ADR/RES cells. It was found that MBO-asGCS increased the levels of nuclear splicing factor SRSF1 in a concentration-dependent manner (Fig. 4B). Nuclear SRSF1 was undetectable in NCI/ADR-RES cells, but its levels were significantly increased to 2-fold and 6-fold after treatments with 100 nM and 200 nM of MBO-asGCS, compared to 50 nM treatment (Fig. 4B). Accompanying SRSF1 increase, the levels pp53 that represent wild-type p53, were significantly induced in cells treated with MBO-asGCS (Fig. 4B). Consistently, RT-PCR analyses indicate that MBO-asGCS substantially induced exon-5 mRNA in NCI/ADR-RES cells, even though it did not have effects on the panel p53 mRNA (Fig. 4C). Immunostaining of SRSF1 and pp53 indicated that translocation of SRSF1 from the cytoplasm to the nucleus correlated well with nuclear pp53 in most of cells treated with MBO-asGCS (Fig. 4D). Together, these data indicate that PP1 and SRSF1 are highly associated with the effects of MBO-asGCS and ceramide in modulating restoration of wild-type p53.
3.4. PP1 and SRSF1 execute the effects of ceramide on restoring wild-type p53 expression
We utilized enzyme inhibition as well as gene silencing to determine the roles of PP1 and SRSF1 in p53 restoration. In addition to Cal A and OA, NCI/ADR-RES cells were pretreated with siSRSF1 (100 nM) to silence SRSF1 expression [22]. Subsequently, the cells were treated with MBO-asGCS (100 nM) or with C6-Cer (5 μM). MBO-asGCS induced the expression of the intact exon-5 mRNA and phosphorylated p53 (pp53), which consequently increased the expression of p21 and Bax in NCI/ADR-RES cells, as compared to vehicle control (Fig. 5A, B). In contrast, Cal A (5 nM) and siSRSF1, which inhibits PP1 and knocks down SRSF1, completely diminished the inductive effects of MBO-asGCS on the expression of p53 exon-5 mRNA, pp53, p21 and Bax in NCI/ADR-RES cells. Consistently, Cal A and siSRSF1 also diminished the effects of C6-Cer on p53 exon-5 mRNA, pp53, p21 and Bax as well. However, neither OA nor siRNA-SC prevented the restoration of wild-type p53 and p53-responsive gene expression in NCI/ADR-RES cells treated with MBO-asGCS and C6-Cer (Fig. 5A, B). Furthermore, introduction of human SRSF1 gene into NCI/ADR-RES cells confers that SRSF1 is essential for ceramide induced p53 restoration. Overexpression of SRSF1 sufficiently restored exon-5 mRNA (Fig. 5C) and pp53 (Fig. 5D) in NCI/ADR-RES cells transfected with pSRSF1, even though no treatment of MBO-asGCS or C6-ceramide. Cal A treatment did not prevent the expression of wild-type p53 and p53 responsive proteins of p21 and Bax in SRSF1 transfected cells (Fig. 5C, 5D).
Fig. 5.

PP1 and SRSF1 determine wild-type p53 expression. NCI/ADR-RES cells were pretreated with PP inhibitors (5 nM Cal A; 10 nM OA) and transfection of siSRSF1 (100 nM) or pSRSF1, and then treated with MBO-asGCS and C6-Cer followed by Dox exposure (2.5 μM, 48 h). A) RT-PCR analysis of p53 exon-5. B) Western blotting for wild-type p53. C) Effect of SRSF1 on p53 exon-5 mRNA, and D) p53 proteins. Total RNA and cellular protein were extracted from NCI/ADR-RES cells transfected with pEGFP-SRSF1 (pSRSF1) or pEFGP-C1 plasmids (mock) and then treated with MBO-asGCS (100 nM), C6-ceramide (5 μM) and Cal A (5 nM), respectively.
We further examined whether PP1 and SRSF1 affect p53-dependent cell death. NCI/ADR-RE cells were pretreated with PP1 inhibitor or transfected with siSRSF1 and pSRSF1 plasmids, and then treated with MBO-asGCS and Dox. As shown in Fig. 6A, MBO-asGCS substantially decreased viability of cells exposed to Dox by 4.5-fold (100% vs. 18.2% of control, p < 0.001) (Fig. 6A). Conversely, inhibition of PP1 with Cal A and knockdown of SRSF1 with siSRSF1 allowed cell survival when they were exposed to MBO-asGCS with Dox, and the cell viability was increased by 3.7-fold (18.2% vs. 86.5% of control, p < 0.001) and 3.4-fold (18.2% vs. 80.3% of control, p < 0.001), respectively. Transfection of SRSF1 significantly decreased cell viability (12% vs. 80.3%, p < 0.001) in NCI/ADR-RES cells, compared to siSRSF1 transfection or mock (Fig. 6A). However, OA and siRNA-SC or mock transfection, which did not affect either PP1 activity or SRSF1 expression, could not diminish the cytotoxic effect of MBO-asGCS with Dox on cells (Fig. 6A). These findings were confirmed by Geimsa staining of cells after indicated treatments (Fig. 6B). On flow cytometry analysis, we found that MBO-asGCS treatment (100 nM, 48 h) substantially increased apoptosis in cells exposed to Dox (2.5 μM), by approximately 11-fold (81% vs. 6.7% of total cells, p < 0.001) (Fig. 6C). SRSF1 transfection increased apoptosis (92% vs. 25%, p < 0.001) in cancer cells, compared to siSRSF1 transfection or mock (Fig. 6C). Conversely, inhibition of PP1 with Cal A and knockdown of SRSF1 with siSRSF1 allowed survival when cells were exposed to MBO-asGCS with Dox; the apoptotic cells were decreased by approximately 3-fold (81% vs. 20.3% of total cells, p < 0.001) and 2-fold (81% vs. 25.9% of total cells, p < 0.001), respectively (Fig. 6C). However, OA and siRNA-SC or mock transfection, which did not affect PP1 activity or SRSF1 expression, could not diminish the cytotoxic effect of MBO-asGCS with Dox on NCI/ADR-RES cells (Fig. 6C).
Fig. 6.

Restored p53 leads cancer cells to apoptosis. NCI/ADR-RES cells were pretreated with PP inhibitors (5 nM Cal A; 10 nM OA), and transfection of siSRSF1 (100 nM) or pSRSF1, and then treated with MBO-asGCS (100 nM) and Dox (2.5 μM). A) Cell viability. *, p < 0.001 compared to control (Dox alone); **, p < 0.001 compared to MBO-asGCS + OA or MBO-asGCS + siRNA-SC; #, p < 0.001 compared to Mock. B) Geimsa staining. Cells were treated with indicated conditions mentioned in Fig. 6A (×100 magnification). C) Apoptosis. *, p < 001 compared to control (Dox alone); **, p < 0.001 compared to MBO-asGCS + OA or MBO-asGCS + siRNA-SC; #, p < 0.001 compared to MBO-asGCS + mock or MBO-asGCS + siSRSF1.
4. Discussion
A previous report shows that suppression of ceramide glycosylation restores wild-type p53 expression and p53-dependent apoptosis in p53 deletion-mutant cells [19]. Our present study elaborates the molecular mechanism, demonstrating that ceramide modulates pre-mRNA splicing to restore p53 expression.
Besides modulating transcription factors and transactivation of gene expression [14,16], recent studies show that ceramide mediates alternative splicing to preferentially express apoptotic isoforms of bcl-x and caspase-9 [17,18]. Little is known about the role of ceramide in restoration of wild-type gene expression in mutant cells. In our present study, suppression of ceramide glycosylation with MBO-asGCS, which restores wild-type p53 in mutant cells, significantly increased endogenous ceramide species, particularly C18- and C24-ceramide, rather than other sphingolipid molecules (Fig. 1). Inhibition of ceramide synthesis in de novo pathway by FB1 diminished the restorative effect of MBO-asGCS on p53 expression; conversely, enhancing cellular ceramide levels, by GCS inhibitor PDMP, SMase and C6-Cer, lead to p53 restoration (Figs. 1, 2). All these results indicate that ceramide is an essential cellular signal in modulating the expression of wild-type p53 versus mutant p53 in these cells. Ceramide, particularly long-carbon chain ceramide species, such as C18-ceramide, can activate protein phosphatases including PP1 and PP2a to mediate cellular signal transduction [41,44,46–48]. We found here that MBO-asGCS treatment via ceramide mainly increased PP1 activity. Further, the nuclear level of SRSF1 was increased in a concentration-dependent manner (Fig. 4). Ceramide-activated PP1 and induced nuclear SRSF1 after MBO-asGCS treatment restored the expression of wild-type p53 and p53-responsive genes (p21, Bax) (Fig. 5), consequently leading cells to apoptosis (Fig. 6).
SRSF1 is a serine/arginine-rich protein involved in pre-mRNA splicing, and it also functions in multiple steps of gene expression, including chromatin remodeling, transcription, non-sense-mediated mRNA decay, mRNA export and stability and translation [49–52]. SRSF1 contains two RNA recognition motifs (RRM) in the N-terminus and one arginine/serine-rich (RS) domain in the C-terminus. In pre-mRNA splicing, early spliceosome assembly begins with U1 small nuclear ribonucleoprotein (U1 snRNP) binding to the 5′ splice site of pre-mRNA, which is assisted by SR proteins in mammalian cells. In this process, the RS domain of SR proteins is thought to directly interact with the RS motif of U1-70K snRNP, which is subject to regulation by RS domain phosphorylation [49,50]. Recent study shows that interaction between the RRM of SRSF1 and U1-70K snRNP protein determines early spliceosome assembly [49]. SR protein kinase (SRPK1) and nuclear protein kinase Clk/Sty (CDC-like kinase) phosphorylate the RS domains [53]; in contrast, PP1 and PP2a dephosphorylate the RS domains, thus modulating SRSF1 cellular translocation from the cytoplasm to the nucleus, and spliceosome recruitment [54,55]. Hyperphosphorylated SR proteins are driven out of the nucleus to the cytoplasm where they are sequestered; the dephosphorylated or hypophosphorylated SR proteins are translocated to the nucleus, increasing their concentrations inside this organelle [56,57]. The phosphorylation/dephosphorylation cycle of SRSF1 has an important effect in alternative splicing, rather than in constitutive splicing [58]. Endogenous ceramide has been found to activate PP1 and dephosphorylate of SRSF1, regulating the selection of exons of caspase-9 isoforms [17,42]. In the present study, we found that after MBO-asGCS treatments, endogenous ceramide increased nuclear SRSF1 and wild-type p53 expression in a concentration-dependent manner (Fig. 4). Inhibition of PP1 or silencing SRSF1 diminished the restorative effect of ceramide on p53 expression; overexpression of SRSF1 confers the effect of ceramide on p53 restoration (Fig. 5). These demonstrate that nuclear SRSF1 plays a critical role in regulating the expression of wild-type p53 versus mutants.
SRSF1 can be oncogenic, as it is reported to be overexpressed in breast cancer cells, driving transformation of murine immoral fibroblasts and mammary epithelial cell transformation [59,60]. The oncogenic activity of SRSF1 is partly medicated by controlling alternative splicing of tumor suppressor BIN1 [60] and potentiating eIF4E activation through mTOR [59]. However, several studies also indicate that SRSF1 has anticancer effect. Dephosphorylated SRSF1 by ceramide preferentially expresses the apoptotic isoforms of bcl-x and caspase-9 in cancer cells [17,18]. The alterative splicing of caspase 9 by SRSF1 regulates the synergistic effects of chemotherapeutic agents in non-small cell lung cancer (NSCLC) [61,62]. Recent work from Krainer’s group shows that SRSF1 stabilizes p53 protein by abrogating its MDM2-dependent proteasomal degradation and induces cellular senescence in primary human fibroblasts [63]. We found that SRSF1 restored wild-type p53 expression and induces apoptosis in deletion-mutant cancer cells. These suggest that the effects of SRSF1 rely on the SRSF1-spliced genes, and their expression and functional profiles determine whether SRSF1 is oncogenic protein or vice versa in particular types of cells and cancers. In p53 mutant cancer cells, SRSF1 can act as tumor suppressor, if it is able to restore wild-type p53 expression.
Supplementary Material
Acknowledgments
The authors thank Dr. Daniel Peeper (Division of Molecular Genetics, the Netherlands Cancer Institute) for human p53 promoter reporters, p53-Luc and its truncated ones. This work was supported by National Institutes of Health Grants P20 GM103424-11 from the National Institute of General Medical Sciences, and R15CA167476 from the National Cancer Institute (to Y.Y.L.). This work was also partially supported by funds from the Board of Regents Louisiana (LEQSF-EPS2012-PFUND-299), the Louisiana Campuses Research Initiative (LaCRI-UL/Liu) and the Mizutani Foundation for Glycoscience, Japan (to Y.Y.L.).
Abbreviations
- SRSF1
serine/arginine-rich splicing factor1
- PP1
protein phosphatase 1
- GCS
glucosylceramide synthase
- MBO-asGCS
mixed backbone oligonucleotide against glucosylceramide synthase
- PDMP
D-threo-1-phenyl-2-decanoylamino-3-morpholino-1-propanol
- SMase
sphingomyelinase
- FB1
fumonisin 1
- CalA
calyculin A
- siRNA
small interfering RNA
- RT-PCR
reverse transcription polymerase chain reaction
- ESI/MS/MS
electrospray ionization-tandem mass spectrometry
Appendix A. Supplementary data
Supplementary data to this article can be found online at http://dx.doi.org/10.1016/j.bbalip.2014.08.017.
References
- 1.Lane DP. Cancer. p53, guardian of the genome. Nature. 1992;358:15–16. doi: 10.1038/358015a0. [DOI] [PubMed] [Google Scholar]
- 2.Brosh R, Rotter V. When mutants gain new powers: news from the mutant p53 field. Nat Rev Cancer. 2009;9:701–713. doi: 10.1038/nrc2693. [DOI] [PubMed] [Google Scholar]
- 3.Hollstein M, Sidransky D, Vogelstein B, Harris CC. p53 mutations in human cancers. Science. 1991;253:49–53. doi: 10.1126/science.1905840. [DOI] [PubMed] [Google Scholar]
- 4.Greenblatt MS, Bennett WP, Hollstein M, Harris CC. Mutations in the p53 tumor suppressor gene: clues to cancer etiology and molecular pathogenesis. Cancer Res. 1994;54:4855–4878. [PubMed] [Google Scholar]
- 5.Petitjean A, Achatz MI, Borresen-Dale AL, Hainaut P, Olivier M. TP53 mutations in human cancers: functional selection and impact on cancer prognosis and outcomes. Oncogene. 2007;26:2157–2165. doi: 10.1038/sj.onc.1210302. [DOI] [PubMed] [Google Scholar]
- 6.Wiman KG. Pharmacological reactivation of mutant p53: from protein structure to the cancer patient. Oncogene. 2010;29:4245–4252. doi: 10.1038/onc.2010.188. [DOI] [PubMed] [Google Scholar]
- 7.Chen F, Wang W, El-Deiry WS. Current strategies to target p53 in cancer. Biochem Pharmacol. 2010;80:724–730. doi: 10.1016/j.bcp.2010.04.031. [DOI] [PubMed] [Google Scholar]
- 8.Liu YY. Resuscitating wild-type p53 expression by disrupting ceramide glycosylation: a novel approach to target mutant p53 tumors. Cancer Res. 2011;71:6295–6299. doi: 10.1158/0008-5472.CAN-11-0700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zeimet AG, Marth C. Why did p53 gene therapy fail in ovarian cancer? Lancet Oncol. 2003;4:415–422. doi: 10.1016/s1470-2045(03)01139-2. [DOI] [PubMed] [Google Scholar]
- 10.Hannun YA, Obeid LM. Principles of bioactive lipid signalling: lessons from sphingolipids. Nat Rev Mol Cell Biol. 2008;9:139–150. doi: 10.1038/nrm2329. [DOI] [PubMed] [Google Scholar]
- 11.Alesse E, Zazzeroni F, Angelucci A, Giannini G, Di Marcotullio L, Gulino A. The growth arrest and downregulation of c-myc transcription induced by ceramide are related events dependent on p21 induction, Rb underphosphorylation and E2F sequestering. Cell Death Differ. 1998;5:381–389. doi: 10.1038/sj.cdd.4400358. [DOI] [PubMed] [Google Scholar]
- 12.Kim WH, Kang KH, Kim MY, Choi KH. Induction of p53-independent p21 during ceramide-induced G1 arrest in human hepatocarcinoma cells. Biochem Cell Biol. 2000;78:127–135. [PubMed] [Google Scholar]
- 13.Patwardhan GA, Liu YY. Sphingolipids and expression regulation of genes in cancer. Prog Lipid Res. 2011;50:104–114. doi: 10.1016/j.plipres.2010.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Liu YY, Yu JY, Yin D, Patwardhan GA, Gupta V, Hirabayashi Y, Holleran WM, Giuliano AE, Jazwinski SM, Gouaze-Andersson V, Consoli DP, Cabot MC. A role for ceramide in driving cancer cell resistance to doxorubicin. FASEB J. 2008;22:2541–2551. doi: 10.1096/fj.07-092981. [DOI] [PubMed] [Google Scholar]
- 15.Wu D, Marko M, Claycombe K, Paulson KE, Meydani SN. Ceramide-induced and age-associated increase in macrophage COX-2 expression is mediated through up-regulation of NF-kappa B activity. J Biol Chem. 2003;278:10983–10992. doi: 10.1074/jbc.M207470200. [DOI] [PubMed] [Google Scholar]
- 16.Ogretmen B, Kraveka JM, Schady D, Usta J, Hannun YA, Obeid LM. Molecular mechanisms of ceramide-mediated telomerase inhibition in the A549 human lung adenocarcinoma cell line. J Biol Chem. 2001;276:32506–32514. doi: 10.1074/jbc.M101350200. [DOI] [PubMed] [Google Scholar]
- 17.Chalfant CE, Rathman K, Pinkerman RL, Wood RE, Obeid LM, Ogretmen B, Hannun YA. De novo ceramide regulates the alternative splicing of caspase 9 and Bcl-x in A549 lung adenocarcinoma cells. Dependence on protein phosphatase-1. J Biol Chem. 2002;277:12587–12595. doi: 10.1074/jbc.M112010200. [DOI] [PubMed] [Google Scholar]
- 18.Massiello A, Chalfant CE. SRp30a (ASF/SF2) regulates the alternative splicing of caspase-9 pre-mRNA and is required for ceramide-responsiveness. J Lipid Res. 2006;47:892–897. doi: 10.1194/jlr.C600003-JLR200. [DOI] [PubMed] [Google Scholar]
- 19.Liu YY, Patwardhan GA, Bhinge K, Gupta V, Gu X, Jazwinski SM. Suppression of glucosylceramide synthase restores p53-dependent apoptosis in mutant p53 cancer cells. Cancer Res. 2011;71:2276–2285. doi: 10.1158/0008-5472.CAN-10-3107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ogretmen B, Safa AR. Expression of the mutated p53 tumor suppressor protein and its molecular and biochemical characterization in multidrug resistant MCF-7/Adr human breast cancer cells. Oncogene. 1997;14:499–506. doi: 10.1038/sj.onc.1200855. [DOI] [PubMed] [Google Scholar]
- 21.Patwardhan GA, Zhang QJ, Yin D, Gupta V, Bao J, Senkal CE, Ogretmen B, Cabot MC, Shah GV, Sylvester PW, Jazwinski SM, Liu YY. A new mixed-backbone oligonucleotide against glucosylceramide synthase sensitizes multidrug-resistant tumors to apoptosis. PLoS ONE. 2009;4:e6938. doi: 10.1371/journal.pone.0006938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Munoz U, Puche JE, Hannivoort R, Lang UE, Cohen-Naftaly M, Friedman SL. Hepatocyte growth factor enhances alternative splicing of the Kruppel-like factor 6 (KLF6) tumor suppressor to promote growth through SRSF1. Mol Cancer Res. 2012;10:1216–1227. doi: 10.1158/1541-7786.MCR-12-0213. [DOI] [PubMed] [Google Scholar]
- 23.Phair RD, Misteli T. High mobility of proteins in the mammalian cell nucleus. Nature. 2000;404:604–609. doi: 10.1038/35007077. [DOI] [PubMed] [Google Scholar]
- 24.Bielawski J, Szulc ZM, Hannun YA, Bielawska A. Simultaneous quantitative analysis of bioactive sphingolipids by high-performance liquid chromatography-tandem mass spectrometry. Methods. 2006;39:82–91. doi: 10.1016/j.ymeth.2006.05.004. [DOI] [PubMed] [Google Scholar]
- 25.Bielawski J, Pierce JS, Snider J, Rembiesa B, Szulc ZM, Bielawska A. Comprehensive quantitative analysis of bioactive sphingolipids by high-performance liquid chromatography-tandem mass spectrometry. Methods Mol Biol. 2009;579:443–467. doi: 10.1007/978-1-60761-322-0_22. [DOI] [PubMed] [Google Scholar]
- 26.Liu YY, Gupta V, Patwardhan GA, Bhinge K, Zhao Y, Bao J, Mehendale H, Cabot MC, Li YT, Jazwinski SM. Glucosylceramide synthase upregulates MDR1 expression in the regulation of cancer drug resistance through cSrc and beta-catenin signaling. Mol Cancer. 2010;9:145. doi: 10.1186/1476-4598-9-145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Klenova E, Chernukhin I, Inoue T, Shamsuddin S, Norton J. Immunoprecipitation techniques for the analysis of transcription factor complexes. Methods. 2002;26:254–259. doi: 10.1016/S1046-2023(02)00029-4. [DOI] [PubMed] [Google Scholar]
- 28.Diaz-Montero CM, McIntyre BW. Acquisition of anoikis resistance in human osteosarcoma cells does not alter sensitivity to chemotherapeutic agents. BMC Cancer. 2005;5:39. doi: 10.1186/1471-2407-5-39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.King TD, Gandy JC, Bijur GN. The protein phosphatase-1/inhibitor-2 complex differentially regulates GSK3 dephosphorylation and increases sarcoplasmic/endoplasmic reticulum calcium ATPase 2 levels. Exp Cell Res. 2006;312:3693–3700. doi: 10.1016/j.yexcr.2006.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Merrill AH., Jr Sphingolipid and glycosphingolipid metabolic pathways in the era of sphingolipidomics. Chem Rev. 2011;111:6387–6422. doi: 10.1021/cr2002917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Liu YY, Hill RA, Li YT. Ceramide glycosylation catalyzed by glucosylceramide synthase and cancer drug resistance. Adv Cancer Res. 2013;117:59–89. doi: 10.1016/B978-0-12-394274-6.00003-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hannun YA, Obeid LM. Many ceramides. J Biol Chem. 2011;286:27855–27862. doi: 10.1074/jbc.R111.254359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pewzner-Jung Y, Ben-Dor S, Futerman AH. When do Lasses (longevity assurance genes) become CerS (ceramide synthases)?: insights into the regulation of ceramide synthesis. J Biol Chem. 2006;281:25001–25005. doi: 10.1074/jbc.R600010200. [DOI] [PubMed] [Google Scholar]
- 34.Andrieu-Abadie N, Levade T. Sphingomyelin hydrolysis during apoptosis. Biochim Biophys Acta. 2002;1585:126–134. doi: 10.1016/s1388-1981(02)00332-3. [DOI] [PubMed] [Google Scholar]
- 35.Bose R, Verheij M, Haimovitz-Friedman A, Scotto K, Fuks Z, Kolesnick R. Ceramide synthase mediates daunorubicin-induced apoptosis: an alternative mechanism for generating death signals. Cell. 1995;82:405–414. doi: 10.1016/0092-8674(95)90429-8. [DOI] [PubMed] [Google Scholar]
- 36.Yang A, McKeon F. P63 and P73: P53 mimics, menaces and more. Nat Rev Mol Cell Biol. 2000;1:199–207. doi: 10.1038/35043127. [DOI] [PubMed] [Google Scholar]
- 37.Flores ER, Tsai KY, Crowley D, Sengupta S, Yang A, McKeon F, Jacks T. p63 and p73 are required for p53-dependent apoptosis in response to DNA damage. Nature. 2002;416:560–564. doi: 10.1038/416560a. [DOI] [PubMed] [Google Scholar]
- 38.Deyoung MP, Ellisen LW. p63 and p73 in human cancer: defining the network. Oncogene. 2007;26:5169–5183. doi: 10.1038/sj.onc.1210337. [DOI] [PubMed] [Google Scholar]
- 39.Rowland BD, Bernards R, Peeper DS. The KLF4 tumour suppressor is a transcriptional repressor of p53 that acts as a context-dependent oncogene. Nat Cell Biol. 2005;7:1074–1082. doi: 10.1038/ncb1314. [DOI] [PubMed] [Google Scholar]
- 40.Ruvolo PP. Ceramide regulates cellular homeostasis via diverse stress signaling pathways. Leukemia. 2001;15:1153–1160. doi: 10.1038/sj.leu.2402197. [DOI] [PubMed] [Google Scholar]
- 41.Chalfant CE, Kishikawa K, Mumby MC, Kamibayashi C, Bielawska A, Hannun YA. Long chain ceramides activate protein phosphatase-1 and protein phosphatase-2A. Activation is stereospecific and regulated by phosphatidic acid. J Biol Chem. 1999;274:20313–20317. doi: 10.1074/jbc.274.29.20313. [DOI] [PubMed] [Google Scholar]
- 42.Chalfant CE, Ogretmen B, Galadari S, Kroesen BJ, Pettus BJ, Hannun YA. FAS activation induces dephosphorylation of SR proteins; dependence on the de novo generation of ceramide and activation of protein phosphatase 1. J Biol Chem. 2001;276:44848–44855. doi: 10.1074/jbc.M106291200. [DOI] [PubMed] [Google Scholar]
- 43.Stamm S. Regulation of alternative splicing by reversible protein phosphorylation. J Biol Chem. 2008;283:1223–1227. doi: 10.1074/jbc.R700034200. [DOI] [PubMed] [Google Scholar]
- 44.Kishikawa K, Chalfant CE, Perry DK, Bielawska A, Hannun YA. Phosphatidic acid is a potent and selective inhibitor of protein phosphatase 1 and an inhibitor of ceramide-mediated responses. J Biol Chem. 1999;274:21335–21341. doi: 10.1074/jbc.274.30.21335. [DOI] [PubMed] [Google Scholar]
- 45.Wright SC, Zheng H, Zhong J. Tumor cell resistance to apoptosis due to a defect in the activation of sphingomyelinase and the 24 kDa apoptotic protease (AP24) FASEB J. 1996;10:325–332. doi: 10.1096/fasebj.10.2.8641566. [DOI] [PubMed] [Google Scholar]
- 46.Chalfant CE, Szulc Z, Roddy P, Bielawska A, Hannun YA. The structural requirements for ceramide activation of serine–threonine protein phosphatases. J Lipid Res. 2004;45:496–506. doi: 10.1194/jlr.M300347-JLR200. [DOI] [PubMed] [Google Scholar]
- 47.Leoni LM, Shih HC, Deng L, Tuey C, Walter G, Carson DA, Cottam HB. Modulation of ceramide-activated protein phosphatase 2A activity by low molecular weight aromatic compounds. Biochem Pharmacol. 1998;55:1105–1111. doi: 10.1016/s0006-2952(97)00685-0. [DOI] [PubMed] [Google Scholar]
- 48.Wolff RA, Dobrowsky RT, Bielawska A, Obeid LM, Hannun YA. Role of ceramide-activated protein phosphatase in ceramide-mediated signal transduction. J Biol Chem. 1994;269:19605–19609. [PubMed] [Google Scholar]
- 49.Cho S, Hoang A, Sinha R, Zhong XY, Fu XD, Krainer AR, Ghosh G. Interaction between the RNA binding domains of Ser–Arg splicing factor 1 and U1-70K snRNP protein determines early spliceosome assembly. Proc Natl Acad Sci U S A. 2011;108:8233–8238. doi: 10.1073/pnas.1017700108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Xiao SH, Manley JL. Phosphorylation–dephosphorylation differentially affects activities of splicing factor ASF/SF2. EMBO J. 1998;17:6359–6367. doi: 10.1093/emboj/17.21.6359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Das R, Yu J, Zhang Z, Gygi MP, Krainer AR, Gygi SP, Reed R. SR proteins function in coupling RNAP II transcription to pre-mRNA splicing. Mol Cell. 2007;26:867–881. doi: 10.1016/j.molcel.2007.05.036. [DOI] [PubMed] [Google Scholar]
- 52.Michlewski G, Sanford JR, Caceres JF. The splicing factor SF2/ASF regulates translation initiation by enhancing phosphorylation of 4E-BP1. Mol Cell. 2008;30:179–189. doi: 10.1016/j.molcel.2008.03.013. [DOI] [PubMed] [Google Scholar]
- 53.Ngo JC, Chakrabarti S, Ding JH, Velazquez-Dones A, Nolen B, Aubol BE, Adams JA, Fu XD, Ghosh G. Interplay between SRPK and Clk/Sty kinases in phosphorylation of the splicing factor ASF/SF2 is regulated by a docking motif in ASF/SF2. Mol Cell. 2005;20:77–89. doi: 10.1016/j.molcel.2005.08.025. [DOI] [PubMed] [Google Scholar]
- 54.Ma CT, Ghosh G, Fu XD, Adams JA. Mechanism of dephosphorylation of the SR protein ASF/SF2 by protein phosphatase 1. J Mol Biol. 2010;403:386–404. doi: 10.1016/j.jmb.2010.08.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Misteli T, Spector DL. Serine/threonine phosphatase 1 modulates the subnuclear distribution of pre-mRNA splicing factors. Mol Biol Cell. 1996;7:1559–1572. doi: 10.1091/mbc.7.10.1559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sanford JR, Bruzik JP. Regulation of SR protein localization during development. Proc Natl Acad Sci U S A. 2001;98:10184–10189. doi: 10.1073/pnas.181340498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lin S, Xiao R, Sun P, Xu X, Fu XD. Dephosphorylation-dependent sorting of SR splicing factors during mRNP maturation. Mol Cell. 2005;20:413–425. doi: 10.1016/j.molcel.2005.09.015. [DOI] [PubMed] [Google Scholar]
- 58.Tenenbaum SA, Aguirre-Ghiso J. Dephosphorylation shows SR proteins the way out. Mol Cell. 2005;20:499–501. doi: 10.1016/j.molcel.2005.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Karni R, de Stanchina E, Lowe SW, Sinha R, Mu D, Krainer AR. The gene encoding the splicing factor SF2/ASF is a proto-oncogene. Nat Struct Mol Biol. 2007;14:185–193. doi: 10.1038/nsmb1209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Anczukow O, Rosenberg AZ, Akerman M, Das S, Zhan L, Karni R, Muthuswamy SK, Krainer AR. The splicing factor SRSF1 regulates apoptosis and proliferation to promote mammary epithelial cell transformation. Nat Struct Mol Biol. 2012;19:220–228. doi: 10.1038/nsmb.2207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Shultz JC, Goehe RW, Murudkar CS, Wijesinghe DS, Mayton EK, Massiello A, Hawkins AJ, Mukerjee P, Pinkerman RL, Park MA, Chalfant CE. SRSF1 regulates the alternative splicing of caspase 9 via a novel intronic splicing enhancer affecting the chemotherapeutic sensitivity of non-small cell lung cancer cells. Mol Cancer Res. 2011;9:889–900. doi: 10.1158/1541-7786.MCR-11-0061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Goehe RW, Shultz JC, Murudkar C, Usanovic S, Lamour NF, Massey DH, Zhang L, Camidge DR, Shay JW, Minna JD, Chalfant CE. hnRNP L regulates the tumorigenic capacity of lung cancer xenografts in mice via caspase-9 pre-mRNA processing. J Clin Invest. 2010;120:3923–3939. doi: 10.1172/JCI43552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Fregoso OI, Das S, Akerman M, Krainer AR. Splicing-factor oncoprotein SRSF1 stabilizes p53 via RPL5 and induces cellular senescence. Mol Cell. 2013;50:56–66. doi: 10.1016/j.molcel.2013.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.