

Abstract
Tumor necrosis factor (TNF)-α inhibitors and thiopurines are among the most important classes of medications utilized in the clinical management of Crohn’s disease and ulcerative colitis. A significant proportion of patients loses response to these agents or develops adverse effects during the course of the treatment. Monitoring of drug levels and anti-drug antibodies (for TNF-α inhibitors) and metabolite levels (for thiopurines) can provide valuable insight into the possible etiology of unfavorable outcomes and allow for an appropriate management strategy for these patients. This review summarizes the current knowledge on the clinical implications of therapeutic drug monitoring in inflammatory bowel disease patients treated with TNF-α inhibitors and thiopurines.
Keywords: Inflammatory bowel disease, Crohn’s disease, ulcerative colitis, biologics, thiopurines
Introduction
Several classes of medications are available for treatment of inflammatory bowel disease (IBD). The rate of clinical and endoscopic response varies greatly between and within the medication classes. Measurement of levels of the medications (or its metabolites) and anti-drug antibodies (ATI) may in many cases provide insight into the mechanism of the evolving loss of response (LOR), as well as suggest a possible salvage strategy. In this review, we will focus on therapeutic monitoring of two main classes of IBD medications, i.e. thiopurines, including azathioprine (AZA) and 6-mercaptopurine (6-MP), and tumor necrosis factor (TNF)-α inhibitors, including infliximab (IFX), adalimumab (ADA), certolizumab pegol (CZP) and golimumab (GLM).
TNF-α inhibitors
Anti-TNFs have been the mainstay of anti-inflammatory treatment in IBD for the past decade. Four agents are currently available in the US (IFX, ADA, CZP, and GLM), and three in Europe and Canada (IFX, ADA, and GLM). In addition, biosimilars are emerging and will provide additional therapeutic options in the near future. Assays for assessment of serum levels of IFX and ADA as well as corresponding anti-drug antibodies (ATI) are available commercially. The majority of clinical experience in therapeutic drug monitoring stems from IFX data, reviewed below in greater detail.
Definitions of LOR
Primary non-response is generally defined as a failure to achieve clinical response following an induction phase of treatment [1]. Importantly, some patients may take longer to achieve initial response. Primary non-response occurs in approximately one third of the patients started on anti-TNFs [2]. Secondary loss of response occurs at any point during the treatment after an initial response has occurred. However, there is no consensus regarding the criteria identifying response. While the European Crohn’s and Colitis Organization guidelines suggest a definition based on clinical scores (Crohn’s disease activity index, CDAI) [3], multiple different definitions varying from endoscopic healing to a need for dose intensification have been proposed. The incidence of secondary loss of response is variable. For IFX, an annual risk of loss of response of 13% was reported [4]. However, the risk is not distributed evenly, with roughly 2/3 of the patients developing LOR with the first 12 months, and the rest losing response at a significantly slower pace. For ADA, annual LOR incidence of 10%-24% has recently been reported [5,6].
Risk factors for primary non-response
Several factors have been demonstrated to be associated with a primary loss of response to anti-TNF in IBD, including longer (>2 years) disease duration, extensive small bowel disease in CD, smoking and normal C-reactive protein (CRP) on initiation of treatment [7,8]. Polymorphisms in the apoptosis-related genes such as FAS-L and Caspase 9, as well as in the IBD5 locus, were also associated with an increased risk of primary LOR in CD [8,9]. In UC, increased age, anti-neutrophil cytoplasmic antibody-positive status and anti-Saccharomyces cerevisiae antibody-) negative status, and prior anti-TNF exposure were identified as risk factors for primary nonresponse [10].
Risk factors for secondary LOR
Although immunogenicity is by far the most studied, it is not the sole mechanism responsible for loss of response. Multiple additional etiologies, including non-adherence, fecal drug loss, non-immune clearance and non-TNF-driven disease, have been implicated in the pathogenesis of secondary LOR (Fig. 1, adapted from Ben-Horin et al).
Figure 1.
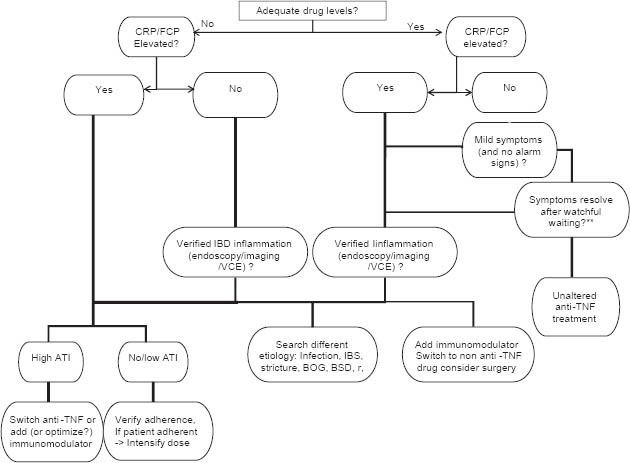
TDM based algorithm for management of loss of response to TNFα inhibitors. Adapted from Ben Horin, et al [87]
LOR, loss of response; CRP, C-reactive protein; FCP, fecal calprotectin; VCE, videocapsule endoscopy; BOG, bacterial overgrowth; BSD, bile salt diarrhea; ATI, anti-drug antibodies
Several risk factors are associated with increased risk of loss of response, including episodic treatment, non-inflammatory symptoms, symptomatic stricture and smoking [2]. In randomized controlled trials, concomitant treatment with AZA was shown to be protective against LOR [11]. In contrast, concomitant treatment with methotrexate failed to improve clinical response rates, possibly owing to a large proportion of patients being on corticosteroids at treatment onset in this particular trial [12]. Nonetheless, the latter study found higher trough levels of IFX and lower rates of antibody formation with methotrexate co-treatment [12].
Multiple studies have confirmed a correlation between clinical response and trough serum levels of anti-TNF medications [13-17]. Moreover, such correlation was recently established not only for clinical response but for endoscopic outcomes (mucosal healing) and decline of inflammation markers [18-21].
Currently, there is no clear consensus on the trough level values that correspond to clinical response. Recently, a cut-off trough level of 3 µg/mL has been suggested to have the optimal discriminatory accuracy for response to IFX in CD [22]. Trough levels of 3-7 µg/mL [23] and 5-10 µg/mL [24] have recently been suggested as target levels for maintenance therapy for both UC and CD. In addition, post-induction (week 14) trough levels of IFX were correlated with long-term (week 54) clinical response in a subgroup analysis of the ACCENT 1 study [25]. Moreover, serum levels at non-trough time points have also correlated with clinical response. For example, a serum level of IFX of 12.0 µg/mL at 4 weeks from the last infusion was independently correlated with clinical response [15]. For ADA, a cut-off drug level of 5.85 µg/mL yielded optimal sensitivity, specificity and positive likelihood ratio for prediction of clinical response [26].
Importantly, identification of a uniform target level for serum IFX is challenging as the detection assays vary significantly between different centers. It also remains to be determined whether the trough levels associated with optimal response are similar for CD and UC.
Antibodies
ATI directed against the FAB fragment of the molecule [27] develop against both chimeric and fully humanized anti-TNFs. ATI interfere with the biologic activity by inhibiting the binding of the TNF-α inhibitors to both serum and membrane-bound TNF-α molecules, and by creating immune complexes that are eliminated by the reticuloendothelial system [28,29]. Formation of ATIs has been demonstrated to be correlated with decreased levels of anti-TNFs and diminished clinical response, although not all studies support that [13-16,30]. This discrepancy may result from several factors, such as different sensitivity of the employed assays (see below), non-neutralizing antibodies, non-anti-TNF-driven disease and alternative methods of elimination of anti-TNFs [1]. Moreover, serum anti-TNF levels and ATI most likely represent a continuous process, which may frequently start with low-titer antibodies that do not hamper the serum levels of the drug significantly, progressing to high-titer antibodies leading to a complete elimination of the drug. Frequently detection of ATI will precede the development of LOR by several weeks, or alternatively, will be detected after LOR has developed [31]. Moreover, transient (appearing on a single measurement without recurrence) ATI are a frequent phenomenon, described in up to 28% of patients [32]. In contrast to persistent ATI that rarely (<10%) appear after 1 year of treatment, these transient antibodies may be detected at any point during the treatment without a significant impact on LOR-free survival [31]. The risk of ATI formation has been repeatedly demonstrated to be lower in patients receiving concomitant immunomodulatory therapy [11,12,30]. Premedication with intravenous corticosteroids was reported to be associated with a lower rate of ATI formation by Farrell et al [33].
Evaluation of serum levels and ATIs
Several techniques are available for the purpose of measurement of serum levels and antibodies. The most common is a solid phase double-antigen ELISA, in which IFX serves as both the capture antigen and the detection antibody. This technique is relatively simple, reproducible and inexpensive [34]. This method has some important drawbacks, the main one being an inability to detect ATI in the presence of IFX in the serum. These results, reported as “inconclusive”, are very commonly reported. For instance, 72% of measurements of ATI in the SONIC trial were deemed inconclusive [11]. In addition, this method is also incapable of detection of immunoglobulin 4 (IgG4) ATI. A modified ELISA, employing anti-human λ antigen detection antibody (AHLC), has an improved capacity for detection of ATI in presence of IFX, a well as for detection of IgG4 ATI [35]. Such “double positive” results were reported in 9% of patients, and were associated with a trend towards a higher future risk of development of LOR [36]. An opposite state of “double negativity” (IFX-ATI-), frequently reported by an early generation ELISA assay, is significantly less frequently detected with the AHLC assay (13 vs 35.5%, P<0.001). If an alternative dilution (1:10) was employed, almost all of the patients still double negative on AHLC ELISA were successfully reclassified as either ATI+, IFX+ or ATI+IFX+ [37].
Additional detection methods, including homogenous mobility shift assay [38], and radioimmunoassay [39], are used in clinical practice. Both techniques, despite the presumable superior analytical accuracy, did not demonstrate superior diagnostic value on direct comparison with ELISA techniques [34,40].
Management of LOR to anti-TNFs guided by IFX and ATI levels
Measurement of drug levels and ATIs can guide important clinical decisions in IBD patients on anti-TNFs. However, they must always be considered within the context of the patient’s clinical status and correlated with objective evidence of ongoing mucosal inflammation. In all cases of LOR, verification of the true inflammatory nature of the symptoms is mandated. Clinical assessment in many cases is unreliable, as patients with irritable bowel syndrome (IBS) or IBS-related symptoms common in IBD may have a CDAI value similar to an active CD patient [41]. Disease activity should also be accessed using an inflammatory marker [fecal calprotectin usually being more accurate than CRP, especially in ulcerative colitis (UC)] [42] and endoscopy. In addition, non-adherence to medications may occur in up to 29% in IBD patients on anti-TNFs [43].
In some cases, an expectant management may lead to a regain of response [9]. However, when the true inflammatory nature of the symptoms and their persistence are ascertained, immunopharmacological considerations should be taken into account while selecting the appropriate strategy for management of LOR.
Therapeutic drug monitoring (TDM) for primary non-response
In the majority of cases, primary non-response is not associated with subtherapeutic IFX levels [44]. In specific clinical situations, such as acute UC, increased clearance of IFX through fecal loss and accelerated formation of ATI [45,46] result in low serum levels of IFX accompanied by non-response. A strategy based on an accelerated dosing scheme based on patient’s clinical response for patients with acute UC has been suggested; however it is yet unclear how the immunopharmacological data should be incorporated in such algorithm [47]. Patients with a primary loss of response may still respond to another anti-TNF [9].
TDM for secondary LOR (Fig. 1)
When drug levels are adequate and there are no signs of active inflammation (CRP, fecal biomarkers, ileocolonoscopy, capsule endoscopy), alternative explanation for the symptoms (underlying IBS, bacterial overgrowth, bile salt diarrhea, infection, etc) should be sought.
When active inflammation is present and drug levels are adequate, the patient is unlikely to respond to dose escalation, and a switch to a different medication class should be considered [2,48].
In the presence of active inflammation with absent or low IFX levels, the determination of ATI should be undertaken. Initial reports had suggested that in presence of detectable ATI, dose escalation was generally ineffective (86% in patients without ATI vs. 17% in patients with detectible ATI, P=0.001) [49]. However, this study utilized a double-antigen assay ELISA unable to simultaneously detect IFX and ATI. Thus, all patients with detectable ATI had no detectable serum IFX. In a French study utilizing a modified ELISA, 6/10 patients with ATIs responded to dose escalation [50]. In a recent study evaluating the utility of IFX and ADA levels ATIs for prediction of response to intervention in LOR, only high-level antibodies (antibodies-to-adalimumab >4 mcg/mL-eq and antibodies-to-IFX >9 mcg/mL-eq) were 90% specific for failure to respond to dose intensification [51]. This study utilized an AHLC ELISA that permits the simultaneous detection of IFX/ADA and ATI. It is likely that an emergence of ATI is not an “all-or-none” phenomenon but rather a continuous phenomenon, with higher levels associated with a lower risk of a successful dose escalation.
An additional, relatively underexplored therapeutic option is the addition of an immunomodulator in patients on anti-TNF monotherapy. Combination therapy with IFX and AZA in the SONIC study was associated with a superior clinical response, as well as higher 4-week IFX levels and lower prevalence of antibodies [11]. A combination of IFX with methotrexate resulted in a significantly lower prevalence of ATI and a trend for higher serum IFX levels, although without a significant difference in clinical efficacy [12]. In these studies, an immunomodulator was initiated simultaneously with and anti-TNF. In a recent report in CD patients who have developed LOR to IFX accompanied by ATI, the addition of an immunomodulator in patients on monotherapy (AZA in 3 patients and methotrexate in 2 patients) resulted in a gradual restoration of clinical response, decrease in ATI titers and augmentation of IFX levels [52].
When clinical LOR appears in the setting of inadequate anti-TNF levels and low/undetectable ATI, dose escalation is suggested after verification of adherence. No consensus regarding the optimal escalation strategy exists. A pharmacokinetic modeling study in patients with rheumatoid arthritis suggested that interval reduction would provide a more effective drug level AUC compared to the equivalent dose increase [53]. However, retrospective studies comparing dose augmentation with shortening of the interval did not demonstrate any significant difference in the rates of regained response in CD [36,54] or UC [55].
In patients with controlled IBD activity on IFX, dose adjustment can also be beneficial. In the TAXIT study, CD patients in clinical remission were randomized to 2 strategies: 1) dose adjustment to trough levels of 3-7 μg/mL; 2) dose optimization as a response to LOR. Patients on the level-optimized strategy had superior disease control without an increase in a total cost [23].
Thiopurines
Thiopurine medications (6-MP and AZA) have long constituted the mainstay of IBD immunomodulator therapy. A randomized controlled trial in 1980 by Present et al [56] observed a 67% response rate to 6-MP (1.5 mg/kg) in CD. A meta-analysis by Pearson et al [57] reported an odds ratio of 3.09 for response to therapy over placebo. In the more recent meta-analyses conducted for the Cochrane database, a pooled efficacy of 54% and 71% for induction and maintenance of CD remission was reported for AZA [58,59]. Although the data are less robust for UC, AZA was shown to be superior for the maintenance of remission compared to placebo (failure to maintain remission: OR 0.41; 95% CI 0.24 to 0.70) [60]. Thiopurine treatment is associated with a decreased risk of surgery in CD patients [61]. Recent reviews suggest that approximately 50% of all IBD patients are treated with a thiopurine [62,63].
Currently recommended dosing of the thiopurines is 1.5-2.5mg/kg/day for AZA or 0.75-1.5mg/kg/day for 6-MP, according to the European Crohn’s and Colitis Organization guidelines [64]. The American Gastroenterology Association (AGA) guidelines recommend 2.0-3.0 mg/kg/day for AZA and 1-1.5 mg/kg/day for 6-MP [65].
Thiopurine treatment is associated with adverse effects in 15-39% of patients, potentially leading to their discontinuation [63,66]. At least 9% of IBD patients are primary non-responders to thiopurines [67]. In addition, treatment with thiopurines is associated with loss of response over time in some cases. In a recent retrospective study including 363 IBD patients followed up for up to 8 years, the proportion of patients still using thiopurines at 3, 6, 12, 24, and 60 months was 73%, 69%, 63%, 51% and 42%, respectively [68]. The main reported reasons for discontinuation reported were refractoriness and adverse effects.
The therapeutic benefit and toxicity of thiopurines are mediated by the levels of their principal intracellular metabolites (Fig. 2). The purine analogue 6-TGNis the primary biologically active metabolite that is incorporated into cellular nucleic acids, resulting in the inhibition of lymphocyte proliferation and T-cell apoptosis. In addition, 6-TGN was reported to inhibit TNF-related apoptosis-inducing ligand, TNF receptor superfamily member 7, and α4-integrin in activated T-lymphocytes [69]. The slow onset of the action of the (up to 3 months) may be explained by the depletion of antigen-specific memory cells [70]. 6-methylmercaptopurine metabolites (6-MMP) are associated with hepatotoxicity [71], but may also exert an independent anti-proliferative effect on T-lymphocytes [63].
Figure 2.
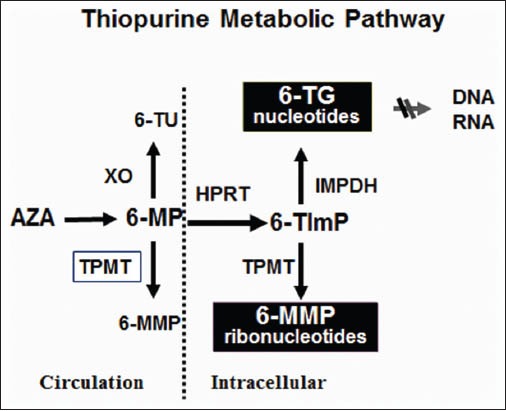
Principal thiopurine metabolic pathways. Azathioprine (AZA) is rapidly converted to 6-mercaptopurine (6-MP) by a non-enzymatic process. 6-MP is subsequently metabolized to immunologically inactive 6-methylmercaptopurine metabolite ribonucleotides (6-MMP) by thiopurine methyltransferase (TPMT). Th e alternative competing pathway is conversion to 6-thioinosine 5-monophosphate (6-TImP) by intracellular hypoxanthineguanine phosphoribosyltransferase (HPRT) and then further enzymatic transformation by 2 separate metabolic pathways to produce either 6-thianoguanine metabolites (6-TGN) through an enzymatic cascade including inosine monophosphate dehydrogenase (IMPDH) and guanosine monophosphate synthase (GMPS) or, alternatively, by TPMT to 6-MMP
Thiopurine metabolism
Once absorbed, AZA is converted to 6-MP by a non-enzymatic pathway. 6-MP can be metabolized through 3 main enzymatic pathways: to 6-thiouric acid (6-TU) by xanthine oxidase (XO), activated to 6-MMP by the key enzyme thiopurine methyltransferase (TMPT), or to the biologically active 6-thioguanine nucleotides (6-TGN) by hypoxanthine phosphoribosyl transferase (HPRT), inosine monophosphate dehydrogenase (IMPDH), and guanosine monophosphate synthetase (GMPS) [72]. The gene for TPMT is located on the short arm of chromosome 6. Several variant alleles (genotypes) have been reported to decrease TPMT activity. These polymorphisms result in a trimodal distribution of TPMT activity in the general population, with absent to very low activity in 0.3% of individuals with a homozygous mutation. Intermediate enzyme activity among heterozygotes is seen in 11% and normal or high activity is observed in about 89%. Patients with low or intermediate TPMT activity phenotypes who are treated with standard doses of AZA or 6-MP are at risk of myelosuppression caused by excess accumulation of 6-TGN [73]. Intermediate or low TPMT activity is most frequently associated with TPMT*2, TPMT*3A or TPMT*3C alleles in Caucasians [73], and TPMT*3C in African-Americans [74].
In patients with homozygous normal phenotype or normal enzymatic activity thiopurines can be initiated at a normal dose, while in heterozygotes or in patients with intermediate enzymatic activity the initial dose should be reduced by 30-70%; in the rare cases of mutant homozygotes with low or absent enzymatic activity, this class of medications should be avoided [75].
Clinical utility of TPMT assessment
TPMT can be assessed either by identification of mutated alleles (genotype) or by measuring the biologic activity (phenotype), usually in erythrocytes [76]. An important limitation of the genotyping is that less frequent mutations mostly relevant to the non-Caucasian population will be missed on standard testing; on the other hand, phenotypical assessment is not reliable (up to 90 days) after a recent blood transfusion [76].
FDA recommendations suggest assessment of TPMT (either genotype or phenotype) before initiation of thiopurines [65]. However, this is not mandated by the European guidelines [64]. TPMT assessment is associated with several benefits in addition to avoiding rare but potentially fatal severe bone marrow suppression [77]. When TPMT genotype/phenotype is unknown, a common practice is to initiate thiopurine treatment at a low dose with a graduate escalation to a therapeutic dosing range. However, this practice is both unsafe and impractical, as it may take up to 6 months to achieve therapeutic metabolite levels. This “start low go slow” strategy is unnecessary in the majority of patients with normal TPMT activity. Guiding the dosing by TPMT and thiopurine metabolite levels will lead to a faster onset of response and reduction of costs [78]. TPMT deficiency explains approximately 25% of all the cases of myelosuppression on thiopurine treatment [60]; it does not account for all the possible factors contributing to the risk of leukopenia and does not preclude the need for continuous blood count monitoring. Moreover, TPMT testing does not prevent long-term myelosuppression that may occur at any time, appearing after one year of treatment in 25% of the patients [79]. In addition, TPMT testing does not predict the risk of idiosyncratic adverse events such as fever, arthralgias, hepatitis or pancreatitis [76]. An association between TPMT activity and response to thiopurines has been suggested. TPMT activity below 35 pmol/h/mg of hemoglobin correlated with a greater chance of clinical response (81% vs. 43%; P<0.001) [81]. In an additional study TPMT activity below 15.3 U/mL RBC is associated with a six fold higher response rate to AZA [82]. The risk of resistance is increased in patients with high TPMT activity (over 14 U/mL RBC (OR, 0.21; 95% CI, 0.06-0.71; P=0.009) [63].
Thiopurine metabolites for monitoring of therapy in IBD
6-TGN levels have been significantly and independently associated with therapeutic response in IBD. The initial assessment of thiopurine metabolites should be performed at least 3weeks after initiation of treatment in patients with normal TPMT activity. Dubinsky and colleagues first reported that patients with 6-TGN level between 235 and 450 pmol/8×108 erythrocytes are 5-times more likely to be in clinical remission in comparison to patients with lower 6-TGN levels; in the same patient cohort, 6-MMP levels above 5,700 pmol/8×108 erythrocytes were associated with a threefold risk of hepatotoxicity [71,80]. A meta-analysis showed that the pooled odds ratio for achieving therapeutic response for a 6-TGN level of >235 pmol/8×108 erythrocytes was 3.3 (95% confidence interval - 1.7-6.3) in comparison to levels <235 (P<0.001)[81]. 6-TGN levels exceeding 450 pmol/8×108 erythrocytes are associated with myelotoxicity [82]. Several additional cut-off 6-TGN values have been proposed to optimize therapeutic response [63]. In patients without clinical response and “subtherapeutic” 6-TGN levels, optimization of the levels was significantly associated with improved response rates [83]. Importantly, there is a poor correlation between thiopurine dose and metabolite levels [72]. In a recent trial comparing conventional weight-based dosing of AZA to individualized strategy aimed at 6-TGN levels of 250-400 pmol/8×108 RBC, the rates at week 16 were 40% in the individualized arm vs. 16% in the weight-based arm [84].
In up to 20% of patients failing therapy, thiopurine metabolism is skewed towards excessive production of 6-MMP (“excessive TPMT”) [76]. In these patients, further escalation of a thiopurine dose frequently results in a disproportional escalation of 6-MMP levels [83], leading to discontinuation of these medications due to lack of efficacy or hepatotoxicity [67]. Prompt identification of this unique subgroup is crucially important for both safety and efficacy considerations, as these patients may still benefit from thiopurines if combined with medications that impact TPMT activity such as allopurinol or 5-aminosalicylates [85,86]. If a thiopurine is combined with allopurinol, the initial dose should be reduced by approximately 65%, and careful complete blood count (CBC) monitoring is absolutely necessary. These patients can be identified by low 6-TGN levels in presence of high 6-MMP levels, with 6-MMP/6-TGN levels usually exceeding 24 [76]. In addition, thiopurine metabolites can easily identify non-compliant patients, characterized by low (<100 pmol/8×108) levels and normal 6-MMP/6-TGN ratio (4-24). A suggested approach to metabolite-guided management of thiopurine therapy is summarized in Table 1.
Table 1.
Th iopurine metabolite levels and ratios help explain therapeutic failures in IBD
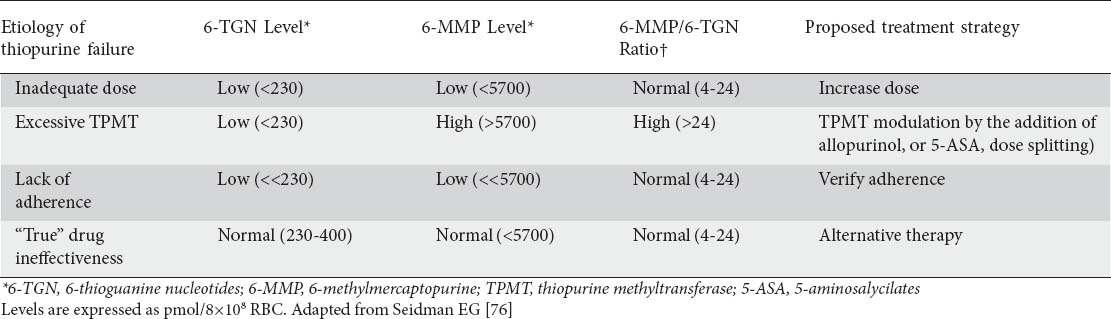
Laboratory monitoring for patients treated with thiopurines
CBC and transaminases should be assessed before onset of treatment and at 2, 4 and 8 weeks after initiating therapy, irrespective of TPMT status. Baseline and follow-up pancreatic enzymes should also be followed, as in some cases elevated amylase and lipase may precede the clinical presentation of pancreatitis [76]. Blood counts should then be repeated every 3 months, or 2 weeks after dose adjustment. Thiopurine metabolite levels can be determined after 2-3 weeks on therapy or after dose adjustment; the levels should be reassessed when facing a loss of response, adverse effect or when a medication with a potential effect on the thiopurine metabolism (such as 5-ASA, allopurinol, furosemide etc) is added. In addition, it is advisable to assess metabolite levels twice yearly for the purposes of routine monitoring and verification of adherence [76].
Concluding remarks
A large body of evidence supports the clinical utility of therapeutic drug monitoring in IBD for patients treated with thiopurines or TNF-α inhibitors. Timely assessment of drug/metabolite levels and anti-drug antibodies may result in an improved clinical outcome and minimization of preventable complications. In cases of loss of response, therapeutic drug monitoring can guide the selection of the appropriate management strategy, combined with clinical, laboratory and endoscopic data.
Biography
McGill University Health Center, McGill University Montreal, QC, Canada; Sheba Medical Center, Tel-Hashomer, Tel-Aviv University, Israel
Footnotes
Conflict of Interest: Uri Kopylov: None; Shomron Ben-Horin: Consultant to Abbot and Schering-Plough and has received unrestricted educational grant from Janssen; Ernest Seidman: Received research support and is a member of the Advisory Board and Speakers Bureau of AbbVie, Janssen Inc., and Prometheus Labs
References
- 1.Ben-Horin S, Chowers Y. Review article: loss of response to anti-TNF treatments in Crohn's disease. Alimen Pharmacol Ther. 2011;33:987–995. doi: 10.1111/j.1365-2036.2011.04612.x. [DOI] [PubMed] [Google Scholar]
- 2.Yanai H, Hanauer SB. Assessing response and loss of response to biological therapies in IBD. Am J Gastroenterol. 2011;106:685–698. doi: 10.1038/ajg.2011.103. [DOI] [PubMed] [Google Scholar]
- 3.Allez M, Karmiris K, Louis E, et al. Report of the ECCO pathogenesis workshop on anti-TNF therapy failures in inflammatory bowel diseases:Definitions, frequency and pharmacological aspects. J Crohns Colitis. 2010;4:355–366. doi: 10.1016/j.crohns.2010.04.004. [DOI] [PubMed] [Google Scholar]
- 4.Gisbert JP, Panes J. Loss of response and requirement of infliximab dose intensification in Crohn's disease: a review. Am J Gastroenterol. 2009;104:760–767. doi: 10.1038/ajg.2008.88. [DOI] [PubMed] [Google Scholar]
- 5.Peters CP, Eshuis EJ, Toxopeus FM, et al. Adalimumab for Crohn's disease: long-term sustained benefit in a population-based cohort of 438 patients. J Crohns Colitis. 2014;8:866–875. doi: 10.1016/j.crohns.2014.01.012. [DOI] [PubMed] [Google Scholar]
- 6.Billioud V, Sandborn WJ, Peyrin-Biroulet L. Loss of response and need for adalimumab dose intensification in Crohn's disease: a systematic review. Am J Gastroenterol. 2011;106:674–684. doi: 10.1038/ajg.2011.60. [DOI] [PubMed] [Google Scholar]
- 7.D’Haens GR, Panaccione R, Higgins PDR, et al. The London position statement of the world congress of Gastroenterology on Biological therapy for IBD with the European Crohn's and Colitis Organization: when to start, when to stop, which drug to choose, and how to predict response. Am J Gastroenterol. 2011;106:199–212. doi: 10.1038/ajg.2010.392. [DOI] [PubMed] [Google Scholar]
- 8.Siegel CA, Melmed GY. Predicting response to anti-TNF agents for the treatment of crohn's disease. Therap Adv Gastroenterol. 2009;2:245–251. doi: 10.1177/1756283X09336364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ben-Horin S, Kopylov U, Chowers Y. Optimizing anti-TNF treatments in inflammatory bowel disease. Autoimmun Rev. 2014;13:24–30. doi: 10.1016/j.autrev.2013.06.002. [DOI] [PubMed] [Google Scholar]
- 10.Ferrante M, Vermeire S, Katsanos KH, et al. Predictors of early response to infliximab in patients with ulcerative colitis. Inflamm Bowel Dis. 2007;13:123–128. doi: 10.1002/ibd.20054. [DOI] [PubMed] [Google Scholar]
- 11.Colombel JF, Sandborn WJ, Reinisch W, et al. Infliximab, azathioprine, or combination therapy for Crohn's disease. N Engl J Med. 2010;362:1383–1395. doi: 10.1056/NEJMoa0904492. [DOI] [PubMed] [Google Scholar]
- 12.Feagan BG, McDonald JW, Panaccione R, et al. Methotrexate in combination with infliximab is no more effective than infliximab alone in patients with Crohn's disease. Gastroenterology. 2014;146:681–688. doi: 10.1053/j.gastro.2013.11.024. e1. [DOI] [PubMed] [Google Scholar]
- 13.Maser EA, Villela R, Silverberg MS, Greenberg GR. Association of trough serum infliximab to clinical outcome after scheduled maintenance treatment for Crohn's disease. Clin Gastroenterol Hepatol. 2006;4:1248–1254. doi: 10.1016/j.cgh.2006.06.025. [DOI] [PubMed] [Google Scholar]
- 14.Seow CH, Newman A, Irwin SP, Steinhart AH, Silverberg MS, Greenberg GR. Trough serum infliximab: a predictive factor of clinical outcome for infliximab treatment in acute ulcerative colitis. Gut. 2010;59:49–54. doi: 10.1136/gut.2009.183095. [DOI] [PubMed] [Google Scholar]
- 15.Baert F, Noman M, Vermeire S, et al. Influence of immunogenicity on the long-term efficacy of infliximab in Crohn's disease. N Engl J Med. 2003;348:601–608. doi: 10.1056/NEJMoa020888. [DOI] [PubMed] [Google Scholar]
- 16.Karmiris K, Paintaud G, Noman M, et al. Influence of trough serum levels and immunogenicity on long-term outcome of adalimumab therapy in Crohn's disease. Gastroenterology. 2009;137:1628–1640. doi: 10.1053/j.gastro.2009.07.062. [DOI] [PubMed] [Google Scholar]
- 17.Colombel JF, Sandborn WJ, Allez M, et al. Association between plasma concentrations of certolizumab pegol and endoscopic outcomes of patients with Crohn's disease. Clin Gastroenterol Hepatol. 2014;12:423–431. doi: 10.1016/j.cgh.2013.10.025. e1. [DOI] [PubMed] [Google Scholar]
- 18.Roblin X, Marotte H, Rinaudo M, et al. Association between pharmacokinetics of adalimumab and mucosal healing in patients with inflammatory bowel diseases. Clin Gastroenterol Hepatol. 2014;12:80–84. doi: 10.1016/j.cgh.2013.07.010. e2. [DOI] [PubMed] [Google Scholar]
- 19.Peyrin-Biroulet L, Reinisch W, Colombel JF, et al. Clinical disease activity, C-reactive protein normalisation and mucosal healing in Crohn's disease in the SONIC trial. Gut. 2014;63:88–95. doi: 10.1136/gutjnl-2013-304984. [DOI] [PubMed] [Google Scholar]
- 20.Moss AC. Therapeutic drug monitoring, mucosal healing, deep remission: the path to nirvana in Crohn's disease? Clin Gastroenterol Hepatol. 2014;12:432–433. doi: 10.1016/j.cgh.2013.12.022. [DOI] [PubMed] [Google Scholar]
- 21.Paul S, Del Tedesco E, Marotte H, et al. Therapeutic drug monitoring of infliximab and mucosal healing in inflammatory bowel disease: a prospective study. Inflamm Bowel Dis. 2013;19:2568–2576. doi: 10.1097/MIB.0b013e3182a77b41. [DOI] [PubMed] [Google Scholar]
- 22.Feagan BG, Singh S, Lockton S, et al. Novel infliximab (IFX) and antibody-to-infliximab (ATI) assays are predictive of disease activity in patients with Crohn's disease (CD) Gastroenterology. 2012;142:S114–S. [Google Scholar]
- 23.Vande Casteele N, Compernolle G, Ballet V, et al. Individualised infliximab treatment using therapeutic drug monitoring: a prospective controlled trough level adapted infliXImab treatment (TAXIT) trial. J Crohns Colitis. 2012;6:S6. [Google Scholar]
- 24.Vaughn BlM-VM, Patwardhan V, et al. Prospective therapeutic drug monitoring to optimizing infliximab (IFX) maintenance therapy in patients with inflammatory bowel disease. Gastroenterology. 2014;146:5. S-54. [Google Scholar]
- 25.Cornillie F, Hanauer SB, Diamond RH, et al. Postinduction serum infliximab trough level and decrease of C-reactive protein level are associated with durable sustained response to infliximab: a retrospective analysis of the ACCENT I trial. Gut. 2014 Mar 4; doi: 10.1136/gutjnl-2012-304094. doi:10.1136/gutjnl-2012-304094. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mazor Y, Kopylov U, Ben Hur D, et al. Evaluating adalimumab drug and antibody levels as predictors of clinical and laboratory response in Crohn's disease patients. Gastroenterology. 2013;144:S778–S. doi: 10.1111/apt.12869. [DOI] [PubMed] [Google Scholar]
- 27.Ben-Horin S, Yavzori M, Katz L, et al. The immunogenic part of infliximab is the F(ab’)(2), but measuring antibodies to the intact infliximab molecule is more clinically useful. Gut. 2011;60:41–48. doi: 10.1136/gut.2009.201533. [DOI] [PubMed] [Google Scholar]
- 28.Yamada A, Sono K, Hosoe N, Takada N, Suzuki Y. Monitoring functional serum antitumor necrosis factor antibody level in Crohn's disease patients who maintained and those who lost response to anti-TNF. Inflamm Bowel Dis. 2010;16:1898–1904. doi: 10.1002/ibd.21259. [DOI] [PubMed] [Google Scholar]
- 29.Rojas JR, Taylor RP, Cunningham MR, et al. Formation, distribution, and elimination of infliximab and anti-infliximab immune complexes in cynomolgus monkeys. J Pharm Exp Ther. 2005;313:578–585. doi: 10.1124/jpet.104.079277. [DOI] [PubMed] [Google Scholar]
- 30.Vermeire S, Noman M, Van Assche G, Baert F, D’Haens G, Rutgeerts P. Effectiveness of concomitant immunosuppressive therapy in suppressing the formation of antibodies to infliximab in Crohn's disease. Gut. 2007;56:1226–1231. doi: 10.1136/gut.2006.099978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ungar B, Chowers Y, Yavzori M, et al. The temporal evolution of antidrug antibodies in patients with inflammatory bowel disease treated with infliximab. Gut. 2013 doi: 10.1136/gutjnl-2013-305259. doi:10.1136/gutjnl-2013-305259. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 32.Vande Casteele N, Gils A, Singh S, et al. Antibody response to infliximab and its impact on pharmacokinetics can be transient. Am J Gastroenterol. 2013;108:962–971. doi: 10.1038/ajg.2013.12. [DOI] [PubMed] [Google Scholar]
- 33.Farrell RJ, Alsahli M, Jeen YT, Falchuk KR, Peppercorn MA, Michetti P. Intravenous hydrocortisone premedication reduces antibodies to infliximab in Crohn's disease: a randomized controlled trial. Gastroenterology. 2003;124:917–924. doi: 10.1053/gast.2003.50145. [DOI] [PubMed] [Google Scholar]
- 34.Vande Casteele N, Buurman DJ, Sturkenboom MG, et al. Detection of infliximab levels and anti-infliximab antibodies: a comparison of three different assays. Aliment Pharmacol Ther. 2012;36:765–771. doi: 10.1111/apt.12030. [DOI] [PubMed] [Google Scholar]
- 35.Kopylov U, Mazor Y, Yavzori M, et al. Clinical utility of antihuman lambda chain-based enzyme-linked immunosorbent assay (ELISA) versus double antigen ELISA for the detection of anti-infliximab antibodies. Inflamm Bowel Dis. 2012;18:1628–1633. doi: 10.1002/ibd.21919. [DOI] [PubMed] [Google Scholar]
- 36.Kopylov U, Mantzaris GJ, Katsanos KH, et al. The efficacy of shortening the dosing interval to once every six weeks in Crohn's patients losing response to maintenance dose of infliximab. Aliment Pharmacol Ther. 2011;33:349–357. doi: 10.1111/j.1365-2036.2010.04523.x. [DOI] [PubMed] [Google Scholar]
- 37.Ungar AA, Yavzori M, Picard O, et al. The clinical and immunological significance of low level of infliximab in the absence of anti-infliximab antibodies in patients with IBD. J Crohns Colitis. 2014;2:s113. [Google Scholar]
- 38.Wang SL, Ohrmund L, Hauenstein S, et al. Development and validation of a homogeneous mobility shift assay for the measurement of infliximab and antibodies-to-infliximab levels in patient serum. J Immunol Methods. 2012;382:177–188. doi: 10.1016/j.jim.2012.06.002. [DOI] [PubMed] [Google Scholar]
- 39.Ainsworth CS, Klaus B, Jørn B, Ole Østergaard T, Mark A. Cut-off levels and diagnostic accuracy of infliximab trough levels and anti-infliximab antibodies in Crohn's disease. Scand J Gastroenterol. 2011;46:310–318. doi: 10.3109/00365521.2010.536254. [DOI] [PubMed] [Google Scholar]
- 40.Steenholdt C, Ainsworth MA, Tovey M, et al. Comparison of techniques for monitoring infliximab and antibodies against infliximab in Crohn's disease. Ther Drug Monit. 2013;35:530–538. doi: 10.1097/FTD.0b013e31828d23c3. [DOI] [PubMed] [Google Scholar]
- 41.Lahiff C, Safaie P, Awais A, et al. The Crohn's disease activity index (CDAI) is similarly elevated in patients with Crohn's disease and in patients with irritable bowel syndrome. Aliment Pharmacol Ther. 2013;37:786–794. doi: 10.1111/apt.12262. [DOI] [PubMed] [Google Scholar]
- 42.Kopylov U, Rosenfeld G, Bressler B, Seidman E. Clinical utility of fecal biomarkers for the diagnosis and management of inflammatory bowel disease. Inflamm Bowel Dis. 2014;20:742–756. doi: 10.1097/01.MIB.0000442681.85545.31. [DOI] [PubMed] [Google Scholar]
- 43.Lopez A, Billioud V, Peyrin-Biroulet C, Peyrin-Biroulet L. Adherence to anti-TNF therapy in inflammatory bowel diseases: a systematic review. Inflamm Bowel Dis. 2013;19:1528–1533. doi: 10.1097/MIB.0b013e31828132cb. [DOI] [PubMed] [Google Scholar]
- 44.Ainsworth MA, Bendtzen K, Brynskov J. Tumor necrosis factor-alpha binding capacity and anti-infliximab antibodies measured by fluid-phase radioimmunoassays as predictors of clinical efficacy of infliximab in Crohn's disease. Am J Gastroenterol. 2008;103:944–948. doi: 10.1111/j.1572-0241.2007.01638.x. [DOI] [PubMed] [Google Scholar]
- 45.Kevans D, Murthy S, Iacono A, Silverberg MS, Greenberg GR. Accelerated clearance of serum infliximab during induction therapy for acute ulcerative colitis is associated with treatment failure. Gastroenterology. 2012:142, S384–S385. [Google Scholar]
- 46.Brandse JF, Wildenberg M, de Bruyn JR, et al. Fecal loss of infliximab as a cause of lack of response in severe inflammatory bowel disease. Gastroenterology. 2013;144:S36. [Google Scholar]
- 47.Gibson D, Heetun Z, Byrne D. An accelerated infliximab dosing strategy for rescue therapy in acute severe colitis is associated with reduced early colectomy rate. (S55-S56).J Crohns Colitis. 2014;8:S1. [Google Scholar]
- 48.Ordas I, Feagan BG, Sandborn WJ. Therapeutic drug monitoring of tumor necrosis factor antagonists in inflammatory bowel disease. Clin Gastroenterol Hepatol. 2012;10:1079–1087. doi: 10.1016/j.cgh.2012.06.032. [DOI] [PubMed] [Google Scholar]
- 49.Afif W, Loftus EV, Jr, Faubion WA, et al. Clinical utility of measuring infliximab and human anti-chimeric antibody concentrations in patients with inflammatory bowel disease. Am J Gastroenterol. 2010;105:1133–1139. doi: 10.1038/ajg.2010.9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Pariente B, de Chambrun GP, Krzysiek R, et al. Trough levels and antibodies to infliximab may not predict response to intensification of infliximab therapy in patients with inflammatory bowel disease. Inflamm Bowel Dis. 2012;18:1199–1206. doi: 10.1002/ibd.21839. [DOI] [PubMed] [Google Scholar]
- 51.Yanai HLL, Assa A, Mazor Y, et al. Anti-TNF and anti-drug antibodies levels to predict the outcomes of interventions for loss of response to adalimumab and infliximab. Gastroenterology. 2014;146:s1. 381. [Google Scholar]
- 52.Ben-Horin S, Waterman M, Kopylov U, et al. Addition of an immunomodulator to infliximab therapy eliminates antidrug antibodies in serum and restores clinical response of patients with inflammatory bowel disease. Clin Gastroenterol Hepatol. 2013;11:444–447. doi: 10.1016/j.cgh.2012.10.020. [DOI] [PubMed] [Google Scholar]
- 53.St Clair EW, Wagner CL, Fasanmade AA, et al. The relationship of serum infliximab concentrations to clinical improvement in rheumatoid arthritis: results from ATTRACT, a multicenter, randomized, double-blind, placebo-controlled trial. Arthritis Rheum. 2002;46:1451–1459. doi: 10.1002/art.10302. [DOI] [PubMed] [Google Scholar]
- 54.Katz L, Gisbert JP, Manoogian B, et al. Doubling the infliximab dose versus halving the infusion intervals in Crohn's disease patients with loss of response. Inflamm Bowel Dis. 2012;18:2026–2033. doi: 10.1002/ibd.22902. [DOI] [PubMed] [Google Scholar]
- 55.Cesarini M, Katsanos K, Papamichael K, et al. Dose optimization is effective in ulcerative colitis patients losing response to infliximab: A collaborative multicentre retrospective study. Dig Liver Dis. 2014;46:135–139. doi: 10.1016/j.dld.2013.10.007. [DOI] [PubMed] [Google Scholar]
- 56.Present DH, Korelitz BI, Wisch N, Glass JL, Sachar DB, Pasternack BS. Treatment of Crohn's disease with 6-mercaptopurine. A long-term, randomized, double-blind study. N Engl J Med. 1980;302:981–987. doi: 10.1056/NEJM198005013021801. [DOI] [PubMed] [Google Scholar]
- 57.Pearson DC, May GR, Fick GH, Sutherland LR. Azathioprine and 6-mercaptopurine in Crohn disease. A meta-analysis. Ann Intern Med. 1995;123:132–142. doi: 10.7326/0003-4819-123-2-199507150-00009. [DOI] [PubMed] [Google Scholar]
- 58.Prefontaine E, Sutherland LR, Macdonald JK, Cepoiu M. Azathioprine or 6-mercaptopurine for maintenance of remission in Crohn's disease. Cochrane Database Syst Rev. 2009:CD000067. doi: 10.1002/14651858.CD000067.pub2. [DOI] [PubMed] [Google Scholar]
- 59.Prefontaine E, Macdonald JK, Sutherland LR. Azathioprine or 6-mercaptopurine for induction of remission in Crohn's disease. Cochrane Database Syst Rev. 2010:CD000545. doi: 10.1002/14651858.CD000545.pub3. [DOI] [PubMed] [Google Scholar]
- 60.Timmer A, McDonald JW, Macdonald JK. Azathioprine and 6-mercaptopurine for maintenance of remission in ulcerative colitis. Cochrane Database Syst Rev. 2007:CD000478. doi: 10.1002/14651858.CD000478.pub2. [DOI] [PubMed] [Google Scholar]
- 61.Chatu S, Subramanian V, Saxena S, Pollok RC. The role of thiopurines in reducing the need for surgical resection in Crohn's disease: a systematic review and meta-analysis. Am J Gastroenterol. 2014;109:23–34. doi: 10.1038/ajg.2013.402. quiz 5. [DOI] [PubMed] [Google Scholar]
- 62.Cosnes J, Gower-Rousseau C, Seksik P, Cortot A. Epidemiology and natural history of inflammatory bowel diseases. Gastroenterology. 2011;140:1785–U118. doi: 10.1053/j.gastro.2011.01.055. [DOI] [PubMed] [Google Scholar]
- 63.Chouchana L, Narjoz C, Beaune P, Loriot MA, Roblin X. Review article: the benefits of pharmacogenetics for improving thiopurine therapy in inflammatory bowel disease. Aliment Pharmacol Ther. 2012;35:15–36. doi: 10.1111/j.1365-2036.2011.04905.x. [DOI] [PubMed] [Google Scholar]
- 64.Dignass A, Van Assche G, Lindsay JO, et al. The second European evidence-based Consensus on the diagnosis and management of Crohn's disease: Current management. J Crohns Colitis. 2010;4:28–62. doi: 10.1016/j.crohns.2009.12.002. [DOI] [PubMed] [Google Scholar]
- 65.Lichtenstein GR, Abreu MT, Cohen R, Tremaine W. American Gastroenterological Association Institute medical position statement on corticosteroids, immunomodulators, and infliximab in inflammatory bowel disease. Gastroenterology. 2006;130:935–939. doi: 10.1053/j.gastro.2006.01.047. [DOI] [PubMed] [Google Scholar]
- 66.Ansari A, Hassan C, Duley J, et al. Thiopurine methyltransferase activity and the use of azathioprine in inflammatory bowel disease. Aliment Pharmacol Ther. 2002;16:1743–1750. doi: 10.1046/j.1365-2036.2002.01353.x. [DOI] [PubMed] [Google Scholar]
- 67.Dubinsky MC. Azathioprine, 6-mercaptopurine in inflammatory bowel disease: pharmacology, efficacy, and safety. Clin Gastroenterol Hepatol. 2004;2:731–743. doi: 10.1016/s1542-3565(04)00344-1. [DOI] [PubMed] [Google Scholar]
- 68.Jharap B, Seinen ML, de Boer NK, et al. Thiopurine therapy in inflammatory bowel disease patients: analyses of two 8-year intercept cohorts. Inflamm Bowel Dis. 2010;16:1541–1549. doi: 10.1002/ibd.21221. [DOI] [PubMed] [Google Scholar]
- 69.Thomas CW, Myhre GM, Tschumper R, et al. Selective inhibition of inflammatory gene expression in activated T lymphocytes: a mechanism of immune suppression by thiopurines. J Pharmacol Exp Ther. 2005;312:537–545. doi: 10.1124/jpet.104.074815. [DOI] [PubMed] [Google Scholar]
- 70.Ben-Horin S, Goldstein I, Fudim E, et al. Early preservation of effector functions followed by eventual T cell memory depletion: a model for the delayed onset of the effect of thiopurines. Gut. 2009;58:396–403. doi: 10.1136/gut.2008.157339. [DOI] [PubMed] [Google Scholar]
- 71.Dubinsky MC, Lamothe S, Yang HY, et al. Pharmacogenomics and metabolite measurement for 6-mercaptopurine therapy in inflammatory bowel disease. Gastroenterology. 2000;118:705–713. doi: 10.1016/s0016-5085(00)70140-5. [DOI] [PubMed] [Google Scholar]
- 72.Bradford K, Shih DQ. Optimizing 6-mercaptopurine and azathioprine therapy in the management of inflammatory bowel disease. World J Gastroenterol. 2011;17:4166–4173. doi: 10.3748/wjg.v17.i37.4166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Weinshilboum RM, Sladek SL. Mercaptopurine pharmacogenetics: monogenic inheritance of erythrocyte thiopurine methyltransferase activity. Am J Hum Genet. 1980;32:651–662. [PMC free article] [PubMed] [Google Scholar]
- 74.Hon YY, Fessing MY, Pui CH, Relling MV, Krynetski EY, Evans WE. Polymorphism of the thiopurine S-methyltransferase gene in African-Americans. Hum Mol Gen. 1999;8:371–376. doi: 10.1093/hmg/8.2.371. [DOI] [PubMed] [Google Scholar]
- 75.Relling MV, Gardner EE, Sandborn WJ, et al. Clinical pharmacogenetics implementation consortium guidelines for thiopurine methyltransferase genotype and thiopurine dosing. Clin Pharmacol Ther. 2011;89:387–391. doi: 10.1038/clpt.2010.320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Seidman EG. Clinical use and practical application of TPMT enzyme and 6-mercaptopurine metabolite monitoring in IBD. Rev Gastroenterol Dis. 2003;3(Suppl 1):S30–S38. [PubMed] [Google Scholar]
- 77.Naughton MA, Battaglia E, O’Brien S, Walport MJ, Botto M. Identification of thiopurine methyltransferase (TPMT) polymorphisms cannot predict myelosuppression in systemic lupus erythematosus patients taking azathioprine. Rheumatology. 1999;38:640–644. doi: 10.1093/rheumatology/38.7.640. [DOI] [PubMed] [Google Scholar]
- 78.Dubinsky MC, Reyes E, Ofman J, Chiou CF, Wade S, Sandborn WJ. A cost-effectiveness analysis of alternative disease management strategies in patients with Crohn's disease treated with azathioprine or 6-mercaptopurine. Am J Gastroenterol. 2005;100:2239–2247. doi: 10.1111/j.1572-0241.2005.41900.x. [DOI] [PubMed] [Google Scholar]
- 79.Frei P, Biedermann L, Nielsen OH, Rogler G. Use of thiopurines in inflammatory bowel disease. World J Gastroenterol. 2013;19:1040–1048. doi: 10.3748/wjg.v19.i7.1040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Cuffari C, Theoret Y, Latour S, Seidman G. 6-Mercaptopurine metabolism in Crohn's disease: correlation with efficacy and toxicity. Gut. 1996;39:401–406. doi: 10.1136/gut.39.3.401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Osterman MT, Kundu R, Lichtenstein GR, Lewis JD. Association of 6-thioguanine nucleotide levels and inflammatory bowel disease activity: a meta-analysis. Gastroenterology. 2006;130:1047–1053. doi: 10.1053/j.gastro.2006.01.046. [DOI] [PubMed] [Google Scholar]
- 82.Lennard L, Van Loon JA, Lilleyman JS, Weinshilboum RM. Thiopurine pharmacogenetics in leukemia: correlation of erythrocyte thiopurine methyltransferase activity and 6-thioguanine nucleotide concentrations. Clin Pharmacol Ther. 1987;41:18–25. doi: 10.1038/clpt.1987.4. [DOI] [PubMed] [Google Scholar]
- 83.Dubinsky MC, Yang H, Hassard PV, et al. 6-MP metabolite profiles provide a biochemical explanation for 6-MP resistance in patients with inflammatory bowel disease. Gastroenterology. 2002;122:904–915. doi: 10.1053/gast.2002.32420. [DOI] [PubMed] [Google Scholar]
- 84.Dassopoulos T, Dubinsky MC, Bentsen JL, et al. Randomised clinical trial: individualised vs. weight-based dosing of azathioprine in Crohn's disease. Aliment Pharmacol Ther. 2014;39:163–175. doi: 10.1111/apt.12555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Leung Y, Sparrow MP, Schwartz M, Hanauer SB. Long term efficacy and safety of allopurinol and azathioprine or 6-mercaptopurine in patients with inflammatory bowel disease. J Crohns Colitis. 2009;3:162–167. doi: 10.1016/j.crohns.2009.02.003. [DOI] [PubMed] [Google Scholar]
- 86.Sparrow MP, Hande SA, Friedman S, Cao D, Hanauer SB. Effect of allopurinol on clinical outcomes in inflammatory bowel disease nonresponders to azathioprine or 6-mercaptopurine. Clin Gastroenterol Hepatol. 2007;5:209–214. doi: 10.1016/j.cgh.2006.11.020. [DOI] [PubMed] [Google Scholar]
- 87.Ben-Horin S, Chowers Y. Tailoring anti-TNF therapy in IBD: drug levels and disease activity. Nat Rev Gastroenterol Hepatol. 2014;11:243–255. doi: 10.1038/nrgastro.2013.253. [DOI] [PubMed] [Google Scholar]