

Abstract
Several largazole analogues with modified surface recognition cap groups were synthesized and their HDAC inhibitory activities were determined. The C7-epimer 12 caused negligible inhibition of HDAC activity, failed to induce global histone 3 (H3) acetylation in the HCT116 colorectal cancer cell line and demonstrated minimal effect on growth. Although previous studies have shown some degree of tolerance of structural changes at C7 position of largazole, these data show the negative effect of conformational change accompanying change of configuration at this position. Similarly, analogue 16a with D-1-naphthylmethyl side chain at C2 too had negligible inhibition of HDAC activity, failed to induce global histone 3 (H3) acetylation in the HCT116 colorectal cancer cell line and demonstrated minimal effect on growth. In contrast, the L-allyl analogue 16b and the L-1-naphthylmethyl analogue 16c were potent HDAC inhibitors, showing robust induction of global H3 acetylation and significant effect on cell growth. The data suggest that even bulky substituents are tolerated at this position, provided the stereochemistry at C2 is retained. With bulky substituents, inversion of configuration at C2 results in loss of inhibitory activity. The activity profiles of 16b and 16c on Class I HDAC1 vs Class II HDAC6 are similar to those of largazole and, taken together with x-ray crystallography information of HDAC8-largazole complex, may suggest that the C2 position of largazole is not a suitable target for structural optimization to achieve isoform selectivity. The results of these studies may guide the synthesis of more potent and selective HDAC inhibitors.
Keywords: Antitumor agents, biological activity, antiproliferation, histone deacetylase inhibitors, molecular modeling
INTRODUCTION
Aberrant epigenetic silencing of tumor suppressor genes can be reversed; thus targeting the enzymes responsible for these modifications has become an attractive therapeutic approach in cancer [1]. Mechanisms of epigenetic silencing include DNA methylation and modification of key residues of histone tails. Lysine residues of histone tails can be acetylated/deacetylated, which can alter gene expression [2]. Histone acetylation status is regulated by two enzymes, histone acetyl transferases (HAT), which acetylates lysine tails of histone proteins and histone deacetylases (HDAC), which deacetylate them [3]. FDA approval of two HDAC inhibitors (HDACi) suberoylanilide hydroxamic acid (1, SAHA, vorinostat) [4] and FK228 (2, romidepsin) [4] (Figure 1) for the treatment of cutaneous T-cell lymphoma (CTCL) has stimulated extensive research to find more potent and isoform selective HDACis which, in addition to their therapeutic potential, will also be useful as probes to determine the physiological/pathogenic role of individual HDAC isoforms [5]. Three major structural motifs characteristic of a histone deacetylase inhibitor are: i) a metal binding domain which chelates with the active site Zn2+ cation ii) a linker, which occupies the hydrophobic channel iii) a surface recognition cap group, which interacts with hydrophobic residues on the rim of the active site [6]. All these three sectors may be targeted for structural alteration to achieve HDAC class and/isoform selectivity. The 14 Å internal cavity at the bottom of the active site [7–14] and the hydrophobic rim at the entrance to the active site [15] are being specially targeted for developing isoform selective HDAC inhibitors by exploiting variations in the amino acid residues in these two regions.
Figure 1.
HDAC inhibitors
Largazole 3 (Figure 1), a potent and selective HDACi recently isolated from a marine cyanobacterium, has attracted the attention of medicinal chemists as an important lead molecule for chemical modification to develop anticancer agents [16]. As HDACs proteins are associated with many basic cellular processes and aberrant HDAC activity is also linked to human disorders other than cancer, the effect of largazole on other disease states such as inflammation and rheumatoid arthritis too are being explored [17–19]. A number of largazole analogues have been synthesized and their HDAC inhibitory activities determined revealing some of the structure-activity relationship (SAR) requirements of the molecule [20–38],[39]. Changes in all three sectors of the molecule have been effected. We recently reported the synthesis and biological activity of several largazole analogues incorporating multiple heteroatoms in the metal binding domain [39]. In continuation of our efforts in the search for selective and potent anticancer agents [39–43], we report here the synthesis and biological activity of several largazole analogues with modifications in the depsipeptide ring, which serves as the cap group that interacts with the hydrophobic rim of the active site. These modifications include inversion of stereochemistry at C7 of largazole to generate the C7-epimer 12 as well as structural and stereochemical changes in the valine side chain at C2 to generate analogues 16a–c.
CHEMISTRY
The synthetic strategy we previously employed in the assembly of largazole [39] was conveniently adopted in the synthesis of these analogues. Using (S)-α-methylcysteine HCl (6), instead of (R)-α-methylcysteine HCl used in largazole synthesis, gave the C7-epimer 12 (Scheme 1). (S)-α-Methylcysteine HCl 6 formed as a product during the synthesis of (R)-α-methylcysteine HCl reported earlier was used in this synthesis [39]. The nitrile 5 [39] was condensed with (S)-α-methylcysteine 6 to obtain the thiazole-thiazoline carboxylic acid 7 [22]. After conversion to methyl ester and removal of Boc protecting group, the acyl group was transferred [44] from 8 to obtain the alcohol 9. Compound 8 was synthesized by stereoselective aldol reaction as previously reported [39]. The conversion of the nitrile 5 to the carboxylic acid 7 and the one-pot conversion of the latter to alcohol 9 gave 67% yield for 3 steps. Yamaguchi esterification [20, 45] of 9 with Fmoc-valine afforded the acyclic precursor 10 in 93% yield. After removal of protecting groups, macrocyclization with HOAt, HATU, and Hunig’s base yielded the cyclized product 11 in 31% yield over the 3 steps [20]. The cyclized core 11, upon removal of trityl group and thioesterification with octanoyl chloride, produced analogue 12 in 72% overall yield for two steps [21].
Scheme 1.

Synthesis of analogue 12 (C7-epimer of largazole)
Synthesis of analogues with C2 side chain modification is shown in Scheme 2. Yamaguchi coupling of alcohol 13, the synthesis of which was reported earlier [39], with Fmoc-protected amino acids D-1-naphthylmethylglycine, L-allylglycine and L-1-naphthylmethylglycine produced the acyclic precursors 14a–c, respectively. Macrocyclization, after removal of protecting groups, gave the cyclic cores 15a–c which were converted to analogues 16a–c as described earlier [39].
Scheme 2.

Synthesis of analogues 16a–c
BIOLOGICAL EVALUATION
The effects of these compounds on cytoproliferation were measured in the colorectal cancer cell line HCT116 using a standard 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium (MTS) reduction assay (Promega) as previously reported [46]. Cells were treated with compounds 12, 16a, 16b, 16c and largazole as a positive control for 96 hours and cytoproliferation was quantified. Varying effects on cell growth were observed indicating these structural modifications have gross differences on activities from the parent compound (Figure 2). Compounds 16b and 16c showed the greatest effect with GI50’s of 10 nM and 56 nM, respectively. No significant effect on cytoproliferation was observed for compounds 16b and 16c at lower concentrations (0.01, 0.1 and 1.0 nM) (data not shown). Additionally, analogues 12 and 16a demonstrated little effect on cell growth even at the highest concentration tested, 5 μM.
Figure 2.
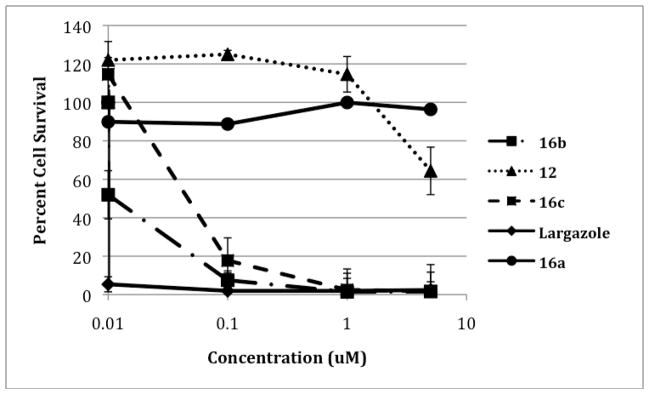
Growth inhibition of HCT116 colon carcinoma cells by largazole and analogues. Each point represents the mean of two experiments, each with 4 replicates. The error bars indicate the SEM.
Analogues were also evaluated for their in vitro effect on HDAC activity measured by a fluorimetric assay using either recombinant HDAC1 or HDAC6 with a substrate specific for class I and class II HDAC activity respectively (Figure 3). Largazole showed the greatest inhibition of the recombinant enzymes, but both analogues 16b and 16c showed significant inhibition of HDAC1. These results were similar to what was observed with the cytoproliferation assay, with both these analogues showing significant activity, but slightly less than the parent compound. Largazole, as well as 16b and 16c showed more than 90% inhibition of HDAC1 activity at 1 μM. Interestingly, there was a dramatic reduction in their inhibitory activity on HDAC1 at 0.1 μM, with both largazole and 16c inhibiting HDAC1 activity by only about 15% while 16b had hardly any effect. Analogues 16a and 12 inhibited HDAC1 activity only by about 15% or less at 1 μM. Importantly, both 16b (~55%) and 16c (~50%) inhibited HDAC6 to a lesser extent than largazole (~75%) at 5 μM, suggesting somewhat greater selectivity of the newer analogues for HDAC1 as compared to largazole. However, at more moderate concentration of 1 uM, HDAC6 inhibitory activity became comparable for all three compounds (~50%). Analogue 16a showed the least amount of HDAC6 specific inhibition, but was inactive in the HCT116 cell growth inhibition assay as well. These data suggest that given the lack of activity against HDAC6, these structural modifications have no effect on the ability of largazole to inhibit class II HDACs like HDAC6.
Figure 3.
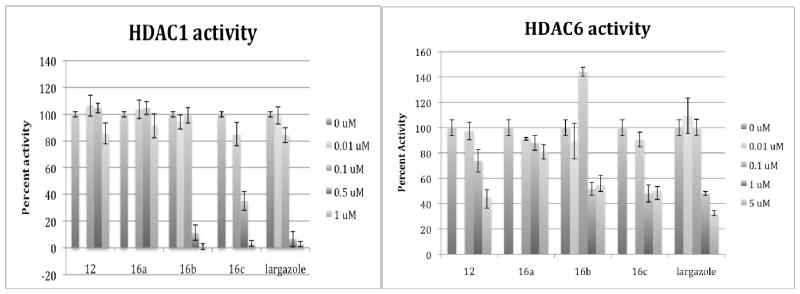
In vitro HDAC inhibitory activity – Recombinant HDAC1 and HDAC6 were incubated with and without analogues at varying concentrations. Reactions were carried out for 1 hour at a concentration of 0–1 μM (for HDAC1) and 0–5 μM (for HDAC6) for all analogues. Analogues 16b and 16c showed the greatest similarity to largazole in its inhibition of HDAC1 and HDAC6. However, none of the compounds showed more than a 50% inhibition of HDAC6 activity, except at the highest concentration of 5 μM.
To determine how these compounds affected cellular HDAC activity in vitro, the colorectal cancer cell line HCT116 was treated with increasing concentrations of each of the compounds for 24 hours. Global histone H3 lysine 9 (H3K9) acetylation was assayed as an indicator of the efficacy of these compounds in global HDAC inhibition. Consistent with what was observed with the in vitro enzyme assays, analogues 12 and 16a produced very little induction of global H3K9 acetylation. Analogue 12 showed a slight increase of global H3K9 acetylation at high concentration, which corresponds to the results of the cell growth assay. Of the analogues synthesized, analogues 16b and 16c showed the greatest induction of global H3K9 acetylation at the lower concentrations (Figure 4). Analogue 16c showed similar levels of induction of global H3K9 acetylation as largazole at the lower concentrations, consistent with the in vitro analysis.
Figure 4.

Western blot analysis of global acetylated H3K9 following the treatment of HCT116 cells with largazole and analogues at 10 nM, 100 nM and 1 μM concentrations after 24 hours of treatment, with beta-actin as a loading control. Increase of global H3K9 acetylation was observed with largazole as early as 10 nM similar to analogue 16c. Analogue 16b showed increase at 100 nm, while 12 and 16a showed no change.
DISCUSSION
Targeting histone deacetylase enzymes for reversible epigenetic regulation of gene expression as a means of therapeutic intervention is an attractive strategy to treat many human disorders, especially cancer. At least 18 different isoforms of human HDAC proteins have been identified, but their specific role in cellular function and physiology are still not well understood [5]. However, specific HDAC isoforms over-expressed in some types of cancer can be targeted to develop selective anticancer agents. The USFDA approved the pan HDACi SAHA (vorinostat) for the treatment of cutaneous T-cell lymphoma (CTCL) in 2006. A second drug FK 228 (romidepsin) approved by the FDA in 2009 for the same human condition is a class I specific inhibitor of HDAC. Both molecules incorporate the essential structural features of HDAC inhibitors and consist of a metal binding domain, a surface recognition cap and a hydrophobic linker. The metal binding domain of SAHA is a strongly metal-chelating hydroxamic acid moiety while that of FK 228 is a thiol group generated by in situ reduction of a disulfide bridge. The large hydrophobic depsipeptide cap group of FK 228 may contribute to its more class I selective properties by interacting with less conserved regions located further away than those reachable by the smaller benzamide cap group of SAHA in the rim of the active site [47]. Interestingly, tubacin and related compounds with structural similarities to SAHA, but with a large cap group, are more selective for HDAC6 than HDAC1, underscoring the importance of the nature of the cap group in discriminating the HDAC isoforms [48, 49]. The interest in the recently isolated marine natural product largazole as a potent HDAC inhibitor stems from many common structural features that it shares with FK 228. Both are depispeptides, the depsipeptide ring constituting the surface recognition cap group of both inhibitors. They have identical linker groups and masked thiol groups as metal-binding motiffs. A number of largazole analogues have been synthesized and their biological properties determined to unravel some SAR requirements of the molecule. The importance of thiol as the metal binding motif is evident from the lack or reduced activity of analogues in which the thiol group was replaced with other groups such as benzamide, thiobenzamide, ketone and esters functionalities [22, 26] and with thioether-containing dual hetero atom combinations. [39] The linker between the metal binding motif and the depsipeptide ring has to be four atoms in length [23, 27] and should preferably have a trans vs cis double bond for optimal activity [31]. Importantly, some structural alterations in the depsipeptide ring were well tolerated. Particularly noteworthy is the generation of the most potent HDACi known by replacing the thiazole ring with a pyridine ring [26]. L-valine could be replaced with other amino acids without affecting affinity. Substitution of L-tyrosine, L-alanine and glycine for L-valine was well tolerated [27, 31, 34, 36]. The C-2 epimer of largazole retained reduced, but sub-micromolar inhibitory potency against HDAC isoforms [26]. Interestingly, the C-2 epimer was found to be more potent than largazole in inhibiting the viability of prostate cancer cell lines LNCaP and PC-3 and both upregulated the expression of p21 [33]. These subtle variations in activity accompanying changes at C2 prompted us to further explore this space by replacing the valine side chain with other groups.
We adopted the convergent approach that we earlier reported for the synthesis of the new analogues 16a–c [39] using the respective amino acids in place of L-valine. The analogues 16a, 16b and 16c with valine replaced by D-1-naphthylmethylglycine, L-allylglycine and L-1-naphthylmethylglycine, respectively were synthesized as shown in scheme 2. The intermediate 13 was esterified with the respective amino acids to obtain 14a–c. Hydrolysis of methyl ester with LiOH and removal of Fmoc group with Et2NH, followed by macrolactamization with HATU and HOAt in the presence of DIEA, gave the cyclic intermediates 15a–c. The trityl protecting group was removed with TFA and the thiols were esterified with octanoyl chloride to obtain analogues 16a–c. The HDAC inhibitory potency of largazole appears to be highly sensitive to electronic, conformational and stereochemical perturbations in the macrocycle. Subtle changes in the chemical structure of the macrocycle have been suggested to produce significant conformational changes resulting in changes in inhibitory potency of the molecule [25]. An isosteric analogue of largazole thiol incorporating a single atom substitution of thiazole sulfur atom for an oxygen atom showed substantial activity on Class IIB enzymes HDAC7 and HDAC8, which were not appreciably sensitive to largazole thiol [37]. It has been suggested that subtle changes in electronic properties resulting from this substitution has a significant effect on conformational and related biological properties. Conformational change associated with replacing the ester bond in the ring with an amide bond had a deleterious effect on the binding interaction with HDAC proteins [25]. The enantiomer of largazole was three times less potent than largazole in inhibiting HDAC isoforms [26]. Therefore, we synthesized the C-7 epimer of largazole 12 (Scheme 1) to probe the effect of change in configuration at this position on activity. The synthesis was accomplished by using (S)-α-methylcysteine HCl (5) instead of (R)-α-methylcysteine HCl used in largazole synthesis reported earlier [39].
The L-naphthylmethyl analogue 16c and the L-allyl analogue 16b inhibited the growth of HCT116 cells with GI50 values of 56 nM and10 nM, respectively compared to a GI50 value of <10 nM for largazole. In contrast, the D-naphthylmethyl analogue 16a had no effect on the growth of HCT116 cells even at 5 μM, the highest concentration tested. The L-amino acid modified analogues 16b and 16c retained their ability to induce global H3 acetylation, however, to a slightly lesser degree than the parent compound, largazole. Analogue 16c showed very similar activity to largazole as measured by the in vitro fluorescent assay of HDAC activity and similar to largazole it was also able to induce global H3K9 acetylation at the lowest concentration tested (10 nM), indicating that L-1-naphthylmethylglycine modification most closely mimics the original. Interestingly, while largazole and analogues 16b and 16c inhibited HDAC1 by more than 90% at 1 μM, there was a sharp reduction in their inhibitory competency at 0.1 μM. Analogues 16b and 16c and largazole had comparable levels of inhibitory activity on HDAC6 at 1 μM which may suggest that structural changes at this position may not have an effect on their class selectivity. The antiproliferative and HDAC inhibitory activity data are consistent with those previously reported for other valine-substituted analogues. Replacement of L-valine with L-alanine was reported to be well tolerated with only 2–3 fold decrease in antiproliferative and HDAC inhbitory activty and without appreciable change in HDAC1 vs HDAC6 selectivity [27]. L-tyrosine analogue too was found to be only slightly less potent than largazole. Interestingly this analogue was 100 fold more selective toward human cancer cells over normal cells [31]. Similarly, replacement of L-valine with glycine in largazole thiol was tolerated and cuased only a slight decrease in HDAC inhibitory activity and nearly a ten fold decrease in antiproliferative activity [34]. Isoform selectivity data for L-tyrosine and glycine analgues are not availble [31]. These results are consistent with the x-ray crystal structure of largazole-HDAC8 complex which shows that the valine side chain of largazole points directly out towards the solvent and has no effect on HDAC-largazole interaction [50]. Therefore it has been suggested that the substitution of L-valine with other amino acids or even D-amino acids and disubstitued amino acids may be well tolerated, provided they do not perturb the conformation of the macrocycle [50]. Infact, the C2 epimer of largazole retained reduced, but sub-micromolar inhibitory potency against HDAC isoforms [26]. However, the lack of antiproliferative and HDAC inhibitory activity and the inability to induce global H3K9 acetylation by 16a suggests that D-amino acids with bulky side chains are not tolerated at this position. The largazole C7-epimer 12 had minimal effect on cell growth. This corresponds to the lack of induction of AcH3K9 levels by analogue 12 in treated colorectal cells. Only a slight induction of acetylation was observed at the highest dose tested. Even the in vitro analysis of recombinant HDAC activity showed relatively minimal effects on class I and II activity for this analogue. Previous studies have shown some degree of tolerance of structural changes at this position; replacement of C7 methyl with a hydrogen atom, or an ethyl or a benzyl group having no signifcant effect on activity[32]. This is not surprising as the methyl group has been shown not to have any interaction with the protein surface in the HDAC8-largazole complex. However, loss of activity in the C7-epimer suggests that conformational change associated with change in stereochemistry at C7 position has a deleterious effect on activity. We used molecular modeling studies to study the effect of inverting configuration at C7 on the molecular shape of analogue 12, which may account for its reduced binding affinity compared to largazole.
MOLECULAR MODELING
Geometry Optimizations
Largazole thiol and the thiol of C7-epimer 12 were energy minimized using Gaussian09-C01 [51] with the use of cc-pVTZ basis set at B3LYP level. The optimized structures and some interatomic distances are shown in Figures 5 and 6. When the two optimized structures of the thiols of largazole and the C7-epimer 12 were compared, the largest deviations were seen around the central ring near the site of epimerization (Figure 5). The sulfur atoms of the thiazole-thiazoline rings pointed upward in largazole thiol. Two strong hydrogen bonds involving the proton attached to N2 (see Figure 6c for numbering used); one with O17 (at a distance of 2.24 Å) and the other with N7 (at a distance of 2.13 Å) are found in largazole thiol (as shown by dotted bonds marked in purple labels in Figure 6a). The geometry of the macrocyclic skeleton may be strongly influenced by these intramolecular hydrogen bonds to acquire the specific shape required for HDAC binding. However, in the optimized configuration of the C7-epimer (Figure 5b middle and bottom), the thiazoline ring which incorporates the C7 atom has its N7 and S8 atoms in somewhat inverted positions compared to the corresponding atom positions of largazole thiol. The N2 proton and O17 are separated by a distance of 2.74 Å, which does not favor hydrogen bonding (Figure 6b). However, the hydrogen bond between the N2 proton and N7 at a distance of 2.2 Å remains intact. Another consequence of the inversion of configuration at C7 is that the N2 proton which is directed towards the interior of the ring in largazole is pointing outward in the C7-epimer. The C7-epimerization seems to have caused a slight expansion of the ring as evidenced by the distances shown in Figure 6. The C3 is rotated outward by almost 60° angle, thereby making the outer width of the ring in the C2-C17 direction expanded by over 2.5 Å. Further, the change in electronic charge distribution associated with the inverted geometry of the thiazoline ring containing N7 and S8 atoms changes the direction of its electric dipole. The position of the more negative atom N7 (charge = −0.7e; the electronic partial charges are estimated using the CHELPG procedure [52] in Gaussian09-C01) is now occupied by a less negative S8 (charge = −0.3e). Also, the methyl group at C7 is pointing away from the ring compared to that of largazole thiol and it is found to be directed towards the general direction of the isopropyl side chain of the valine. These changes in the spatial orientation of groups in the C7-epimer may affect its binding to HDAC and account for its reduced HDAC inhibitory activity. We further evaluated this information in the context of the binding (or lack of it) of largazole thiol and its C7-epimer to HDAC.
Figure 5.

Optimized structures of largazole thiol and the thiol of its C7-epimer. (a) Three different views of the optimized largazole thiol structure; and to facilitate direct comparison, (b) corresponding views of the C7-epimer are displayed. The hydrocarbon chain connecting the macrocyclic skeleton and the thiol S is seen to be unaltered in all views. The directionality of the thiazoline ring is altered (shown in the middle panel). Also, a change in directionality of C7 methyl group is observed in the top left corner of the middle panel. The isopropyl group is rotated (top right corner of the bottom panel) outward and the outer width of the ring around this area is increased substantially.
Figure 6.

The distances important for the assessment of changes in the macrocyclic skeleton due to C7- epimerization are shown for (a) largazole thiol and (b) the thiol of its C7-epimer. Also shown (in (c)) is the numbering of atoms used in the modeling. Possible hydrogen bonds are labeled in purple.
In the X-ray crystal structure [50] the tail segment connecting atom C17 of the central ring down to S21were found to be embedded in the binding site as a column where S21 found at the end of the column is in direct contact with the HDAC bound Zn2+. This hydrocarbon linkage between the central ring and the sulfur bound to Zn2+ is in a hydrophobic environment provided by HDAC (H143, F152, H180, F208, M274 and Y306 residues of HDAC). In addition to its biding to Zn2+, S21 is in contact with D178, D267, and G304 of HDAC. As shown in Figure 5, both largazole and its C7-epimer show very similar spatial arrangements and charge distributions in their hydrocarbon tail segments. In addition, in the X-ray crystal structure, N14 makes a hydrogen bond with D101 and C15 is in contact with F208. In fact, this side of the two molecules does not seem to deviate much from each other. If we were to use the binding features of largazole thiol directly from the X-ray crystal structure, we would undoubtedly arrive at the conclusion that the thiol of the C7-epimer should bind at the HDAC binding site quite similar to largazole thiol. What leads to different HDAC binding behavior of thiols of largazole and its C7-epimer is further characterized using the results of molecular dynamics (MD) simulations of the complex.
Molecular Dynamics Simulations
In the X-ray crystal structure, the asymmetric unit cell contains two HDAC molecules with largazole thiol bound near their Zn2+ binding sites [50]. In the current crystal form, not only are the bound largazole molecules in direct contact with each other (Figure 7a), but also each largazole thiol interacts with residues from the other HDAC molecule thereby influencing the natural binding environment that the molecule can have in a one ligand - one receptor scenario. In the case where structural differences of largazole derivatives are found to be in the areas where the binding environment is modified due to the presence of the second molecule in the crystallographic unit cell, it is difficult to use existing information directly from the X-ray structure for the purpose of predicting the binding or explaining why some derivatives do not bind. Therefore, we have sought help from a MD simulation of HDAC with largazole thiol bound to its active site to obtain an unadulterated binding pose of largazole thiol to HDAC.
Figure 7.

(a) The dimer interactions from the X-ray crystal structure (pdb: 3RQD). (b) superimposition of the X-ray crystal configuration (white ribbon) with the final structure from the molecular dynamics simulation (blue ribbon). Largazole thiols from X-ray (yellow) and simulation (red) are shown in the ball-and-stick and surface representations; (c) Largazole thiol (ball-and-stick) form MD simulation shown with the D101 interaction with N14; (d) C7-epimer docked at the largazole binding site of HDAC showing the steric contacts with the backbone atoms of the loop containing D101. Moving this loop away from the C7-epimer results in the broken hydrogen bond between D101 and N14.
Present simulation system is an all-atom-fully-solvated HDAC molecule with a single largazole thiol bound near its Zn2+ binding site. The initial structure comes directly from the high resolution X-ray crystal structure of Cole at al. [50], and the final structure after sufficiently lengthy molecule dynamics equilibration (over 30ns of dynamics) can reasonably approximate a solution structure of largazole thiol bound HDAC.
In order to assess whether the system is at equilibrium with the surrounding solvent, root mean square deviations (RMSD) of the backbone atoms of the final MD configuration were compared with the X-ray structure. In the final 10ns of the molecular dynamics trajectory, the RMSD values fluctuate around 1Å (results not shown), indicating a reasonably equilibrated system. Comparison of a representative structure from the final stage of MD with the X-ray configuration shown in Figure 7b clearly indicates that most structural segments of the protein are in close alignment with its X-ray structure. As expected, loop regions show movements in their positions due to dynamics, and the position of macrocyclic ring of largazole thiol ligand is shifted towards the area formally occupied by the second largazole thiol in the X-ray crystal structure. Figure 7c displays the ligand binding site and the bound ligand. In its equilibrated position, C2 group from one side and C11 from the opposite side of the central ring are in tight contact with the binding site residues. As pointed out earlier through the optimized structure of C7-epimer, outward rotation of C3 extends the spatial occupation along the C2-C14 direction by over 2.5 Å. If the C7-epimer were to bind (Figure 7d), the binding site needs to be expanded to accommodate the new length. This will require that the loop containing D101 moves away from the substrate, thus leading to a situation that makes N14 interaction with D101 difficult. In addition, in the case of largazole thiol, the C9 containing methyl group is in contact with the methyl group of M274. In the C7-epimer, the corresponding methyl group is shifted up toward the solution and is in the side of isopropyl group of valine in the macrocyclic skeleton (C2-C3-C4) making an extended hydrophobic surface. If the C7-epimer were to adopt a bound conformation similar to largazole thiol, this methyl group in the C7-epimer would become completely solvent exposed in a hydrophobic part of the molecule, and therefore, it would associate with a significant energy cost if it were to bind.
In addition, the top portion of the macrocyclic skeleton of the ligand is solvated entirely by water with no direct contact with HDAC. These water molecules are in a certain hydrogen bonded network to solvate the top portion of the ligand while wetting the residues of HDAC in the cleft of the binding cavity. Since the positions of N7and S8 atoms are switched in the C12 epimer, it would play unfavorably in stabilizing the cooperative network of waters observed over the ligand and the binding site cleft. Note here that in addition to the water molecule found near S21 in the X-ray crustal structure, two additional water molecules are trapped around this region of the solution structure.
CONCLUSION
The C7-epimer and C2 side chain modified analogues of largazole were synthesized using the general synthetic approach to largazole reported earlier [39]; the C7-epimer was synthesized by substituting (S)-α-methylcysteine HCl (6) for (R)-α-methylcysteine HCl and the C2 side chain modified analogues were synthesized using Fmoc-protected amino acids D-1-naphthylmethylglycine, L-allylglycine and L-1-naphthylmethyllglycine instead of Fmoc-valine, in the synthesis of largazole. Replacement of L-valine with other L-amino acids was tolerated. The L-allyl analogue 16b and the L-naphthylmethyl analogue 16c were potent HDAC inhibitors as measured by the induction of global H3 acetylation in treated HCT116 colorectal carcinoma cells. However, these analogues were slightly less potent compared to the parent compound largazole. Previous studies have shown that the replacement of L-valine of largazole with L-tyrosine, L-alanine and glycine caused only a slight decrease in HDAC and antiproliferative activities [27, 31, 34]. These results are consistent with the x-ray crystal structure of largazole-HDAC8 complex in which the valine side chain pionts directly out toward solvent and has no effect on HDAC-largazole interaction [50]. Hence, it has been suggested and that even D-amino acids may be tolreated at this position and not surpisingly, the C2-epimer of largazole retained reduced, but sub-micromolar inhibitory potency against HDAC isoforms. However, the inactive D-naphthylmethyl analogue 16a shows that there is a steric limitation to this tolerance and D-amino acids with bulky substituents are not tolerated at C2. The data suggests that substituents other than isopropyl, and even bulky substituents like naphthylmethyl, are tolerated at this position provided the original stereochemistry at C2 is retained. Inverting configuration, at least with bulky substituents, is detrimental as shown by the loss of inhibitory activity of 16a. The activity profiles of 16b and 16c on Class I HDAC1 vs Class II HDAC6 are similar to those of largazole and, taken together with x-ray crystallography information of HDAC8-largazole complex, may suggest that the C2 position of largazole is not a suitable target for structural optimization to achieve isoform selectivity. Inversion of configuration at C7 as well resulted in loss of HDAC inhibition and can be attributed to conformational changes associated with this stereochemical change affecting binding to HDAC protein. The results of these studies guide our ongoing efforts to improve the isoform selectivity potential of largazole by further structural changes in the depsipeptide ring of the molecule.
EXPERIMENTAL SECTION
THF was freshly distilled from Na and benzophenone prior to use. NMR spectra were recorded on Varian INOVA 600 MHz, Bruker Avance 600 MHz and Varian VXRS 400 MHz spectrometers and calibrated using residual undeuterated solvent as internal reference (CDCl3: 1H NMR at δ 7.26, 13C NMR at δ 77.23 ppm). Optical rotations were recorded on AUTOPOL III and AUTOPOL IV polarimeters. High-resolution mass spectra (HRMS) were recorded on a Micromass LCT Electrospray mass spectrometer at the Central Instrument Facility, the Wayne State University, Detroit, Michigan and on a Micromass Q-Tof II mass spectrometer at Mass Spectrometry and Proteomics Facility, the Ohio State University, Columbus, Ohio. Flash column chromatography was performed on silica gel (32–63 μ, Dynamic Adsorbents Inc.) and on RediSep prepacked silica cartridges on Teledyne ISCO Combiflash Companion chromatography system. 1000 μ Uniplates (Analtech Inc.) were used for preparative thin layer chromatography using commercial solvents as specified. Microwave synthesis was performed using a Biotage Initiator. HPLC analyses were performed on a Waters 1525 Binary Pump HPLC system with Waters 2487 dual wavelength absorbance detector on a Symmetry C18 column (reverse phase, 5 μ, 4.6 mm × 150 mm) using a linear gradient of 10–100% H2O:MeOH over 15–20 min; flow rate of 1 mL/min and UV detection at 254 nm. A Luna 5 μC18 (2) 100A (250 X 10 mm, 5 μ) column by Phenomenex Inc. was used for HPLC separations. Structural integrity and purity of the test compounds were determined by the composite of 1H NMR and 13C NMR, HRMS, and HPLC and were found to be >95% pure.
(S)-2-(2-((tert-Butoxycarbonylamino)methyl)thiazol-4-yl)-4-methyl-4,5-dihydrothiazole-4-carboxylic acid (7)
To the mixture of the nitrile 5 (0.096 g, 0.4 mmol, 1 equiv) and NaHCO3 (0.232 g, 2.76 mmol, 5.6 equiv) in methanol (5 mL) was added, with stirring, (s)-α-methylcysteine hydrochloride 6 (0.084 g, 0.491, 1.23 equiv), followed by phosphate buffer pH 5.95 (2.5 mL). The reaction mixture was degassed by bubbling nitrogen and was stirred under nitrogen at 70 °C for 1 h. It was acidified with 1 M HCl and extracted with ethyl acetate (3 × 15 mL). The combined organic extract was washed with saturated NaCl solution, dried over anhydrous sodium sulfate and concentrated to obtain the carboxylic acid 7 (0.112g). It was used in the next step without further purification.
(S)-Methyl 2-(2-(((S,E)-3-hydroxy-7-(tritylthio)hept-4-enamido)methyl)thiazol-4-yl)-4-methyl-4,5-dihydrothiazole-4-carboxylate (9)
HCl gas was bubbled through a solution of the carboxylic acid 7 (0.056 g, 0.157 mmol, 1 equiv) in anhydrous methanol (5 mL) for 5 minutes. The reaction mixture was stirred overnight at room temperature, concentrated in vacuo and the residue was azeotroped with toluene. A mixture of the residue and DMAP (0.05 g, 0.410 mmol, 2.61 equiv) in dichloromethane (2 mL) was stirred for 5 minutes and a solution of aldol product 8 (0.088, 0.157 mmol, 1 equiv) in dichloromethane (1 mL) was added. The reaction mixture was stirred for 1 h, concentrated in vacuo and purified by flash chromatography on silica gel in ethyl acetate/hexanes (20–100%) to afford the alcohol 9 (0.07 g, 67% from nitrile 5 over 3 steps). [α]D20=−0.77 (c=0.65 in CHCl3). 1H NMR (600 MHz, CDCl3): δ=7.91 (s, 1H), 7.37 (d, J = 7.2, Hz, 6H), 7.26 (t, J = 6.6 Hz, 6H), 7.19 (t, J = 7.2 Hz, 3H), 6.89 (t, J = 6.0 Hz, 1H), 5.52–5.56 (m, 1H), 5.40 (dd, J = 6.6, 15.0 Hz, 1H), 4.71 (m, 2H), 4.43 (m, 1H), 3.86 (d, J = 11.4 Hz, 1H), 3.78 (s, 3H), 3.26 (d, J = 11.4 Hz, 1H), 2.44 (dd, J = 3.0, 15.6 Hz, 1H), 2.38 (dd, J = 8.4, 15.0 Hz, 1H), 2.18 (t, J = 7.8 Hz, 2H), 2.05 (q, J = 7.2 Hz, 2H), 1.62 ppm (s, 3H); 13C NMR (100 MHz, CDCl3): δ=173.8, 172.0, 167.9, 162.9, 148.5, 145.0, 132.4, 130.5, 129.7, 128.1, 126.8, 122.4, 84.7, 69.4, 66.8, 53.2, 42.9, 41.7, 41.0, 31.6, 31.5, 24.2 ppm. HRMS-ESI m/z [M + Na]+ calcd for C36H37N3O4S3Na: 694.1844, found: 694.1851.
(S)-Methyl 2-(2-((5S,8S)-1-(9H-fluoren-9-yl)-5-isopropyl-3,6,10-trioxo-8-((E)-4-(tritylthio)but-1-enyl)-2,7-dioxa-4,11-diazadodecan-12-yl)thiazol-4-yl)-4-methyl-4,5-dihydrothiazole-4-carboxylate (10)
A solution of Fmoc-L-valine (0.071 g, 0.209 mmol, 2 equiv) in THF (1 mL) at 0 °C was treated with Hunig’s base (0.040 g, 0.31 mmol, 2.98 equiv) and 2,4,6-trichlorobenzoyl chloride (0.062 g, 0.255 mmol, 2.45 equiv). The reaction mixture was stirred at 0 °C for 1 h, when TLC indicated the formation of the anhydride. The alcohol 9 (0.07 g, 0.104 mmol, 1 equiv) and DMAP (0.014 g, 0.115 mmol, 1.1 equiv) in THF (1 mL) were added to the reaction mixture at 0 °C. It was stirred overnight at room temperature and was concentrated in vacuo. Flash chromatographic purification of the residue on silica gel in ethyl acetate/hexanes (20–100%) yielded the acyclic precursor 10 (0.095 g, 93%). [α]D20=−4.18 (c 2.75 in CHCl3). 1H NMR (400 MHz, CDCl3): δ=7.88 (s, 1H), 7.75 (d, J = 7.6 Hz, 2H), 7.57 (d, J = 7.6 Hz, 2H), 7.38 (d, J = 7.2 Hz, 8H), 7.25–7.30 (m, 8H), 7.20 (t, J = 7.2 Hz, 3H), 6.91 (t, J = 5.6 Hz, 1H), 5.59–5.71 (m, 2H), 5.39–5.46 (m, 1H), 5.30 (d, J = 8.4 Hz, 1H), 4.69 (d, J = 6.0 Hz, 2H), 4.30–4.40 (m, 3H), 4.19 (q, J = 7.2 Hz, 1H), 4.07 (dd, J = 3.6, 6.0 Hz, 1H), 3.83 (d, J = 11.2 Hz, 1H), 3.77 (s, 3H), 3.24 (d, J = 11.6 Hz, 1H), 2.57 (d, J = 5.6 Hz, 2H), 2.14–2.20 (m, 2H), 2.00–2.07 (m, 2H), 1.62 (s, 3H), 0.89 (d, J = 6.8 Hz, 3H), 0.84 ppm (d, J = 6.8 Hz, 3H); 13C NMR (100 MHz, CDCl3): δ=173.8, 169.3, 168.6, 163.1, 156.6, 148.3, 144.9, 144.0, 143.9, 143.8, 141.4, 141.4, 134.2, 129.7, 127.9, 127.7, 127.3, 126.8, 125.2, 125.2, 84.6, 72.4, 67.2, 66.8, 59.6, 53.1, 47.3, 47.3, 41.6, 41.6, 41.2, 31.5, 31.3, 31.0, 24.1, 19.2, 18.1 ppm. HRMS-ESI m/z [M + Na]+ calcd for C56H56N4O7S3Na: 1015.3209, found: 1015.3203.
Cyclic core 11
To a stirred solution of 10 (0.095 mg, 0.096 mmol, 1 equiv) in THF/H2O (4:1, 3 mL) at 0 °C was added 0.1 M LiOH (0.96 mL, 0.096 mmol, 1.0 equiv) dropwise over a period of 15 minutes. The reaction mixture was stirred at 0 °C for 1 h. It was acidified with 1 M HCl solution and extracted with EtOAc three times. The combined organic extract was washed with brine, dried over anhydrous sodium sulfate, concentrated and purified by preparative TLC in ethyl acetate to give the carboxylic acid (0.052 g, 0.053 mmol, 1 equiv) which was carried to the next step. The starting material ester 10 (0.039 g) was also recovered during this purification. The carboxylic acid was dissolved in dichloromethane (4 mL) and treated with diethylamine (0.213 g, 2.92 mmol, 55.05 equiv). The mixture was stirred at room temperature for 3 h. It was concentrated to dryness to afford the free amino derivative, which was dried azeotropically with toluene and was treated with HATU (0.041 g, 0.108 mmol, 2.04 equiv), HOAt (0.015 g, 0.110 mmol, 2.08 equiv), dichloromethane (60 mL, ~ 1 mM), and Hunig’s base (0.030 g, 0.230 mmol, 4.33 equiv). The mixture was stirred for 30 h at room temperature, concentrated to dryness and purified by flash chromatography on silica gel in ethyl acetate/hexanes (10–60%) to yield the cyclic core 11 (0.013 g, 31%). [α]D20=−17.08 (c 0.65 in CHCl3); 1H NMR (400 MHz, CDCl3): δ=7.55 (s, 1H), 7.49 (d, J = 10.0 Hz, 1H), 7.34 (d, J = 8.0 Hz, 6H), 7.29 (t, J = 6.8 Hz, 6H), 7.23 (t, J = 7.2 Hz, 3H), 6.72 (t, J = 6.0 Hz, 1H), 5.56–5.57 (m, 1H), 5.14–5.20 (m, 1H), 5.03 (dd, J = 6.0, 15.6 Hz, 1H), 4.79 (dd, J = 7.2, 15.6 Hz, 1H), 4.57 (dd, J = 6.8, 14.4 Hz, 1H), 4.17 (d, J = 11.6 Hz, 1H), 3.94 (dd, J = 5.2, 15.6 Hz, 1H), 3.27 (d, J = 11.6 Hz, 1H), 2.64 (dd, J = 4.0, 10.4 Hz, 1H), 2.51 (dd, J = 4.0, 10.8 Hz, 1H), 2.07–2.25 (m, 3H), 1.80 (s, 3H), 1.52–1.58 (m, 1H), 1.00 (d, J = 6.8 Hz, 3H), 0.97 ppm (d, J = 6.8 Hz, 3H); 13C NMR (100 MHz, CDCl3): δ=174.1, 169.3, 167.5, 163.0, 148.6, 144.7, 130.9, 129.8, 129.7, 128.2, 128.1, 127.1, 121.8, 85.2, 71.5, 67.2, 58.3, 42.9, 42.3, 32.7, 31.1, 31.1, 26.6, 19.3, 18.2 ppm; HRMS (ESI) m/z: [M + Na]+ calcd for C40H42N4O4S3Na: 761.2266, found: 761.2266.
Analogue 12 (C7-epimer of largazole)
To a solution of 11 (0.013 g, 0.0176 mmol, 1 equiv) in dichloromethane (2.5 mL) at 0 °C was added triisopropylsilane (0.007 g, 0.044 mmol, 2.5 equiv) followed by trifluoroacetic acid (0.1535 g, 1.35 mmol, 76.5 equiv) and the mixture was stirred for 3 h at room temperature. It was concentrated in vacuo and the crude product was purified by flash chromatography on silica gel in ethyl acetate/hexanes (20%) to first remove impurity followed by 1% methanol in ethyl acetate to obtain the thiol. To a stirred solution of the thiol in dichloromethane (1 mL) at 0 °C were added Hunig’s base (0.012 g, 0.092 mmol, 5.23 equiv) and octanoyl chloride (0.0114 g, 0.0699 mmol, 3.97 equiv). The mixture was stirred for 4 h at room temperature. The reaction was quenched with methanol and the mixture was concentrated in vacuo. Purification of the crude product by preparative thin layer chromatography on silica gel in ethyl acetate: hexanes (30–60%) gave analogue 12 (0.008 g, 72%). [α]D20=−28.57 (c=0.175 in CHCl3); 1H NMR (600 MHz, CDCl3): δ=7.72 (s, 1H), 7.37 (d, J = 10.2 Hz, 1H), 6.65 (t, J = 6.0 Hz, 1H), 5.62 (q, J = 4.2 Hz, 1H), 5.16–5.24 (m, 2H), 4.85 (dd, J = 7.2, 15.6 Hz, 1H), 4.62 (dd, J = 6.6, 10.2 Hz, 1H), 4.56 (dd, J = 4.8, 15.6 Hz, 1H), 4.18 (d, J = 11.4 Hz, 1H), 3.31 (d, J = 11.4 Hz, 1H), 2.51–2.65 (m, 4H), 2.51 (t, J = 7.8 Hz, 2H), 2.14–2.20 (m, 1H), 1.96 (q, J = 7.2 Hz, 2H), 1.84 (s, 3H), 1.62–1.65 (m, 8H), 1.25–1.29 (m, 10 H), 1.03 (d, J = 7.2 Hz, 3H), 1.01 (d, J = 6.6 Hz, 3H), 0.88 ppm (t, J = 7.2 Hz, 3H); 13C NMR (150 MHz, CDCl3): δ=199.6, 174.2, 169.2, 169.1, 167.7, 163.9, 148.4, 130.6, 127.0, 122.7, 84.9, 71.4, 58.4, 44.4, 42.4, 42.2, 40.3, 32.9, 32.3, 31.8, 29.9, 29.1, 27.8, 26.4, 25.9, 22.8, 19.2, 18.3, 14.3 ppm; HRMS-ESI m/z: [M + Na]+ calcd for C29H42N4O5S3Na: 645.2215, found: 645.2197.
(R)-Methyl 2-(2-((5R,8S)-1-(9H-fluoren-9-yl)-5-(naphthalen-1-ylmethyl)-3,6,10-trioxo-8-((E)-4-(tritylthio)but-1-enyl)-2,7-dioxa-4,11-diazadodecan-12-yl)thiazol-4-yl)-4-methyl-4,5-dihydrothiazole-4-carboxylate (14a)
To a solution of Fmoc-D-1-naphthylalanine (0.130 g, 0.297 mmol, 3.06 equiv) in THF (1 mL) at 0 °C were added Hunig’s base (0.045 g, 0.345 mmol, 3.55 equiv) and 2,4,6-trichlorobenzoyl chloride (0.078 g, 0.32 mmol, 3.3 equiv). The reaction mixture was stirred at 0 °C for 1 h, when TLC indicated the formation of the anhydride. The alcohol 13 (0.065 g, 0.097, 1 equiv) and DMAP (0.013 g, 0.107 mmol, 1.1 equiv) in THF (1 mL) were added to the reaction mixture at 0 °C. It was stirred overnight at room temperature. The reaction mixture was concentrated in vacuo and flash chromatography purification on silica gel in ethyl acetate/hexanes (20–100%) yielded the acyclic precursor 14a (0.058 g, 55%); [α]D20 − 1.38 (c=0.65 in CHCl3). 1H NMR (400 MHz, CDCl3): δ=8.07 (d, J = 8.0 Hz, 1H), 7.80–7.85 (m, 2H), 7.75- 7.77 (m, 3H), 7.44–7.51(m, 4H), 7.34–7.41 (m, 9H), 7.23–7.29 (m, 9H), 7.15–7.19 (t, J = 7.2 Hz, 3H), 5.94 (t, J = 5.6 Hz, 1H), 5.55–5.62 (m, 1H), 5.39–5.46 (m, 2H), −5.32 (dd, J = 7.2, 15.6 Hz, 1H), 4.73 (q, J = 7.2 Hz, 1H), 4.52 (dd, J = 6.0, 16.0 Hz, 1H), 4.42 (dd, J = 5.6, 16.0 Hz, 1H), 4.35 (dd, J = 7.2, 10.4 Hz, 1H), 4.26 (dd, J = 7.2, 10.4 Hz, 1H), 4.13 (t, J = 7.2 Hz, 1H), 3.87 (d, J = 11.2 Hz, 1H), 3.79 (s, 3H), 3.49 (d, J = 8.8 Hz, 1H), 3.24 (d, J = 11.2 Hz, 1H), 2.28 (dd, J = 6.0, 14.4 Hz, 1H), 2.15 (t, J = 7.2 Hz, 2H), 2.00–2.09 (m, 2H), 1.63 (s, 3H), 1.26 ppm (t, J = 7.2 Hz, 1H). 13C (100 MHz, CDCl3) δ=173.8, 170.9, 168.9, 167.9, 162.9, 155.8, 148.5, 144.9, 143.9, 143.8, 141.4, 133.9, 133.9, 132.5, 132.1, 129.7, 129.1, 128.2, 128.1, 127.9, 127.8, 127.3, 127.3, 127.2, 126.9, 126.8, 126.3, 125.6, 125.3, 125.2, 123.6, 122.1, 120.2, 84.7, 72.7, 67.3, 66.8, 55.0, 53.2, 47.2, 41.7, 41.3, 41.1, 36.2, 31.5, 31.3, 24.2 ppm. HRMS-ESI m/z [M + Na]+ calcd for C64H58N4O7S3Na: 1113.3365, found: 1113.3379.
Cyclic core 15a
To a stirred solution of 14a (0.058 g, 0.0532 mmol, 1 equiv) in THF/H2O (4:1, 1.6 mL) at 0 °C was added 0.1 M LiOH (0.53 mL, 0.053 mmol, 1.00 equiv) dropwise over a period of 15 minutes. The reaction mixture was stirred at 0 °C for 1 h, acidified with 1 M HCl solution and extracted with EtOAc three times. The combined organic layer was washed with brine, dried over anhydrous sodium sulfate, and concentrated in vacuo. The residue was purified by prep TLC in ethyl acetate to give the carboxylic acid. To a solution of the carboxylic acid in dichloromethane (13 mL) was added diethylamine (0.1917 g, 2.70 mmol, 50.75 equiv). After stirring at room temperature for 3 h, it was concentrated in vacuo to afford the free amino derivative. After drying azeotropically with toluene, it was treated with HATU (0.041 g, 0.108 mmol, 2.02 equiv), HOAt (0.015 g, 0.110 mmol, 2.07 equiv), dichloromethane (55 mL, ~ 1 mM), and Hunig’s base (0.030 g, 0.23 mmol, 4.32 equiv) and the mixture was stirred for 30 h at room temperature. The reaction mixture was concentrated to dryness and purified by flash chromatography on silica gel in ethyl acetate/hexanes (10–60%) to yield the cyclic core 15a (0.0057 g, 14%). [α]D20=+ 39.3 (c 0.112 in CHCl3). 1H NMR (600 MHz, CDCl3): δ=8.36 (d, J = 8.4 Hz, 1H), 7.80 (d, J = 8.4 Hz, 1H) 7.68 (d, J = 9.0 Hz, 1H), 7.67 (s, 1H), 7.55 (t, J = 7.8 Hz, 1H), 7.45 (t, J = 7.8 Hz, 1H), 7.39 (d, J = 8.4 Hz, 6H), 7.32 (d, J = 7.2 Hz, 1H), 7.26 (t, J = 7.8 Hz, 6H), 7.19 (t, J = 7.2 Hz, 3H), 7.16 (d, J = 7.8 Hz, 1H), 7.06 (d, J = 6.6 Hz, 1H), 6.39 (s, 1H), 5.68 (t, J = 9.6 Hz, 1H), 5.61–5.66 (m, 1H), 5.21 (dd, J = 8.4, 15.6 Hz, 1H), 5.05 (dd, J = 8.4, 17.4 Hz, 1H), 4.83 (m, 1H), 4.20–4.24 (m, 2H), 3.68 (dd, J = 5.4, 13.8 Hz, 1H), 3.19–3.22 (m, 2H), 2.81 (dd, J = 9.8, 16.8 Hz, 1H), 2.55 (d, J = 16.2 Hz, 1H), 2.05–2.21 (m, 3H), 2.00–2.03 (m, 1H), 1.72 (s, 3H), 1.21 (dd, J = 6.6, 16.2 Hz, 1H), 0.92–1.02 ppm (m, 2H). 13C (150 MHz, CDCl3): δ=174.2, 169.6, 167.9, 167.4, 162.9, 147.8, 145.0, 134.8, 134.0, 132.5, 132.1, 129.8, 128.8, 128.3, 128.2, 128.1, 126.9, 126.7, 125.9, 124.9, 124.5, 124.3, 84.9, 72.8, 66.9, 55.7, 41.8, 41.5, 40.3, 37.1, 31.6, 31.2, 26.4 ppm. HRMS- ESI m/z[M + H]+ calcd for C48H45N4O4S3: 837.2603, found, 837.2599.
Analogue 16a
A solution of 15a (0.005 g, 0.006 mmol, 1 equiv) in dichloromethane (1 mL) at 0 °C was treated with triisopropylsilane (0.0023 g, 0.0147 mmol, 2.44 equiv), followed by trifluoroacetic acid (0.046 g, 0.404 mmol, 67.25 equiv). The reaction mixture was stirred for 3 h at room temperature and concentrated in vacuo. The crude product was purified by flash chromatography on silica gel in ethyl acetate/hexanes (20%) to first remove impurity, followed by ethyl acetate to obtain the thiol. To a stirred solution of the thiol in dichloromethane (1 mL) at 0 °C were added Hunig’s base (0.006 g, 0.046 mmol, 7.7 equiv) and octanoyl chloride (0.006 g, 0.035 mmol, 5.83 equiv). The mixture was stirred for 4 h at room temperature. The reaction was quenched with methanol and the mixture was concentrated in vacuo. Purification of the crude product by preparative thin layer chromatography on silica gel in ethyl acetate, followed by purification by HPLC on C18 column in methanol: water (20–100%) gave analogue 16a (0.0022 g, 50%); [α]D20=+ 45.0 (c 0.1 in CHCl3): 1H NMR (600 MHz, CDCl3): δ=8.40 (d, J = 8.4 Hz, 1H), 7.82 (d, J = 8.4 Hz, 1H), 7.72 (d, J = 7.8 Hz, 1H), 7.68 (s, 1H), 7.59 (t, J = 7.8 Hz, 1H), 7.47 (t, J = 7.8 Hz, 1H), 7.35 (d, J = 6.6 Hz, 1H), 7.26–7.29 (m, 1H), 7.15 (d, J = 7.2 Hz, 1H), 6.39–6.41 (m, 1H), 5.73–5.78 (m, 2H), 5.35 (dd, J = 8.4, 15.6 Hz, 1H), 5.07 (dd, J = 7.8, 16.8 Hz, 1H), 4.86 (q, J = 7.8 Hz, 1H), 4.25 (dd, J = 4.8, 16.8 Hz, 1H), 4.21 (d, J = 11.4 Hz, 1H), 3.72 (dd, J = 4.8, 13.8 Hz, 1H), 3.25 (dd, J = 9.6, 13.8 Hz, 1H), 3.21 (d, J = 11.4 Hz, 1H), 2.83–2.88 (m, 3H), 2.59 (d, J = 16.8 Hz, 1H), 2.51 (t, J = 7.2 Hz, 2H), 2.22–2.31 (m, 2H), 1.74 (s, 3H), 1.60–1.65 (m, 2H) 1.25 (m, 11H), 0.87 ppm (t, J = 7.2 Hz, 3H). 13C (150 MHz, CDCl3): δ=199.5, 174.2, 169.6, 167.9, 167.4, 162.9, 147.9, 134.2, 134.0, 132.5, 132.2, 128.9, 128.7, 128.3, 128.2, 126.7, 125.9, 124.9, 124.4, 124.3, 114.2, 85.0, 72.8, 55.7, 44.4, 41.8, 41.5, 40.4, 37.1, 32.4, 31.8, 29.9, 29.1, 28.0, 26.4, 25.8, 22.8, 14.2 ppm. HRMS-ESI m/z [M + Na]+ calcd for C37H44N4O5S3Na: 743.2372, found: 743.2372.
(R)-Methyl 2-(2-((5S,8S)-5-allyl-1-(9H-fluoren-9-yl)-3,6,10-trioxo-8-((E)-4-(tritylthio)but-1-enyl)-2,7-dioxa-4,11-diazadodecan-12-yl)thiazol-4-yl)-4-methyl-4,5-dihydrothiazole-4-carboxylate (14b)
To a solution of Fmoc-L-allylglycine (0.046 g, 0.137 mmol, 2 equiv) in THF (1 mL) at 0 °C were added Hunig’s base (0.030 g, 0.230 mmol, 3.38 equiv) and 2,4,6-trichlorobenzoyl chloride (0.047 g, 0.192 mmol, 2.83 equiv). The reaction mixture was stirred at 0 °C for 1 h, when TLC indicated the formation of the anhydride. The alcohol 13 (0.046 g, 0.068 mmol, 1 equiv) and DMAP (0.01 g, 0.082 mmol, 1.2 equiv) in THF (1 mL) were added to the reaction mixture at 0 °C. It was stirred overnight at room temperature. The reaction mixture was concentrated in vacuo and flash chromatography purification on silica gel in ethyl acetate/hexanes (20–100%) yielded the acyclic precursor 14b (0.061 g, 90%); [α]D20=− 12.53 (c 1.7 in CHCl3); 1H NMR (400 MHz, CDCl3): δ=0.88 (s, 1H), 7.75 (d, J = 7.6 Hz, 2H), 7.56 (d, J = 7.2 Hz, 1H), 7.37–7.40 (m, 8H), 7.25–7.31 (m, 3H), 7.20 (t, J = 6.8 Hz, 3H), 6.84 (t, J = 6.0 Hz, 1H), 5.57–5.68 (m, 3H), 5.42 (dd, J = 7.2, 15.2, Hz, 1H), 5.32 (d, J = 7.6 Hz, 1H), 5.06–5.10 (m, 2H), 4.68 (d, J = 6.0 Hz, 2H), 4.31–4.38 (m, 2H), 4.26 (q, J = 6.4 Hz, 1H), 4.19 (t, J = 7.2 Hz, 1H), 3.85 (d, J = 11.2 Hz, 1H), 3.78 (s, 3H), 3.24 (d, J = 11.6 Hz, 1H), 2.58 (d, J = 6.4 Hz, 2H), 2.49–2.54 (m, 1H), 2.38–2.44 (m, 1H), 2.19 (t, J = 6.8 Hz, 2H), 2.05 (q, J = 7.2 Hz, 2H), 1.85 (s, 1H), 1.62 ppm (s, 3H); 13C NMR (100 MHz, CDCl3): δ=173.8, 171.0, 169.2, 168.5, 163.0, 156.1, 148.5, 144.9, 143.9, 143.8, 141.5, 141.4, 134.0, 132.0, 129.7, 128.1, 127.9, 127.6, 127.3, 126.8, 125.3, 125.2, 122.3, 120.2, 119.8, 84.7, 72.7, 67.3, 66.8, 53.6, 53.1, 47.2, 41.7, 41.6, 41.2, 36.3, 31.5, 31.3, 24.1 ppm. HRMS-ESI m/z [M + Na]+ calcd for C56H54N4O7S3Na: 1013.3052, found: 1013.3058.
Cyclic core 15b
To a stirred solution of 14b (0.061 g, 0.0615 mmol, 1 equiv) in THF/H2O (4:1, 2 mL) at 0 °C was added 0.1 M LiOH (0.71 mL, 0.071 mmol, 1.15 equiv) dropwise over a period of 15 minutes. The reaction mixture was stirred at 0 °C for 1 h, acidified with 1 M HCl solution and extracted with EtOAc three times. The combined organic layer was washed with brine, dried over anhydrous sodium sulfate, and concentrated in vacuo. The crude product was purified by preparative TLC in ethyl acetate: hexanes (70%) to give the carboxylic acid (0.043 g, 0.044 mmol, 1 equiv), which was carried to the next step. During purification, starting material 14b (0.01 g) was recovered. The carboxylic acid was dissolved in dichloromethane (4.5 mL) and treated with diethylamine (0.156 g, 2.14 mmol, 48.4 equiv). The reaction mixture was stirred at room temperature for 3 h and concentrated to dryness to afford the free amino derivative. After drying azeotropically with toluene, it was treated with HATU (0.041 g, 0.108 mmol, 2.45 equiv), HOAt (0.015 g, 0.110 mmol, 2.29 equiv), dichloromethane (60 mL, ~ 1 mM), and Hunig’s base (0.030 g, 0.23 mmol, 4.79 equiv) and the mixture was stirred for 30 h at room temperature. It was concentrated to dryness and purified by flash chromatography on silica gel in ethyl acetate/hexanes (10–60%) to yield the cyclic core 15b (5.5 mg, 17%). [α]D20=+20.0 (c 0.28 in CHCl3); 1H NMR (600 MHz, CDCl3): δ=7.69 (s, 1H), 7.37 (d, J = 7.8 Hz, 6H), 7.27 (t, J = 8.4 Hz, 6H), 7.19 (t, J = 8.4 Hz, 3H), 6.42 (dd, J = 3.6, 9.0 Hz, 1H), 5.64–5.70 (m, 2H), 5.36 (dd, J = 6.6, 15.0 Hz, 1H), 5.20 (dd, J = 9.6, 17.4 Hz, 1H), 4.69 (d, J = 16.8 Hz, 1H), 4.62–4.65 (m, 1H), 4.35 (d, J = 10.2 Hz, 1H), 4.16 (dd, J = 3.6, 18.0 Hz, 1H), 4.07 (d, J = 11.4 Hz, 1H), 4.21 (d, J = 11.4 Hz, 1H), 2.81 (dd, J = 10.2, 16.8 Hz, 1H), 2.63 (dd, J = 3.0, 16.8 Hz, 1H), 2.47–2.50 (m, 1H), 2.31–2.36 (m, 1H), 2.15–2.20 (m, 1H), 1.99–2.08 (m, 2H), 1.81 ppm (s, 3H). 13C NMR (100 MHz, CDCl3): δ=173.7, 169.5, 169.1, 167.8, 164.4, 147.9, 145.0, 133.7, 131.5, 129.8, 128.1, 128.0, 126.9, 119.5, 84.5, 72.5, 52.7, 42.7, 41.3, 40.6, 38.0, 31.6, 31.4, 24.8 ppm. HRMS-ESI m/z [M + Na]+ calcd for C40H40N4O4S3Na: 759.2109, found: 759.2039.
Analogue 16b
To a solution of 15b (0.0055 g, 0.0075 mmol, 1 equiv) in dichloromethane (1 mL) at 0 °C was added triisopropylsilane (0.0023 g, 0.0147 mmol, 1.96 equiv), followed by trifluoroacetic acid (0.057 g, 0.498 mmol, 66.78 equiv). The reaction mixture was stirred for 3 h at room temperature and concentrated in vacuo. The crude product was purified by flash chromatography on silica gel in ethyl acetate/hexanes (20%) to first remove impurity, followed by ethyl acetate to obtain the thiol. To a stirred solution of the thiol in dichloromethane (1 mL) at 0 °C were added Hunig’s base (0.006 g, 0.046 mmol, 7.7 equiv) and octanoyl chloride (0.006 g, 0.035 mmol, 5.83 equiv). The reaction mixture was stirred for 4 h at room temperature. The reaction was quenched with methanol and the mixture was concentrated in vacuo. Purification of the crude product by preparative thin layer chromatography on silica gel in ethyl acetate as solvent gave analogue 16b (0.0016 mg, 35%); [α]D20=+ 25.56 (c 0.08 in CHCl3); 1H NMR (600 MHz, CDCl3): δ=7.71 (s, 1H), 7.19 (d, J = 8.4 Hz, 1H), 6.36 (dd, J = 3.0, 9.0 Hz, 1H), 4.79–4.83 (m, 1H), 5.73 (t, J = 8.4 Hz, 1H), 5.50 (dd, J = 7.2, 15.6 Hz, 1H) 5.26–5.32 (m, 3H), 4.67–4.72 (m 2H), 4.35 (d, J = 16.2 Hz, 1H), 4.27 (dd, J = 3.6, 18.0 Hz, 1H), 4.10 (d, J = 11.4 Hz, 1H), 3.22 (d, J = 11.4 Hz, 1H), 2.90 (t, J = 7.2 Hz, 2H), 2.87 (dd, J = 10.8, 16.8 Hz, 1H), 2.68 (dd, J = 2.4, 16.2 Hz, 1H), 2.53 (t, J = 7.2 Hz, 3H), 2.34–2.39 (m, 1H), 2.31 (q, J = 7.2 Hz, 2H), 1.85 (s, 3H), 1.63–1.67 (m, 2H), 1.25–1.32 (m, 13H), 0.88 ppm (t, J = 7.2 Hz, 3H); 13C NMR (150 MHz, CDCl3): δ=199.7, 173.7, 169.5, 169.1, 167.8, 164.4, 147.9, 133.2, 131.5, 128.5, 124.3, 121.8, 119.5, 84.5, 72.6, 52.8, 44.4, 42.7, 41.3, 38.0, 32.5, 31.8, 29.9, 29.1, 28.1, 25.9, 24.8, 22.8, 14.29 ppm. HRMS-ESI m/z [M + Na]+ calcd for C29H40N4O5S3Na: 643.2059, found: 643.2046.
(R)-Methyl 2-(2-((5S,8S)-1-(9H-fluoren-9-yl)-5-(naphthalen-1-ylmethyl)-3,6,10-trioxo-8-((E)-4-(tritylthio)but-1-enyl)-2,7-dioxa-4,11-diazadodecan-12-yl)thiazol-4-yl)-4-methyl-4,5-dihydrothiazole-4-carboxylate (14c)
To a solution of Fmoc-L-1-naphthylalanine (0.052 g, 0.119 mmol, 1.95 equiv) in THF (1 mL) at 0 °C were added Hunig’s base (0.028 g, 0.219 mmol, 3.59 equiv) and 2,4,6-trichlorobenzoyl chloride (0.031 g, 0.128 mmol, 2.09 equiv). The reaction mixture was stirred at 0 °C for 1 h, when TLC indicated the formation of the anhydride. The alcohol 13 (0.041 g, 0.061 mmol, 1 equiv) and DMAP (0.009 g, 0.074 mmol, 1.2 equiv) in THF (1 mL) were added to the reaction mixture at 0 °C. It was stirred overnight at room temperature. The reaction mixture was concentrated in vacuo and flash chromatography purification on silica gel in ethyl acetate/hexanes (20–60%) yielded the acyclic precursor 14c (0.065 g, 98; [α]D20=− 9.05 (c 3.25 in CHCl3); 1H NMR (600 MHz, CDCl3): δ=8.02 (d, J = 8.4 Hz, 1H), 7.80 (m, 2H), 7.73 (dd, J = 2.4, 7.2 Hz, 2H), 7.68 (d, J = 8.4 Hz, 1H), 7.42–7.50 (m, 4H), 7.36–7.39 (m, 8H), 7.24–7.29 (m, 10H), 7.18 (t, J = 7.2 Hz, 4H), 6.61 (t, J = 6.0 Hz, 1H), 5.39–5.43 (m, 2H), 5.30–5.35 (m, 1H), 5.11 (dd, J = 7.2, 15.6 Hz, 1H), 4.53–4.60 (m, 2H), 4.52 (dd, J = 6.0, 16.2 Hz, 1H), 4.28–4.35 (m, 2H), 4.09–4.11 (m, 1H), 3.82 (d, J = 11.4 Hz, 1H), 3.76 (s, 3H), 3.40–3.49 (m, 2H), 3.20 (d, J = 10.8 Hz, 1H), 2.41 (dd, J = 4.2, 14.4 Hz, 1H), 3.30 (dd, J = 6.6, 14.4 Hz, 1H), 2.13 (t, J = 7.8 Hz, 2H) 1.97 (q, J = 7.2 Hz, 2H), 1.60 ppm (s, 3H); 13C NMR (100 MHz, CDCl3) δ=173.9, 171.2, 169.2, 168.8, 163.0, 156.1, 148.5, 145.0, 143.8, 143.8, 141.5, 134.0, 133.6, 132.1, 132.1, 84.7, 72.7, 71.6, 67.3, 66.8, 60.6, 55.1, 53.1, 47.2, 41.6, 41.5, 41.2, 35.5, 31.5, 31.3, 24.1, 21.3, 20.6, 14.4 ppm. HRMS-ESI m/z [M + Na]+ calcd for C64H58N4O7S3Na: 1113.3365, found: 1113.3384.
Cyclic core 15c
To a stirred solution of 14c (0.061 g, 0.0615 mmol, 1 equiv) in THF/H2O (4:1, 2 mL) at 0 °C was added 0.1 M LiOH (0.71 mL, 0.071 mmol, 1.15 equiv) dropwise over a period of 15 minutes. After stirring at 0 °C for 1 h, it was acidified with 1 M HCl solution and extracted with EtOAc three times. The combined organic layer was washed with brine, dried over anhydrous sodium sulfate, and concentrated in vacuo. The reaction mixture was purified by preparative TLC in ethyl acetate: hexanes (70%) to give the carboxylic acid (0.043 g, 0.044 mmol, 1 equiv) which was carried to the next step. During purification, starting material 14c (0.01 g) was recovered. The carboxylic acid was dissolved in dichloromethane (4.5 mL) and treated with diethylamine (0.156 g, 2.14 mmol, 48.4 equiv). After stirring at room temperature for 3 h it was concentrated to dryness to afford the free amino derivative. After drying azeotropically with toluene it was treated with HATU (0.041 g, 0.108 mmol, 2.45 equiv), HOAt (0.015 g, 0.110 mmol, 2.29 equiv), dichloromethane (60 mL, ~ 1 mM), and Hunig’s base (0.030 g, 0.23 mmol, 4.79 equiv) and the mixture was stirred for 30 h at room temperature. The reaction mixture was concentrated to dryness and was purified by flash chromatography on silica gel in ethyl acetate/hexanes (10–60%) to yield the cyclic core 15c (0.0094g, 20%); [α]D20=– 9.57 (c 0.23 in CHCl3; 1H NMR (600 MHz, CDCl3): δ=8.00 (d, J = 8.4 Hz, 1H), 7.70 (d, J = 7.8 Hz, 1H), 7.62 (s, 1H), 7.37–7.44 (m, 4H), 7.29 (d, J = 7.8 Hz, 6H), 7.23 (t, J = 7.2 Hz, 6H), 7.16 (t, J = 6.6 Hz, 3H), 7.00 (d, J = 7.2 Hz, 1H), 6.77 (t, J = 7.2 Hz, 1H), 5.59–5.65 (m, 2H), 5.3–5.5 (m, 1H), 5.37 (dd, J = 6.0, 15.6 Hz, 1H), 5.07–5.10 (m, 1H), 4.41 (dd, J = 5.4, 17.4 Hz, 1H), 4.30 (dd, J = 4.8, 17.4 Hz, 1H), 4.04 (d, J = 11.4 Hz, 1H), 3.66 (dd, J = 4.2, 14.4 Hz, 1H), 3.41 (dd, J = 6.6, 14.4 Hz, 1H), 3.23 (d, J = 11.4 Hz, 1H), 2.35 (dd, J = 5.4, 15.6 Hz, 1H), 2.08–2.11 (m, 2H), 1.90–1.94 (m, 3H), 1.71 ppm (s, 3H); 13C NMR (150 MHz, CDCl3) δ=174.0, 168.8, 168.6, 166.4, 147.5, 145.0, 133.7, 133.1, 132.4, 132.1, 129.8, 128.9, 128.6, 128.1, 128.1, 127.1, 127, 126.8, 126.6, 126.0, 124.0, 124.2, 123.2, 71.9, 66.7, 54.3, 42.8,41.2, 40.5, 35.1, 31.6, 31.4, 29.9, 25.1 ppm; HRMS-ESI m/z: [M + Na]+ calcd for C48H44N4O4S3Na: 859.2422, found: 859.2429.
Analogue 16c
To a solution of 15c (0.0134 g, 0.016 mmol, 1 equiv) in dichloromethane (3 mL) at 0 °C was added triisopropylsilane (0.007 g, 0.044 mmol, 2.78 equiv) followed by trifluoroacetic acid (0.134 g, 1.176 mmol, 73.47 equiv). After stirring for 3 h at room temperature, the reaction mixture was concentrated in vacuo. The crude product was purified by flash chromatography on silica gel in ethyl acetate/hexanes (20%) to first remove impurity followed by ethyl acetate to obtain the thiol. To a stirred solution of the thiol in dichloromethane (1.5 mL) at 0 °C were added Hunig’s base (0.0163 g, 0.127 mmol, 7.9 equiv) and octanoyl chloride (0.0152 g, 0.093 mmol, 5.82 equiv). After stirring for 4 h at room temperature, the reaction was quenched with methanol and the mixture was concentrated in vacuo. Purification of the crude product by preparative thin layer chromatography on silica gel in ethyl acetate and final purification by HPLC on C18 column in methanol: water (20–100%) gave analogue 16c (0.0034 g, 30%). [α]D20=−16.15 (c 0.13 in CHCl3). ); 1H NMR (600 MHz, CDCl3): δ=8.00 (d, J = 9.0 Hz, 1H), 7.71 (d, J = 7.8 Hz, 1H), 7.24–7.45 (m, 3H), 7.01 (d, J = 6.0 Hz, 1H), 6.76 (dd, J = 0.6, 7.0 Hz, 1H), 5.73–5.78 (m, 1H), 5.58–5.60 (m, 1H), 5.49 (dd, J = 6.6, 15.6 Hz, 1H), 5.42 (t, J = 4.0 Hz, 1H), 5.12–5.15 (m, 1H), 4.52 (dd, J = 3.6, 18.0 Hz, 1H), 4.42 (dd, J = 3.6, 18.0 Hz, 1H), 4.05 (d, J = 11.4 Hz, 1H), 3.69 (dd, J = 4.2, 14.4 Hz, 1H), 3.45 (dd, J = 6.6, 15.0 Hz, 1H), 3.24 (d, J = 11.4 Hz, 1H), 2.79–2.83 (m, 1H), 2.72–2.76 (m, 1H), 2.45 (t, J = 7.2 Hz, 2H), 2.37 (dd, J = 6.6, 16.2 Hz, 1H), 2.18–2.21 (m, 2H), 1.87 (dd, J = 3.6, 16.2 Hz, 1H), 1.78 (s, 3H), 1.23–1.28 (m, 12H), 0.84–0.88 ppm (m, 3H). 13C NMR (150 MHz, CDCl3): δ=199.4, 173.9, 168.8, 168.4, 166.2, 163.6, 147.4, 133.5, 132.6, 132.2, 131.9, 128.8, 128.4, 126.8, 126.4, 125.9, 124.8, 124.0, 123.0, 113.9, 84.4, 71.9, 54.1, 44.1, 42.6, 41.0, 40.2, 34.7, 32.2, 31.6, 29.7, 28.9, 27.9, 25.6, 25.0, 22.6, 14.1 ppm; HRMS-ESI m/z [M + Na]+ calcd for C37H44N4O5S3Na: 743.2372, found: 743.2372.
Cytoproliferation assay
Cell proliferation was measured using a 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxy-phenyl)-2-(4-sulfophenyl)-2H-tetrazolium (MTS) reduction assay with the CellTiter 96 One solution MTS assay as described by the manufacturer. (Promega, Madison, WI) Briefly, cells (3×104 cells/well) were seeded in quadruplicate in 96- well plates and allowed to attach overnight. Media was replaced with 100 μL of fresh medium containing the appropriate concentration of the indicated compounds. Following incubation at 37 °C, 5% CO2, 20 μL of CellTiter 96 One Solution was added per well and incubated for 1.5 h at 37 °C and absorbance measured at 490 nm.
HDAC activity assays
HDAC activity was assayed using the Enzo Life Sciences (Plymouth Meeting, PA) fluorimetric drug discovery kits with specificity for HDAC1 or HDAC6 enzymatic activity. Experiments were performed as described by the protocol supplied by the manufacturer. Briefly, the indicated inhibitor, enzyme, and Fluor de Lys substrate were incubated for 1 hour when the developing solution was added and incubated for the designated time prior to reading. Experiments were repeated 3 times and done in triplicate each time. Analogues were incubated with the Fluor de Lys substrate and developer in the absence of enzyme as a control and it showed that the analogues had no influence on the fluorescent signal.
Evaluation of global histone acetylation levels
HCT116 cells were treated for 24 hours with 10 nM, 100 nM, or 1 μM of test compounds or DMSO. Cell lysates were extracted using RIPA buffer (50 mmol/L Tris-HCl, 1% NP-40, 0.25% sodium deoxycholate, 150 mmol/L NaCl, 50 mmol/L sodium fluoride), protein were quantified by a DC assay (Biorad) and equal protein amounts (30 μg/Lane) were loaded onto an SDS-PAGE gel. Standard Western blotting protocols were used with the Invitrogen NuPAGE Western blotting system. Primary antibodies used were AcH3 (Millipore) and β-actin (Sigma). Dye-conjugated secondary antibodies from Li-Cor Biosciences were used for detection and scanned using the Odyssey Infrared Detection System (LI-COR Biosciences, Lincoln, NE).
Computational Methods
Several configurations of largazole thiol and the thiol of its C7-epimer were optimized with Gaussian09-C01 [51]. The cc-pVTZ basis set was used with B3LYP functionals and the CHELPG procedure [52] was used in the charge derivation. S21 in thiol was kept in its S− form so that the results are comparable to the Zn2+ bound form.
The initial structure for molecular dynamics was taken from the X-ray crystal structure of largazole thiol bound HDAC8 (pdb ID: 3RQD) [50]. Missing residues in HDAC were reintroduced using the X-ray crystal structure of HDAC8 complexed with APHA (pdb ID:3F07)[53]. Largazole thiol charges were derived following the RESP procedure in the Amber program suite. All X-ray water molecules are kept in the system along with the Zn2+ and two K+ ions. Additional water molecules were introduced to solvate the system yielding a total of 28838 water molecules. Sander and PMEMD modules of the Amber-11 program suite [54] were used for optimizations and dynamic trajectory calculations. The FF03 force field was used to represent the HDAC atoms. Following our standard protocol, with particle mesh Ewald summation [55] for long range interactions and 1 fs time step, a 10ns NVT equilibration and a 30ns NPT production run were carried out at 300K. Configurations from the final 10 ns of dynamics were used for analysis.
Supplementary Material
Acknowledgments
This work was supported, in part, by the NIH grant CA51085 (to RAC) and by the Intramural Research Program of the NIH-National Institute of Environmental Health Sciences (Z01 ES125392) (to LP). The authors thank Dr. Yong Wah Kim of the Department of Chemistry, University of Toledo for assistance with NMR spectroscopy.
Footnotes
Supporting information for this article can be found at http://
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Atadja PW. HDAC inhibitors and cancer therapy. Prog Drug Res. 2011;67:175–195. doi: 10.1007/978-3-7643-8989-5_9. [DOI] [PubMed] [Google Scholar]
- 2.Bertrand P. Inside HDAC with HDAC inhibitors. Eur J Med Chem. 2010;45:2095–2116. doi: 10.1016/j.ejmech.2010.02.030. [DOI] [PubMed] [Google Scholar]
- 3.Fouladi M. Histone deacetylase inhibitors in cancer therapy. Cancer Invest. 2006;24:521–527. doi: 10.1080/07357900600814979. [DOI] [PubMed] [Google Scholar]
- 4.Wagner JM, Hackanson B, Lubbert M, Jung M. Histone deacetylase (HDAC) inhibitors in recent clinical trials for cancer therapy. Clin Epigenetics. 2010;1:117–136. doi: 10.1007/s13148-010-0012-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Muraglia E, Altamura S, Branca D, Cecchetti O, Ferrigno F, Orsale MV, Palumbi MC, Rowley M, Scarpelli R, Steinkuhler C, Jones P. 2-Trifluoroacetylthiophene oxadiazoles as potent and selective class II human histone deacetylase inhibitors. Bioorg Med Chem Lett. 2008;18:6083–6087. doi: 10.1016/j.bmcl.2008.09.076. [DOI] [PubMed] [Google Scholar]
- 6.Miller TA, Witter DJ, Belvedere S. Histone deacetylase inhibitors. J Med Chem. 2003;46:5097–5116. doi: 10.1021/jm0303094. [DOI] [PubMed] [Google Scholar]
- 7.Vannini A, Volpari C, Filocamo G, Casavola EC, Brunetti M, Renzoni D, Chakravarty P, Paolini C, De Francesco R, Gallinari P, Steinkuhler C, Di Marco S. Crystal structure of a eukaryotic zinc-dependent histone deacetylase, human HDAC8, complexed with a hydroxamic acid inhibitor. Proc Natl Acad Sci U S A. 2004;101:15064–15069. doi: 10.1073/pnas.0404603101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wang DF, Wiest O, Helquist P, Lan-Hargest HY, Wiech NL. On the function of the 14 A long internal cavity of histone deacetylase-like protein: implications for the design of histone deacetylase inhibitors. J Med Chem. 2004;47:3409–3417. doi: 10.1021/jm0498497. [DOI] [PubMed] [Google Scholar]
- 9.Finnin MS, Donigian JR, Cohen A, Richon VM, Rifkind RA, Marks PA, Breslow R, Pavletich NP. Structures of a histone deacetylase homologue bound to the TSA and SAHA inhibitors. Nature. 1999;401:188–193. doi: 10.1038/43710. [DOI] [PubMed] [Google Scholar]
- 10.Whitehead L, Dobler MR, Radetich B, Zhu Y, Atadja PW, Claiborne T, Grob JE, McRiner A, Pancost MR, Patnaik A, Shao W, Shultz M, Tichkule R, Tommasi RA, Vash B, Wang P, Stams T. Human HDAC isoform selectivity achieved via exploitation of the acetate release channel with structurally unique small molecule inhibitors. Bioorg Med Chem. 2011;19:4626–4634. doi: 10.1016/j.bmc.2011.06.030. [DOI] [PubMed] [Google Scholar]
- 11.Moradei OM, Mallais TC, Frechette S, Paquin I, Tessier PE, Leit SM, Fournel M, Bonfils C, Trachy-Bourget MC, Liu J, Yan TP, Lu AH, Rahil J, Wang J, Lefebvre S, Li Z, Vaisburg AF, Besterman JM. Novel aminophenyl benzamide-type histone deacetylase inhibitors with enhanced potency and selectivity. J Med Chem. 2007;50:5543–5546. doi: 10.1021/jm701079h. [DOI] [PubMed] [Google Scholar]
- 12.Methot JL, Chakravarty PK, Chenard M, Close J, Cruz JC, Dahlberg WK, Fleming J, Hamblett CL, Hamill JE, Harrington P, Harsch A, Heidebrecht R, Hughes B, Jung J, Kenific CM, Kral AM, Meinke PT, Middleton RE, Ozerova N, Sloman DL, Stanton MG, Szewczak AA, Tyagarajan S, Witter DJ, Secrist JP, Miller TA. Exploration of the internal cavity of histone deacetylase (HDAC) with selective HDAC1/HDAC2 inhibitors (SHI-1:2) Bioorg Med Chem Lett. 2008;18:973–978. doi: 10.1016/j.bmcl.2007.12.031. [DOI] [PubMed] [Google Scholar]
- 13.Witter DJ, Harrington P, Wilson KJ, Chenard M, Fleming JC, Haines B, Kral AM, Secrist JP, Miller TA. Optimization of biaryl Selective HDAC1&2 Inhibitors (SHI-1:2) Bioorg Med Chem Lett. 2008;18:726–731. doi: 10.1016/j.bmcl.2007.11.047. [DOI] [PubMed] [Google Scholar]
- 14.Wambua MK, Nalawansha DA, Negmeldin AT, Pflum MK. Mutagenesis studies of the 14 A internal cavity of histone deacetylase 1: insights toward the acetate-escape hypothesis and selective inhibitor design. J Med Chem. 2014;57:642–650. doi: 10.1021/jm401837e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Butler KV, Kozikowski AP. Chemical origins of isoform selectivity in histone deacetylase inhibitors. Curr Pharm Des. 2008;14:505–528. doi: 10.2174/138161208783885353. [DOI] [PubMed] [Google Scholar]
- 16.Taori K, Paul VJ, Luesch H. Structure and Activity of Largazole, a Potent Antiproliferative Agent from the Floridian Marine Cyanobacterium Symploca sp. J Am Chem Soc. 2008;130:1806–1807. doi: 10.1021/ja7110064. [DOI] [PubMed] [Google Scholar]
- 17.Ahmed S, Riegsecker S, Beamer M, Rahman A, Bellini JV, Bhansali P, Tillekeratne LM. Largazole, a class I histone deacetylase inhibitor, enhances TNF-alpha-induced ICAM-1 and VCAM-1 expression in rheumatoid arthritis synovial fibroblasts. Toxicol Appl Pharmacol. 2013;270:87–96. doi: 10.1016/j.taap.2013.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ungermannova D, Parker SJ, Nasveschuk CG, Wang W, Quade B, Zhang G, Kuchta RD, Phillips AJ, Liu X. Largazole and its derivatives selectively inhibit ubiquitin activating enzyme (e1) PLoS One. 2012;7:e29208. doi: 10.1371/journal.pone.0029208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Liu Y, Wang Z, Wang J, Lam W, Kwong S, Li F, Friedman SL, Zhou S, Ren Q, Xu Z, Wang X, Ji L, Tang S, Zhang H, Lui EL, Ye T. A histone deacetylase inhibitor, largazole, decreases liver fibrosis and angiogenesis by inhibiting transforming growth factor-beta and vascular endothelial growth factor signalling. Liver Int. 2013;33:504–515. doi: 10.1111/liv.12034. [DOI] [PubMed] [Google Scholar]
- 20.Ying Y, Taori K, Kim H, Hong J, Luesch H. Total Synthesis and Molecular Target of Largazole, a Histone Deacetylase Inhibitor. J Am Chem Soc. 2008;130:8455–8459. doi: 10.1021/ja8013727. [DOI] [PubMed] [Google Scholar]
- 21.Bowers A, West N, Taunton J, Schreiber SL, Bradner JE, Williams RM. Total synthesis and biological mode of action of largazole: A potent Class I histone deacetylase inhibitor. J Am Chem Soc. 2008;130:11219–11222. doi: 10.1021/ja8033763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nasveschuk CG, Ungermannova D, Liu X, Phillips AJ. A Concise Total Synthesis of Largazole, Solution Structure, and Some Preliminary Structure Activity Relationships. Org Lett. 2008;10:3595–3598. doi: 10.1021/ol8013478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Seiser T, Kamena F, Cramer N. Synthesis and biological activity of largazole and derivatives. Angew Chem, Int Ed. 2008;47:6483–6485. doi: 10.1002/anie.200802043. [DOI] [PubMed] [Google Scholar]
- 24.Ghosh AK, Kulkarni S. Enantioselective Total Synthesis of (+)-Largazole, a Potent Inhibitor of Histone Deacetylase. Org Lett. 2008;10:3907–3909. doi: 10.1021/ol8014623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bowers AA, Greshock TJ, West N, Estiu G, Schreiber SL, Wiest O, Williams RM, Bradner JE. Synthesis and Conformation-Activity Relationships of the Peptide Isosteres of FK228 and Largazole. J Am Chem Soc. 2009;131:2900–2905. doi: 10.1021/ja807772w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bowers AA, West N, Newkirk TL, Troutman-Youngman AE, Schreiber SL, Wiest O, Bradner JE, Williams RM. Synthesis and Histone Deacetylase Inhibitory Activity of Largazole Analogs: Alteration of the Zinc-Binding Domain and Macrocyclic Scaffold. Org Lett. 2009;11:1301–1304. doi: 10.1021/ol900078k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ying Y, Liu Y, Byeon SR, Kim H, Luesch H, Hong J. Synthesis and Activity of Largazole Analogues with Linker and Macrocycle Modification. Org Lett. 2008;10:4021–4024. doi: 10.1021/ol801532s. [DOI] [PubMed] [Google Scholar]
- 28.Ren Q, Dai L, Zhang H, Tan W, Xu Z, Ye T. Total synthesis of largazole. Synlett. 2008:2379–2383. [Google Scholar]
- 29.Wang B, Forsyth CJ. Total synthesis of largazole - devolution of a novel synthetic strategy. Synthesis. 2009:2873–2880. [Google Scholar]
- 30.Chen F, Gao A-H, Li J, Nan F-J. Synthesis and Biological Evaluation of C7-Demethyl Largazole Analogues. ChemMedChem. 2009;4:1269–1272. doi: 10.1002/cmdc.200900125. [DOI] [PubMed] [Google Scholar]
- 31.Zeng X, Yin B, Hu Z, Liao C, Liu J, Li S, Li Z, Nicklaus MC, Zhou G, Jiang S. Total synthesis and biological evaluation of Largazole and derivatives with promising selectivity for cancers cells. Org Lett. 2010;12:1368–1371. doi: 10.1021/ol100308a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Souto JA, Vaz E, Lepore I, Poppler A-C, Franci G, Alvarez R, Altucci L, de Lera AR. Synthesis and biological characterization of the histone deacetylase inhibitor largazole and C7-modified analogues. J Med Chem. 2010;53:4654–4667. doi: 10.1021/jm100244y. [DOI] [PubMed] [Google Scholar]
- 33.Wang B, Huang P-H, Chen C-S, Forsyth CJ. Total Syntheses of the Histone Deacetylase Inhibitors Largazole and 2-epi-Largazole: Application of N-Heterocyclic Carbene Mediated Acylations in Complex Molecule Synthesis. J Org Chem. 2011;76:1140–1150. doi: 10.1021/jo102478x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Benelkebir H, Marie S, Hayden AL, Lyle J, Loadman PM, Crabb SJ, Packham G, Ganesan A. Total synthesis of largazole and analogues: HDAC inhibition, antiproliferative activity and metabolic stability. Bioorg Med Chem. 2011;19:3650–3658. doi: 10.1016/j.bmc.2011.02.024. [DOI] [PubMed] [Google Scholar]
- 35.Hong J, Luesch H. Largazole: from discovery to broad-spectrum therapy. Nat Prod Rep. 2012;29:449–456. doi: 10.1039/c2np00066k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Liu Y, Salvador LA, Byeon S, Ying Y, Kwan JC, Law BK, Hong J, Luesch H. Anticolon cancer activity of largazole, a marine-derived tunable histone deacetylase inhibitor. J Pharmacol Exp Ther. 2010;335:351–361. doi: 10.1124/jpet.110.172387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Guerra-Bubb JM, Bowers AA, Smith WB, Paranal R, Estiu G, Wiest O, Bradner JE, Williams RM. Synthesis and HDAC inhibitory activity of isosteric thiazoline-oxazole largazole analogs. Bioorg Med Chem Lett. 2013;23:6025–6028. doi: 10.1016/j.bmcl.2013.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Schotes C, Ostrovskyi D, Senger J, Schmidtkunz K, Jung M, Breit B. Total synthesis of (18S)- and (18R)-homolargazole by rhodium-catalyzed hydrocarboxylation. Chemistry. 2014;20:2164–2168. doi: 10.1002/chem.201303300. [DOI] [PubMed] [Google Scholar]
- 39.Bhansali P, Hanigan CL, Casero RA, Tillekeratne LM. Largazole and analogues with modified metal-binding motifs targeting histone deacetylases: synthesis and biological evaluation. J Med Chem. 2011;54:7453–7463. doi: 10.1021/jm200432a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Alhamadsheh MM, Hudson RA, Tillekeratne LMV. Design, Total Synthesis, and Evaluation of Novel Open-Chain Epothilone Analogues. Org Lett. 2006;8:685–688. doi: 10.1021/ol0528787. [DOI] [PubMed] [Google Scholar]
- 41.Gupta S, Rajagopalan M, Alhamadsheh MM, Tillekeratne LMV, Hudson RA. First total synthesis and absolute configuration of the styryl lactone gonioheptolide A. Synthesis. 2007:3512–3518. [Google Scholar]
- 42.Alhamadsheh MM, Gupta S, Hudson RA, Perera L, Tillekeratne LMV. Total synthesis and selective activity of a new class of conformationally restrained epothilones. Chem--Eur J. 2008;14:570–581. doi: 10.1002/chem.200701143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gupta S, Poeppelman L, Hinman CL, Bretz J, Hudson RA, Tillekeratne LMV. Apoptotic activities in closely related styryllactone stereoisomers toward human tumor cell lines: Investigation of synergism of styryllactone-induced apoptosis with TRAIL. Bioorg Med Chem. 2010;18:849–854. doi: 10.1016/j.bmc.2009.11.045. [DOI] [PubMed] [Google Scholar]
- 44.Su D-W, Wang Y-C, Yan T-H. A novel DMAP-promoted oxazolidinethione deacylation. Application for the direct conversion of the initial chiral thioimide aldols to various ester protecting groups. Tetrahedron Lett. 1999;40:4197–4198. [Google Scholar]
- 45.Inanaga J, Hirata K, Saeki H, Katsuki T, Yamaguchi M. A rapid esterification by mixed anhydride and its application to large-ring lactonization. Bull Chem Soc Jpn. 1979;52:1989–1993. [Google Scholar]
- 46.Sharma SK, Wu Y, Steinbergs N, Crowley ML, Hanson AS, Casero RA, Woster PM. (Bis)urea and (bis)thiourea inhibitors of lysine-specific demethylase 1 as epigenetic modulators. J Med Chem. 2010;53:5197–5212. doi: 10.1021/jm100217a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Crabb SJ, Howell M, Rogers H, Ishfaq M, Yurek-George A, Carey K, Pickering BM, East P, Mitter R, Maeda S, Johnson PW, Townsend P, Shin-ya K, Yoshida M, Ganesan A, Packham G. Characterisation of the in vitro activity of the depsipeptide histone deacetylase inhibitor spiruchostatin A. Biochem Pharmacol. 2008;76:463–475. doi: 10.1016/j.bcp.2008.06.004. [DOI] [PubMed] [Google Scholar]
- 48.Mai A, Massa S, Pezzi R, Simeoni S, Rotili D, Nebbioso A, Scognamiglio A, Altucci L, Loidl P, Brosch G. Class II (IIa)-selective histone deacetylase inhibitors. 1. Synthesis and biological evaluation of novel (aryloxopropenyl)pyrrolyl hydroxyamides. J Med Chem. 2005;48:3344–3353. doi: 10.1021/jm049002a. [DOI] [PubMed] [Google Scholar]
- 49.Siliphaivanh P, Harrington P, Witter DJ, Otte K, Tempest P, Kattar S, Kral AM, Fleming JC, Deshmukh SV, Harsch A, Secrist PJ, Miller TA. Design of novel histone deacetylase inhibitors. Bioorg Med Chem Lett. 2007;17:4619–4624. doi: 10.1016/j.bmcl.2007.05.080. [DOI] [PubMed] [Google Scholar]
- 50.Cole KE, Dowling DP, Boone MA, Phillips AJ, Christianson DW. Structural basis of the antiproliferative activity of largazole, a depsipeptide inhibitor of the histone deacetylases. J Am Chem Soc. 2011;133:12474–12477. doi: 10.1021/ja205972n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Frisch MJ, Trucks GW, Schlegel HB, SGE, Robb MA, Cheeseman JR, Scalmani G, Barone V, Mennucci B, Petersson GA, Nakatsuji H, Caricato M, Li X, Hratchian HP, Izmaylov AF, Bloino J, Zheng G, Sonnenberg JL, Hada M, Ehara M, Toyota K, Fukuda R, Hasegawa J, Ishida M, Nakajima T, Honda Y, Kitao O, Nakai H, VT, Montgomery JAJ, Peralta JE, Ogliaro F, BM, Heyd JJ, Brothers E, Kudin KN, Staroverov VN, Kobayashi R, Normand J, Raghavachari K, Rendell A, Burant JC, Iyengar SS, Tomasi J, Cossi M, Rega N, Millam JM, Klene M, Knox JE, Cross JB, Bakken V, Adamo C, Jaramillo J, Gomperts R, Stratmann RE, Yazyev O, Austin AJ, Cammi RP, Ochterski CJW, Martin RL, MK, Zakrzewski VG, Voth GA, Salvador P, Dannenberg JJ, Dapprich S, Daniels AD, Farkas Ö, Foresman JB, Ortiz JV, Cioslowski J, Fox DJ. Gaussian 09, Revision C1. Gaussian, Inc; Wallingford CT: 2009. [Google Scholar]
- 52.Chirlian LE, Francl MM. Atomic charges derived from electrostatic potentials: A detailed study. J Comput Chem. 1987;8:894–905. [Google Scholar]
- 53.Dowling DP, Gantt SL, Gattis SG, Fierke CA, Christianson DW. Structural studies of human histone deacetylase 8 and its site-specific variants complexed with substrate and inhibitors. Biochemistry. 2008;47:13554–13563. doi: 10.1021/bi801610c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Case DADTA, Cheatham TE, III, Simmerling CL, Wang J, Duke RE, Luo R, Walker RC, Zhang W, Merz KM, Roberts B, Wang B, Hayik S, Roitberg A, Seabra G, Kolossváry I, Wong KF, Paesani F, Vanicek J, Wu X, Brozell SR, Steinbrecher T, Gohlke H, Cai Q, Ye X, Wang J, Hsieh M-J, Cui G, Roe DR, Mathews DH, Seetin MG, Sagui C, Babin V, Luchko T, Gusarov S, Kovalenko A, Kollman PA. AMBER 11. University of California; San Francisco: 2010. [Google Scholar]
- 55.Essmann U, Perera L, Berkowitz ML, Darden T, Lee H, Pedersen LG. A smooth particle mesh Ewald method. J Chem Phys. 1995;103:8577–8593. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.