

Abstract
Viral double-stranded RNA (dsRNA) is the most important viral structure recognized by cytosolic pattern-recognition receptors of the innate immune system, and its recognition results in the activation of signaling cascades that stimulate the production of antiviral cytokines and apoptosis of infected cells. 14-3-3 proteins are ubiquitously expressed regulatory molecules that participate in a variety of cellular processes, and 14-3-3 protein-mediated signaling pathways are activated by cytoplasmic dsRNA in human keratinocytes. However, the functional role of 14-3-3 protein-mediated interactions during viral dsRNA stimulation has remained uncharacterized. Here, we used functional proteomics to identify proteins whose phosphorylation and interaction with 14-3-3 is modulated by dsRNA and to characterize the signaling pathways activated during cytosolic dsRNA-induced innate immune response in human HaCaT keratinocytes. Phosphoproteome analysis showed that several MAPK- and immune-response-related signaling pathways were activated after dsRNA stimulation. Interactome analysis identified RelA-associated inhibitor, high-mobility group proteins, and several proteins associated with host responses to viral infection as novel 14-3-3 target proteins. Functional studies showed that RelA-associated inhibitor regulated dsRNA-induced apoptosis and TNF production. Integrated network analyses of proteomic data revealed that sirtuin1 was a central molecule regulated by 14-3-3s during dsRNA stimulation. Further experiments showed that sirtuin 1 negatively regulated dsRNA-induced NFκB transcriptional activity, suppressed expression of antiviral cytokines, and protected cells from apoptosis in dsRNA-stimulated and encephalomyocarditis-virus-infected keratinocytes. In conclusion, our data highlight the importance of 14-3-3 proteins in antiviral responses and identify RelA-associated inhibitor and sirtuin 1 as novel regulators of antiviral innate immune responses.
Keratinocytes are the predominant cell type in the epidermis, the outermost layer of the skin. Although the primary function of these cells is to provide the structural integrity and barrier capability of the epidermis, it is now well accepted that they play an important role in skin inflammatory and immunological reactions (1). During skin infections, viral genomic RNA and its replication intermediates serve as molecular signatures that can be recognized by the specific pattern recognition receptor proteins of the host. The membrane-associated Toll-like receptor 3 (TLR3)1 recognizes viral nucleic acids in the extracellular milieu or within endosomes (2), and RIG-I like receptors (RLRs) are cytoplasmic pattern recognition receptor proteins that can detect cytoplasmic viral double-stranded RNA (dsRNA) (3). The RLR family comprises three receptors, RIG-I, melanoma differentiation-associated gene 5 (MDA5), and laboratory of genetics and physiology 2 (4). RIG-I recognizes 5′-triphosphate RNA and short forms of the synthetic dsRNA analog polyinosic-polycytidylic acid (pI:C), whereas MDA5 is mainly responsible for recognizing longer dsRNA species (5–7). Laboratory of genetics and physiology 2 has also been shown to exert antiviral properties (8). Activated RLRs induce downstream signaling by binding to the mitochondrial antiviral-signaling protein MAVS, an adaptor protein that transmits a signal through serine-kinase signaling cascades leading to the activation of NFκB and interferon regulatory transcription factor 3 transcription factors (9). These transcription factors are essential for the production of type I and type III interferon and other cytokines that initiate antiviral immunity. The recognition of foreign RNA by RLRs also activates programmed cell death, or apoptosis, of virus-infected keratinocytes (10).
The 14-3-3 proteins are a family of acidic regulatory proteins that exist primarily as homodimers and mixed heterodimers within all eukaryotic cells (11). 14-3-3 dimers interact with hundreds of phosphoproteins inside cells, thereby regulating target protein activity, conformation, subcellular distribution, and stability. In most cases, 14-3-3 protein dimers bind to target proteins via pairs of conserved phosphoserine/-threonine binding motifs. Two high-affinity 14-3-3 binding motifs have been described as optimal for 14-3-3 binding to phosphopeptides, namely, RSXpSXP and RXXXpSXP, where pS represents phosphoserine (12). However, variations on these 14-3-3-binding phosphomotifs have been identified in physiological targets (13), and unphosphorylated 14-3-3 binding sites have been identified in proteins that are injected into host cells by pathogenic bacteria (14). Interactome studies of 14-3-3 proteins have demonstrated the importance of 14-3-3 interactions in a variety of cellular processes, including metabolism, protein trafficking, apoptosis, signal transduction, and cell–cycle regulation (15). Recently, 14-3-3s have also been shown to modulate TLR-mediated innate immune response (16–18).
We have previously shown that 14-3-3 signaling pathways are activated by cytoplasmic dsRNA in human keratinocytes (19). However, the functional role of 14-3-3 protein-mediated interactions in RLR-mediated signaling has not been defined. Here, we identified changes in the 14-3-3 interactome and wider phosphoproteome during the cytosolic dsRNA-induced innate immune response, and these changes led us to elucidate 14-3-3-mediated signaling mechanisms that operate during viral infection. Our results underscore the importance of 14-3-3-mediated signaling in host response to viral infection and reveal a novel important role for RelA-associated inhibitor (RAI) and sirtuin 1 in antiviral response.
EXPERIMENTAL PROCEDURES
Cell Culture and Stimulations, Antibodies, and Reagents
Human keratinocyte HaCaT cells (American Type Culture Collection) were cultured in DMEM supplemented with 10% FCS, L-glutamate, and antibiotics. Cells were transfected with a mimetic of dsRNA, pI:C (Sigma-Aldrich), using LipofectamineTM 2000 transfection reagent (Invitrogen) according to the manufacturer's instructions. Control samples were transfected without pI:C. Encephalomyocarditis virus (EMCV) was propagated in L929 cells grown in DMEM with 2% FCS and antibiotics as previously described (19). HaCaT cells were infected with a multiplicity of infection of 1 for 15 h. The following antibodies were used for immunoblotting: p-Erk1/2 (p-p44/42, Thr202/Tyr204), p44/42 MAPK, p-p38 MAPK (Thr180/Tyr182), p38 MAPK, caspase 3, cleaved caspase-3 (Asp175), Bid, ISG15, and IκBα from Cell Signaling Technology (Danvers, MA); and RAI (iASPP, 2808C5a) and SIRT1 (H-300) from Santa Cruz Biotechnology (Heidelberg, Germany). To confirm equal loading and transfer of the protein, membranes were stripped and GADPH (0411, Santa Cruz) was detected.
MAPK inhibitors SB203580 (p38), SP600125 (JNK), and UO126 (MEK1/2 inhibitor that inhibits Erk1/2) were purchased from Sigma and were used at 10 μm final concentration. SIRT1 inhibitor sirtinol (final concentration = 15 μm) and activator SRT1720 (8 μm) were from Sigma. All inhibitors were added to cells 1 h before stimulation.
Phosphoproteomic Analysis and Mass Spectrometry
HaCaT cells were transfected with and without pI:C for 4 h, after which the cells were lysed with HEPES lysis buffer (50 mm HEPES, 150 mm NaCl, 1 mm EDTA, 1% Nonidet P-40, pH 7.4) including protease and phosphatase inhibitor cocktails (Sigma-Aldrich). The cell lysates were centrifuged, and the protein contents were measured using the Bio-Rad DC™ protein assay. The proteins in cell lysates (10 mg per sample) were reduced, alkylated, and digested in-solution with trypsin (Promega, Madison, WI). The undigested proteins and cell debris were removed via centrifugation, and the samples were desalted with Sep-Pak Vac RP C18 cartridges (Waters, Milford, MA). For phosphoproteome analysis, strong cation exchange (SCX) fractionation of the peptides was followed by further phosphopeptide enrichment with immobilized metal affinity chromatography, and the resulting peptides were analyzed via LC-MS/MS as in a previously published protocol (20). The peptides were fractionated by SCX using an ÄKTApurifier™ instrument (Amersham Biosciences). The peptides were separated on a 200 × 4.6 mm, 5-μm, 200-Å PolySULFOETHYL A™ column (PolyLC, Columbia, MD) by a gradient run with increasing salt concentration. The A buffer contained 10 mm KH2PO4, 20% acetonitrile with a pH < 3. The gradient was 0% to 50% buffer B (buffer A + 0.4 m KCl) in 25 min followed by 50% to 100% buffer B in 15 min. The flow rate was 1 ml/min, and 1-ml fractions were collected by an autosampler. The SCX fractions containing the phosphopeptides were collected and desalted. The phosphopeptide enrichment was performed with immobilized metal affinity chromatography using PHOS-Select Iron Affinity Gel (Sigma Aldrich) according to the manufacturer's instructions. The enriched phosphopeptides were analyzed with an Ultimate 3000 nano-LC (Dionex, Thermo Fisher Scientific, San Jose, CA) coupled to a QSTAR Elite hybrid quadrupole TOF-MS (AB Sciex, Framingham, MA) with nano-electrospray ionization. The samples were loaded on a ProteCol C18-Trap column (SGE, Trajan Scientific, Milton Keynes, UK) and separated on a PepMap C18 analytical column (15 cm × 75 μm, 5 μm, 100 Å) (LC Packings/Dionex) at 200 nl/min with a linear gradient of 0% to 40% acetonitrile in 120 min. The MS data were acquired with Analyst QS 2.0 software. The information-dependent acquisition method consisted of a 0.5-s TOF-MS survey scan of m/z 400–1400. From every survey scan, the two most abundant ions with charge states of +2 to +4 were selected for product ion scans, and each selected target ion was dynamically excluded for 60 s. Smart IDA (information dependent acquisition) was activated with automatic collision energy and automatic MS/MS accumulation. Three independent biological experiments were performed.
14-3-3 Capture and iTRAQ Labeling
HaCaT cells (∼100 × 106 cells) were left untreated or transfected with pI:C for 4 to 5 h, after which the cells were extracted in 5 ml of lysis buffer containing 50 mm Tris, 1% Tx-100, 350 mm NaCl, 1 mm EDTA, and 50 mm NaF supplemented with Protease Inhibitor Mixture and Phosphatase Inhibitor Mixture 1 and 3 (Sigma). The clarified solutions were pre-cleared with blocked NHS-activated CH-Sepharose 4B (GE Healthcare) for 1 h at 4 °C. Purified BMH1 and BMH2 were coupled to Sepharose (21), and HaCaT cell lysates were mixed with the affinity matrix for 1 h at 4 °C. As a control, we used BSA-coupled Sepharose. The mixture was washed extensively with 500 mm NaCl, and proteins that bound to the phosphopeptide-binding site of 14-3-3s were eluted with 1 mm ARAApSAPA phosphopeptide (22). The eluted phosphoproteins were concentrated to 200 μl with Amicon-4 centrifugal filter units (cutoff: 3 kDa) and precipitated with a 2-D Clean-Up Kit (GE Healthcare).
The 14-3-3-affinity-purified proteins were resuspended with iTRAQ dissolution buffer and then subjected to protein alkylation, trypsin digestion, and iTRAQ labeling of the resulting peptides according to the manufacturer's instructions (Applied Biosystems). After labeling, the samples were pooled and dried, and the peptides were fractionated by SCX chromatography. Each SCX fraction containing labeled peptides was analyzed twice with LC-MS/MS as described with phosphoproteomic analysis. MS data were acquired automatically using Analyst QS 2.0 software. Three independent biological experiments with two technical replicates were performed.
Proteomics Data Analysis
The phosphopeptide identification was performed through the ProteinPilot 4.0 interface (Applied Biosystems/MDS Sciex) using an in-house Mascot database search engine (version 4.0, Matrix Science, London, UK) (23) and the ProteinPilot 4.0 algorithm Paragon (24). The data were searched against human canonical sequences in the UniProt database (version 08/2012 with 20,232 human sequences). The criteria for the Paragon search were cysteine alkylation with iodoacetamide, trypsin digestion, biological modifications, and phosphorylation emphasis allowed, with a thorough search and detected protein threshold of 95% confidence (unused ProtScore > 1.3), and the criteria for the Mascot search were cysteine alkylation with iodoacetamide, trypsin digestion, methionine oxidation, and serine/threonine/tyrosine phosphorylation allowed; identification threshold p < 0.05; one missed cleavage allowed; mass tolerance for precursor ions of 50 ppm; and mass tolerance for fragment ions of 0.2 Da. The final list of phosphopeptides was generated using an in-house-developed software tool, PhosFox,2 using a cutoff score value of 30 for Mascot peptide ion score and 80 for Paragon peptide confidence. The false discovery rates were determined for the identifications and calculated using the target-decoy strategy with concatenated normal and reversed sequence databases (25) and were less than 1%. The phosphoproteomics data have been deposited to the ProteomeXchange Consortium (www.proteomexchange.org) via the partner repository (26) with the dataset identifier PXD000670.
Protein identification and relative quantitation of 14-3-3 interacting proteins were performed using the ProteinPilot 4.0 interface (Applied Biosystems/MDS Sciex) with the Paragon algorithm. Data files from both technical replicates of an iTRAQ sample set were processed together. Database searching was done against UniProt human protein sequences (version 08/2012 with 20,232 human sequences). The search criteria were cysteine alkylation with methyl methanethiosulfonate, trypsin digestion, biological modifications allowed, and thorough search and detected protein threshold of 95% confidence (unused ProtScore > 1.3). Automatic bias correction was used in quantitation. The false discovery rates were less than 1% for all three biological replicates. The original iTRAQ data have been deposited to the ProteomeXchange Consortium with the dataset identifier PXD000677. Results from three independent biological replicates were combined. Only proteins showing significant (p value ≤ 0.05 for at least one of the biological replicates) and reproducible (showing similar modulation in the biological replicates) differences between control and pI:C-treated samples were considered. The fold-change used for altered binding was 1.3; it was chosen based on a study by Keller et al. (27), who showed that the lowest iTRAQ quantification ratio they could confirm via Western blotting to be differentially regulated was fold-change 1.220, and also because iTRAQ is known to underestimate the fold change (reviewed in Ref. 28). The 14-3-3 interacting proteins identified were classified based on their Gene Ontology annotations using GeneTrail (29) with UniProt as the data scource and Homo sapiens as the organism. Canonical pathway analysis of phosphoprotein datasets and network analysis of 14-3-3 interacting proteins were performed with Ingenuity Pathway Analysis (IPA).
Lactate Dehydrogenase Cytotoxicity Assay
A lactate dehydrogenase (LDH) assay was used to quantify LDH release into cell culture media during cell death. The LDH assay was performed according to the manufacturer's instructions (Roche Diagnostics GmbH). The experiments were done twice, with similar results.
RT-PCR
Total cellular RNA was isolated using the RNeasy Plus Mini Kit (Qiagen, Hilden, Germany) and was reverse transcribed using the High Capacity cDNA Reverse Transcription Kit (Applied Biosystems) according to the manufacturer's instructions. Quantitative real-time PCR was performed with an ABI PRISM 7500 Sequence Detection System and Applied Biosystems 7500 Fast Real-Time PCR System applying TaqMan chemistry including predeveloped TaqMan assay primers and probes (18S (endogenous control): Hs99999901_s1; TNF: Hs99999043_m1; IFNβ: Hs01077958_s1; IL-29: Hs00601677_g1; NFKB1A: Hs00355671_g1) TaqMan® Fast Advanced Master Mix (Applied Biosystems). The RT-PCR data were processed and quantified as described before (19). The results are expressed as relative units. The experiments were done twice, with similar results.
siRNA Silencing
HaCaT cells were reverse transfected with 100 nm non-targeting control siRNA (AllStars Negative Control siRNA, Qiagen) and with 50 nmol of each of two different siRNAs (Hs_SIRT1_1/Hs_SIRT1_2 or Hs_PPP1R13L_8/Hs_PPP1R13L_10) using HiPerFect Transfection Reagent (Qiagen) according to the manufacturer's instructions. Cells were stimulated 24 h after siRNA silencing.
Statistical Analysis
Data are presented as the mean ± S.E. Student's t test was used in the statistical analysis with the significance threshold set at p ≤ 0.05.
RESULTS
Phosphoproteomic Analysis Reveals That Several MAPK and Immune-response-related Signaling Pathways Are Activated after dsRNA Stimulation in HaCaT Keratinocytes
In the present study, we used phosphoproteomics combined with quantitative 14-3-3-affinity capture, bioinformatics, and functional studies to define phosphorylation- and 14-3-3-protein-dependent signaling events regulated during RLR activation in human keratinocytes (Fig. 1A). First, the protein expression of the key components of the RLR signaling pathway was assessed in untreated and dsRNA-stimulated HaCaT keratinocytes (Fig. 1B). This showed that RIG-I was weakly expressed in untreated cells and up-regulated by dsRNA, whereas MDA5 was constitutively expressed. In contrast, the amount of MAVS decreased during stimulation. Next, the general phosphorylation events in dsRNA-induced HaCaT keratinocytes were characterized using phosphoproteomic analysis. Cells were transfected with pI:C, a synthetic analog of viral dsRNA, which is widely used to mimic viral infection via activation of RLRs. As a negative control, transfection was done with lipofectamine alone. After 4 h of transfection, cells were lysed, proteins were digested with trypsin, phosphopeptides were enriched from lysates using SCX and immobilized metal affinity chromatography, and peptides were analyzed via LC-MS/MS. Phosphoproteomic analysis was performed on results from three independent biological experiments, and the detailed phosphopeptide and protein identification results for each biological replicate are shown in supplemental Fig. S1 and supplemental Table S1. We identified a total of 1614 phosphoproteins with 5380 phosphorylated peptides and 5214 distinct phosphorylation sites (Fig. 2A). Of these phosphorylation sites, 668 were previously unreported in UniProt, PHOSIDA, and PhosphoSite Plus databases. Peptide-level data showed that 1641 phosphopeptides were uniquely identified in the control sample, and 1391 in the pI:C sample, corresponding to 850 and 758 unique phosphoproteins, respectively (Fig. 2B, supplemental Table S1). The ratio of phosphoamino acids was similar in both samples—85% pSer, 14% pThr, and <1% pTyr—and was in accordance with the literature (30).
Fig. 1.
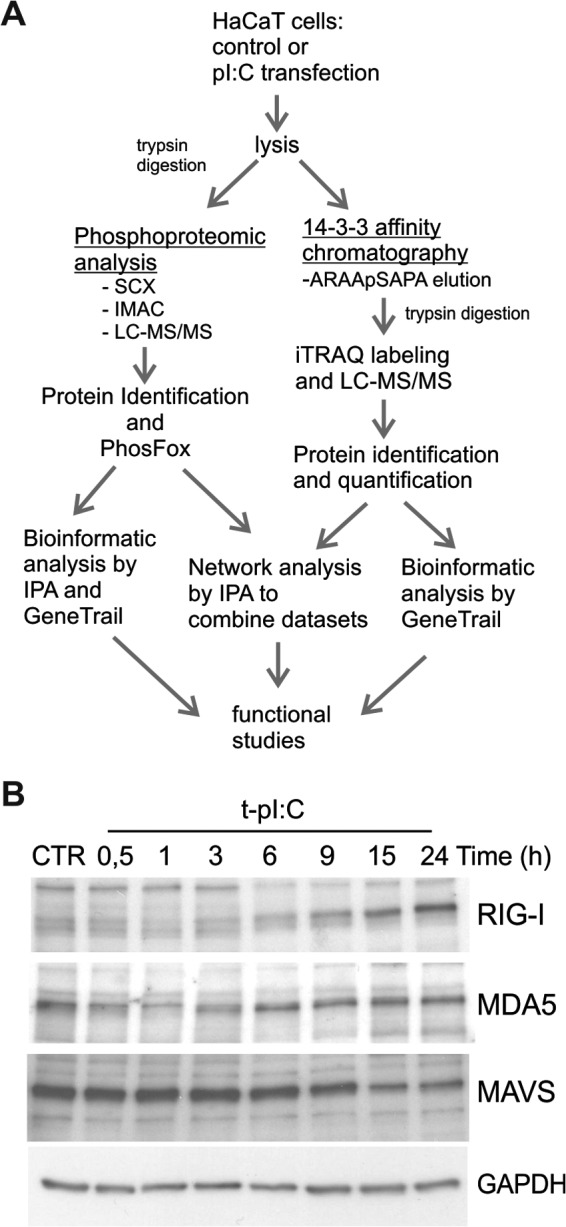
Experimental workflow to identify proteins whose phosphorylation and interaction with 14-3-3 proteins is modulated by dsRNA in human keratinocytes. A, samples were analyzed using phosphoproteomics and quantitative 14-3-3 affinity chromatography, and bioinformatic analyses were done with Ingenuity Pathway Analysis (IPA) and GeneTrail software. Details of experiments are described in “Experimental Procedures.” B, HaCaT keratinocytes were transfected with pI:C for the indicated times, and the expression of RIG-I, MDA5, and MAVS was analyzed from cell lysates via immunoblotting. GAPDH detection was used to confirm the equal loading.
Fig. 2.

Phosphoproteome analysis of dsRNA-stimulated human keratinocytes. A, identified phosphoproteins and phosphopeptides and distinct phosphorylation sites. B, the distributions of all identified phosphopeptides (upper panel) and uniquely phosphorylated proteins (lower panel) in control and dsRNA-stimulated samples. C, most significant biological functions of uniquely phosphorylated proteins in control (850) and dsRNA-induced (758) samples were determined using IPA. D, selected canonical pathways activated during viral dsRNA stimulation (complete list in supplemental Table S2). IPA analysis was done with the 419 uniquely phosphorylated proteins identified only after pI:C transfection. Highly similar canonical pathways were identified when the phosphoproteins identified in at least two out of three biological replicates were taken into analysis (supplemental Table S2). E, HaCaT keratinocytes were transfected with pI:C for the indicated times, after which cell lysates were subjected to SDS-PAGE and immunoblotting using antibodies against phosphorylated p38 MAPK (Thr180/Tyr182), total p38, phosphorylated Erk1/2 (Thr202/Tyr204), total Erk1/2, or IκBα. GAPDH detection was used to confirm the equal loading.
To understand the biological function of the identified proteins, we performed core analysis of the uniquely phosphorylated proteins using the IPA software. The proteins whose phosphopeptides were uniquely identified in control cells were involved in basic cellular functions, such as gene expression, cellular maintenance, cell cycle, and cellular movement (Fig. 2C). In addition, IPA analysis indicated that phosphopeptides identified only after dsRNA stimulation included proteins involved in cell death and survival (especially apoptosis), cell signaling (especially protein kinase cascades and the NFκB pathway), and post-translational modifications. Moreover, IPA analysis of the phosphoproteome data showed that multiple canonical signaling pathways were activated during infection (Fig. 2D, supplemental Table S2). The most relevant dsRNA-induced signaling pathway was Erk/MAPK signaling. The data were also consistent with the activation of other MAPK pathways, ERK5, p38, and JNK signaling, known to respond to dsRNA stimulation. In addition, the data indicate that dsRNA modulates signaling related to cell morphology and motility, including regulation of Rho GTPases, ILK, FAK, actin cytoskeleton, and cell junctions. Furthermore, signaling related to calcium function, cell cycle, and the endocrine system was represented in the phosphopeptides identified during dsRNA stimulation. Importantly, several signaling pathways involved in immune response were also represented, including interleukin-1 (IL-1) signaling, NFκB signaling, and apoptosis.
Activation of Erk and p38 MAPKs and NFκB signaling were confirmed by Western blotting (Fig. 2E). The Erk1/2 pathway was activated already at 3 h after dsRNA transfection, whereas p38 activation was seen at 6 h after stimulation. The degradation of the inhibitory IκB subunit of the NFκB complex was apparent at 1 h after dsRNA stimulation, demonstrating early activation of NFκB.
p38 and JNK Pathways Are the Major MAPK Signaling Pathways Involved in dsRNA-induced Innate Immune Response in Human Keratinocytes
MAPK family members are responsible for signaling that regulates cell proliferation, differentiation, apoptosis, and immune response (31). To determine the specific roles of different MAPKs in antiviral innate immune response in human keratinocytes, we studied the effects of specific inhibitors of the respective MAPK pathways on cytokine expression and apoptosis. HaCaT cells were treated with SP600125, SB203580, and UO126, which inhibit JNK, p38, and the Erk1/2 activators MEK1 and -2, respectively, and cells were subsequently transfected with dsRNA. MAPK inhibitors had only marginal effects on RIG-I, MDA5, and MAVS expression, indicating that the possible effect of MAPKs is on downstream components of the RLR signaling pathway (supplemental Fig. S2A). Inhibition of p38 resulted in a statistically significant reduction in the expression of the pro-inflammatory cytokine TNF, whereas the JNK inhibitor had a minor opposite effect on TNF expression (Fig. 3A). Similarly, the expression of the type I and III interferons IFNβ and IL-29 was reduced by p38 inhibition. In contrast, JNK inhibitor had no effect on IFNβ mRNA expression. To further analyze the IFN response, we monitored the expression levels of a well-known IFN-inducible protein, interferon-stimulated ubiquitin-like protein ISG15, from cell lysate 17 h after pI:C transfection, in the presence and absence of MAPK pathway inhibitors. Similarly, both JNK and p38 inhibitors reduced ISG15 protein expression, although the reduction was more pronounced with the p38 inhibitor than the JNK inhibitor (Fig. 3B). It has been shown previously that type III IFNs, including IL-29, can activate ISG15 gene expression (32), and it might be that the decrease in ISG15 expression upon JNK inhibition is due to the decrease in IL-29 expression. In addition, these results showed that Erk1/2 activation is not required for dsRNA-induced expression of antiviral cytokines.
Fig. 3.

p38 and JNK pathways are the major MAPK signaling pathways involved in dsRNA-induced innate immune response in human keratinocytes. A, HaCaT keratinocytes were pretreated with MAPK inhibitors 1 h before stimulation with cytoplasmic pI:C for 5 h, and the effect of inhibitors on cytokine expression was detected using quantitative RT-PCR. SB203580 = p38 inhibitor, SP600125 = JNK inhibitor, and UO126 = MEK1/2 inhibitor that therefore inhibits Erk1/2 activation. Comparable data were obtained from two separate experiments including six samples. *p < 0.05 versus pI:C-transfected DMSO-sample. B, cells were activated overnight in the presence or absence of MAPK inhibitors, and the expression of ISG15 was detected. C, the effect of MAPK inhibitors on dsRNA-induced cell death was studied by analyzing the formation of cleaved caspase-3 in HaCaT cells after 17 h of stimulation. Silver stained gel was used to confirm the equal loading. D, LDH cytotoxicity assay was performed after 8 h of pI:C stimulation in the presence of MAPK inhibitors. The result is representative of two separate experiments with four independent samples.
Next we studied the role of MAPK pathways in dsRNA-induced programmed cell death in HaCaT cells. Cell were transfected with pI:C for 17 in the presence and absence of MAPK inhibitors, and the expression of procaspase 3 and biologically active p19/17 fragments of caspase 3 was analyzed with Western blotting. JNK, p38, and Erk1/2 inhibitors had little effect on procaspase-3 expression (Fig. 3C). Transfection of the cells with pI:C induced proteolytical processing of procaspase-3 into the biologically active p19/17 forms of caspase-3, indicating activation of apoptosis. JNK and p38 inhibitors markedly decreased the appearance of p19/17 forms of caspase-3, whereas Erk1/2 inhibitor had no influence on caspase-3 activation (Fig. 3C and supplemental Fig. S3). In addition, we performed an LDH cytotoxicity assay to analyze the effect of MAPK inhibitors on dsRNA-induced cell death in HaCaT keratinocytes (Fig. 3D). JNK inhibitor almost completely abolished LDH release in response to dsRNA stimulation, and also p38 inhibitor reduced LDH release in dsRNA-activated cells. In contrast, Erk1/2 inhibitor had no effect on dsRNA-induced cell death. In conclusion, our results suggest that the p38 pathway is the major MAPK signaling component regulating dsRNA-induced cytokine production, whereas both p38 and JNK pathways have a key role in dsRNA-activated apoptosis and cell death in human keratinocytes.
Interactome Studies Reveal RAI, HMGs, and Several Proteins Associated with Host Response to Viral Infection as Novel 14-3-3 Target Proteins
We have previously shown that phosphorylation of 14-3-3 target proteins is increased by dsRNA stimulation in human keratinocytes (19). In the current study we performed quantitative 14-3-3 protein affinity capture to identify 14-3-3 interacting proteins whose binding to 14-3-3 proteins changes after dsRNA stimulation. HaCaT cells were transfected with dsRNA for 4 h or left untreated, and phosphorylated 14-3-3 target proteins were purified from cell lysate using 14-3-3 affinity chromatography. Proteins were eluted with a 14-3-3-binding phosphopeptide to selectively purify those proteins that are bound to the phosphopeptide-binding site on the 14-3-3s. The eluted proteins were analyzed by means of 4plex iTRAQ labeling (33) combined with LC-MS/MS analysis as previously described (34). iTRAQ analysis was performed on three independent biological experiments. We could identify and quantify 646 distinct proteins with high confidence (supplemental Table S3), of which 209 of displayed altered binding to 14-3-3s after dsRNA transfection: 147 were more abundant (fold change > 1.3) and 62 were less abundant (fold change < 0.77) in the 14-3-3-affinity captured protein pool (Fig. 4A). Of the identified 14-3-3 targets, 272 proteins (42%) have not been previously identified as 14-3-3 interacting proteins (15, 31). Parallel preparations using BSA-column showed only modest background (supplemental Table S4), and only three proteins—serum album, cytokeratin 10, and cytokeratin 2—were more abundant in the eluate from BSA-column than from the 14-3-3 column.
Fig. 4.

Quantitative 14-3-3 protein capture identified RAI as a novel 14-3-3 target protein that regulates dsRNA-induced apoptosis and TNF production. A, 646 distinct 14-3-3 target proteins were identified and quantified from HaCaT cell lysate using quantitative 14-3-3 protein capture, and of these 209 proteins had changed affinity to 14-3-3s after dsRNA transfection (left panel). The proteins whose 14-3-3 capture was altered by dsRNA were classified based on biological function using GeneTrail (right panel). B, top 10 proteins whose binding to 14-3-3 was increased most after dsRNA stimulation. Access no. = Swiss-Prot access number; FC = fold change in 14-3-3 binding relative to control. “Published” column shows whether 14-3-3 interaction had been published previously. C, HaCaT keratinocytes were transfected with pI:C for the indicated times, and the expression of RAI was analyzed from cell lysates via immunoblotting. The size of the intact RAI protein is approximately 90 kDa. D, the cells were transfected with control siRNA and RAI (gene name PPP1R13L) specific siRNA molecules for 24 h before stimulation with dsRNA for 5 h, after which the expression of RAI was detected via immunoblotting. The effect on RAI silencing on dsRNA-induced caspase-3 activation, tBid, and IκBα expression was detected. GAPDH detection was used to confirm the equal loading. E, the effect of RAI silencing on dsRNA-induced TNF cytokine expression was detected using quantitative RT-PCR. Comparable data were obtained from two separate experiments. *p < 0.05 versus pI:C-transfected ctr-siRNA-sample.
14-3-3 target proteins having altered affinity to 14-3-3 were classified according to biological function using the GeneTrail tool. The functions with the most obvious relevance to innate immune responses were intracellular transport, immune system, cytoskeleton organization, apoptosis, and oxidation-reduction (Fig. 4A and supplemental Table S5). The proteins whose binding to 14-3-3 proteins was increased the most after dsRNA stimulation included structural proteins such as keratins and tubulins, which are reported 14-3-3 target proteins (Fig. 4B). RAI (also known as PPP1R13L or iASPP) was previously unidentified as a 14-3-3 target, but here its binding to 14-3-3s increased more than 4-fold during dsRNA stimulation. Our phosphoproteomic data show that RAI had five phosphorylated sites that were uniquely identified after dsRNA stimulation (supplemental Table S1), and one of these, phosphoSer102, conformed to a potential recognition motif for 14-3-3 proteins. This indicates that RAI is a novel target of 14-3-3 regulation. Another interesting new 14-3-3 target protein family identified comprised HMGs. HMGs are universal sensors of nucleic acids and are required for the induction of transmembrane and cytoplasmic receptor-mediated innate immune responses. We identified four members of this family—HMGB1, HMGB2, HMGB3, and HMGA1—as novel interactors with 14-3-3s. Furthermore, several proteins that are directly linked to virus response were also identified as 14-3-3 interactors, including stathmin, IL-1 receptor antagonist protein, and ubiquitin-like protein ISG15, suggesting that 14-3-3 proteins participate in antiviral innate immune response.
RAI Regulates dsRNA-induced Apoptosis and TNF Production
To study the regulation of RAI expression in response to dsRNA stimulation, we performed Western blot analyses with anti-RAI antibody (Fig. 4C). dsRNA stimulation induced a marked decrease in the amount of RAI protein expression, indicating that increased binding to 14-3-3s is not due to increased protein expression. RAI has been shown to have a central role in the regulation of apoptosis and NFκB signaling, which are also activated by RLRs, via its interaction with p53 proteins and NFκB subunit p65RelA (35, 36). The NFκB complex plays a key role in regulating cell survival and cytokine production. To determine the contribution of RAI in dsRNA-induced apoptosis, we decreased the expression of endogenous RAI protein with siRNA (Fig. 4D). The decrease in RAI expression did not induce apoptosis in untreated cells, but it significantly increased caspase-3 activation after 5 h of stimulation with dsRNA. In addition, processing of Bid to apoptotic tBid after 5 h of stimulation was enhanced in RAI-depleted cells (Fig. 4D), further suggesting that RAI negatively regulates dsRNA-induced apoptosis. We also analyzed whether silencing of RAI had an effect on RLR expression and found that RAI depletion had no effect on RIG-I, MDA-5, or MAVS expression (supplemental Fig. S2B).
Next, we examined whether RAI regulates dsRNA-induced NFκB signaling. HaCaT keratinocytes were transfected with RAI siRNA, and degradation of NFκB inhibitory IκBα protein was measured via immunoblotting. In untreated cells, siRNA silencing of RAI markedly increased the degradation of IκBα, indicating NFκB activation (Fig. 4D). However, RAI depletion had no effect on NFκB signaling after dsRNA stimulation. These results suggest that RAI inhibits dsRNA-induced apoptosis independently of NFκB signaling. Because NFκB signaling also contributes to the production of interferons and other cytokines that initiate antiviral immunity, we investigated the effect on RAI silencing on cytokine production. RAI depletion had no effect on dsRNA-induced interferon production (data not shown). Interestingly, however, reduced RAI expression inhibited the expression of pro-inflammatory cytokine TNF in dsRNA-stimulated cells (Fig. 4E), suggesting a regulatory role for RAI in cytokine response.
SIRT1 Is a Central Molecule Regulated by 14-3-3 Proteins and Negatively Regulates dsRNA-induced NFβκB Transcriptional Activity in Human Keratinocytes
We then performed network analysis on the 14-3-3-affinity-captured proteins and combined the results from phosphoproteomic analysis with this network to pinpoint key molecules regulated by 14-3-3 upon dsRNA stimulation. The most significant network of 14-3-3 interacting proteins was related to cell death and survival, cell-to-cell signaling and interaction, and cell morphology (Fig. 5A). In this network, the main targets of 14-3-3-mediated signaling are TNF and NFκB signaling, which have also previously been described in TLR-induced immune response (16–18). The network analysis indicated a central position for sirtuin 1 (SIRT1) in a 14-3-3-regulated TNF–NFκB subnetwork. SIRT1 is an NAD+-dependent deacetylase that participates in the coordination of several separate cellular functions such as cell cycle, response to DNA damage, metabolism, apoptosis, and autophagy (37). It has mainly been linked to neuronal development, but recent studies have also revealed the possibility that SIRT1 has a role in immune response (38). However, no role for SIRT1 in RLR-mediated signaling and viral infection had been defined.
Fig. 5.

SIRT1 negatively regulates dsRNA-induced NFkB transcriptional activity in human keratinocytes. A, the most significant network of 14-3-3 interacting proteins was related to cell death and survival, cell-to-cell signaling and interaction, and cell morphology. Molecules marked with red color have altered affinity to 14-3-3 proteins after dsRNA stimulation, and molecules with red circles are uniquely phosphorylated after dsRNA stimulation. Network analysis of 14-3-3 interaction proteins was made using IPA. B, HaCaT keratinocytes were transfected with pI:C for the indicated times, and the expression of SIRT1 was detected from cell lysate via immunoblotting. C, cells were transfected with control siRNA and SIRT1 specific siRNA molecules for 24 h before stimulation with cytoplasmic pI:C for 5 h, after which the expression of SIRT1 was detected. Silver stained gel was used to confirm the equal loading. D, E, cells were stimulated with cytoplasmic dsRNA for 5 h, and the effect of SIRT1 silencing on NFκB signaling was determined. Quantitative RT-PCR was used to measure the expression of NFκB1A mRNA (D), and immunoblotting was used to detect the degradation of inhibitor subunit IκBα (E). Quantitative RT-PCR data were obtained from two separate experiments. *p < 0.05 versus pI:C-transfected ctr-siRNA sample.
We therefore examined the role of SIRT1 dsRNA-induced innate immune response in human keratinocytes. First, we studied the expression of SIRT1 protein in HaCaT cells. Interestingly, stimulation of the cells with dsRNA decreased SIRT1 protein levels in a time-dependent manner (Fig. 5B). Because the p65RelA subunit of the NFκB complex is one of several nonhistone substrates of the lysine deacetylase activity of SIRT1 (39), we next investigated whether SIRT1 was essential for dsRNA-induced NFκB signaling. The NFκB complex is a heterodimeric protein composed of different combinations of members of the Rel family of transcription factors, including the inhibitory IκB protein, which inactivates NFκB by masking the nuclear localization signals. Activation of NFκB occurs via degradation of IκB. In addition, NFκB activation turns on IκB gene (NFKB1A) expression, forming a negative feedback loop. We used siRNA knockdown to decrease the expression of endogenous SIRT1 protein (Fig. 5C). We found that SIRT1 silencing strongly enhanced the dsRNA-induced degradation of IκB protein (Fig. 5C) and significantly increased the expression level of NFKB1A mRNA (Fig. 5D), indicating that SIRT1 negatively regulates dsRNA-induced NFκB activity in human keratinocytes. We also determined whether SIRT1 regulates the expression of CCL5 gene (Rantes), which is a well-known target of NFκB regulation. CCL5 expression was significantly increased in SIRT1-depleted cells (Fig. 5E), further suggesting that SIRT1 plays an important role in dsRNA-induced innate immune response. We also analyzed whether silencing of SIRT1 has an influence on RLR expression. Decreased SIRT1 protein expression had no effect on RIG-I, MDA5, or MAVS expression (supplemental Fig. S2B).
SIRT1 Suppresses Cytokine Production and Protects Cells from Apoptosis in dsRNA-stimulated and EMCV-infected Keratinocytes
Previous studies have demonstrated that SIRT1 suppresses bacterial lipopolysaccharide-induced inflammatory signaling and the expression of pro-inflammatory cytokines such as TNF (40, 41). Because TNF is also a target of NFκB regulation, we decided to elucidate whether SIRT1 regulates the TNF response upon dsRNA stimulation. SIRT1 silencing clearly enhanced dsRNA-induced expression of TNF (Fig. 6A), which is consistent with earlier studies. SIRT1 silencing also markedly increased the expression of the antiviral cytokines IFNβ and IL-29 (Fig. 6A). Next, we studied the effect of SIRT1 silencing on cytokine production in response to EMCV, which activates the same RLR MDA-5 as cytosolic dsRNA. Similar to dsRNA stimulation, SIRT1 silencing increased the mRNA expression of TNF, IFNβ, and IL-29 in EMCV-infected keratinocytes (Fig. 6B), confirming a negative regulatory role for SIRT1 in virus-induced cytokine production.
Fig. 6.

SIRT1 suppresses virus-induced cytokine production and protects cells from dsRNA-induced apoptosis. HaCaT keratinocytes were transfected with control siRNA and SIRT1 specific siRNA molecules. After 24 h of silencing, the cells were transfected with pI:C for 5 h (A) or infected with EMCV for 15 h (B), and the effect of SIRT1 silencing on induced cytokine expression was detected using quantitative RT-PCR. Comparable data were obtained from two separate experiments. *p < 0.05 versus pI:C-transfected or EMCV-infected ctr-siRNA sample. C, the effect of SIRT1 silencing on dsRNA-induced caspase-3 activation was detected via Western blotting. GAPDH detection was used to confirm the equal loading. HaCaT cells were pretreated with SIRT1 inhibitor (Sirtinol) or activator (SRT1720) for 1 h before stimulation with cytoplasmic pI:C for 17 h (D) or infection with EMCV for 15 h (E). The formation of the active form of caspase-3 and tBid was analyzed via Western blotting. Silver stained gel and GAPDH detection were used to confirm the equal loading.
The role of sirtuins in cell death and survival cannot be easily defined, because the ability of sirtuins to induce either apoptosis or cell survival depends on the apoptotic stimuli and cell type (39). To define the role of SIRT1 in dsRNA-induced apoptosis in human keratinocytes, we silenced the expression of SIRT1 and measured caspase-3 activity. SIRT1 silencing increased caspase-3 activation at 6 h after stimulation (Fig. 6C). Consistent with this result, whereas the SIRT1 inhibitor sirtinol had no clear effect on the levels of cleaved caspase-3 or the tBid fragment after 17 h of stimulation with dsRNA, the SIRT1 activator SRT1720 significantly inhibited apoptosis (Fig. 6D). Apoptosis was also inhibited by SRT1720 in EMCV-infected cells (Fig. 6E), indicating that SIRT1 protects cells from virus-induced apoptosis. Taken together, our results suggest a regulatory role of SIRT1 in virus-induced immune response.
DISCUSSION
Protein phosphorylation is a key regulatory mechanism of cell signaling pathways involved in cell growth, proliferation, and survival in response to both intracellular and extracellular stimuli. Mass-spectrometry-based phosphoproteomics has enabled the detection of thousands of phosphorylation sites from a biological sample in a single experiment, making it a powerful method for studying cell signaling (42). 14-3-3 proteins play critical roles in many cellular processes through interaction with a large number of intracellular proteins. Because 14-3-3 interactions are primarily phosphorylation dependent, the 14-3-3s have emerged as important components of phosphorylation-regulated biological processes, such as signal transduction (43). We showed previously that 14-3-3 signaling pathways are activated by cytoplasmic dsRNA in human keratinocytes (19). In this study, we combined two proteomic approaches, phosphoproteomics and 14-3-3 affinity capture, to examine the signaling pathways in which 14-3-3 proteins participate and to identify new targets of 14-3-3 signaling during dsRNA stimulation.
During viral infection, receptors of the innate immune system recognize specific molecular patterns, leading to transcription of antiviral and inflammatory cytokines and apoptosis of infected cells. In addition, viruses use the host signaling activity to induce changes in the cell that promote viral entry, replication of viral genetic material, and production of viral proteins. Our phosphoproteomic data showed that several host signaling pathways were activated upon dsRNA stimulation in human keratinocytes, including apoptosis signaling and gene expression. We further show that recognition of cytoplasmic dsRNA leads to the activation of p38, Erk1/2, and JNK MAP kinase pathways. However, only the p38 pathway was required for the optimal dsRNA-induced cytokine expression, whereas both JNK and p38 pathways were necessary for dsRNA-induced apoptosis. These observations are in line with previous studies showing that activation of p38 is essential for RIG-I-mediated viral induction of IFNs (44, 45) and that both JNK and p38 promote virus-induced apoptosis (46, 47). Moreover, our data suggest that signaling pathways involved in actin cytoskeleton reorganization, such as Rho and Cdc42 signaling, are modulated during dsRNA activation. Such cytoskeletal proteins are involved in virus particle internalization and later during viral replication and assembly (48). In addition, viral infection alters cell shape and adhesion, which are also regulated by actin cytoskeleton, leading to uncontrolled cell division and invasive phenotype (48).
14-3-3 interactome studies have demonstrated that 14-3-3 proteins regulate a variety of cellular processes through phosphorylation-dependent interactions (15). Here, we show that 14-3-3 interaction partners participate in many events associated with viral infection, such as apoptosis, intracellular transport, and activation of immune response. We were able to identify a number of new putative substrates of 14-3-3, some of which are directly linked to immune responses, including IL-1 receptor antagonist protein, ubiquitin-like protein ISG15, and Rho GDP-dissociation inhibitor 2. Stathmin, an important microtubule-destabilizing protein, was also detected as a new dsRNA-induced binding partner of 14-3-3. Its activity has been recently shown to be regulated through virus-induced phosphorylation (49), consistent with stathmin being a target for 14-3-3 regulation during viral infection. In addition, four HMGs (HMGB1, HMGB2, HMGB3, and HMGA1) were identified as previously unreported binding partners of 14-3-3s. HMGBs are evolutionarily conserved non-histone chromatin-binding proteins that are released into the extracellular space during infection or injury, and at least HMGB1 acts as a danger signal triggering inflammation (50). Interestingly, other danger signal proteins were also detected as dsRNA-induced 14-3-3 targets, such as S100-proteins, annexins, and galectins, which suggests that 14-3-3s regulate the secretion of danger signals during virus infection.
Moreover, we demonstrate here that TNF and NFκB signaling are important targets of 14-3-3-mediated regulation during RLR activation. TNF is a multifunctional pro-inflammatory cytokine that plays important roles in various physiological and pathological processes, especially in inflammation. The NFκB family of transcription factors is involved mainly in stress-induced, immune, and inflammatory responses. NFκB is also an important regulator in cell fate decisions, such as programmed cell death and proliferation control. The role of 14-3-3 proteins in TLR-induced immune response has been investigated in some studies (16–18, 51). In line with our data, these studies emphasize the importance of 14-3-3s in TNF and NFκB signaling. However, the results have been somewhat conflicting depending on different 14-3-3 isoforms and TLR members, and the precise molecular role for 14-3-3 in TLR signaling requires further investigation. The main cytosolic receptor for dsRNA is RLR MDA-5, and our findings demonstrate a novel role for 14-3-3 proteins in MDA-5 signaling. The function of 14-3-3 proteins in TLR and RLR signaling suggests a major regulatory role for 14-3-3 proteins in the antiviral response against viral infections.
One of the major findings of our study was the identification of RAI as a novel viral dsRNA-induced binding partner of 14-3-3s. RAI is a member of the ankyrin repeat-, SH3 domain- and proline-rich region–containing protein (ASPP) family. ASPPs bind to proteins that are key players in controlling apoptosis, including p53 and NFkB p65RelA subunit (35, 36). ASPP1 and ASPP2 have been shown to stimulate p53-dependent apoptosis through the induction of pro-apoptotic target genes in tumor cells. In contrast, RAI, the inhibitory ASPP (also known as iASPP), has the opposite effect (35, 52). Recently, it was shown that besides regulating p53 activity, ASPPs are also involved in the regulation of other cellular signaling pathways. RAI was found to act as a key player in the regulation of epithelial stratification through modulation of the transcriptional activity of p63, which also belongs to the p53 protein family (53, 54), and ASPP1, ASPP2, and RAI (iASPP) also interact with other proteins including the protein phosphatase 1 catalytic subunit (55). However, the importance of RAI in immune response in non-tumorigenic cell lines is much less clear. In the present study, silencing of RAI protein expression clearly increased dsRNA-induced caspase-3 activation, demonstrating that RAI inhibits dsRNA-induced apoptosis in human keratinocytes. The inhibition of apoptosis by RAI was not mediated by NFκB, as silencing of RAI had no effect on dsRNA-induced NFκB signaling. In addition, silencing of RAI protein expression did not have an effect on RNA sensing pathway components RIGI, MDA5, and MAVS, suggesting that RAI does not function upstream of these molecules. Furthermore, RAI silencing decreased TNF cytokine production, demonstrating a previously unrecognized link between RAI and TNF expression. However, our data do not rule out the possibility that the reduced TNF expression was a result of increased apoptosis, seen in RAI-depleted cells. Taken together, our findings demonstrate that RAI is a novel regulator of antiviral innate immune response.
The other major observation in our analysis was the central role of SIRT1 in antiviral innate immune response. Sirtuins are a family of class III histone deacetylases that catalyze the cleavage of acetyl groups from target lysine residues. Sirtuins, especially SIRT1, have both histone and nonhistone substrates, and some of these have recently been linked to immune response (38). The role of sirtuins in host response to viral infections has remained largely uncharacterized. Recent studies have shown that SIRT1 modulates HIV-1 transcription and may contribute to the chronic immune activation state of HIV-infected individuals (56). Therefore, it is important to understand in more detail the role of SIRT1 in antiviral host response, as well as in regulation of the virus life cycle. Silencing of SIRT1 had no effect on RIG-I, MDA5, or MAVS expression, suggesting that SIRT1 regulates downstream signaling components of the RIG-I/MDA5 RNA sensing pathway. One of the targets for SIRT1 deacetylation is the p65RelA subunit of NFκB, which is a key component of the intracellular inflammatory response (39). Inhibition of SIRT1 expression or activity leads to the accumulation of acetylated forms of p65RelA and increases NFκB activity in several cell types in vitro and in vivo (40, 57, 58). In accordance, our results show that SIRT1 silencing increased the activity of NFκB and its target gene expression in human keratinocytes during virus infection. It has also been demonstrated the SIRT1 regulates cell apoptosis by deacetylating apoptosis-related proteins, including NFκB, p53, Ku70, and FoxO1 (39, 59–61). Noticeably, our data show that SIRT1 protected cells from dsRNA-induced apoptosis, underlining the importance of SIRT1 in antiviral innate immune response.
The activity of SIRT1 is controlled tightly through general regulatory mechanisms, including transcriptional, post-transcriptional, and post-translational regulation (62). Our phosphoproteomic analysis showed that SIRT1 became phosphorylated on Ser27 and Ser47 after dsRNA stimulation (supplemental Table S1). It has been shown previously that SIRT1 can be phosphorylated by JNK on three sites, Ser27, Ser47, and Ser530, and these phosphorylations enhance SIRT1 protein stability and specific activity (63, 64). In contrast, a recent study showed that SIRT1 phosphorylation induces a brief activation of SIRT1 function and degradation of SIRT1 thereafter by the proteasome (65). Our data show that activation of the cytosolic dsRNA recognition pathway led to the degradation of SIRT1 protein. Similar decreases in SIRT1 levels were also observed in several cell types, including in inflammatory cells of the lungs of rats exposed to cigarette smoke, lipopolysaccharide-treated murine macrophages, and TNF-induced vascular adventitial fibroblasts (40, 57, 61), and this decreased SIRT1 level correlates with increased NFκB-dependent pro-inflammatory mediator release. Therefore, we suggest that SIRT1 negatively regulates inflammatory response in human keratinocytes and that during skin infection, the level and activity of SIRT1 decreases, which enables the activation of immune response against viral infection.
In conclusion, the data presented herein enhance our understanding of the role of 14-3-3 interactions in antiviral immune responses and identify RAI and SIRT1 as novel regulators of antiviral innate immune response.
Supplementary Material
Footnotes
Author contributions: T.O., S.M., and T.A.N. designed research; T.O., S.S., E.V., and N.L. performed research; P.H., C.M., and T.A. contributed new reagents or analytic tools; T.O. and T.A.N. analyzed data; T.O., S.M., and T.A.N. wrote the paper.
* This work was supported by grants from the Academy of Finland (Grant Nos. 135628, 140950, 272931, 255842, 120569, 133227, 140880, 269862, and 272437), the Integrative Life Science Doctoral Program (S.S.), the National Doctoral Programme in Informational and Structural Biologythe (E.V.), the Sigrid Jusélius Foundation, and the Magnus Ehrnrooth Foundation.
 This article contains supplemental material.
This article contains supplemental material.
2 Söderholm, S., Hintsanen, P., Öhman, T., Aittokallio, T., and Nyman, T. A. PhosFox: a bioinformatics tool for peptide-level processing of LC-MS/MS-based phosphoproteomic data (Published: Proteome Sci. 12:36.)
1 The abbreviations used are:
- TLR
- Toll-like receptor
- ASPP
- ankyrin repeat-, SH3 domain- and proline-rich region-containing protein
- dsRNA
- double-stranded RNA
- EMCV
- encephalomyocarditis virus
- HMG
- high-mobility group protein
- IL
- interleukin
- IPA
- Ingenuity Pathway Analysis
- ISG15
- interferon-stimulated ubiquitin-like protein ISG15
- iTRAQ
- isobaric tags for relative and absolute quantitation
- MDA5
- melanoma differentiation-associated gene 5
- LDH
- lactate dehydrogenase
- NFκB
- nuclear factor κ-light-chain-enhancer of activated B cells
- pI:C
- polyinosic-polycytidylic acid
- RAI
- RelA-associated inhibitor
- RLR
- RIG-I like receptor
- RT-PCR
- real-time polymerase chain reaction
- SCX
- strong cation exchange
- SIRT1
- sirtuin 1
- TNF
- tumor necrosis factor.
REFERENCES
- 1. Albanesi C., Scarponi C., Giustizieri M. L., Girolomoni G. (2005) Keratinocytes in inflammatory skin diseases. Curr. Drug Targets Inflamm. Allergy 4, 329–334 [DOI] [PubMed] [Google Scholar]
- 2. Kawai T., Akira S. (2011) Toll-like receptors and their crosstalk with other innate receptors in infection and immunity. Immunity 34, 637–650 [DOI] [PubMed] [Google Scholar]
- 3. Kato H., Sato S., Yoneyama M., Yamamoto M., Uematsu S., Matsui K., Tsujimura T., Takeda K., Fujita T., Takeuchi O., Akira S. (2005) Cell type-specific involvement of RIG-I in antiviral response. Immunity 23, 19–28 [DOI] [PubMed] [Google Scholar]
- 4. Yoneyama M., Kikuchi M., Natsukawa T., Shinobu N., Imaizumi T., Miyagishi M., Taira K., Akira S., Fujita T. (2004) The RNA helicase RIG-I has an essential function in double-stranded RNA-induced innate antiviral responses. Nat. Immunol. 5, 730–737 [DOI] [PubMed] [Google Scholar]
- 5. Hornung V., Ellegast J., Kim S., Brzozka K., Jung A., Kato H., Poeck H., Akira S., Conzelmann K. K., Schlee M., Endres S., Hartmann G. (2006) 5′-triphosphate RNA is the ligand for RIG-I. Science 314, 994–997 [DOI] [PubMed] [Google Scholar]
- 6. Pichlmair A., Schulz O., Tan C. P., Naslund T. I., Liljestrom P., Weber F., Reis e Sousa C. (2006) RIG-I-mediated antiviral responses to single-stranded RNA bearing 5′-phosphates. Science 314, 997–1001 [DOI] [PubMed] [Google Scholar]
- 7. Kato H., Takeuchi O., Sato S., Yoneyama M., Yamamoto M., Matsui K., Uematsu S., Jung A., Kawai T., Ishii K. J., Yamaguchi O., Otsu K., Tsujimura T., Koh C. S., Reis e Sousa C., Matsuura Y., Fujita T., Akira S. (2006) Differential roles of MDA5 and RIG-I helicases in the recognition of RNA viruses. Nature 441, 101–105 [DOI] [PubMed] [Google Scholar]
- 8. Venkataraman T., Valdes M., Elsby R., Kakuta S., Caceres G., Saijo S., Iwakura Y., Barber G. N. (2007) Loss of DExD/H box RNA helicase LGP2 manifests disparate antiviral responses. J. Immunol. 178, 6444–6455 [DOI] [PubMed] [Google Scholar]
- 9. Bruns A. M., Horvath C. M. (2012) Activation of RIG-I-like receptor signal transduction. Crit. Rev. Biochem. Mol. Biol. 47, 194–206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Rintahaka J., Wiik D., Kovanen P. E., Alenius H., Matikainen S. (2008) Cytosolic antiviral RNA recognition pathway activates caspases 1 and 3. J. Immunol. 180, 1749–1757 [DOI] [PubMed] [Google Scholar]
- 11. Fu H., Subramanian R. R., Masters S. C. (2000) 14-3-3 proteins: structure, function, and regulation. Annu. Rev. Pharmacol. Toxicol. 40, 617–647 [DOI] [PubMed] [Google Scholar]
- 12. Yaffe M. B., Rittinger K., Volinia S., Caron P. R., Aitken A., Leffers H., Gamblin S. J., Smerdon S. J., Cantley L. C. (1997) The structural basis for 14-3-3:phosphopeptide binding specificity. Cell 91, 961–971 [DOI] [PubMed] [Google Scholar]
- 13. Johnson C., Crowther S., Stafford M. J., Campbell D. G., Toth R., MacKintosh C. (2010) Bioinformatic and experimental survey of 14-3-3-binding sites. Biochem. J. 427, 69–78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Ottmann C., Yasmin L., Weyand M., Veesenmeyer J. L., Diaz M. H., Palmer R. H., Francis M. S., Hauser A. R., Wittinghofer A., Hallberg B. (2007) Phosphorylation-independent interaction between 14-3-3 and exoenzyme S: from structure to pathogenesis. EMBO J. 26, 902–913 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Johnson C., Tinti M., Wood N. T., Campbell D. G., Toth R., Dubois F., Geraghty K. M., Wong B. H., Brown L. J., Tyler J., Gernez A., Chen S., Synowsky S., MacKintosh C. (2011) Visualization and biochemical analyses of the emerging mammalian 14-3-3-phosphoproteome. Mol. Cell. Proteomics 10, M110.005751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Faisal A., Saurin A., Gregory B., Foxwell B., Parker P. J. (2008) The scaffold MyD88 acts to couple protein kinase cepsilon to toll-like receptors. J. Biol. Chem. 283, 18591–18600 [DOI] [PubMed] [Google Scholar]
- 17. Schuster T. B., Costina V., Findeisen P., Neumaier M., Ahmad-Nejad P. (2011) Identification and functional characterization of 14-3-3 in TLR2 signaling. J. Proteome Res. 10, 4661–4670 [DOI] [PubMed] [Google Scholar]
- 18. Butt A. Q., Ahmed S., Maratha A., Miggin S. M. (2012) 14-3-3epsilon and 14-3-3sigma inhibit toll-like receptor (TLR)-mediated proinflammatory cytokine induction. J. Biol. Chem. 287, 38665–38679 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 19. Ohman T., Lietzen N., Valimaki E., Melchjorsen J., Matikainen S., Nyman T. A. (2010) Cytosolic RNA recognition pathway activates 14-3-3 protein mediated signaling and caspase-dependent disruption of cytokeratin network in human keratinocytes. J. Proteome Res. 9, 1549–1564 [DOI] [PubMed] [Google Scholar]
- 20. Villen J., Gygi S. P. (2008) The SCX/IMAC enrichment approach for global phosphorylation analysis by mass spectrometry. Nat. Protoc. 3, 1630–1638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Moorhead G., Douglas P., Cotelle V., Harthill J., Morrice N., Meek S., Deiting U., Stitt M., Scarabel M., Aitken A., MacKintosh C. (1999) Phosphorylation-dependent interactions between enzymes of plant metabolism and 14-3-3 proteins. Plant J. 18, 1–12 [DOI] [PubMed] [Google Scholar]
- 22. Pozuelo Rubio M., Geraghty K. M., Wong B. H., Wood N. T., Campbell D. G., Morrice N., Mackintosh C. (2004) 14-3-3-affinity purification of over 200 human phosphoproteins reveals new links to regulation of cellular metabolism, proliferation and trafficking. Biochem. J. 379, 395–408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Perkins D. N., Pappin D. J., Creasy D. M., Cottrell J. S. (1999) Probability-based protein identification by searching sequence databases using mass spectrometry data. Electrophoresis 20, 3551–3567 [DOI] [PubMed] [Google Scholar]
- 24. Shilov I. V., Seymour S. L., Patel A. A., Loboda A., Tang W. H., Keating S. P., Hunter C. L., Nuwaysir L. M., Schaeffer D. A. (2007) The paragon algorithm, a next generation search engine that uses sequence temperature values and feature probabilities to identify peptides from tandem mass spectra. Mol. Cell. Proteomics 6, 1638–1655 [DOI] [PubMed] [Google Scholar]
- 25. Elias J. E., Gygi S. P. (2010) Target-decoy search strategy for mass spectrometry-based proteomics. Methods Mol. Biol. 604, 55–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Vizcaino J. A., Cote R. G., Csordas A., Dianes J. A., Fabregat A., Foster J. M., Griss J., Alpi E., Birim M., Contell J., O'Kelly G., Schoenegger A., Ovelleiro D., Perez-Riverol Y., Reisinger F., Rios D., Wang R., Hermjakob H. (2013) The PRoteomics IDEntifications (PRIDE) database and associated tools: status in 2013. Nucleic Acids Res. 41, D1063–D1069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Keller M., Ruegg A., Werner S., Beer H. D. (2008) Active caspase-1 is a regulator of unconventional protein secretion. Cell 132, 818–831 [DOI] [PubMed] [Google Scholar]
- 28. Evans C., Noirel J., Ow S. Y., Salim M., Pereira-Medrano A. G., Couto N., Pandhal J., Smith D., Pham T. K., Karunakaran E., Zou X., Biggs C. A., Wright P. C. (2012) An insight into iTRAQ: where do we stand now? Anal. Bioanal. Chem. 404, 1011–1027 [DOI] [PubMed] [Google Scholar]
- 29. Backes C., Keller A., Kuentzer J., Kneissl B., Comtesse N., Elnakady Y. A., Muller R., Meese E., Lenhof H. P. (2007) GeneTrail—advanced gene set enrichment analysis. Nucleic Acids Res. 35, W186–W192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Mann M., Ong S. E., Gronborg M., Steen H., Jensen O. N., Pandey A. (2002) Analysis of protein phosphorylation using mass spectrometry: deciphering the phosphoproteome. Trends Biotechnol. 20, 261–268 [DOI] [PubMed] [Google Scholar]
- 31. Kyriakis J. M., Avruch J. (2012) Mammalian MAPK signal transduction pathways activated by stress and inflammation: a 10-year update. Physiol. Rev. 92, 689–737 [DOI] [PubMed] [Google Scholar]
- 32. Zhang D., Zhang D. E. (2011) Interferon-stimulated gene 15 and the protein ISGylation system. J. Interferon Cytokine Res. 31, 119–130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Ross P. L., Huang Y. N., Marchese J. N., Williamson B., Parker K., Hattan S., Khainovski N., Pillai S., Dey S., Daniels S., Purkayastha S., Juhasz P., Martin S., Bartlet-Jones M., He F., Jacobson A., Pappin D. J. (2004) Multiplexed protein quantitation in Saccharomyces cerevisiae using amine-reactive isobaric tagging reagents. Mol. Cell. Proteomics 3, 1154–1169 [DOI] [PubMed] [Google Scholar]
- 34. Lietzen N., Ohman T., Rintahaka J., Julkunen I., Aittokallio T., Matikainen S., Nyman T. A. (2011) Quantitative subcellular proteome and secretome profiling of influenza A virus-infected human primary macrophages. PLoS Pathog. 7, e1001340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Bergamaschi D., Samuels Y., O'Neil N. J., Trigiante G., Crook T., Hsieh J. K., O'Connor D. J., Zhong S., Campargue I., Tomlinson M. L., Kuwabara P. E., Lu X. (2003) iASPP oncoprotein is a key inhibitor of p53 conserved from worm to human. Nat. Genet. 33, 162–167 [DOI] [PubMed] [Google Scholar]
- 36. Yang J. P., Hori M., Sanda T., Okamoto T. (1999) Identification of a novel inhibitor of nuclear factor-kappaB, RelA-associated inhibitor. J. Biol. Chem. 274, 15662–15670 [DOI] [PubMed] [Google Scholar]
- 37. Michan S., Sinclair D. (2007) Sirtuins in mammals: insights into their biological function. Biochem. J. 404, 1–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Kong S., McBurney M. W., Fang D. (2012) Sirtuin 1 in immune regulation and autoimmunity. Immunol. Cell Biol. 90, 6–13 [DOI] [PubMed] [Google Scholar]
- 39. Yeung F., Hoberg J. E., Ramsey C. S., Keller M. D., Jones D. R., Frye R. A., Mayo M. W. (2004) Modulation of NF-kappaB-dependent transcription and cell survival by the SIRT1 deacetylase. EMBO J. 23, 2369–2380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Shen Z., Ajmo J. M., Rogers C. Q., Liang X., Le L., Murr M. M., Peng Y., You M. (2009) Role of SIRT1 in regulation of LPS- or two ethanol metabolites-induced TNF-alpha production in cultured macrophage cell lines. Am. J. Physiol. Gastrointest. Liver Physiol. 296, G1047–G1053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Yoshizaki T., Schenk S., Imamura T., Babendure J. L., Sonoda N., Bae E. J., Oh D. Y., Lu M., Milne J. C., Westphal C., Bandyopadhyay G., Olefsky J. M. (2010) SIRT1 inhibits inflammatory pathways in macrophages and modulates insulin sensitivity. Am. J. Physiol. Endocrinol. Metab. 298, E419–E428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Roux P. P., Thibault P. (2013) The coming of age of phosphoproteomics—from large data sets to inference of protein functions. Mol. Cell. Proteomics 12, 3453–3464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Gardino A. K., Yaffe M. B. (2011) 14-3-3 proteins as signaling integration points for cell cycle control and apoptosis. Semin. Cell Dev. Biol. 22, 688–695 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Mikkelsen S. S., Jensen S. B., Chiliveru S., Melchjorsen J., Julkunen I., Gaestel M., Arthur J. S., Flavell R. A., Ghosh S., Paludan S. R. (2009) RIG-I-mediated activation of p38 MAPK is essential for viral induction of interferon and activation of dendritic cells: dependence on TRAF2 and TAK1. J. Biol. Chem. 284, 10774–10782 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Hui K. P., Lee S. M., Cheung C. Y., Ng I. H., Poon L. L., Guan Y., Ip N. Y., Lau A. S., Peiris J. S. (2009) Induction of proinflammatory cytokines in primary human macrophages by influenza A virus (H5N1) is selectively regulated by IFN regulatory factor 3 and p38 MAPK. J. Immunol. 182, 1088–1098 [DOI] [PubMed] [Google Scholar]
- 46. Huang Y., Liu H., Li S., Tang Y., Wei B., Yu H., Wang C. (2014) MAVS-MKK7-JNK2 defines a novel apoptotic signaling pathway during viral infection. PLoS Pathog. 10, e1004020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Mosallanejad K., Sekine Y., Ishikura-Kinoshita S., Kumagai K., Nagano T., Matsuzawa A., Takeda K., Naguro I., Ichijo H. (2014) The DEAH-box RNA helicase DHX15 activates NF-kappaB and MAPK signaling downstream of MAVS during antiviral responses. Sci. Signal. 7, ra40. [DOI] [PubMed] [Google Scholar]
- 48. Taylor M. P., Koyuncu O. O., Enquist L. W. (2011) Subversion of the actin cytoskeleton during viral infection. Nat. Rev. Microbiol. 9, 427–439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Chen P. W., Lin S. J., Tsai S. C., Lin J. H., Chen M. R., Wang J. T., Lee C. P., Tsai C. H. (2010) Regulation of microtubule dynamics through phosphorylation on stathmin by epstein-barr virus kinase BGLF4. J. Biol. Chem. 285, 10053–10063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Lu B., Wang H., Andersson U., Tracey K. J. (2013) Regulation of HMGB1 release by inflammasomes. Protein Cell 4, 163–167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Zuo S., Xue Y., Tang S., Yao J., Du R., Yang P., Chen X. (2010) 14-3-3 epsilon dynamically interacts with key components of mitogen-activated protein kinase signal module for selective modulation of the TNF-alpha-induced time course-dependent NF-kappaB activity. J. Proteome Res. 9, 3465–3478 [DOI] [PubMed] [Google Scholar]
- 52. Samuels-Lev Y., O'Connor D. J., Bergamaschi D., Trigiante G., Hsieh J. K., Zhong S., Campargue I., Naumovski L., Crook T., Lu X. (2001) ASPP proteins specifically stimulate the apoptotic function of p53. Mol. Cell 8, 781–794 [DOI] [PubMed] [Google Scholar]
- 53. Chikh A., Matin R. N., Senatore V., Hufbauer M., Lavery D., Raimondi C., Ostano P., Mello-Grand M., Ghimenti C., Bahta A., Khalaf S., Akgul B., Braun K. M., Chiorino G., Philpott M. P., Harwood C. A., Bergamaschi D. (2011) iASPP/p63 autoregulatory feedback loop is required for the homeostasis of stratified epithelia. EMBO J. 30, 4261–4273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Notari M., Hu Y., Koch S., Lu M., Ratnayaka I., Zhong S., Baer C., Pagotto A., Goldin R., Salter V., Candi E., Melino G., Lu X. (2011) Inhibitor of apoptosis-stimulating protein of p53 (iASPP) prevents senescence and is required for epithelial stratification. Proc. Natl. Acad. Sci. U.S.A. 108, 16645–16650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Skene-Arnold T. D., Luu H. A., Uhrig R. G., De Wever V., Nimick M., Maynes J., Fong A., James M. N., Trinkle-Mulcahy L., Moorhead G. B., Holmes C. F. (2013) Molecular mechanisms underlying the interaction of protein phosphatase-1c with ASPP proteins. Biochem. J. 449, 649–659 [DOI] [PubMed] [Google Scholar]
- 56. Pinzone M. R., Cacopardo B., Condorelli F., Di Rosa M., Nunnari G. (2013) Sirtuin-1 and HIV-1: an overview. Curr. Drug Targets 14, 648–652 [DOI] [PubMed] [Google Scholar]
- 57. Yang S. R., Wright J., Bauter M., Seweryniak K., Kode A., Rahman I. (2007) Sirtuin regulates cigarette smoke-induced proinflammatory mediator release via RelA/p65 NF-kappaB in macrophages in vitro and in rat lungs in vivo: implications for chronic inflammation and aging. Am. J. Physiol. Lung Cell. Mol. Physiol. 292, L567–L576 [DOI] [PubMed] [Google Scholar]
- 58. Schug T. T., Xu Q., Gao H., Peres-da-Silva A., Draper D. W., Fessler M. B., Purushotham A., Li X. (2010) Myeloid deletion of SIRT1 induces inflammatory signaling in response to environmental stress. Mol. Cell. Biol. 30, 4712–4721 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Vaziri H., Dessain S. K., Ng Eaton E., Imai S. I., Frye R. A., Pandita T. K., Guarente L., Weinberg R. A. (2001) hSIR2(SIRT1) functions as an NAD-dependent p53 deacetylase. Cell 107, 149–159 [DOI] [PubMed] [Google Scholar]
- 60. Cohen H. Y., Lavu S., Bitterman K. J., Hekking B., Imahiyerobo T. A., Miller C., Frye R., Ploegh H., Kessler B. M., Sinclair D. A. (2004) Acetylation of the C terminus of Ku70 by CBP and PCAF controls bax-mediated apoptosis. Mol. Cell 13, 627–638 [DOI] [PubMed] [Google Scholar]
- 61. Wang W., Yan C., Zhang J., Lin R., Lin Q., Yang L., Ren F., Zhang J., Ji M., Li Y. (2013) SIRT1 inhibits TNF-alpha-induced apoptosis of vascular adventitial fibroblasts partly through the deacetylation of FoxO1. Apoptosis 18, 689–701 [DOI] [PubMed] [Google Scholar]
- 62. Preyat N., Leo O. (2013) Sirtuin deacylases: a molecular link between metabolism and immunity. J. Leukoc. Biol. 93, 669–680 [DOI] [PubMed] [Google Scholar]
- 63. Ford J., Ahmed S., Allison S., Jiang M., Milner J. (2008) JNK2-dependent regulation of SIRT1 protein stability. Cell Cycle 7, 3091–3097 [DOI] [PubMed] [Google Scholar]
- 64. Nasrin N., Kaushik V. K., Fortier E., Wall D., Pearson K. J., de Cabo R., Bordone L. (2009) JNK1 phosphorylates SIRT1 and promotes its enzymatic activity. PLoS One 4, e8414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Gao Z., Zhang J., Kheterpal I., Kennedy N., Davis R. J., Ye J. (2011) Sirtuin 1 (SIRT1) protein degradation in response to persistent c-jun N-terminal kinase 1 (JNK1) activation contributes to hepatic steatosis in obesity. J. Biol. Chem. 286, 22227–22234 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.