

Abstract
Malaria remains one of the most prevalent and lethal human infectious diseases worldwide. A comprehensive characterization of antibody responses to blood stage malaria is essential to support the development of future vaccines, sero-diagnostic tests, and sero-surveillance methods. We constructed a proteome array containing 4441 recombinant proteins expressed by the blood stages of the two most common human malaria parasites, P. falciparum (Pf) and P. vivax (Pv), and used this array to screen sera of Papua New Guinea children infected with Pf, Pv, or both (Pf/Pv) that were either symptomatic (febrile), or asymptomatic but had parasitemia detectable via microscopy or PCR. We hypothesized that asymptomatic children would develop antigen-specific antibody profiles associated with antidisease immunity, as compared with symptomatic children. The sera from these children recognized hundreds of the arrayed recombinant Pf and Pv proteins. In general, responses in asymptomatic children were highest in those with high parasitemia, suggesting that antibody levels are associated with parasite burden. In contrast, symptomatic children carried fewer antibodies than asymptomatic children with infections detectable by microscopy, particularly in Pv and Pf/Pv groups, suggesting that antibody production may be impaired during symptomatic infections. We used machine-learning algorithms to investigate the relationship between antibody responses and symptoms, and we identified antibody responses to sets of Plasmodium proteins that could predict clinical status of the donors. Several of these antibody responses were identified by multiple comparisons, including those against members of the serine enriched repeat antigen family and merozoite protein 4. Interestingly, both P. falciparum serine enriched repeat antigen-5 and merozoite protein 4 have been previously investigated for use in vaccines. This machine learning approach, never previously applied to proteome arrays, can be used to generate a list of potential seroprotective and/or diagnostic antigens candidates that can be further evaluated in longitudinal studies.
Of the five species of malaria parasites that infect humans, Plasmodium falciparum (Pf)1 and P. vivax (Pv) are the most common. Interventions aimed at reducing transmission and improving diagnosis and treatment have led to a dramatic reduction in morbidity and mortality (1, 2). For example, Pf fatalities have declined from an estimated one million to 655,000 annually (2). Although Pv is now recognized as the most widespread species worldwide and a significant cause of severe disease, this parasite, which can relapse months to years after the initial blood stage infection, is still largely ignored (3, 4). Furthermore, mixed-species infections, most commonly of Pf and Pv, are more frequent than previously thought. Although blood smears suggest that <2% of cases are mixed-species infections, PCR-based diagnoses suggest that 55–65% of infections in Thailand, Papua New Guinea (PNG), and other countries in south-east Asia (5–7) are mixed-species infections.
Natural immunity can be subdivided into antidisease immunity and antiparasitic immunity. Antidisease immunity (defined as the absence of symptoms) develops quickly, sometimes requiring only one or two infections in high transmission areas (8–11). However, individuals living in high transmission areas develop non-sterile antiparasite immunity, resulting in low-level parasitemia and asymptomatic infections. This immunity is acquired much more slowly than antidisease immunity, may require repeated infections depending on the transmission rate, and is rarely sterilizing (12). Parasite densities in individuals that have acquired antiparasite immunity are average 104- to 106-fold lower than those in non-immune individuals (13).
Blood stage parasites activate innate responses, which in turn lead to significant levels of humoral and cellular adaptive immunity (reviewed in (14)). Antiparasitic immunity appears to be mediated primarily by antibody responses against blood stage antigens (15, 16). Specific antimalarial antibodies can block invasion of host erythrocytes in vitro by both Pf (17) and Pv merozoites (18–21). Additionally, certain antibody isotypes, in particular IgG3, can induce antibody-dependent cellular inhibition (ADCI) of parasite invasion and development in erythrocytes, which is strongly associated with protection against malaria parasites (13, 22). Moreover, passive transfer of Pf antimalarial antibodies to infected patients can result in parasite clearance (15, 16). Evidence from field studies suggests that the slow acquisition of antibodies to genetically variant circulating strains over several years is associated with antidisease immunity to Pf (23), but to a lesser extent to Pv (24). Cell-mediated immune responses also play a role in protection, particularly early in the immune response. A strong pro-inflammatory response mediated primarily by interferon-gamma (IFN-γ) and tumor necrosis factor-α (TNF-α) contributes to the initial killing and clearance of parasite-infected red blood cells (25).
Identifying antibody targets that are associated with infection, disease, or immunity will support the development of vaccines, diagnostics, and tools for sero-surveillance. By comparing the humoral response profiles of defined populations possessing varying degrees of antidisease and/or antiparasite immunity, it may be possible to identify combinations or responses that are associated with protection against clinical disease and/or parasitemia. These responses could guide selection of antigen(s) for blood stage vaccines. Here, we applied Plasmodium genome sequence, proteomics, bioinformatics, and proteome array fabrication technologies to construct a Pf/Pv blood stage proteome array. The Plasmodium genome encodes over 5000 proteins (5538 and 5435 in Pf and Pv, respectively (26)), nearly half of which have been identified via proteomics at the different stages of malaria parasite life cycle (27, 28). A total of 4441 recombinant proteins, representing 1922 Pf and 1936 Pv native proteins previously reported or predicted to be expressed by the blood stages of these parasites were included on the Pf/Pv proteome arrays, which were then used to analyze antibody responses to both Plasmodium species in naturally-exposed individuals with clinically characterized infections.
The resulting data were interrogated using machine-learning algorithms to identify antibody responses that associated with disease status. We identified sets of antigen-specific antibody responses that can be used to distinguish between asymptomatic donors with parasitemias detectable by light microscopy or PCR and asymptomatic donors, some of which were identified by multiple comparisons. This study is a proof-of-concept of the power of applying machine learning algorithms to biomarker discovery, and paves the way for future, more robust studies to identify novel malaria vaccine targets.
EXPERIMENTAL PROCEDURES
Serological Samples
Serum samples from children aged 6 months to 10 years in the Madang area on the north coast of Papua New Guinea (PNG) were used for this study. The study sites have a tropical, humid climate year-round, with a rainy season from Nov. to May; malaria is hyperendemic with limited seasonality (29). Both P. falciparum (Pf) and P. vivax (Pv) are common, and the disease burden is mainly in children under 10 years of age (29, 30). To establish parasitemia, two thick and two thin smears per participant were evaluated under 100× oil immersion and parasitemia was calculated from the number of asexual parasites per 200 leukocytes, assuming mean leukocyte counts of 8000/μl. Two independent reads were performed, and a third of the first two reads disagreed. This was later confirmed by PCR: whole blood genomic DNA containing parasite DNA was prepared for species-specific PCR/LDR-based diagnosis of Plasmodium infection (31) to confirm parasitemia and species identification. Among samples that were positive for light microscopy, there were very few discrepancies between microscopy and PCR regarding the identified species; in these cases the species identification was changed to reflect the species-specific PCR result. Plasma was collected and stored for future studies.
Samples from symptomatic (clinical) cases (n = 108) were collected as part of a morbidity surveillance study of children aged 0–5 at three health centers near Madang (Yagaum, Mugil, and Alexishafen) in 2005–2006 (32, 33), and were defined as those individuals presenting at local health clinics with parasitemia over 1000 Pf asexual stage parasites per μl of whole blood, or 250 Pv asexual stage parasites per μl of whole blood, and fever within the last 24 h but without any severe malaria symptoms (32). Samples from asymptomatic children used in this study (n = 116) were collected as part of a larger (n = 1275) cross-sectional community survey in 2005 that included individuals of all ages (34, 35) presenting with no fever or history of fever in the last 48 h, but with parasitemias detectable by light microscopy (LM) or PCR. For this study, we selected a subset of sera from children aged 0–10 with PCR-confirmed, asymptomatic infection that were either positive (LM, n = 55) or negative (PCR, n = 61) by LM. Because of the rapid acquisition of antidisease immunity, in particular to P. vivax (36), a full age-matching to the symptomatic cases was not possible in the study population. However, in order to minimize the difference in age between the groups, samples from the youngest children with asymptomatic infection were preferentially selected. These groups were further subdivided into nine final groups according to the parasite species detected (Pf, Pv, or Pf/Pv infections) (Table I). 18 US malaria-naïve control donors were used to determine the malaria-specificity of the responses.
Table I. Description of clinical groups of P. falciparum and P. vivax infections.
The characteristics of the 9 clinical groups and the non-exposed donors are indicated, including the Plasmodium species detected, the abbreviation used to designate the group throughout the manuscript and a short description of the clinical group (PCR: asymptomatic donors with parasites detected by PCR, LM: asymptomatic donors with parasites detected by light microscopy, S: symptomatic donors, Pf:donors with detectable P. falciparum infection; Pv:donors with detectable P. vivax infection; Pf/Pv: donors with detectable P. falciparum and P. vivax infection); as well the mean and range of parasitemia values observed by LM, the result of the PCR test for parasitemia, the presence or absence of fever, the mean and range of the age of donors, the percentage of donors older than 5 years, and the number of donors in each group (N). * - all symptomatic samples are from a study that includes only children younger than 5 years; ^ - all malaria naïve controls are older than 18 years.
| Abbreviation | Clinical group | Mean parasitemia (range) | Parasitemia (by PCR) | Fever | Mean age, yrs (range) | % of donors > 5 Yrs | N | |
|---|---|---|---|---|---|---|---|---|
| Pf infection | Pf.PCR | Asymptomatic Pf infection, PCR+ | 0 (0–0) | Yes | No | 5.83 (2–9) | 65.5 | 29 |
| Pf.LM | Asymptomatic Pf infection, LM+ | 798.8 (40–2,015) | Yes | No | 6.21 (0–10) | 73.7 | 19 | |
| Pf.S | Symptomatic Pf infection | 85,480.0 (3,000–378,000) | Yes | Yes | 2.83 (1–5) | 0* | 40 | |
| Pv infection | Pv.PCR | Asymptomatic Pv infection, PCR+ | 0 (0–0) | Yes | No | 5.32 (0.5–10) | 50 | 20 |
| Pv.LM | Asymptomatic Pv infection, LM+ | 544.5 (80–1,482) | Yes | No | 4.90 (2–6) | 65 | 20 | |
| Pv.S | Symptomatic Pv infection | 21,622.40 (280–223,040) | Yes | Yes | 1.32 (0–4) | 0* | 50 | |
| Pf&Pv co-infection | Pf/Pv.PCR | Asymptomatic mixed infection, PCR+ | 0 (0–0) | Yes | No | 7.25 (2–10) | 83.3 | 12 |
| Pf/Pv.LM | Asymptomatic mixed infection, LM+ | Pf: 604.5 (40–2,180) | Yes | No | 5.87 (2–9) | 75 | 16 | |
| Pv: 381.9 (65–1,612) | ||||||||
| Pf/Pv.S | Symptomatic mixed infection | Pf: 112,073.3 (5,160–326,720) | Yes | Yes | 2.56 (1–4) | 0* | 18 | |
| Pv: 1966.7 (360–10,160) | ||||||||
| Non-exposed controls | Malaria naïve | 0 (0–0) | No | No | Adults | 100̂ | 18 | |
Study Approval
All samples were collected in previous studies. Written informed consent for use of samples in future studies were obtained from a parent or guardian of all subjects. All studies were approved by the PNG Institute of Medical Research (IMR) Internal Review Board (IMR IRB 0901) and the PNG Medical Advisory Committee (MRAC 09.03).
Construction of Pf and Pv Blood Stage Proteome Array
We designed a proteome array containing 2208 Pf and 2233 Pv recombinant proteins, representing 1922 Pf and 1936 Pv native proteins, respectively. Blood stage proteins predicted to be secreted or presented on the surface of the parasite were selected. Pf genes had evidence for blood stage expression by microarray, proteomics, or expressed sequence tags (ESTs), or were predicted to be in the blood stage secretome (37, 38). Pv genes had evidence for blood stage expression by microarray (39, 40). These included both unique Pv proteins and Pv orthologs of Pf proteins. Proteins predicted to be on the surface of the parasite were defined as genes with signal peptides and/or transmembrane domain(s) according to PlasmoDB (26). Putative cytoplasmic proteins, lacking both signal peptides and transmembrane domains, were also selected (pI < 9.0 for Pf genes and pI < 7.6 for Pv genes, respectively). Genes encoding antigenically variant proteins, such as the Pf gene families PfEMP1s, rifins, stevors, surfins, the Pv vir genes, as well as pseudogenes were excluded. Both single and multi-exon genes were included, but exons of multi-exon genes were cloned into separate plasmids. Furthermore, large genes or exons were further divided into overlapping segments in order to limit amplicon length to between 300 and 3000 nt. For example, a 5000-nt exon would be amplified as two 3000-nt segments that overlapped by 1000 nt. Amplicons were labeled with the exon number and the total number of exons, such as “1o2” for exon 1 of a 2-exon gene. Genes that were further divided into segments were labeled s1, s2, etc.
The array was fabricated as previously described (41). Briefly, coding sequences were PCR-amplified from Pv (Sal I strain [MRA-552, MR4]) or Pf (clone 3D7, [MRA-102, MR4]) genomic DNAs and cloned into the PXT7 plasmid with a T7 transcription terminator, and tagged with 5′ polyhistidine (HIS) and 3′ hemagglutinin (HA) epitopes. Recombinant proteins were expressed using E. coli cell-free in vitro transcription and translation reactions (RTS 100 HY kits from 5 PRIME, Gaithersburg, MD) according to the manufacturer's instructions. Proteome arrays were printed as previously described, with each recombinant protein spotted once in each array (41). Each array contained 256 negative control spots made with an in vitro transcription translation master mix without plasmid DNA. Once printed, recombinant protein expression was verified using antipolyhistidine (clone His-1, Sigma) and antihemagglutinin (clone 3F10, Roche) monoclonal antibodies, as previously described (41). All signal intensities were corrected for spot-specific background. A recombinant protein was deemed to be present on the slide if its mean fluorescence intensity was that of the mean of the “no DNA” control spots plus two standard deviations.
Overall, 1767 Pf recombinant proteins (80.02%) and 1470 Pv recombinant proteins (65.83%) were expressed with both the His and HA tags, confirming that the majority of recombinant proteins were fully expressed. These represented a total of 1558 Pf native proteins and 1328 Pv native proteins. Furthermore, 2026 Pf recombinant proteins (91.76%) and 1949 Pv recombinant proteins (87.28%) were expressed with at least one tag (representing 1781 and 1725 native proteins respectively), suggesting that these recombinant proteins were at least partially expressed.
Probing the Proteome Array with Human Sera
Sera were from symptomatic and asymptomatic children diagnosed with Pf, Pv, or both (i.e. Pf/Pv infection) by LM or PCR, as previously described (41). Sera were diluted to 1/100 in 1x Blocking Buffer containing 1 mg/ml E. coli lysate and incubated at room temperature for 30 min with constant mixing to remove E. coli-specific antibodies. Anti-EBNA1 or anti-human IgG were used as controls for the primary antibodies, and serially diluted anti-human IgG was used as a control for the secondary antibody. After rehydration for 30 min, the arrays were probed in triplicate with the pre-treated sera overnight at 4C with constant agitation, then washed and incubated with Cy3-labeled antihuman immunoglobulin. The slides were washed seven times, air-dried, and analyzed using a Perkin Elmer ScanArray Express HT microarray scanner. Intensities were quantified using QuantArray software and corrected for spot-specific background. Statistical analyses were performed as described previously (42). Briefly, the triplicate data points were averaged, and the data were calibrated and transformed using the variance stabilizing normalization (vsn) package (43) in the R statistical environment (www.r-project.org).
Computational Methods
To identify antibody responses that discriminated between the different groups, we first filtered the dataset. First, we removed antibody responses with signal intensities that were not significantly greater in malaria-exposed children than in the 256 empty wells. To do this, we fit the signals arising from the 256 empty wells to a normal distribution. Next, we assigned the response to each recombinant protein a p value by testing it against the right tail of this distribution. These p values were subjected to a standard FDR correction and those signal intensities with a resulting q-value > 0.01 (44) were set to zero. Any recombinant protein that did not elicit significant signal intensity by this analysis was removed. Next, we filtered out responses that were not significantly different in exposed children and the 18 malaria-naïve US donors. For each recombinant protein, we used a Welch's two-tailed t test to test the hypothesis that the signal intensities in 224 infected children were significantly different than those of the 18 US controls. The resulting p values were converted to q-values using Benjamini and Hochberg false discovery rate (FDR) correction (n = 4441)(44). Those recombinant proteins with a q-value > 0.01 were removed. Next, we performed clustering analysis and replaced clusters of proteins that elicited similar responses in all exposed donors with a representative reactivity profile for each cluster. In brief, we clustered proteins that were had positive signals in more than 75% of children into groups, and we replaced the reactivities to individual recombinant proteins with a logical “or”ing representative signal of the reactivity ratios.
We further reduced the remaining antibody responses using an age filter to select for antibody responses that were associated with asymptomatic infections regardless of age group. Briefly, as asymptomatic children tended to be older than symptomatic children, and as the breadth and intensity of reactivities are associated with increasing pathogen exposure, age was significantly correlated with mean reactivity before filtering and clustering (for the entire data set, Pearson's rho = .15, p value = 0.026, n = 224) and this was exacerbated by the filtering (rho = .18, p value = 0.006, n = 224). We calculated the mutual information (45) between the reactivity to each recombinant protein and the individual's age and the disease status of the individual. Those recombinant proteins that shared more information with the disease status than with age (i.e. those that showed a stronger association with the individual's phenotype than with the individual's age (resulting rho = 0.10, p value = 0.120, n = 224) were preserved, whereas the others were filtered out. In other words, if a recombinant protein was more predictive of age than of disease status, it was removed. Because this filtering required the algorithm to use the individual disease status, it was performed inside the cross-validation loop as necessary. Next, we used machine-learning approaches to identify biomarkers that associated with disease status by comparing each of the six different asymptomatic groups separately to the corresponding symptomatic control. We first used cross-validation to estimate whether a classifier was effective at predicting a given clinical category. We constructed evolutionary trees using the evTree algorithm (46), and performed eightfold cross-validation to measure the agreement and the Pearson's correlation between the predicted and actual categories. These metrics were reported alongside the final decision tree as a general measure of classifier accuracy. Finally, to select which recombinant proteins would be most informative as biomarkers associated with disease status when working in concert with other biomarkers, we used the mProbes (47) feature ranking algorithm with Random Forest (48) feature selection. mProbes estimates a false discovery rate by repeatedly running feature selection after adding noise features, generated by assigning the signal intensities for a real protein to random patients, to the data set. The reported false discover rate is the fraction of times that a noise feature was ranked as being more informative that the signal intensity to the recombinant protein in question. It should be noted that this latter analysis using the mProbes algorithm was not performed for the Pf/Pv.LM versus Pf/Pv.S comparison because the cross-validation analysis revealed it did not possess sufficient information to predict donor status.
RESULTS
Children Rapidly Acquire Immunity Against Development of Fever
In order to define the serological responses to P. falciparum (Pf) and P. vivax (Pv) infection in relation to host immunity, we obtained samples from 224 children in PNG, where both parasite species are endemic (29, 30). Symptomatic cases (n = 108) correspond to donors <5 years of age presenting at local health clinics with positive parasitemia, defined as having over 1000 Pf and/or 250 Pv asexual blood stage parasites per μl of whole blood, and fever within the last 24 h, but without any severe malaria symptoms (32). Asymptomatic cases (n = 116) were selected from a wider cross-sectional field study that included all age groups, and were defined as children <10 years of age with no fever or history of fever in the last 48 h, but with Pf and/or Pv parasitemia detectable by PCR or light microscopy (LM) (33). Although symptomatic and asymptomatic samples were collected in different studies, and were not matched for age or parasitemia, this data set allowed us to query the natural immune response to both Pf and Pv simultaneously.
Nine groups were defined based on the combination of clinical status (i.e. symptomatic (S), asymptomatic but with parasitemia detectable by light microscopy (LM), or asymptomatic but with parasitemia detectable only by PCR (PCR)), and the parasite species detected (i.e. Pf, Pv, or both, indicated as Pf/Pv) (Table I). The parasitemias of all samples from LM and S children are shown in Fig. 1A; parasitemia levels were similar in all LM asymptomatic children, regardless of the parasite species detected (Kruskal-Wallis, p value = 0.24), and were lower than in symptomatic cases, although this difference was less pronounced in Pv infections. Mean parasitemias in symptomatic Pf infections were sevenfold higher than in symptomatic Pv infections (Kruskal-Wallis, p value <0.0001). These high Pf parasitemia levels are due in part to Pf's ability to invade both mature erythrocytes and reticulocytes, whereas Pv preferentially invades reticulocytes (49) and generally produces lower parasitemias than Pf. Moreover, symptomatic children who were infected only with Pv carried higher Pv parasite loads than symptomatic children with Pf/Pv infections (Fig. 1A).
Fig. 1.

Mean parasitemia, age distribution, and cumulative incidence of symptoms in smear positive children infected with Pf, Pv or both. A, Parasitemia (number of parasites detected by light microscopy per μl of whole blood) is shown for the six clinical groups with parasites detectable by smear: Pf. LM (asymptomatic children positive only for P. falciparum; filled red circles); Pf.S (symptomatic children positive only for P. falciparum; open red circles); Pv.LM (asymptomatic children positive only for P. vivax; filled blue squares); Pv.S (symptomatic children positive only for P. vivax; open blue squares); Pf/Pv.LM (asymptomatic children positive for P. falciparum and P. vivax; red and blue filled triangles, respectively); and Pf/Pv.S (symptomatic children positive for P. falciparum and P. vivax; red and blue open triangles, respectively). Solid black bars show the median for each group. B, Relationship between parasitemia and age (in years) for all six clinical groups included in part A. Symbols are as in part A. For mixed Pf/Pv infections, each child is represented by two values (one for Pf and one for Pv parasitemia). C, Cumulative incidence of symptomatic malaria cases. Shown are the cumulative fractions of symptomatic children that are positive for Pf (open red circles), Pv (open blue squares), or Pf and Pv (open black triangles), as a function of age (in years).
Among the symptomatic children (all under the age of 5 years as per the original study design), those infected only with Pv were significantly younger than all other infected children (one-way ANOVA, p value <0.0001), with an average age of 1.3 years, compared with 2.8 and 2.6 years for those with Pf and Pf/Pv infections, respectively (Table I). This suggests that children acquire anti-Pv disease immunity (i.e. fever) more rapidly than anti-Pf disease immunity, in agreement with previous findings in PNG (36). However, it should be noted that the age distribution of the asymptomatic samples does not correspond to a random sample of asymptomatic infections in the general population because we selected samples from the youngest children in this group for our study. Despite this, the mean age of the children in the asymptomatic groups was higher than that of children in the symptomatic groups (5.6 and 5.9 years for the LM and the PCR children, respectively, one-way ANOVA, p value <0.0001). Among the asymptomatic children, there was no significant difference in the age range of those infected with different species of Plasmodium (one-way ANOVA, p value = 0.15).
In general, younger children were more likely to carry high parasitemias, whereas older children had lower parasitemias (Fig. 1B). Although we selected the youngest children from the asymptomatic study, between 65 and 83.3% of the asymptomatic LM cases, depending on the infecting Plasmodium species, occurred in children that were between 5 and 10 years of age (Table I and Fig. 1B), suggesting that children in this endemic site rapidly acquired antidisease immunity to the development of fever. Finally, we compared the cumulative incidence of symptomatic Pv, Pf, and Pf/Pv cases over time (Fig. 1C). We saw that in this cohort, the majority of symptomatic Pv infections are detected earlier in life than symptomatic Pf or Pf/Pv infections. For example, 85% of the symptomatic Pv cases were detected by age 2, whereas only 40% of the symptomatic Pf and Pf/Pv cases had occurred by that age. Although our dataset does not allow us to evaluate the effect of previous exposure to malaria parasites on the development of immunity, this result suggests that children acquire antidisease immunity to Pv more rapidly than they do to Pf (30, 36, 50).
Antibody Responses Against Parasite Proteins Depend on Symptomatic Status and the Infecting Parasite Species
Next, we assessed the reactivity of the children's sera to Pf and Pv proteins using a proteome array that contains 4441 recombinant polypeptides (2208 from Pf and 2233 from Pv) representing 1922 Pf and 1936 Pv native proteins, respectively. Proteins with evidence or prediction of expression during blood stages and secretion or surface localization were selected for inclusion in the array. We probed the arrays with sera from the 224 PNG children, as well as sera from 18 US malaria naïve controls (Table I), and quantified the resulting signals as previously described (41). The strategy used to analyze antibody responses to the Plasmodium recombinant proteins contained in the array is illustrated in Fig. 2. We first selected antibody responses against arrayed proteins (or indicators) that were significantly higher in sera from malaria-exposed children than in US adult controls or in empty wells, using both z and t-tests, with a cut-off of q = 0.01 (Fig. 2A). This step excluded 635 (14.3%) antibody responses that did not differ between exposed children and empty wells, as well as 1586 (35.7%) antibody responses that were similar in malaria-exposed children and malaria-naïve US controls. The 2220 responses that remained were directed against 1026 Pf recombinant proteins (out of 2208 Pf proteins in the array, or 46.5%) and 1194 Pv recombinant proteins (out of 2233 in the array, or 53.5%)) derived from 2021 native proteins (928 Pf and 1093 Pv, respectively). Next, we performed clustering analysis, and identified sets of antibody responses that were highly similar among all exposed donors. These 373 antibody responses (8.4%) grouped into 16 clusters, each of which was replaced in the final dataset with a signature profile, resulting in the net removal of 357 antibody responses. The remaining 1863 antibody responses were then passed through an age filter that removed those that associated more strongly with the age of the donor than with their symptomatic status (n = 1206, Fig. 2A). After applying these filters, 657 antibody responses (directed against 344 Pf and 313 Pv recombinant proteins, respectively, Fig. 2B) were retained for further analysis.
Fig. 2.

Data filtering strategy and global view of antibody responses against arrayed proteins. A, Depiction of the filtering strategy used to eliminate uninformative responses. Shown are the number of responses removed by the “Empty Well” and “US Control” filters, the clustering algorithm and the “Age” filter for the complete dataset of antibody responses from 224 donors to 4441 arrayed proteins. B, Heatmap showing the antibody responses of all donors against arrayed proteins after applying the dataset filtering pipeline described in part A. Responses were sorted by clinical groups (in columns) and by the Plasmodium species of the arrayed protein (in rows). The scale shows the relative reactivity of each spot, with red and yellow corresponding to the lowest and highest reactivity, respectively. C, Box plots showing the fraction of arrayed Pf (top) and Pv (bottom) proteins recognized by serum of children in each of the nine clinical groups.
As shown in Fig. 2B and 2C, the nine groups defined on the basis of the infecting parasite specie(s) and symptomatic status exhibited distinct serological profiles. For example, among symptomatic children, those infected only with Pf (Pf.S) recognized significantly more of the arrayed Pf and Pv polypeptides than Pv.S or Pf/Pv.S children (Fig. 2B and 2C and supplemental Table S1). On the other hand, there was no significant difference in the number of polypeptides recognized by sera from Pv.S or Pf/Pv.S children. Moreover, the mean numbers of polypeptides recognized by Pv.S or Pf/Pv.S children were not significantly different from those recognized by Pv.PCR or Pf/Pv.PCR children, but was significantly lower than those recognized by Pv.LM or Pf/Pv.LM children. However, this was not the case for children infected only with Pf, because Pf.S children recognized significantly more polypeptides than Pf.PCR children, but not Pf.LM children.
The number of polypeptides recognized by Pf.PCR, Pv.PCR, and Pf/Pv.PCR children did not differ significantly. Among LM children, those with Pf/Pv recognized significantly more Pf and Pv proteins than those infected only with Pv, but curiously, those infected with Pf recognized significantly more Pv proteins than those infected with Pv. The mean fraction of Pf polypeptides recognized was 13.40% for Pf.LM, 7.28% for Pv.LM, and 24.06% for Pf/Pv.LM children (Fig. 2C). Overall, fewer Pv polypeptides were recognized by sera from LM children (9.22% for Pf.LM, 4.59% for Pv.LM, and 18.17% for Pf/Pv.LM children; Fig. 2C and supplemental Table S1; two-tailed Wilcoxon p value = .0313, t test p value = .0017, n = 6). Finally, for both Pf and Pf/Pv asymptomatic infections, the mean fraction of positive responses was significantly higher in LM cases than in PCR cases.
Our findings suggest that older, asymptomatic children with infections detectable by LM have broader serological responses than younger, symptomatic children, and that the breadth of these responses may be linked to protection against fever. Furthermore, although the interpretation of these data is limited by the fact that we cannot distinguish between infections detectable only by PCR that may reflect pre-existing antiparasite immunity resulting from previous episodes of parasitemia or resolving infections in which antibodies have waned, our results suggest that asymptomatic infections detectable by LM may associate with an active and effective immunological response against fever.
A Machine-learning Approach Can Classify Children into Different Disease Groups Based on Antibody Responses Determined via Proteome Arrays
Having used the proteome arrays to profile antibody responses against Pf and Pv proteins in PNG children infected with Pf, Pv, or both species, next we asked whether these reactivity profiles could be used to accurately classify children into the different disease groups described in Table I. First, we used the evTree decision tree construction algorithm (46), to assess the dataset and determine whether the observed antibody responses to the arrayed proteins could be used to accurately predict whether any individual donor belonged to a symptomatic or asymptomatic group. To do this, we compared the antibody responses of donors in each of the six asymptomatic groups to those in the corresponding symptomatic groups (i.e. Pf.LM versus Pf.S; Pf.PCR versus Pf.S; Pv.LM versus Pv.S; Pv.PCR versus Pv.S; Pf/Pfv.LM versus Pf/Pv.S; and Pf/Pfv.PCR versus Pf/Pv.S). We applied the filters described previously to eliminate responses that were similar to those observed in empty wells, as well as those that were not significantly different between PNG children and naïve U.S. donors or were nearly identical for all PNG donors. Then, to estimate whether or not the donors in each dataset could be accurately assigned to the symptomatic and asymptomatic groups, we used the evTree machine-learning algorithm with 8-fold cross validation (46), as illustrated in Fig. 3A. In brief, we divided the dataset into an 88% training set and a 12% validation set, and applied the age filter described above to remove responses that associated more strongly with the age of the donor than with their clinical status. Next, a decision tree was built from the training dataset and used to classify the validation dataset into the symptomatic and asymptomatic categories. This process was repeated eight times until the status of each donor was predicted exactly once in the validation dataset. Each cycle of cross-validation produced a different tree that predicted the symptomatic and asymptomatic status of each donor. These assignments were then used to calculate the cross-validated accuracy and correlation of each classifier, as shown in Fig. 3B. With the exception of the Pf/Pv.LM versus Pf/Pv.S comparison, all the pairwise comparisons classified donors into the symptomatic and asymptomatic groups with cross-validated accuracy (XV Acc.) greater than 65% and significant p values (XV Acc.p). Finally, each undivided dataset was passed through the age filter to determine the final number of significant features and the evTree algorithm to generate a final decision tree. For all datasets, the evTree was able to identify antibody responses to 1 to 3 polypeptides that were sufficient to classify donors into the S and A groups (supplemental Table S2). For example, for the Pf.LM versus Pf.S comparison, positive antibody responses against PVX_115450_1o2 and Pf11_0292_2o3 classified donors into the Pf.LM group with an accuracy of 96.6%. However, because there were many more polypeptide responses than donors, the system remained undetermined, such that many other combinations of antibody responses could be used to predict whether a child belonged to a symptomatic or asymptomatic group with similar accuracy.
Fig. 3.

Machine-learning approach for biomarker selection. A, evTree cross-validation strategy used to compare antibody responses in each of the six asymptomatic groups to those in the corresponding symptomatic groups. After dividing each dataset into an 88% training set and a 12% validation set and applying the age filter, the evTree algorithm was trained with 8-fold cross-validation to generate decision trees that predicted whether donors were symptomatic or asymptomatic based on their antibody responses to subsets of arrayed proteins. B, Results of filtering and evTree cross-validation parameters for each of the six pairwise comparisons. The table shows the number of donors in each comparison (#D); the number of responses removed by the empty well (#EW), U.S. naïve donor (#US), and clustering (#CL) filters; the number of responses that remained after these filters were applied (I1); the number of responses removed by the age filter (#Age) and the number of responses that remained after it was applied (I2); the cross-validated accuracy (XV Acc.) and p value (XV Acc. p) of the resulting classifier; as well as the corresponding Matthew's correlation coefficient (XV Corr.) and p value (XV Corr. p) for the classifier. C, Overview of mProbes with Random Forest feature selection strategy used to identify antibody responses that discriminate between symptomatic and asymptomatic donors. After adding noise by randomizing labels for indicators that remain rafter the Age filter was applied (I2), the algorithm identifies features that distinguish between symptomatic and asymptomatic donors. Shown is a representative subset of features selected from the Pf. LM versus Pf.S pairwise comparison.
Therefore, we next used the mProbes algorithm with Random Forest feature selection (47, 48) to identify antibody responses against arrayed proteins that distinguished between symptomatic and asymptomatic donors for the five pairwise comparisons that were determined by the cross-validation analysis to contain sufficient information to predict donor status (i.e. with the exception of the Pf/Pv.LM versus Pf/Pv.S comparison, which did not comply with this criteria based on the results of applying the evTree analysis because the accuracy and correlation p values were > 0.05). mProbes with Random Forest built thousands of decision trees by repeatedly running feature selection after adding noise features generated by shuffling the labels within the dataset, and report a false discovery rate that corresponds to the fraction of times that a noise feature is ranked as being more informative than the actual data. This process selected features (i.e. antibody responses against arrayed polypeptides) that were most informative in distinguishing between symptomatic and asymptomatic donors within each dataset (Fig. 3C). The complete list of the most informative responses selected by the mProbes algorithm for each of the five comparisons is included in supplemental Table S3.
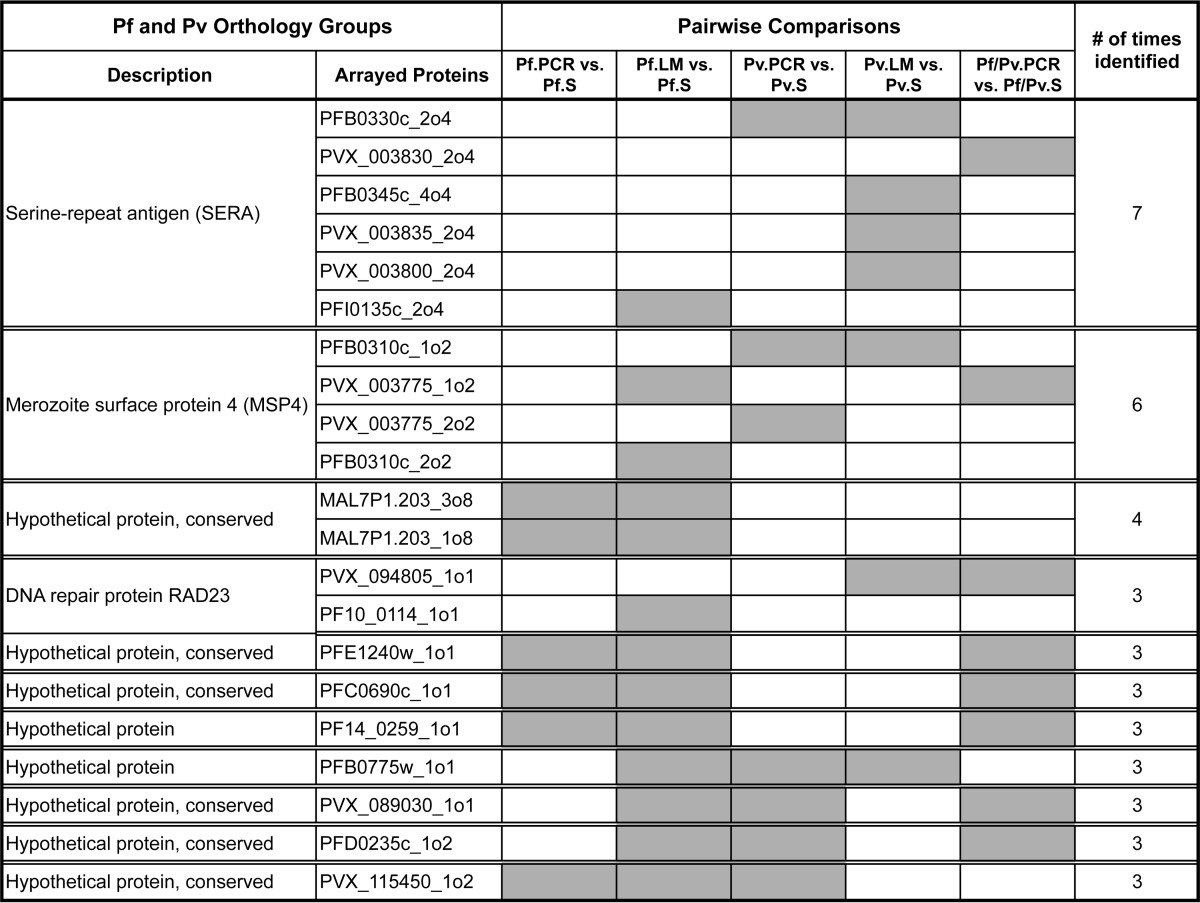
We noticed that several polypeptides were selected in more than one of the pairwise comparisons, including several instances in which sera from Pv donors recognized Pf proteins, and vice versa (supplemental Table S4). To further investigate this observation, we grouped the polypeptides targeted by the antibody responses based on orthology between Pf and Pv proteins, as defined by Ortho_MCL (26). These data are included in supplemental Table S5. Ortholog groups that were selected at least three times by different pairwise comparisons are shown in Table II, ranked by the number of times they were identified across multiple comparisons. The top two candidates correspond to the papain family of SERA proteins, and to MSP4. All other proteins in the table are annotated as hypothetical with the exception of RAD23.
Table II. Pf and Pv protein orthology groups identified by computational analysis. Orthology groups containing arrayed proteins identified by mProbes in three or more pairwise comparisons between clinical groups are shown, including the description of the orthology group, the identity of the individual arrayed proteins, the pairwise comparisons that identified each arrayed protein (shaded), and the number of times the orthology group was identified.
DISCUSSION
We developed a P. falciparum (Pf)-P. vivax (Pv) proteome array containing 4441 proteins to characterize antibody responses to both parasite species in children under 10 years old living in endemic areas in PNG. Sera were obtained from symptomatic children who had attended local health clinics, and from asymptomatic but infected children identified in cross-sectional surveys. We found that asymptomatic children with high parasite loads detectable by light microscopy had antibody responses to more antigens than asymptomatic children with very low parasite loads detectable only by PCR, suggesting that antibody responses in the asymptomatic children were associated with parasite load. Conversely, children with symptoms exhibited very few humoral responses, suggesting a possible disregulation of the antibody response during acute infection. Using a machine-learning approach, we identified humoral responses against subsets of parasite proteins that can be used to predict whether a child is symptomatic or asymptomatic. The approaches we describe may be applied to a well-characterized study population to identify potential antigens for vaccine research or serological surveillance.
The proteome array technology has been used to identify novel antigens in several infectious agents, including Francisella tularensis (51), Burkholderia pseudomallei (52), Pf (53–56), and Pv (41). The array described here detects antibody responses to both Pf and Pv proteins simultaneously, enabling the analysis of sera from malaria endemic areas where both parasites are transmitted. Combining whole proteome arrays with modern analytical tools is a very effective strategy to discover novel antigens for vaccines or diagnostics. Despite the advantages offered by proteome array technology, it is important to note that folding, multimerization, and post-translational modifications such as phosphorylation or glycosylation of arrayed proteins synthesized via in vitro transcription–translation will differ from the native proteins. However, the ability to screen thousands of potential antigens simultaneously is a substantial advantage over conventional approaches.
The samples used in this work were collected in two different studies: one hospital-based and the other field-based. As a result of this, all children in the symptomatic group were younger than 5 years of age, whereas those in the asymptomatic group were older. In fact, although we selected the youngest children among donors in the asymptomatic group, we were unable to age-match the symptomatic and asymptomatic cases because of the fact that there were few asymptomatic children younger than 6 years of age in the asymptomatic cohort. Unsurprisingly, symptomatic cases also carried higher parasitemia than asymptomatic cases. Furthermore, although we utilized a powerful algorithm to analyze antibody responses in the sera from 224 children to >4000 parasite proteins, this study relied on a limited number of banked samples collected in previous studies and we did not know the donors' sex, malaria history, or other factors that impact infection. Future studies with larger sample sizes will be required to increase statistical power and confirm that the responses we identified here with banked serum samples are associated with asymptomatic infections. These caveats, along with the cross-sectional nature of this study, do not allow for the detection of immune correlates of protection. However, this dataset allowed us to describe humoral responses to both Pf and Pv simultaneously, and to develop an analysis pipeline for use in future studies.
It has been previously shown in PNG (57–59) and other locations (23, 60), that antibody titers to Pf proteins are higher in children with LM-detectable parasitemia than in children with LM-undetectable or no parasitemia. We confirmed this finding in Pf and Pf/Pv infections, for which the percentage of positive responses was significantly higher in the LM children than the PCR children. The high antibody titers in asymptomatic children with high parasitemia (LM children) compared with asymptomatic children with very low parasitemia (PCR children) suggests that in asymptomatic donors, the breadth of the antibody response correlates with antigen load, potentially because of the critical mass of antigen required to elicit a response. This may also indicate poor boosting of the memory response, as the very few parasites detectable by PCR in these children may not have been sufficient to boost antibody responses. Alternatively, these children may have developed cellular immunity through past infections that is able to control parasitemia independently of antibodies (61).
Long-term IgG production is maintained by short-lived plasma cells derived from a memory B cell population, or by long-lived plasma cells, which secrete antibodies for as long as several months after immunization (62, 63). Studies in mice have shown that younger individuals develop fewer and shorter-lived plasma cells than adults (64, 65). If this result is applicable to humans, it is possible that young children with malaria may require constant antigen stimulation in order to produce antibodies until they are able to develop long-lived plasma cells. This view is supported by several studies showing that antibody responses against Pf antigens are more persistent in adults than in children (66–68). Furthermore, a more recent study showed that antibody titers declined more slowly with time in both older children and asymptomatic children than they did in younger children, suggesting roles for both antigen persistence and immunological memory in determining the longevity of the antibody response (69). Young symptomatic children may therefore have low antibodies because they have not been sufficiently exposed to the parasite and cannot sustain an antibody response. Alternatively, acute malaria has also been associated with a decreased response to tetanus toxoids, meningococcal polysaccharide, Hib conjugate, and whole cell vaccines for typhoid fever (70–72). Acute malaria itself may therefore disregulate the immune response and prevent generation of antibody responses of sufficient magnitude for protection. However, it may also be the case that younger children rapidly acquire antidisease immunity independently of antibodies, such as a strong cellular response (61) and then slowly acquire antiparasite immunity as they grow older.
In this study we attempted to identify antibody responses that could be used to predict whether a child was asymptomatic, as these antibody responses could protect against development of clinical disease. For this, we turned to machine learning algorithms to identify sets of antibody responses that could discriminate between symptomatic (febrile) and asymptomatic (afebrile) cases detectable by LM or PCR for subsets of children infected with P. falciparum, P. vivax, or both. The machine learning community has long grappled with how to best determine which features, in this case antibody responses, are most informative. Typically these methods focus on selecting a subset of features (e.g. biomarkers) that maximize predictive accuracy, such as support vector machine (SVM) with backward feature selection (73) or partial area under the curve (AUC) (74), algorithms. However, recent research using simulated data implies that for biomarker discovery the best feature-ranking methods also estimate false discovery rates, which are not calculated by SVM and AUC algorithms. For example, the mProbes algorithm with Random Forest (47, 48) seemed especially well suited for problems such as the one discussed here where the accumulated evidence suggests that immunity to malaria only develops after exposure to numerous different antigens, as mProbes with Random Forest can detect groups of antibody responses that work well in concert. However, to our knowledge this is the first time that mProbes has been used in the context of biomarker discovery.
Using this approach, we compared each asymptomatic group to the corresponding symptomatic group, and identified responses to subsets of Pf and Pv proteins that distinguished between asymptomatic and symptomatic donors within each pairwise comparison. In general, the absence of a response was indicative of a symptomatic infection, as symptomatic donors had on average very few responses. Interestingly, several proteins were identified by multiple pairwise comparisons. Moreover, when we classified these shared responses based on Pf and Pv protein orthology, we observed overlapping responses, for example, instances where sera from P. falciparum-exposed donors recognized P. vivax proteins and vice versa. This suggests that some Pf and Pv orthologs may share linear or conformational epitopes that elicit cross-reactive antibodies able to protect against symptomatic Pf, Pv, or Pf/Pv infections. Naturally-acquired cross-reactive antibody responses to MSP5 (75) and CLAG9 (76) have been observed in other endemic settings. Alternatively, it is possible that these apparent cross-reactive responses do not stem from shared epitopes between P. vivax and P. falciparum orthologous proteins, but instead could result from previous infections with the other Plasmodium species. Importantly, because this study did not track malaria infection, our data cannot distinguish between these two possibilities.
Two of the orthology groups identified by multiple analyses contain proteins that have been previously studied as vaccine candidates. MSP4 is a 40 kDa GPI-anchored membrane protein expressed on the merozoite surface that appears to be essential because it is refractory to genetic deletion (77, 78). Unlike most other known merozoite proteins, MSP4 is taken into the invaded erythrocyte without proteolytic processing and is detectable for several hours postinvasion (79). In a recent cross-sectional study in malaria-exposed individuals in the Brazilian Amazon (80), plasma from asymptomatic individuals reacted more strongly to recombinant MSP4 protein than those from symptomatic cases, but anti-MSP4 antibodies could not be independently associated with asymptomatic status. However, polyclonal antisera raised in rabbits inhibited the growth of asexual P. falciparum parasites in vitro, and in rodent models, MSP4 recombinant protein plus adjuvant (81–83) and MSP4 DNA vaccines (84, 85) provided partial protection against blood stage challenge.
The serine-repeat antigens SERAs (86) are soluble parasitophorous vacuolar proteins that are co-expressed in late trophozoite and schizont stages, released upon schizont rupture, and appear to facilitate merozoite egress from rupturing schizonts (87). P. falciparum possesses nine SERA proteins, all but one of which are encoded in an 8-gene cluster on chromosome 2; the ninth gene encoding SERA9 is on chromosome 9. All SERAs have been classified as cysteine-like proteases because in several paralogs the catalytic active site Cys residue has been substituted by Ser. Six SERA proteins, three each from P. falciparum and P. vivax, respectively, were preferentially recognized by the PNG sera (Table II). PfSERA5, the most well-characterized SERA, is abundantly expressed and is refractory to deletion in asexual stages (86). Because antibodies to SERA5 have growth-inhibitory activity in vitro, including those isolated from sera of individuals naturally exposed to P. falciparum (88), SERA5 has been studied extensively as a potential antigen for blood stage vaccines. Although this study did not find that asymptomatic status was associated with antibody responses against PfSERA5, it did identify two putative P. vivax orthologs, PVX_003830 and PVX003800, to which antibody responses discriminated between the Pf/Pv.PCR and Pf/Pv.S groups, and Pv.LM and Pv.S groups, respectively (Table II). Responses to other Pf or Pv SERAs were also associated with asymptomatic status, suggesting that antibodies to multiple SERA family proteins provide protection against symptomatic malaria, perhaps by interfering with release of infectious merozoites from mature schizonts.
The other proteins that were identified by more than three analyses are all annotated as “hypothetical proteins.” Our data showing that antibody responses against them associate with asymptomatic infections in naturally exposed donors suggests that they should be studied in greater detail.
Longitudinal studies in endemic areas are required to validate our findings from this cross-sectional study and to better assess the role of these antibody responses in immunity to malaria. Extending this study to distinct geographical locations might also allow protective antibody responses that are observed in multiple endemic sites to be identified. Furthermore, the use of this technology on longitudinal samples from areas of declining malaria incidence could provide a unique opportunity to identify markers of recent exposure that could be used as surveillance markers to support malaria elimination programs.
In conclusion, we have combined high-throughput laboratory methods with machine-learning analytical tools, providing a proof-of-concept for a novel approach to identify globally relevant novel antigens for vaccine research. This approach could be extended to screen the entire Pf and Pv proteome, and accelerate the search for new vaccine candidate antigens, diagnostic antigens, and serological surveillance markers for malaria.
Supplementary Material
Acknowledgments
We thank the volunteers and PNG IMR field teams that collected the samples, without whom this study could not have been possible. Pascal Michon and Harin Karunajeewa assisted with the collection of samples from clinical cases, Nicolas Senn coordinated the collection of the asymptomatic samples. We also thank Phil Felgner for developing the prototype of the proteome array that made this study possible. We also thank Gowthaman Ramasamy for bioinformatics support to select the proteins used on the array.
Footnotes
Author contributions: O.C.F., S.A.D., D.M.M., X.L., J.D.A., M.J.G., and R.W. designed research; O.C.F., S.A.D., D.M.M., A.T., D.I.S., X.L., and R.W. performed research; D.M.M., P.M.S., X.L., and J.D.A. contributed new reagents or analytic tools; O.C.F., S.A.D., D.M.M., M.V., M.J., P.M.S., J.D.A., I.M., M.J.G., and R.W. analyzed data; O.C.F., S.A.D., M.V., J.D.A., M.J.G., and R.W. wrote the paper.
* This work was supported by NIH/NIAID SBIR award 5R43AI75692.
 This article contains supplemental Tables S1 to S5.
This article contains supplemental Tables S1 to S5.
Conflict of interest: The authors have declared that no conflict of interest exists.
1 The abbreviations used are:
- Pf
- P. falciparum
- ADCI
- antibody-dependent cellular inhibition
- EBNA1
- Epstein-Barr nuclear antigen 1
- EST
- expressed sequence tag
- HA
- hemagglutinin
- HIS
- polyhistidine
- IFN-γ
- interferon gamma
- LM
- asymptomatic donors with parasites detected by light microscopy
- PCR
- asymptomatic donors with parasites detectable only by PCR
- Pf/Pv
- donors infected with both P. falciparum and P. vivax
- PNG
- Papua New Guinea
- Pv
- P. vivax
- S
- symptomatic donors.
REFERENCES
- 1. Murray C. J., Rosenfeld L. C., Lim S. S., Andrews K. G., Foreman K. J., Haring D., Fullman N., Naghavi M., Lozano R., Lopez A. D. (2012) Global malaria mortality between 1980 and 2010: A systematic analysis. Lancet 379, 413–431 [DOI] [PubMed] [Google Scholar]
- 2. WHO (2011) World Malaria Report 2011. World Health Organization, Geneva [Google Scholar]
- 3. Price R. N., Douglas N. M., Anstey N. M. (2009) New developments in Plasmodium vivax malaria: severe disease and the rise of chloroquine resistance. Curr. Opin. Inf. Dis. 22, 430–435 [DOI] [PubMed] [Google Scholar]
- 4. Baird J. K. (2013) Evidence and implications of mortality associated with acute Plasmodium vivax malaria. Clin. Microbiol. Rev. 26, 36–57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Snounou G., Viriyakosol S., Jarra W., Thaithong S., Brown K. N. (1993) Identification of the four human malaria parasite species in field samples by the polymerase chain reaction and detection of a high prevalence of mixed infections. Mol. Biochem. Parasitol. 58, 283–292 [DOI] [PubMed] [Google Scholar]
- 6. Snounou G., White N. J. (2004) The co-existence of Plasmodium: Sidelights from falciparum and vivax malaria in Thailand. Trends Parasitol. 20, 333–339 [DOI] [PubMed] [Google Scholar]
- 7. Zimmerman P. A., Mehlotra R. K., Kasehagen L. J., Kazura J. W. (2004) Why do we need to know more about mixed Plasmodium species infections in humans? Trends Parasitol. 20, 440–447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Marsh K., Kinyanjui S. (2006) Immune effector mechanisms in malaria. Parasite Immunol. 28, 51–60 [DOI] [PubMed] [Google Scholar]
- 9. Gupta S., Snow R. W., Donnelly C. A., Marsh K., Newbold C. (1999) Immunity to non-cerebral severe malaria is acquired after one or two infections. Nat. Med. 5, 340–343 [DOI] [PubMed] [Google Scholar]
- 10. Barcus M. J., Krisin, Elyazar I. R., Marwoto H., Richie T. L., Basri H., Wiady I., Fryauff D. J., Maguire J. D., Bangs M. J., Baird J. K. (2003) Primary infection by Plasmodium falciparum or P. vivax in a cohort of Javanese migrants to Indonesian Papua. Ann. Trop. Med. Parasitol. 97, 565–574 [DOI] [PubMed] [Google Scholar]
- 11. Kleinschmidt I., Sharp B. (2001) Patterns in age-specific malaria incidence in a population exposed to low levels of malaria transmission intensity. Trop. Med. Int. Health 6, 986–991 [DOI] [PubMed] [Google Scholar]
- 12. Schofield L., Mueller I. (2006) Clinical immunity to malaria. Curr. Mol. Med. 6, 205–221 [DOI] [PubMed] [Google Scholar]
- 13. Druilhe P., Perignon J. L. (1994) Mechanisms of defense against P. falciparum asexual blood stages in humans. Immunol. Lett. 41, 115–120 [DOI] [PubMed] [Google Scholar]
- 14. Stevenson M. M., Riley E. M. (2004) Innate immunity to malaria. Nat. Rev. Immunol. 4, 169–180 [DOI] [PubMed] [Google Scholar]
- 15. Cohen S., McGregor I. A., Carrington S. (1961) Gamma-globulin and acquired immunity to human malaria. Nature 192, 733–737 [DOI] [PubMed] [Google Scholar]
- 16. Sabchareon A., Burnouf T., Ouattara D., Attanath P., Bouharoun-Tayoun H., Chantavanich P., Foucault C., Chongsuphajaisiddhi T., Druilhe P. (1991) Parasitologic and clinical human response to immunoglobulin administration in falciparum malaria. Am. J. Trop. Med. Hyg. 45, 297–308 [DOI] [PubMed] [Google Scholar]
- 17. Holder A. A., Guevara Patino J. A., Uthaipibull C., Syed S. E., Ling I. T., Scott-Finnigan T., Blackman M. J. (1999) Merozoite surface protein 1, immune evasion, and vaccines against asexual blood stage malaria. Parassitologia 41, 409–414 [PubMed] [Google Scholar]
- 18. Chitnis C. E., Miller L. H. (1994) Identification of the erythrocyte binding domains of Plasmodium vivax and Plasmodium knowlesi proteins involved in erythrocyte invasion. J. Exp. Med. 180, 497–506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ceravolo I. P., Souza-Silva F. A., Fontes C. J., Braga E. M., Madureira A. P., Krettli A. U., Souza J. M., Brito C. F., Adams J. H., Carvalho L. H. (2008) Inhibitory properties of the antibody response to Plasmodium vivax Duffy binding protein in an area with unstable malaria transmission. Scand. J. Immunol. 67, 270–278 [DOI] [PubMed] [Google Scholar]
- 20. Grimberg B. T., Udomsangpetch R., Xainli J., McHenry A., Panichakul T., Sattabongkot J., Cui L., Bockarie M., Chitnis C., Adams J., Zimmerman P. A., King C. L. (2007) Plasmodium vivax invasion of human erythrocytes inhibited by antibodies directed against the Duffy binding protein. PLoS Med. 4, e337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Michon P., Fraser T., Adams J. H. (2000) Naturally acquired and vaccine-elicited antibodies block erythrocyte cytoadherence of the Plasmodium vivax Duffy binding protein. Inf. Immun. 68, 3164–3171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Tebo A. E., Kremsner P. G., Luty A. J. (2001) Plasmodium falciparum: a major role for IgG3 in antibody-dependent monocyte-mediated cellular inhibition of parasite growth in vitro. Exp. Parasitol. 98, 20–28 [DOI] [PubMed] [Google Scholar]
- 23. Osier F. H., Fegan G., Polley S. D., Murungi L., Verra F., Tetteh K. K., Lowe B., Mwangi T., Bull P. C., Thomas A. W., Cavanagh D. R., McBride J. S., Lanar D. E., Mackinnon M. J., Conway D. J., Marsh K. (2008) Breadth and magnitude of antibody responses to multiple Plasmodium falciparum merozoite antigens are associated with protection from clinical malaria. Infect. Immun. 76, 2240–2248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Cole-Tobian J. L., Michon P., Biasor M., Richards J. S., Beeson J. G., Mueller I., King C. L. (2009) Strain-specific Duffy binding protein antibodies correlate with protection against infection with homologous compared to heterologous Plasmodium vivax strains in Papua New Guinean children. Infect. Immun. 77, 4009–4017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Good M. F., Doolan D. L. (1999) Immune effector mechanisms in malaria. Curr. Opin. Immunol. 11, 412–419 [DOI] [PubMed] [Google Scholar]
- 26. Aurrecoechea C., Brestelli J., Brunk B. P., Dommer J., Fischer S., Gajria B., Gao X., Gingle A., Grant G., Harb O. S., Heiges M., Innamorato F., Iodice J., Kissinger J. C., Kraemer E., Li W., Miller J. A., Nayak V., Pennington C., Pinney D. F., Roos D. S., Ross C., Stoeckert C. J., Jr., Treatman C., Wang H. (2009) PlasmoDB: A functional genomic database for malaria parasites. Nucleic Acids Res. 37, D539–D543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Florens L., Washburn M. P., Raine J. D., Anthony R. M., Grainger M., Haynes J. D., Moch J. K., Muster N., Sacci J. B., Tabb D. L., Witney A. A., Wolters D., Wu Y., Gardner M. J., Holder A. A., Sinden R. E., Yates J. R., Carucci D. J. (2002) A proteomic view of the Plasmodium falciparum life cycle. Nature 419, 520–526 [DOI] [PubMed] [Google Scholar]
- 28. Gardner M. J., Hall N., Fung E., White O., Berriman M., Hyman R. W., Carlton J. M., Pain A., Nelson K. E., Bowman S., Paulsen I. T., James K., Eisen J. A., Rutherford K., Salzberg S. L., Craig A., Kyes S., Chan M. S., Nene V., Shallom S. J., Suh B., Peterson J., Angiuoli S., Pertea M., Allen J., Selengut J., Haft D., Mather M. W., Vaidya A. B., Martin D., Fairlamb A. H., Fraunholz M. J., Roos D. S., Ralph S. A., McFadden G. I., Cummings L. M., Subramanian G. M., Mungall C., Venter J. C., Carucci D. J., Hoffman S. L., Newbold C., Davis R. W., Fraser C. M., Barrell B. (2002) Genome sequence of the human malaria parasite Plasmodium falciparum. Nature 419, 498–511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Cattani J. A., Tulloch J. L., Vrbova H., Jolley D., Gibson F. D., Moir J. S., Heywood P. F., Alpers M. P., Stevenson A., Clancy R. (1986) The epidemiology of malaria in a population surrounding Madang, Papua New Guinea. Am. J. Trop. Med. Hyg. 35, 3–15 [DOI] [PubMed] [Google Scholar]
- 30. Michon P., Cole-Tobian J. L., Dabod E., Schoepflin S., Igu J., Susapu M., Tarongka N., Zimmerman P. A., Reeder J. C., Beeson J. G., Schofield L., King C. L., Mueller I. (2007) The risk of malarial infections and disease in Papua New Guinean children. Am. J. Trop. Med. Hyg. 76, 997–1008 [PMC free article] [PubMed] [Google Scholar]
- 31. Kasehagen L. J., Mueller I., McNamara D. T., Bockarie M. J., Kiniboro B., Rare L., Lorry K., Kastens W., Reeder J. C., Kazura J. W., Zimmerman P. A. (2006) Changing patterns of Plasmodium blood-stage infections in the Wosera region of Papua New Guinea monitored by light microscopy and high throughput PCR diagnosis. Am. J. Trop. Med. Hyg. 75, 588–596 [PMC free article] [PubMed] [Google Scholar]
- 32. Karunajeewa H. A., Mueller I., Senn M., Lin E., Law I., Gomorrai P. S., Oa O., Griffin S., Kotab K., Suano P., Tarongka N., Ura A., Lautu D., Page-Sharp M., Wong R., Salman S., Siba P., Ilett K. F., Davis T. M. (2008) A trial of combination antimalarial therapies in children from Papua New Guinea. N. Engl. J. Med. 359, 2545–2557 [DOI] [PubMed] [Google Scholar]
- 33. Davy C., Sicuri E., Ome M., Lawrence-Wood E., Siba P., Warvi G., Mueller I., Conteh L. (2010) Seeking treatment for symptomatic malaria in Papua New Guinea. Malar. J. 9, 268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Schultz L., Wapling J., Mueller I., Ntsuke P. O., Senn N., Nale J., Kiniboro B., Buckee C. O., Tavul L., Siba P. M., Reeder J. C., Barry A. E. (2010) Multilocus haplotypes reveal variable levels of diversity and population structure of Plasmodium falciparum in Papua New Guinea, a region of intense perennial transmission. Malar. J. 9, 336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Arnott A., Barnadas C., Senn N., Siba P., Mueller I., Reeder J. C., Barry A. E. (2013) High genetic diversity of Plasmodium vivax on the north coast of Papua New Guinea. Am. J. Trop. Med. Hyg. 89, 188–194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Lin E., Kiniboro B., Gray L., Dobbie S., Robinson L., Laumaea A., Schopflin S., Stanisic D., Betuela I., Blood-Zikursh M., Siba P., Felger I., Schofield L., Zimmerman P., Mueller I. (2010) Differential patterns of infection and disease with P. falciparum and P. vivax in young Papua New Guinean children. PLoS ONE 5, e9047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Hiller N. L., Bhattacharjee S., van Ooij C., Liolios K., Harrison T., Lopez-Estrano C., Haldar K. (2004) A host-targeting signal in virulence proteins reveals a secretome in malarial infection. Science 306, 1934–1937 [DOI] [PubMed] [Google Scholar]
- 38. van Ooij C., Tamez P., Bhattacharjee S., Hiller N. L., Harrison T., Liolios K., Kooij T., Ramesar J., Balu B., Adams J., Waters A., Janse C. J., Janse C., Haldar K. (2008) The malaria secretome: from algorithms to essential function in blood stage infection. PLoS Pathog. 4, e1000084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Bozdech Z., Mok S., Hu G., Imwong M., Jaidee A., Russell B., Ginsburg H., Nosten F., Day N. P., White N. J., Carlton J. M., Preiser P. R. (2008) The transcriptome of Plasmodium vivax reveals divergence and diversity of transcriptional regulation in malaria parasites. Proc. Natl. Acad. Sci. U. S. A. 105, 16290–16295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Westenberger S. J., McClean C. M., Chattopadhyay R., Dharia N. V., Carlton J. M., Barnwell J. W., Collins W. E., Hoffman S. L., Zhou Y., Vinetz J. M., Winzeler E. A. (2010) A systems-based analysis of Plasmodium vivax lifecycle transcription from human to mosquito. PLoS Negl. Trop. Dis. 4, e653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Molina D. M., Finney O. C., Aravelo-Herrera M., Herrera S., Felgner P. L., Gardner M. J., Liang X., Wang R. (2012) Plasmodium vivax pre-erythrocytic-stage antigen discovery: exploiting naturally acquired humoral responses. Am. J. Trop. Med. Hyg. 87, 460–469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Sundaresh S., Randall A., Unal B., Petersen J. M., Belisle J. T., Hartley M. G., Duffield M., Titball R. W., Davies D. H., Felgner P. L., Baldi P. (2007) From protein microarrays to diagnostic antigen discovery: a study of the pathogen Francisella tularensis. Bioinformatics 23, i508–i518 [DOI] [PubMed] [Google Scholar]
- 43. Huber W., von Heydebreck A., Sultmann H., Poustka A., Vingron M. (2002) Variance stabilization applied to microarray data calibration and to the quantification of differential expression. Bioinformatics 18, S96–S104 [DOI] [PubMed] [Google Scholar]
- 44. Storey J. D. (2002) A direct approach to false discovery rates. J. R. Stat. Soc. Series B Stat. Methodol. 64, 479–498 [Google Scholar]
- 45. Hausser J., Strimmer K. (2009) Entropy inference and the James-Stein estimator, with application to nonlinear gene association networks. J. Mach. Learn. Res. 10, 1469–1484 [Google Scholar]
- 46. Grubinger T., Zeileis A., Pfeiffer K.-P. (2011) evtree: evolutionary learning of globally optimal classification and regression trees in R. Working Papers 2011–20 Ed., Faculty of Economics and Statistics, University of Innsbruck [Google Scholar]
- 47. Huynh-Thu V. A., Saeys Y., Wehenkel L., Geurts P. (2012) Statistical interpretation of machine learning-based feature importance scores for biomarker discovery. Bioinformatics 28, 1766–1774 [DOI] [PubMed] [Google Scholar]
- 48. Liaw A., Wiener M. (2002) Classification and regression by randomForest. R. News 2, 18–22 [Google Scholar]
- 49. Kitchen S. F. (1938) The infection of reticulocytes by Plasmodium vivax. Am. J. Trop. Med. Hyg. 18, 347–353 [Google Scholar]
- 50. Koepfli C., Colborn K. L., Kiniboro B., Lin E., Speed T. P., Siba P. M., Felger I., Mueller I. (2013) A high force of Plasmodium vivax blood-stage infection drives the rapid acquisition of immunity in Papua New Guinean children. PLoS Negl. Trop. Dis. 7, e2403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Eyles J. E., Unal B., Hartley M. G., Newstead S. L., Flick-Smith H., Prior J. L., Oyston P. C., Randall A., Mu Y., Hirst S., Molina D. M., Davies D. H., Milne T., Griffin K. F., Baldi P., Titball R. W., Felgner P. L. (2007) Immunodominant Francisella tularensis antigens identified using proteome microarray. Proteomics 7, 2172–2183 [DOI] [PubMed] [Google Scholar]
- 52. Felgner P. L., Kayala M. A., Vigil A., Burk C., Nakajima-Sasaki R., Pablo J., Molina D. M., Hirst S., Chew J. S., Wang D., Tan G., Duffield M., Yang R., Neel J., Chantratita N., Bancroft G., Lertmemongkolchai G., Davies D. H., Baldi P., Peacock S., Titball R. W. (2009) A Burkholderia pseudomallei protein microarray reveals serodiagnostic and cross-reactive antigens. Proc. Natl. Acad. Sci. U. S. A. 106, 13499–13504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Crompton P. D., Kayala M. A., Traore B., Kayentao K., Ongoiba A., Weiss G. E., Molina D. M., Burk C. R., Waisberg M., Jasinskas A., Tan X., Doumbo S., Doumtabe D., Kone Y., Narum D. L., Liang X., Doumbo O. K., Miller L. H., Doolan D. L., Baldi P., Felgner P. L., Pierce S. K. (2010) A prospective analysis of the Ab response to Plasmodium falciparum before and after a malaria season by protein microarray. Proc. Natl. Acad. Sci. U. S. A. 107, 6958–6963 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Trieu A., Kayala M. A., Burk C., Molina D. M., Freilich D. A., Richie T. L., Baldi P., Felgner P. L., Doolan D. L. (2011) Sterile protective immunity to malaria is associated with a panel of novel P. falciparum antigens. Mol. Cell. Proteomics 10, M111 007948 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Barry A. E., Trieu A., Fowkes F. J., Pablo J., Kalantari-Dehaghi M., Jasinskas A., Tan X., Kayala M. A., Tavul L., Siba P. M., Day K. P., Baldi P., Felgner P. L., Doolan D. L. (2011) The stability and complexity of antibody responses to the major surface antigen of Plasmodium falciparum are associated with age in a malaria endemic area. Mol. Cell. Proteomics 10, M111.008326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Doolan D. L., Mu Y., Unal B., Sundaresh S., Hirst S., Valdez C., Randall A., Molina D., Liang X., Freilich D. A., Oloo J. A., Blair P. L., Aguiar J. C., Baldi P., Davies D. H., Felgner P. L. (2008) Profiling humoral immune responses to P. falciparum infection with protein microarrays. Proteomics 8, 4680–4694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. al-Yaman F., Genton B., Falk M., Anders R. F., Lewis D., Hii J., Beck H. P., Alpers M. P. (1995) Humoral response to Plasmodium falciparum ring-infected erythrocyte surface antigen in a highly endemic area of Papua New Guinea. Am. J. Trop. Med. Hyg. 52, 66–71 [DOI] [PubMed] [Google Scholar]
- 58. Stanisic D. I., Richards J. S., McCallum F. J., Michon P., King C. L., Schoepflin S., Gilson P. R., Murphy V. J., Anders R. F., Mueller I., Beeson J. G. (2009) Immunoglobulin G subclass-specific responses against Plasmodium falciparum merozoite antigens are associated with control of parasitemia and protection from symptomatic illness. Infect. Immun. 77, 1165–1174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Richards J. S., Stanisic D. I., Fowkes F. J., Tavul L., Dabod E., Thompson J. K., Kumar S., Chitnis C. E., Narum D. L., Michon P., Siba P. M., Cowman A. F., Mueller I., Beeson J. G. (2010) Association between naturally acquired antibodies to erythrocyte-binding antigens of Plasmodium falciparum and protection from malaria and high-density parasitemia. Clin. Infect. Dis. 51, e50–e60 [DOI] [PubMed] [Google Scholar]
- 60. Kinyanjui S. M., Mwangi T., Bull P. C., Newbold C. I., Marsh K. (2004) Protection against clinical malaria by heterologous immunoglobulin G antibodies against malaria-infected erythrocyte variant surface antigens requires interaction with asymptomatic infections. J. Infect. Dis. 190, 1527–1533 [DOI] [PubMed] [Google Scholar]
- 61. Pombo D. J., Lawrence G., Hirunpetcharat C., Rzepczyk C., Bryden M., Cloonan N., Anderson K., Mahakunkijcharoen Y., Martin L. B., Wilson D., Elliott S., Elliott S., Eisen D. P., Weinberg J. B., Saul A., Good M. F. (2002) Immunity to malaria after administration of ultra-low doses of red cells infected with Plasmodium falciparum. Lancet 360, 610–617 [DOI] [PubMed] [Google Scholar]
- 62. Slifka M. K., Antia R., Whitmire J. K., Ahmed R. (1998) Humoral immunity due to long-lived plasma cells. Immunity 8, 363–372 [DOI] [PubMed] [Google Scholar]
- 63. Manz R. A., Hauser A. E., Hiepe F., Radbruch A. (2005) Maintenance of serum antibody levels. Ann. Rev. Immunol. 23, 367–386 [DOI] [PubMed] [Google Scholar]
- 64. Pihlgren M., Schallert N., Tougne C., Bozzotti P., Kovarik J., Fulurija A., Kosco-Vilbois M., Lambert P. H., Siegrist C. A. (2001) Delayed and deficient establishment of the long-term bone marrow plasma cell pool during early life. Eur. J. Immunol. 31, 939–946 [DOI] [PubMed] [Google Scholar]
- 65. Pihlgren M., Friedli M., Tougne C., Rochat A. F., Lambert P. H., Siegrist C. A. (2006) Reduced ability of neonatal and early-life bone marrow stromal cells to support plasmablast survival. J. Immunol. 176, 165–172 [DOI] [PubMed] [Google Scholar]
- 66. Branch O. H., Oloo A. J., Nahlen B. L., Kaslow D., Lal A. A. (2000) Anti-merozoite surface protein-1 19-kDa IgG in mother-infant pairs naturally exposed to Plasmodium falciparum: subclass analysis with age, exposure to asexual parasitemia, and protection against malaria. V. The Asembo Bay Cohort Project. J. Infect. Dis. 181, 1746–1752 [DOI] [PubMed] [Google Scholar]
- 67. Branch O. H., Udhayakumar V., Hightower A. W., Oloo A. J., Hawley W. A., Nahlen B. L., Bloland P. B., Kaslow D. C., Lal A. A. (1998) A longitudinal investigation of IgG and IgM antibody responses to the merozoite surface protein-1 19-kiloDalton domain of Plasmodium falciparum in pregnant women and infants: associations with febrile illness, parasitemia, and anemia. Am. J. Trop. Med. Hyg. 58, 211–219 [DOI] [PubMed] [Google Scholar]
- 68. Taylor R. R., Egan A., McGuinness D., Jepson A., Adair R., Drakely C., Riley E. (1996) Selective recognition of malaria antigens by human serum antibodies is not genetically determined but demonstrates some features of clonal imprinting. Int. Immunol. 8, 905–915 [DOI] [PubMed] [Google Scholar]
- 69. Akpogheneta O. J., Duah N. O., Tetteh K. K., Dunyo S., Lanar D. E., Pinder M., Conway D. J. (2008) Duration of naturally acquired antibody responses to blood-stage Plasmodium falciparum is age dependent and antigen specific. Infect. Immun. 76, 1748–1755 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Greenwood B. M., Bradley-Moore A. M., Bryceson A. D., Palit A. (1972) Immunosuppression in children with malaria. Lancet 1, 169–172 [DOI] [PubMed] [Google Scholar]
- 71. Williamson W. A., Greenwood B. M. (1978) Impairment of the immune response to vaccination after acute malaria. Lancet 1, 1328–1329 [DOI] [PubMed] [Google Scholar]
- 72. Usen S., Milligan P., Ethevenaux C., Greenwood B., Mulholland K. (2000) Effect of fever on the serum antibody response of Gambian children to Haemophilus influenzae type b conjugate vaccine. Pediatr. Infect. Dis. J. 19, 444–449 [DOI] [PubMed] [Google Scholar]
- 73. Wingren C., Sandstrom A., Segersvard R., Carlsson A., Andersson R., Lohr M., Borrebaeck C. A. (2012) Identification of serum biomarker signatures associated with pancreatic cancer. Cancer Res. 72, 2481–2490 [DOI] [PubMed] [Google Scholar]
- 74. Anderson K. S., Sibani S., Wallstrom G., Qiu J., Mendoza E. A., Raphael J., Hainsworth E., Montor W. R., Wong J., Park J. G., Lokko N., Logvinenko T., Ramachandran N., Godwin A. K., Marks J., Engstrom P., Labaer J. (2011) Protein microarray signature of autoantibody biomarkers for the early detection of breast cancer. J. Prot. Res. 10, 85–96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Woodberry T., Minigo G., Piera K. A., Hanley J. C., de Silva H. D., Salwati E., Kenangalem E., Tjitra E., Coppel R. L., Price R. N., Anstey N. M., Plebanski M. (2008) Antibodies to Plasmodium falciparum and Plasmodium vivax merozoite surface protein 5 in Indonesia: species-specific and cross-reactive responses. J. Infect. Dis. 198, 134–142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Costa J. D., Zanchi F. B., Rodrigues F. L., Honda E. R., Katsuragawa T. H., Pereira D. B., Taborda R. L., Tada M. S., Ferreira Rde G., Pereira-da-Silva L. H. (2013) Cross-reactive anti-PfCLAG9 antibodies in the sera of asymptomatic parasite carriers of Plasmodium vivax. Mem. Inst. Oswaldo Cruz. 108, 98–105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Marshall V. M., Silva A., Foley M., Cranmer S., Wang L., McColl D. J., Kemp D. J., Coppel R. L. (1997) A second merozoite surface protein (MSP-4) of Plasmodium falciparum that contains an epidermal growth factor-like domain. Infect. Immun. 65, 4460–4467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Sanders P. R., Kats L. M., Drew D. R., O'Donnell R. A., O'Neill M., Maier A. G., Coppel R. L., Crabb B. S. (2006) A set of glycosylphosphatidyl inositol-anchored membrane proteins of Plasmodium falciparum is refractory to genetic deletion. Infect. Immun. 74, 4330–4338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Boyle M. J., Langer C., Chan J. A., Hodder A. N., Coppel R. L., Anders R. F., Beeson J. G. (2014) Sequential processing of merozoite surface proteins during and after erythrocyte invasion by Plasmodium falciparum. Infect. Immun. 82, 924–936 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Medeiros M. M., Fotoran W. L., dalla Martha R. C., Katsuragawa T. H., Pereira da Silva L. H., Wunderlich G. (2013) Natural antibody response to Plasmodium falciparum merozoite antigens MSP5, MSP9, and EBA175 is associated to clinical protection in the Brazilian Amazon. BMC Infect. Dis. 13, 608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Goschnick M. W., Black C. G., Kedzierski L., Holder A. A., Coppel R. L. (2004) Merozoite surface protein 4/5 provides protection against lethal challenge with a heterologous malaria parasite strain. Infect. Immun. 72, 5840–5849 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Kedzierski L., Black C. G., Goschnick M. W., Stowers A. W., Coppel R. L. (2002) Immunization with a combination of merozoite surface proteins 4/5 and 1 enhances protection against lethal challenge with Plasmodium yoelii. Infect. Immun. 70, 6606–6613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Kedzierski L., Black C. G., Stowers A. W., Goschnick M. W., Kaslow D. C., Coppel R. L. (2001) Comparison of the protective efficacy of yeast-derived and Escherichia coli-derived recombinant merozoite surface protein 4/5 against lethal challenge by Plasmodium yoelii. Vaccine 19, 4661–4668 [DOI] [PubMed] [Google Scholar]
- 84. Rainczuk A., Scorza T., Spithill T. W., Smooker P. M. (2004) A bicistronic DNA vaccine containing apical membrane antigen 1 and merozoite surface protein 4/5 can prime humoral and cellular immune responses and partially protect mice against virulent Plasmodium chabaudi adami DS malaria. Infect. Immun. 72, 5565–5573 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Rainczuk A., Smooker P. M., Kedzierski L., Black C. G., Coppel R. L., Spithill T. W. (2003) The protective efficacy of MSP4/5 against lethal Plasmodium chabaudi adami challenge is dependent on the type of DNA vaccine vector and vaccination protocol. Vaccine 21, 3030–3042 [DOI] [PubMed] [Google Scholar]
- 86. McCoubrie J. E., Miller S. K., Sargeant T., Good R. T., Hodder A. N., Speed T. P., de Koning-Ward T. F., Crabb B. S. (2007) Evidence for a common role for the serine-type Plasmodium falciparum serine repeat antigen proteases: implications for vaccine and drug design. Infect. Immun. 75, 5565–5574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Palacpac N. M., Arisue N., Tougan T., Ishii K. J., Horii T. (2011) Plasmodium falciparum serine repeat antigen 5 (SE36) as a malaria vaccine candidate. Vaccine 29, 5837–5845 [DOI] [PubMed] [Google Scholar]
- 88. Aoki S., Li J., Itagaki S., Okech B. A., Egwang T. G., Matsuoka H., Palacpac N. M., Mitamura T., Horii T. (2002) Serine repeat antigen (SERA5) is predominantly expressed among the SERA multigene family of Plasmodium falciparum, and the acquired antibody titers correlate with serum inhibition of the parasite growth. J. Biol. Chem. 277, 47533–47540 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.