

Abstract
Mutations in the RP1 gene can cause retinitis pigmentosa. We identified a spontaneous L66P mutation caused by two adjacent point mutations in the Rp1 gene in a colony of C57BL/6J mice. Mice homozygous for the L66P mutation exhibited slow, progressive photoreceptor degeneration throughout their lifespan. Optical coherence tomography imaging found abnormal photoreceptor reflectivity at 1 month of age. Histology found shortening and disorganization of the photoreceptor inner and outer segments and progressive thinning of the outer nuclear layer. Electroretinogram a- and b-wave amplitudes were decreased with age. Western blot analysis found that the quantity and size of the mutated retinitis pigmentosa 1 (RP1) protein were normal. However, immunohistochemistry found that the mutant Rp1 protein partially mislocalized to the transition zone of the shortened axonemes. This mutation disrupted colocalization with cytoplasmic microtubules in vitro. In conclusion, the L66P mutation in the first doublecortin domain of the Rp1 gene impairs Rp1 protein localization and function, leading to abnormalities in photoreceptor outer segment structure and progressive photoreceptor degeneration. This is the first missense mutation in Rp1 shown to cause retinal degeneration. It provides a unique, slowly progressive photoreceptor degeneration model that mirrors the slow degeneration kinetics in most patients with retinitis pigmentosa.
As the most common inherited form of blindness, retinitis pigmentosa (RP) affects >100,000 persons in the United States and 1.5 million worldwide.1 Clinically, it is characterized by night blindness, progressive loss of peripheral vision, and bone spicule-shaped pigmentary retinopathy. Mutations in the RP1 gene are a common cause of autosomal dominant RP and a less common cause of autosomal recessive RP.2–5 Most pathological mutations in RP1 are either nonsense or frameshift mutations and are located at the beginning of exon 4 of the RP1 gene. A few missense variants in RP1, such as T373I,6 A669T,6 K663N,7 L1808P,7 D984G,8 R1652L,9 and K1370E,9 have been reported in patients with RP, but it is unclear whether they are disease-causing mutations.
The RP1 gene located on chromosome 8q12 consists of four exons with an open reading frame of 6468 bp, which is primarily contained within exon 4 (788 to 6468 bp), and it encodes a predicted protein of 2156 amino acids. It has been determined by Northern blot analysis3,10,11 and in situ hybridization3 that RP1 is expressed exclusively in rod and cone photoreceptor cells of the retina. In mice, retinitis pigmentosa 1 (Rp1) is localized to the axoneme of both rod and cone photoreceptors.12 Targeted disruption of the Rp1 gene in mice results in disorganization of outer segments (OS) with progressive degeneration of photoreceptors.13 Further studies reported that Rp1 plays a role in controlling the orientation and organization of disks in the OS.14 The photoreceptor axoneme begins at the basal body in the inner segment (IS), passes through the transition zone between the IS and OS, and continues into the OS.15 It was suggested that the axoneme has a role in stabilizing the stack of disk membranes.15
The N-terminus of RP1 shares significant homology with the protein doublecortin (DCX), whose mutation is associated with cerebral cortical abnormalities.16,17 This domain binds microtubules and promotes their assembly.18 Our data show that the homozygous L66P substitution in the first DCX domain of the Rp1 gene alters axoneme binding and causes axoneme shortening, disorganization of OS, and slowly progressive photoreceptor death.
Materials and Methods
Animals
Experimental procedures were performed in accordance with the statement for the use of animals in ophthalmology and vision research by the Association for Research in Vision and Ophthalmology. All protocols were approved by the animal care review board of the University of Pennsylvania.
Mutation Mapping
Our colony of C57BL/6J mice that contained the IL4 knockout (stock number 002518; The Jackson Laboratory, Bar Harbor, ME) maintained for several years at the University of Pennsylvania were found to have abnormal retinal morphology, detected by optical coherence tomography (OCT) (see Spectral Domain OCT Imaging). Reacquisition of the line from The Jackson Laboratory found that the retinal phenotype was not present; therefore, the phenotype was assumed to have been caused by a de novo mutation that became fixed in the University of Pennsylvania colony. To localize the mutation, mice with the retinal phenotype were out-crossed to wild-type (WT) DBA/2J mice (The Jackson Laboratory), which do not have the retinal degeneration phenotype detected by OCT, and F1 heterozygotes were intercrossed. The phenotype segregated independently from the IL4 knockout allele (detected by PCR) in mice that had been intercrossed with DBA/2J or back-crossed to WT C57BL/6J mice followed by a cross of F1s, which resulted in retinal degeneration in a quarter of the F2 mice. Whole genomic DNA was extracted from mouse tails, and DNA pools from affected and unaffected mice were genotyped with simple sequence length polymorphism markers by The Jackson Laboratory Fine Mapping Service. The sequences of WT and mutant retinal cDNA amplified with primers encompassing Rp1, a candidate gene mapping to the region, were compared for nucleotide differences.
Morphological Analysis
Eyes enucleated from both mutant mice and controls were immersion-fixed in 2% paraformaldehyde/2% glutaraldehyde overnight. For standard histology, plastic sections 3-μm thick were cut in the sagittal plane and stained with toluidine blue as previously described.19 The number of photoreceptor nuclei per column in the outer nuclear layer (ONL) was counted by an observer masked to the genotype, in triplicate, 800 μmol/L superior to the optic nerve head with the use of ImagePro Plus version 4.1 software (Media Cybernetics; Bethesda, MD) to calculate distances from manually set lengths. Comparison of the number of ONL nuclei per row was performed with analysis of variance for repeated measures.
Spectral Domain OCT Imaging
Before imaging, mice were anesthetized, and their pupils were dilated with 1% tropicamide. Artificial tears were used throughout the procedure to maintain corneal clarity, and mice were seated in the Bioptigen AIM-RAS holder. Spectral domain OCT images were obtained with the Envisu R2200-HR SD-OCT device (Bioptigen, Durham, NC) with the reference arm path length set at 950 mm. Image acquisition software was provided by the vendor. Both the averaged single B scan and volume scans were obtained with images centered on the optic nerve head.
Electron Microscopy
After animals were sacrificed, eyes were enucleated and fixed in 2% paraformaldeyhyde/2% glutaraldehyde overnight. After removal of the anterior segment, the posterior portion of each eye was cut into small wedge-shaped pieces and postfixed in 1% osmium tetroxide/0.1 mol/L sodium cacodylate buffer, dehydrated, and embedded in EMbed-812 (Electron Microscopy Sciences, Hatfield, PA). Ultrathin sections (60 to 80 nm thick) were stained and examined with a JEOL1010 transmission electron microscope (JEOL Ltd., Tokyo, Japan). Images were acquired with Advanced Microscopy Techniques Image Capture software version 602 (Advanced Microscopy Techniques Corp., Woburn, MA) and were rotated and cropped with Adobe Photoshop CS5 (Adobe Systems Incorporated, San Jose, CA).
qPCR
Neurosensory retinas were acquired after removing the anterior segment and ciliary body and detaching the neurosensory retina from the underlying retinal pigment epithelium/choroid tissue as described previously.19 RNA was isolated (RNeasy Mini Kit; Qiagen, Inc., Valencia, CA) according to the manufacturer's protocol. The RNA was quantified with a spectrophotometer and stored at −80°C. cDNA was synthesized (TaqMan Reverse Transcription Reagents; Applied Biosystems, Inc., Carlsbad, CA), according to the manufacturer's protocol. Gene expression in the neurosensory retina from mutants and controls was analyzed by real-time quantitative PCR (qPCR). Probes used were rhodopsin (Rho, Mm00520345_m1) and m-cone opsin (Opn1mw, Mm00433560_m1). Eukaryotic 18S rRNA (Hs99999901_s1) was used as an endogenous control. qPCR (Taqman; Applied Biosystems, Inc.) was performed on a sequence detection system (Prism Model 7500; Applied Biosystems, Inc.) by using the ΔΔCT method, which provides normalized expression values. The amount of target mRNA was compared among the groups of interest. All reactions were performed in biological and technical (three qPCR replicates per biological sample) triplicates.
Electroretinography
Electroretinography (ERG) recordings followed procedures described previously.20 In brief, mice were dark-adapted overnight and then anesthetized with a cocktail that contained ketamine (25 mg/kg body weight), xylazine (10 mg/kg body weight), and urethane (1000 mg/kg body weight). Pupils were dilated with 1% tropicamide saline solution (Mydriacil; Alconox, New York, NY), and mice were placed on a stage maintained at 37°C. Two electrodes made of UV-transparent plastic with embedded platinum wires were placed in electrical contact with the corneas. A platinum wire loop placed in the mouth served as the reference and ground electrode. ERGs were then recorded (Espion Electrophysiology System; Diagnosys LLC, Lowell, MA). The apparatus was modified by the manufacturer for experiments with mice by substituting light-emitting diodes with emission maximum at 365 nm for standard blue ones. A stage with the mouse was positioned so the animal's head was located inside the stimulator (ColorDome; Diagnosys LLC), ensuring uniform full-field illumination. The a- and b-wave amplitudes are reported for saturating light stimuli.
Immunofluorescence
After the globes were fixed in 4% paraformaldehyde, eyecups were generated by removing the anterior segment. The eyecups were infiltrated in 30% sucrose overnight and embedded in Tissue-Tek O.C.T. compound (Sakura Finetek, Torrance, CA). Immunofluorescence was performed on sections 10 μm thick as previously described.21 The primary antibodies against C-terminal Rp1,12 α-microtubule (ab7291; Abcam, Cambridge, MA), and RP GTPase regulator (RPGR; gift from Dr. Tiansen Li; National Eye Institute, Bethesda, MD) were detected with fluorophore-labeled secondary antibodies (Jackson ImmunoResearch Laboratories, Inc., West Grove, PA). Control sections were treated identically but without primary antibody. The sections were imaged by Zeiss LSM-510 Meta confocal microscope (Carl Zeiss Inc., Thornwood, NY). The lengths of axonemes were measured with the Zeiss Meta 510 software (Carl Zeiss Inc.).
Western Blot Analysis of Rp1
Mouse retinas were dissected and frozen immediately at −80°C. For Western blot analysis, the retinas were lysed in RIPA buffer plus Complete, EDTA-free protease inhibitor (Roche Diagnostics, Indianapolis, IN). One hundred micrograms of total protein was used per lane. The samples were incubated in SDS sample buffer for 10 minutes at 70°C. Protein lysates were separated on a 4% to 12% gradient SDS-PAGE gel and transferred to nitrocellulose membrane. Blocking was achieved by incubation for 1 hour in Tris-buffered saline that contained 5% milk and 0.1% Tween 20. Membranes were incubated overnight at 4°C with 2.5 μg/mL chicken anti–C-terminal RP1 antibodies. After washes, membranes were incubated with anti-chicken secondary antibody at 1:5000 dilution (Jackson ImmunoResearch Laboratories, Inc.) and were developed with ECL Plus enhanced chemiluminescence reagent (GE Healthcare, Chalfont St. Giles, UK). Anti–α-tubulin antibody (Sigma-Aldrich, St. Louis, MO) was used as loading control. Images were acquired with a Typhoon 9400 variable mode imager (GE Healthcare), and densitometry analysis was performed with ImageQuant TL software version 2005 (GE Healthcare).
Expression of Human RP1 Proteins in COS-7 Cells
The cDNA fragment corresponding to codons 1 to 682 (N1) of the human RP1 coding sequence was cloned into a pcDNA3.1/V5-His vector (Life Technologies, Grand Island, NY), as described previously.18 The vector that contained the corresponding human mutation (L67P) was generated by using N1 as the template with the Q5 Site-Directed Mutagenesis Kit (New England BioLabs, Ipswich, MA), according to the manufacturer's instructions. COS-7 cells were cultured on fibronectin-coated glass coverslips (BD Biosciences, Bedford, MA) in six-well plates by using Dulbecco’s modified Eagle’s medium (Life Technologies) supplemented with 10% fetal bovine serum (HyClone, Logan, UT) at 37°C with 10% CO2. To express the proteins, 2 μg of each RP1 construct was transfected into 2 × 105 cells with the use of Lipofectamine LTX (Life Technologies). After 48 hours, the cells were prepared for imaging as described previously,18 with the exception that the antibodies, mouse monoclonal anti–V5-Cy3 (Sigma-Aldrich) and fluorescein isothiocyanate/anti–α-tubulin clone DM1A (Sigma-Aldrich), were incubated together. Fluorescent signals were visualized with a Zeiss LSM-510 Meta confocal microscope (Carl Zeiss Inc.). To verify comparable expression levels of the RP1 and RP1-L67P proteins, Western blot analysis of transfected COS-7 cell lysates was performed with mouse anti-V5 antibody (Life Technologies) at 1:1000 dilution by standard procedures as described above.
Statistical Analysis
The means ± SEM were calculated for each comparison pair. Statistical analyses for ERG, axoneme length measurement, qPCR, and densitometry of Western blot analysis in the present study were performed in Prism version 5.03 (GraphPad Inc., San Diego, CA) by using the Student's two-group, two-sided t-test. P < 0.05 was considered statistically significant. Comparison of ONL thickness (nuclei) was performed with one-way analysis of variance with post hoc pairwise comparisons by using Bonferroni adjustment.
Results
Identification of an L66P Mutation in Rp1m1Jdun
The causative mutation mapped to proximal chromosome 1 cosegregating with marker D1Mit427, located near the Rp1 gene. We therefore sequenced the Rp1 coding sequence in retinal cDNA prepared from mice with retinal degeneration or their normal littermates (all phenotyped by OCT). Two adjacent missense mutations T402C and G403T, resulting in a leucine-to-proline change in amino acid 66, were identified in the first DCX domain in exon 2 of the Rp1 gene (Figure 1A). The normal nucleotide sequence encoding the first DCX domain and the corresponding translation of the Rp1 gene are shown in Figure 1B.
Figure 1.

A: DNA sequences of the Rp1 gene of homozygous mutant and WT control. Two adjacent point mutations, T402C and G403T, resulting in a L66P amino acid substitution located in the DCX domain in exon 2 of the Rp1 gene, were identified. B: DCX domain (amino acids 35 to 110) is highlighted in part of the nucleotide sequence and corresponding translation of the normal Rp1 gene. The location of the mutations and their corresponding amino acid are underlined and labeled with a double-headed arrow.
Age-Dependent Photoreceptor Degeneration in Rp1m1Jdun Mutant Retinas
Retinas from Rp1m1Jdun mutant mice were morphologically abnormal at an early age, and photoreceptor degeneration progressed slowly. Plastic sections indicated IS/OS shortening and disorganization at 1 month of age (Figure 2B) as compared to age-matched controls (Figure 2A). The ONL of the mutant retina thinned with age. At 26 months, the ONL was dramatically thinner (Figure 2D), as were IS/OS, than in age-matched controls (Figure 2C). Quantification of nuclei in the ONL indicated that Rp1m1Jdun mutant mice had age-dependent significant loss of rod nuclei compared with WT mice (P < 0.05) (Figure 2E).
Figure 2.

Histology of mouse retinas shows photoreceptor degeneration and disorganized outer segments. Photomicrographs of plastic sections 3-μm thick indicates shortening and disorganization of IS and OS (asterisks) of 1-month-old (B) and 26-month-old (D) Rp1m1Jdun mutant mice relative to 1-month-old (A) and 26-month-old (C) controls and age-dependent progressive thinning of the ONL (black arrow) in 26-month-old Rp1m1Jdun mutant retina. E: The thickness of the ONL was measured in numbers of photoreceptor nuclei per column. Measurements were made in triplicate 800 μm from the optic nerve head in superior section of the retina. Rp1m1Jdun mutants exhibit an age-dependent loss of rod nuclei compared with WT. GraphPad Prism was used to fit the data with a nonlinear order polynomial equation. Electron microscopy shows disorganized OS in some photoreceptors in 2-month-old mutant mice (G, white arrowheads) compared with normal controls (F), but the transition zone appears normal (G and F, black arrowheads). OCT indicates thinning of the ONL (I, white double-headed arrow) in mutants at age of 1 month compared with WT control (H, white double-headed arrow). The thin and highly reflective band (H, red arrowheads) shows the two separated dark bands that represent IS (H, inset, upper red bracket) and OS (H, inset, lower red bracket) separately in control mice. However, the mutant mice have one dark band (I, inset, arrowed bracket) and diffuse high reflectivity in the OS region (I, inset, red asterisk). ∗P < 0.05. Scale bars: 10 μm (A–D); 500 nm (F and G). RPE, retinal pigment epithelium.
Disorganized Photoreceptor OS in Rp1m1Jdun Mutant Mice
When imaged by transmission electron microscopy, the transition zone of mutant mice appeared normal (Figure 2, F and G), but OS disks were disorganized in mutant mice at age 2 months (Figure 2G). OCT was used as a noninvasive method to phenotype affected mice. OCT images showed a thinning of ONL at 1 month of age in mutant mice (Figure 2, H and I). A thin, highly reflective IS/OS junction band visible in WT retinas (Figure 2H) separated IS (Figure 2H) from OS (Figure 2H). However, the normally thin, highly reflective IS/OS junction was thick and diffuse in mutant retinas (Figure 2I). The disorganized OS found on electron microscopy are consistent with the thick, highly reflective band in the OS region that was detected in OCT images.
Altered mRNA Levels of Rhodopsin and m-Cone Opsin in Rp1m1Jdun Mutant Retinas
Consistent with shortening of OS and loss of photoreceptors, quantitative PCR found that rhodopsin mRNA levels were diminished in Rp1m1Jdun mutants compared with WT controls at 3, 21, 25, and 27 months of age, but not at 1 month (Figure 3A). Compared with age-matched WT controls, 18-month-old Rp1m1Jdun mutants had higher levels of m-cone opsin mRNA, presumably because cones represent a higher percentage of the photoreceptors in Rp1m1Jdun mice that have lost most of their rods (Figure 3B).
Figure 3.
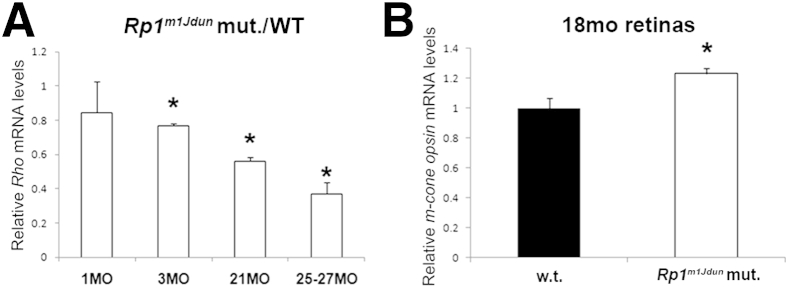
Relative mRNA levels of rhodopsin and m-cone opsin in WT and Rp1m1Jdun mutant retinas. A: Quantitative PCR shows a decrease in the mutant versus WT rhodopsin mRNA ratios with age; 3-, 21, and 25- to 27-month-old Rp1m1Jdun have significantly reduced rhodopsin mRNA levels compared with WT. B:Rp1m1Jdun mutants have significantly higher levels of m-cone opsin mRNA at 18 months than do WT mice. Data are expressed as means ± SD. n = 3. ∗P < 0.05.
ERG Responses in Rp1m1Jdun Mutant Mice Decrease with Age
To study the retinal function of this mutant line, ERG was performed on both Rp1m1Jdun mutants and age-matched WT controls. Corresponding to histological findings, Rp1m1Jdun mice had decreased ERG responses with age. At 1 month of age, no differences were found between mutants and controls in all three parameters of the ERG (Figure 4, A, C, and E). However, by 24 months, Rp1m1Jdun mutants had significantly reduced saturated rod a- and rod b-wave responses compared with controls (Figure 4, B and D). Cone b-wave amplitudes were not significantly diminished for control mice (Figure 4F).
Figure 4.

Rp1m1Jdun mutant mice have decreased ERG response with age. ERG performed on 1-month-old WT and Rp1m1Jdun mutant mice does not show significant differences in saturated rod b- (A), rod a- (C), and cone b- (E) wave responses. Mutant mice at 24 months of age have significantly decreased saturated rod b- (B) and rod a- (D) wave responses compared with age-matched controls, whereas cone b-wave amplitude (F) is not diminished (E). Data are expressed as means ± SD. n = 3. ∗P < 0.05.
Localization of Rp1 Protein Is Altered and Axonemes Shortened in Rp1m1Jdun Mutant Mice
Confocal images found that the mutant RP1 protein was localized to the axoneme in the proximal OS, but there was a subtle abnormality in its localization. In normal control retinas, Rp1 (Figure 5A) did not overlap with the transition zone labeled by RPGR (Figure 5A) and localized to the axoneme distal to RPGR. However, in 2-month-old mice homozygous for the Rp1m1Jdun allele, mutant Rp1 partially colocalized with RPGR (Figure 5B). To determine whether axonemes of the mutant animals were compromised, sections were stained with an anti-acetylated α-tubulin antibody (Figure 5, D and E). The mean axoneme length was shortened in the 2-month-old Rp1m1Jdun retinas compared with WT controls (Figure 5C). To determine whether the Rp1 protein expressed in Rp1m1Jdun mutant retinas was altered either in size, quantity, or localization, Western blot analysis and immunolabeling were performed with an anti–C-terminal RP1 antibody. Western blot analysis found that the size (approximately 240 kDa) and quantity (Figure 5F) of the mutant Rp1 was comparable with its WT counterpart.
Figure 5.

Localization of Rp1 protein is altered, and axenomes are shortened in Rp1m1Jdun mutant mice. A and B: Confocal imaging with anti–C-terminal RP1 antibody (green) indicates that mutated Rp1 protein is expressed and localized in the proximal OS. However, the 2-month-old Rp1m1Jdun mutant protein (B, green) labels a shorter region relative to WT controls. In the control retina, Rp1 signal (A, inset, green) does not overlap with the RPGR signal (A, inset, red) indicated by the gap in between (A, inset, black arrowhead), whereas in mutant retina, Rp1 signal (B, inset, green) partially overlaps with the RPGR signal (B, inset, red) indicated by the yellow signal (B, inset, white arrowhead). D and E: The length of axonemes (E, anti-acetylated α-tubulin, red) traced with white line in 2-month-old Rp1m1Jdun mutant retina is shorter than those of the WT retina (D, anti-acetylated α-tubulin, red). Transition zone is shown by labeling with anti-RPGR antibody (green). F: Western blot analysis with anti–C-terminal RP1 antibody. The Rp1 protein is expressed and has normal size (approximately 240 kDa) in both Rp1m1Jdun mutant and WT mice at 2 months of age. Data are expressed as mean lengths ± SD of the axonemes from WT controls and Rp1m1Jdun mutants (C). n = 3 (C). ∗∗P < 0.01. Scale bars: 5 μm (A, B, D, and E). TZ, Transition zone.
Disrupted RP1-Microtubule Interaction in Vitro Due to L67P Mutation
Given the in vivo findings of altered location of Rp1 and shortened axonemes in Rp1m1Jdun mutant mice, we next asked whether the interaction between RP1 and the microtubules is altered by this mutation. A cDNA fragment corresponding to codons 1 to 682 (N1) of the human RP1 coding sequence was cloned into a pcDNA3.1/V5-His vector. The vector that contained the corresponding human mutation (L67P) was generated with N1 as the template. After WT and L67P mutant RP1 cDNA constructs were transfected into COS-7 cells, the cells were co-immunostained with antibodies to α-tubulin to detect the microtubule cytoskeleton and with anti-V5 antibodies to detect the L67P RP1 protein. In COS-7 cells transfected with WT RP1, the filaments are lineal, consecutive (Figure 6B), and colocalized with α-tubulin (Figure 6, A and C). However, in L67P mutant RP1-transfected COS-7 cells, the RP1 protein displayed a punctate pattern (Figure 6E) which only partially colocalized with α-tubulin (Figure 6, D and F). Western blot analysis that used an anti-V5 antibody detected the recombinant RP1 proteins (Figure 6G). The size and quantity of WT RP1 and L67P mutant RP1 in COS-7 cells were similar (Figure 6H).
Figure 6.

L67P mutation of RP1 alters its colocalization with microtubules. The RP1 cDNA constructs were transfected into COS-7 cells, and the cells were co-immunolabled with anti-V5 antibodies to detect the recombinant RP1 protein (B and E, red) and antibodies to α-tubulin to detect the microtubule cytoskeleton (A and D, green). Merged images (C and F). WT RP1 protein is lineal and colocalizes with cytoplasmic microtubules. E: The localization of the mutant protein is disrupted and displays a punctate pattern. cDNA fragments that contained a C-terminal V5 epitope tag were cloned into COS-7 cells, which facilitates identification of the recombinant RP1 proteins. G: Recombinant RP1 protein detection was performed with Western blot analysis (anti-V5 antibodies). H: Quantification of the levels of WT RP1 and L67P mutant RP1 in COS-7 cells shows similar levels. Data are expressed as means ± SD. n = 3. Scale bars: 10 μm (C and F).
Discussion
RP1 is important for photoreceptor survival; mutations in RP1 account for 5% to 10% of cases of autosomal dominant RP and have also been found to cause autosomal recessive RP.4,22 Our data indicate that the L66P mutation in the Rp1 gene is sufficient to disrupt binding of RP1 to microtubules and its localization in the axoneme. Rp1m1Jdun homozygous mice had OS disorganization and slowly progressive photoreceptor death. To our knowledge, this is the first animal line to carry a degeneration-causing missense mutation in the Rp1 gene. An unusual pathological feature of the Rp1m1Jdun mice is the slowly progressive retinal degeneration, which mirrors the classical phenotype observed in human RP. The Rp1m1Jdun line will serve as a model of RP and can be used for functional studies to further elucidate the role of RP1 in the retina. It is helpful that the phenotype can be detected by OCT at 1 month of age. Similar OCT abnormalities may be found in patients with abnormal axonemes and disrupted photoreceptor disk stacking.
The protein DCX is believed to be involved in directing neuronal migration during development of the central nervous system.16 In addition, the DCX region is known to bind and stabilize microtubules.23,24 The DCX domain of RP1 is required for its correct localization to the OS portion of the axoneme.14 In addition, RP1 is the first photoreceptor-specific microtubule-associated protein to be identified.18 In vivo studies found that RP1 is necessary for controlling the length and stability of the photoreceptor axoneme, and the microtubule-associated protein activities of the protein are primarily associated with the DCX domains.18 It has been hypothesized that RP1 may assist in regulating the length of the axoneme by promoting elongation or suppressing the shortening of microtubules within the axoneme.25 The Rp1m1Jdun mice have a single amino acid change in the DCX region that most likely affects the function of this domain. In vitro data indicated that the L67P mutation in the DCX region of RP1 impairs its microtubule association, suggesting that the underlying mechanism by which the L67P mutation in the Rp1 gene that causes retinal degeneration in Rp1m1Jdun mice may be due to its loss of function as a microtubule-associated protein in photoreceptors. Previous in vitro studies suggested that RP1 is phosphorylated by male germ cell-associated kinase, which regulates ciliary elongation.26 Patients with MAK gene mutations have retinal degeneration that resembles those with RP1 mutation.27 In mouse retinas, male germ cell-associated kinase colocalizes with Rp1 in the photoreceptor OS axonemes and extends into the transition zone.26 It is essential for preventing excessive elongation of the cilia. In addition, overexpression of WT RP1 induces ciliary elongation, and expression of the N-terminal portion of RP1 induces an increased intensity of acetylated α-tubulin labeling in cultured cells.26 These results suggest that excess activation of the microtubule-associated protein RP1 can induce excess ciliary elongation. A previous study reported that RP1 and RPGR are in distinct portions of axoneme. There is a gap of 200 to 300 nm between RP1 and RPGR localization in normal retinas, with RP1 located distally in the OS and RPGR found proximally in the transition zone.18 There is mislocalization to the transition zone with loss of axoneme localization in the mutant RP1 with a targeted deletion of exons 2 and 3 in the Rp1 gene.18 Interestingly, we found a partial mislocalization of RP1-L66P mutant protein to the RPGR-labeled transition zone in the homozygous Rp1m1Jdun mice. However, distinct from the exons 2 to 3 deletion mice, the Rp1m1Jdun mice retained RP1 localization to the shortened axoneme. Our results suggest that the DCX domain of RP1 is necessary to exclude the protein from the transition zone of the axoneme. Perhaps accumulation of mutant Rp1 in the transition zone results in structural abnormalities in this region and/or diminishes Rp1 protein levels in the distal axoneme within the OS where it may be needed for elongation and maintenance of the axoneme.
The Rp1tm1Jnz mutants with a targeted deletion of exons 2 to 3, in which the DCX domain is deleted, indicated that RP1 is required for keeping newly formed OS disks in the correct orientation and stacking of disks into mature OS.13,14 Rhodopsin was also found to be mislocalized in these mice, suggesting a possible transport defect. Similar to the Rp1tm1Jnz mice, the Rp1m1Jdun mice have disoriented OS disks and axoneme shortening. It is possible that exclusion of Rp1 from the transition zone is necessary for proper OS disk orientation. It is also possible that this novel missense mutation in the Rp1 gene disrupts the protein transport function of the transition zone. Defective protein transport would disrupt OS formation and would ultimately lead to photoreceptor death. Yet, rhodopsin localization appears normal in Rp1m1Jdun mice at 6 months of age (data not shown), making this possibility less likely.
To our knowledge, this mutation has not been found in patients as yet. Our finding that an Rp1 missense mutation can cause mouse retinal degeneration provides a rationale to search for additional RP1 missense mutations in patients with RP. Although the retinal degeneration phenotype maps to the Rp1 gene and we observe functional consequences of the mutations for RP1 protein localization, it is formally possible that a linked mutation contributes to the phenotype. Confirmation that the Rp1 T402C and G403T mutations are causative will require generation of knockin mice with these mutations.
This new mutant line provides evidence that an Rp1 missense mutation can cause retinal degeneration and supports the importance of the DCX domain of RP1 for proper localization and function for axoneme length and OS disk orientation. Further, it provides an unusual, slowly progressive model of retinal degeneration that will be useful for testing chronic neuroprotective therapeutics.
Acknowledgments
We thank Xinyu (Jasmine) Zhao, Biao Zuo, and Ray Meade (University of Pennsylvania, Philadelphia, PA) for research support and Dr. Gui-shuang Ying (University of Pennsylvania, PA) for biostatistics analysis. The primary antibodies against RPGR were a gift from Dr. Tiansen Li (National Eye Institute, Bethesda, MD).
Footnotes
Supported by NIH grants R01 EY015240 (J.L.D.), EY016501 (P.M.N.), EY2063 (J.D.L.), A16870 (J.D.L.), and EY12910 (E.A.P.); unrestricted funding from Research to Prevent Blindness; the F.M. Kirby Foundation, United States; a gift in memory of Dr. Lee F. Mauger; the Paul and Evanina Bell Mackall Foundation Trust; a Cancer Center CORE grant 5P30CA034196 (P.M.N.); the Foundation Fighting Blindness USA (Q.L.).
D.S., S.G., and Y.L. contributed equally to this work.
Disclosures: None declared.
References
- 1.Bunker C.H., Berson E.L., Bromley W.C., Hayes R.P., Roderick T.H. Prevalence of retinitis pigmentosa in Maine. Am J Ophthalmol. 1984;97:357–365. doi: 10.1016/0002-9394(84)90636-6. [DOI] [PubMed] [Google Scholar]
- 2.Berson E.L., Grimsby J.L., Adams S.M., McGee T.L., Sweklo E., Pierce E.A., Sandberg M.A., Dryja T.P. Clinical features and mutations in patients with dominant retinitis pigmentosa-1 (RP1) Invest Ophthalmol Vis Sci. 2001;42:2217–2224. [PubMed] [Google Scholar]
- 3.Pierce E.A., Quinn T., Meehan T., McGee T.L., Berson E.L., Dryja T.P. Mutations in a gene encoding a new oxygen-regulated photoreceptor protein cause dominant retinitis pigmentosa. Nat Genet. 1999;22:248–254. doi: 10.1038/10305. [DOI] [PubMed] [Google Scholar]
- 4.Liu Q., Collin R.W., Cremers F.P., den Hollander A.I., van den Born L.I., Pierce E.A. Expression of wild-type Rp1 protein in Rp1 knock-in mice rescues the retinal degeneration phenotype. PloS One. 2012;7:e43251. doi: 10.1371/journal.pone.0043251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Singh H.P., Jalali S., Narayanan R., Kannabiran C. Genetic analysis of Indian families with autosomal recessive retinitis pigmentosa by homozygosity screening. Invest Ophthalmol Vis Sci. 2009;50:4065–4071. doi: 10.1167/iovs.09-3479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Khaliq S., Abid A., Ismail M., Hameed A., Mohyuddin A., Lall P., Aziz A., Anwar K., Mehdi S.Q. Novel association of RP1 gene mutations with autosomal recessive retinitis pigmentosa. J Med Genet. 2005;42:436–438. doi: 10.1136/jmg.2004.024281. (letter to the editor) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bowne S.J., Daiger S.P., Hims M.M., Sohocki M.M., Malone K.A., McKie A.B., Heckenlively J.R., Birch D.G., Inglehearn C.F., Bhattacharya S.S., Bird A., Sullivan L.S. Mutations in the RP1 gene causing autosomal dominant retinitis pigmentosa. Hum Mol Genet. 1999;8:2121–2128. doi: 10.1093/hmg/8.11.2121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chiang S.W., Wang D.Y., Chan W.M., Tam P.O., Chong K.K., Lam D.S., Pang C.P. A novel missense RP1 mutation in retinitis pigmentosa. Eye (Lond) 2006;20:602–605. doi: 10.1038/sj.eye.6701944. [DOI] [PubMed] [Google Scholar]
- 9.Zhang X., Chen L.J., Law J.P., Lai T.Y., Chiang S.W., Tam P.O., Chu K.Y., Wang N., Zhang M., Pang C.P. Differential pattern of RP1 mutations in retinitis pigmentosa. Mol Vis. 2010;16:1353–1360. [PMC free article] [PubMed] [Google Scholar]
- 10.Sullivan L.S., Heckenlively J.R., Bowne S.J., Zuo J., Hide W.A., Gal A., Denton M., Inglehearn C.F., Blanton S.H., Daiger S.P. Mutations in a novel retina-specific gene cause autosomal dominant retinitis pigmentosa. Nat Genet. 1999;22:255–259. doi: 10.1038/10314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Guillonneau X., Piriev N.I., Danciger M., Kozak C.A., Cideciyan A.V., Jacobson S.G., Farber D.B. A nonsense mutation in a novel gene is associated with retinitis pigmentosa in a family linked to the RP1 locus. Hum Mol Genet. 1999;8:1541–1546. doi: 10.1093/hmg/8.8.1541. [DOI] [PubMed] [Google Scholar]
- 12.Liu Q., Zhou J., Daiger S.P., Farber D.B., Heckenlively J.R., Smith J.E., Sullivan L.S., Zuo J., Milam A.H., Pierce E.A. Identification and subcellular localization of the RP1 protein in human and mouse photoreceptors. Invest Ophthalmol Vis Sci. 2002;43:22–32. [PMC free article] [PubMed] [Google Scholar]
- 13.Gao J., Cheon K., Nusinowitz S., Liu Q., Bei D., Atkins K., Azimi A., Daiger S.P., Farber D.B., Heckenlively J.R., Pierce E.A., Sullivan L.S., Zuo J. Progressive photoreceptor degeneration, outer segment dysplasia, and rhodopsin mislocalization in mice with targeted disruption of the retinitis pigmentosa-1 (Rp1) gene. Proc Natl Acad Sci U S A. 2002;99:5698–5703. doi: 10.1073/pnas.042122399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Liu Q., Lyubarsky A., Skalet J.H., Pugh E.N., Jr., Pierce E.A. RP1 is required for the correct stacking of outer segment discs. Invest Ophthalmol Vis Sci. 2003;44:4171–4183. doi: 10.1167/iovs.03-0410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kaplan M.W., Iwata R.T., Sears R.C. Lengths of immunolabeled ciliary microtubules in frog photoreceptor outer segments. Exp Eye Res. 1987;44:623–632. doi: 10.1016/s0014-4835(87)80134-3. [DOI] [PubMed] [Google Scholar]
- 16.Gleeson J.G., Allen K.M., Fox J.W., Lamperti E.D., Berkovic S., Scheffer I., Cooper E.C., Dobyns W.B., Minnerath S.R., Ross M.E., Walsh C.A. Doublecortin, a brain-specific gene mutated in human X-linked lissencephaly and double cortex syndrome, encodes a putative signaling protein. Cell. 1998;92:63–72. doi: 10.1016/s0092-8674(00)80899-5. [DOI] [PubMed] [Google Scholar]
- 17.des Portes V., Pinard J.M., Billuart P., Vinet M.C., Koulakoff A., Carrié A., Gelot A., Dupuis E., Motte J., Berwald-Netter Y., Catala M., Kahn A., Beldjord C., Chelly J. A novel CNS gene required for neuronal migration and involved in X-linked subcortical laminar heterotopia and lissencephaly syndrome. Cell. 1998;92:51–61. doi: 10.1016/s0092-8674(00)80898-3. [DOI] [PubMed] [Google Scholar]
- 18.Liu Q., Zuo J., Pierce E.A. The retinitis pigmentosa 1 protein is a photoreceptor microtubule-associated protein. J Neurosci. 2004;24:6427–6436. doi: 10.1523/JNEUROSCI.1335-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hadziahmetovic M., Song Y., Wolkow N., Iacovelli J., Grieco S., Lee J., Lyubarsky A., Pratico D., Connelly J., Spino M., Harris Z.L., Dunaief J.L. The oral iron chelator deferiprone protects against iron overload-induced retinal degeneration. Invest Ophthalmol Vis Sci. 2011;52:959–968. doi: 10.1167/iovs.10-6207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lyubarsky A.L., Lem J., Chen J., Falsini B., Iannaccone A., Pugh E.N., Jr. Functionally rodless mice: transgenic models for the investigation of cone function in retinal disease and therapy. Vision Res. 2002;42:401–415. doi: 10.1016/s0042-6989(01)00214-0. [DOI] [PubMed] [Google Scholar]
- 21.Dunaief J.L., Dentchev T., Ying G.S., Milam A.H. The role of apoptosis in age-related macular degeneration. Arch Ophthalmol. 2002;120:1435–1442. doi: 10.1001/archopht.120.11.1435. [DOI] [PubMed] [Google Scholar]
- 22.Jacobson S.G., Cideciyan A.V., Iannaccone A., Weleber R.G., Fishman G.A., Maguire A.M., Affatigato L.M., Bennett J., Pierce E.A., Danciger M., Farber D.B., Stone E.M. Disease expression of RP1 mutations causing autosomal dominant retinitis pigmentosa. Invest Ophthalmol Vis Sci. 2000;41:1898–1908. [PubMed] [Google Scholar]
- 23.Gleeson J.G., Lin P.T., Flanagan L.A., Walsh C.A. Doublecortin is a microtubule-associated protein and is expressed widely by migrating neurons. Neuron. 1999;23:257–271. doi: 10.1016/s0896-6273(00)80778-3. [DOI] [PubMed] [Google Scholar]
- 24.Horesh D., Sapir T., Francis F., Wolf S.G., Caspi M., Elbaum M., Chelly J., Reiner O. Doublecortin, a stabilizer of microtubules. Hum Mol Genet. 1999;8:1599–1610. doi: 10.1093/hmg/8.9.1599. [DOI] [PubMed] [Google Scholar]
- 25.Desai A., Mitchison T.J. Microtubule polymerization dynamics. Annu Rev Cell Dev Biol. 1997;13:83–117. doi: 10.1146/annurev.cellbio.13.1.83. [DOI] [PubMed] [Google Scholar]
- 26.Omori Y., Chaya T., Katoh K., Kajimura N., Sato S., Muraoka K., Ueno S., Koyasu T., Kondo M., Furukawa T. Negative regulation of ciliary length by ciliary male germ cell-associated kinase (Mak) is required for retinal photoreceptor survival. Proc Natl Acad Sci U S A. 2010;107:22671–22676. doi: 10.1073/pnas.1009437108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Stone E.M., Luo X., Héon E., Lam B.L., Weleber R.G., Halder J.A., Affatigato L.M., Goldberg J.B., Sumaroka A., Schwartz S.B., Cideciyan A.V., Jacobson S.G. Autosomal recessive retinitis pigmentosa caused by mutations in the MAK gene. Invest Ophthalmol Vis Sci. 2011;52:9665–9673. doi: 10.1167/iovs.11-8527. [DOI] [PMC free article] [PubMed] [Google Scholar]