

Abstract
The HUPO Proteomics Standards Initiative has developed several standardized data formats to facilitate data sharing in mass spectrometry (MS)-based proteomics. These allow researchers to report their complete results in a unified way. However, at present, there is no format to describe the final qualitative and quantitative results for proteomics and metabolomics experiments in a simple tabular format. Many downstream analysis use cases are only concerned with the final results of an experiment and require an easily accessible format, compatible with tools such as Microsoft Excel or R.
We developed the mzTab file format for MS-based proteomics and metabolomics results to meet this need. mzTab is intended as a lightweight supplement to the existing standard XML-based file formats (mzML, mzIdentML, mzQuantML), providing a comprehensive summary, similar in concept to the supplemental material of a scientific publication. mzTab files can contain protein, peptide, and small molecule identifications together with experimental metadata and basic quantitative information. The format is not intended to store the complete experimental evidence but provides mechanisms to report results at different levels of detail. These range from a simple summary of the final results to a representation of the results including the experimental design. This format is ideally suited to make MS-based proteomics and metabolomics results available to a wider biological community outside the field of MS. Several software tools for proteomics and metabolomics have already adapted the format as an output format. The comprehensive mzTab specification document and extensive additional documentation can be found online.
Mass spectrometry (MS)1 has become a major analysis tool in the life sciences (1). It is currently used in different modes for several “omics” approaches, proteomics and metabolomics being the most prominent. In both disciplines, one major burden in the exchange, communication, and large-scale (re-) analysis of MS-based data is the significant number of software pipelines and, consequently, heterogeneous file formats used to process, analyze, and store these experimental results, including both identification and quantification data. Publication guidelines from scientific journals and funding agencies' requirements for public data availability have led to an increasing amount of MS-based proteomics and metabolomics data being submitted to public repositories, such as those of the ProteomeXchange consortium (2) or, in the case of metabolomics, the resources from the nascent COSMOS (Coordination of Standards in Metabolomics) initiative (3).
In the past few years, the Human Proteome Organization Proteomics Standards Initiative (PSI) has developed several vendor-neutral standard data formats to overcome the representation heterogeneity. The Human Proteome Organization PSI promotes the usage of three XML file formats to fully report the data coming from MS-based proteomics experiments (including related metadata): mzML (4) to store the “primary” MS data (the spectra and chromatograms), mzIdentML (5) to report peptide identifications and inferred protein identifications, and mzQuantML (6) to store quantitative information associated with these results.
Even though the existence of the PSI standard data formats represents a huge step forward, these formats cannot address all use cases related to proteomics and metabolomics data exchange and sharing equally well. During the development of mzML, mzIdentML, and mzQuantML, the main focus lay on providing an exact and comprehensive representation of the gathered results. All three formats can be used within analysis pipelines and as interchange formats between independent analysis tools. It is thus vital that these formats be capable of storing the full data and analysis that led to the results. Therefore, all three formats result in relatively complex schemas, a clear necessity for adequate representation of the complexity found in MS-based data.
An inevitable drawback of this approach is that data consumers can find it difficult to quickly retrieve the required information. Several application programming interfaces (APIs) have been developed to simplify software development based on these formats (7–9), but profound proteomics and bioinformatics knowledge still is required in order to use them efficiently and take full advantage of the comprehensive information contained.
The new file format presented here, mzTab, aims to describe the qualitative and quantitative results for MS-based proteomics and metabolomics experiments in a consistent, simpler tabular format, abstracting from the mass spectrometry details. The format contains identifications, basic quantitative information, and related metadata. With mzTab's flexible design, it is possible to report results at different levels ranging from a simple summary or subset of the complete information (e.g. the final results) to fairly comprehensive representation of the results including the experimental design. Many downstream analysis use cases are only concerned with the final results of an experiment in an easily accessible format that is compatible with tools such as Microsoft Excel® or R (10) and can easily be adapted by existing bioinformatics tools. Therefore, mzTab is ideally suited to make MS proteomics and metabolomics results available to the wider biological community, beyond the field of MS.
mzTab follows a similar philosophy as the other tab-delimited format recently developed by the PSI to represent molecular interaction data, MITAB (11). MITAB is a simpler tab-delimited format, whereas PSI-MI XML (12), the more detailed XML-based format, holds the complete evidence. The microarray community makes wide use of the format MAGE-TAB (13), another example of such a solution that can cover the main use cases and, for the sake of simplicity, is often preferred to the XML standard format MAGE-ML (14). Additionally, in MS-based proteomics, several software packages, such as Mascot (15), OMSSA (16), MaxQuant (17), OpenMS/TOPP (18, 19), and SpectraST (20), also support the export of their results in a tab-delimited format next to a more complete and complex default format. These simple formats do not contain the complete information but are nevertheless sufficient for the most frequent use cases.
mzTab has been designed with the same purpose in mind. It can be used alone or in conjunction with mzML (or other related MS data formats such as mzXML (21) or text-based peak list formats such as MGF), mzIdentML, and/or mzQuantML. Several highly successful concepts taken from the development process of mzIdentML and mzQuantML were adapted to the text-based nature of mzTab.
In addition, there is a trend to perform more integrated experimental workflows involving both proteomics and metabolomics data. Thus, we developed a standard format that can represent both types of information in a single file.
EXPERIMENTAL PROCEDURES
The development of mzTab was influenced by existing text-based output formats from several search engines and analysis pipelines. Development of the model started at the beginning of 2011, after the finalization of mzIdentML 1.1. The complete mzTab specification document (version 1.0) can be found online (http://mztab.googlecode.com), along with example files demonstrating the use of mzTab for several scenarios.
mzTab aims to
be simpler than the XML-based standard formats to make MS-based proteomics/metabolomics results accessible to researchers outside the respective fields;
be accessible using standard software such as Microsoft Excel® or Open Office Spreadsheet;
allow the reporting of results at different levels of detail, ranging from the summarized final results to a detailed representation of the results including the experimental design;
safeguard/require a minimal level of information about a proteomics/metabolomics study's results to be used as a standard documentation format for “supplementary material” sections of publications;
support export from mzIdentML and mzQuantML;
support efficient import into statistical tools such as R, SIMCA®, or SPSS®;
make MS-derived results easily accessible to scripting languages;
be suitable as an output format for Web-based services; and
support references back to more detailed sources.
Although mzTab can be used to report a detailed view on data, it explicitly does not aim to capture the whole complexity and evidence trail of a proteomics study. Even the most complex mzTab files still include simplifications/assumptions of the experimental results. This, for instance, is the case in identification (e.g. protein inference/grouping is only supported to a limited extent) and quantification results (e.g. the coordinates for isotope patterns in quantified two-dimensional “features” cannot be fully reported). This missing information can be reported using the existing PSI standard formats mzIdentML and mzQuantML.
The model was developed as a tab-delimited file accompanied by controlled vocabulary (CV) terms and definitions as part of the PSI-Mass Spectrometry CV, also used in other PSI formats and actively maintained by the PSI-Mass Spectrometry and PSI-Proteomics Informatics working groups (22).
mzTab Overview
mzTab is a tab-delimited text file format that can store protein, peptide, peptide-spectrum match (PSM), small-molecule identification, and basic quantification data, as well as different levels of metadata related to the processed samples, experimental design, and used experimental methods. The data are stored in five different sections: “metadata,” “protein,” “peptide,” “PSM,” and “small molecule” (Fig. 1). Every line in an mzTab file starts with a three-letter code indicating the type of information captured in the line: “MTD” for lines from the metadata section, “PRH” for the “protein” section header, “PRT” for protein identifications, “PEH” for the “peptide” section header, “PEP” for peptide identifications, “PSH” for the “PSM” section header, “PSM” for PSMs, “SMH” for the “small molecule” section header, “SML” for small molecule identifications, and “COM” for a comment line. The “metadata” section is mandatory; every other section is optional but must not be present more than once in a single file.
Fig. 1.

A diagrammatic representation of the data model for mzTab.
There are two types of mzTab files: identification and quantification. This is specified in the mandatory metadata field “mzTab-type.” Identification files can be used to report peptide, protein, and small molecule identifications. Quantification files can be used for quantification results, which optionally may contain identification results about the quantified protein/peptide or small molecules.
In addition, there are two levels of detail (called “modes”) of reporting data in mzTab files: “summary” and “complete.” The “summary” mode can be used to report the final results, putting together data coming from, for example, different replicates. The “complete” mode is used if detailed information for each individual assay/replicate is provided. The mode is specified in the mandatory metadata field “mzTab-mode.” This flexible design was needed to support as many different experimental scenarios as possible in a single, relatively simple file format.
Similar to mzIdentML and mzQuantML, mzTab files do not contain the underlying MS data but may contain references to the spectra in external files. mzTab uses exactly the same method to reference external spectra as do mzIdentML and mzQuantML. Additionally, each identification reported in an mzTab file can be linked to its original source.
Modeling an Experimental Design in mzTab
mzTab supports the reporting of technical/biological replicates within experimental designs using an adaptation of the system originally developed for mzQuantML. This is made up of four components:
(i) Study variable. Study variables represent the core final results of the study (e.g. inflammatory response versus control). Often, these will have been derived by averaging the results of a group of replicate measurements (assays). In files where assays are reported, study variables have references to assays. The same concept could be called an “experimental factor.”
(ii) MS run. An MS run is effectively one run (or set of runs on prefractionated samples) on an MS instrument and is referenced from the assay in different contexts.
(iii) Assay—the application of a measurement of the sample (in this case through MS) producing values about small molecules, peptides, or proteins. One assay is typically mapped to one MS run in the case of label-free MS analysis, or multiple assays are mapped to one MS run for multiplexed techniques (e.g. iTRAQ or SILAC), along with a description of the label or tag applied.
(iv) Sample—a biological material that has been analyzed, to which descriptors of species such as cell/tissue type can be attached. In all mzTab files, these can be reported in the metadata section as “sample[1-n]-description.” Samples are not mandatory in mzTab, as many software packages cannot determine what type of sample was analyzed (e.g. whether biological or technical replication was performed).
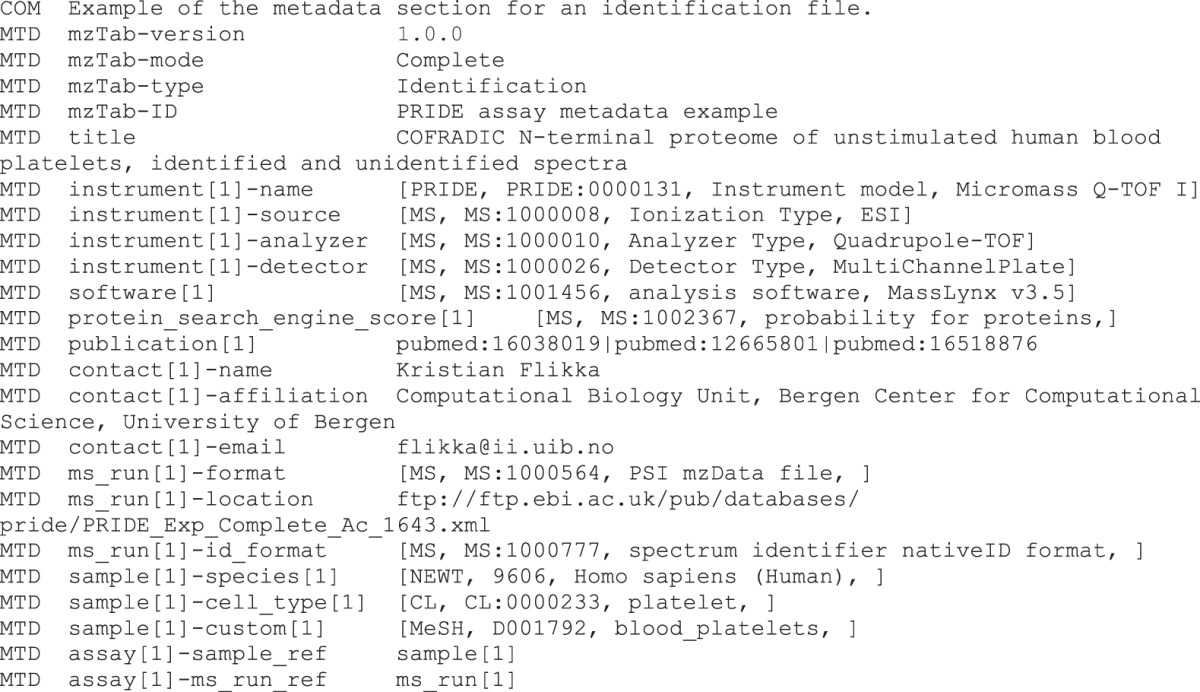
Metadata Section
The “metadata” section in mzTab was deliberately kept flexible, and the majority of fields are optional. Therefore, it is possible to report different levels of experimental annotation depending on the interest of the producer of the files, ranging from basic annotations to the complete metadata defined by the Human Proteome Organization PSI as the minimum information about a proteomics experiment (MIAPE) (23) or the core information for metabolomics reporting (CIMR) guidelines (24).
Whereas the “protein,” “peptide,” “PSM,” and “small molecule” sections are classical table-based structures with a header line, the “metadata” section contains one tab-separated key-value pair per row. This layout was chosen because the “metadata” section can contain considerably more fields than other sections, and this arrangement helps the files remain more readable.
The “metadata” section can hold basic information about the study—for example, a title, a human readable description, contact information, publication references, false discovery rates, basic properties of the analyzed sample(s), assays, MS runs, and study variables. The protocol can be described using CV terms for individual steps. Information about the mass spectrometer(s) used can be stored, as well as details about the software tool(s) (including settings) used to generate the data. Furthermore, it is possible to specify the quantification method used together with the reagents. Similar to the additional columns in the table-based sections (see below), it is possible to specify custom fields in the metadata. These can hold any kind of information as CV or user parameters. Several example snippets are present throughout the manuscript to facilitate the comprehension of the format. Nevertheless, these are not comprehensive examples of mzTab files. Complete examples meant to be taken as references can be found online (http://mztab.googlecode.com). Below is an excerpt from the “metadata” section.
Peptide and Protein Identification Results
Protein/peptide identifications are reported in the “protein” and “PSM” sections, respectively. As mentioned above, the “peptide” section is used to report aggregated quantification data based on several PSMs. Its use is not recommended in identification files. In the “PSM” section, peptide identifications are linked to proteins by the “accession” column. It is also possible to report peptides that do not reference any protein by setting the protein accession to “null”.
Next to the protein accession, the “protein” section can hold a human-readable description (generally the protein's name), the species, and the source protein database. As an overview, three kinds of values can be reported in order to indicate how many PSMs support a protein identification: the total number of PSMs, the number of distinct peptides (based on both the sequence and modifications), and the number of unique peptides (distinct number of peptides that can be unambiguously assigned to the given protein in the sequence database). Furthermore, it is possible to provide the coverage of the protein sequence by the identified peptides; the search engine(s) used, including scores that identified the protein; and a reliability score of the protein identification. In addition, gene ontology terms that are used to annotate the protein (25) can be reported in this section. Below is an example from the “protein” section.

In the “PSM” section, it is possible to indicate whether the peptides were unambiguously assigned to a given protein. Additionally, for proteins, information about the sequence database and the search engines used for the identification can be included. Furthermore, MS-specific information, such as the precursor's m/z ratio, charge state, and retention time, can be stored. Every PSM can be linked to a spectrum in an external MS data file and can be given a reliability score (see below).
In current proteomics pipelines, it has become a common procedure to process the MS data using multiple search engines (26, 27) and post-processing software. mzTab explicitly supports this use case by allowing peptide and protein identifications to be associated with multiple search engines and search engine scores. Thus, the information that a given peptide was, for example, identified by three search engines whereas another peptide was identified by only a single search engine, as well as a possible overall score, can be reported explicitly. Search engine scores are reported in one column per score (search_engine_score[1-n]). The type of score reported in the different sections is defined in the “metadata” section using a CV parameter. Below is an example from the “PSM” section.
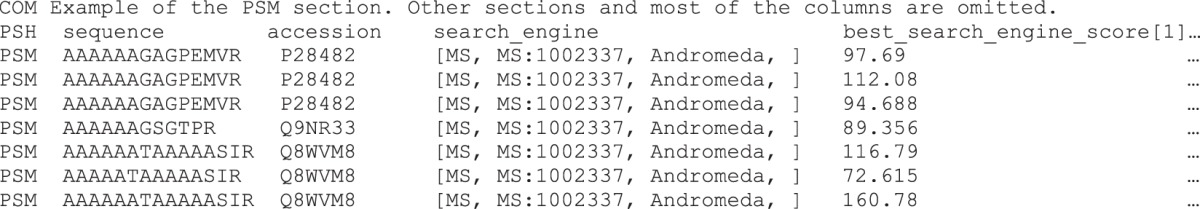
It is common that an identified peptide sequence cannot be unambiguously attributed to a single protein and may originate from several proteins—the so-called protein inference problem (28). It is not possible to model detailed protein inference data without a significant level of complexity, as in mzIdentML (5). Therefore, as a compromise, it was decided to model protein inference in a simple way in mzTab, excluding detailed information on how ambiguity was resolved. Protein entries in mzTab files contain the column “ambiguity_members.” The protein accessions listed in this field should identify proteins that were also identified through the same set of peptides or spectra, or proteins supported by a largely overlapping set of evidence, and could also be a viable candidate for the “true” identification of the entity reported. Therefore, the definition of “indistinguishable proteins” and thus also of the “ambiguity_members” is up to the generating software and will vary between mzTab files generated by different resources. The mapping of a single PSM to multiple accessions is supported through the reporting of the same PSM on multiple rows of the PSM section, as exemplified below.

Metabolomics Identification Results
There is currently no widely used standardized file format available to store MS-based metabolomics data. The COSMOS initiative (3) aims to address this issue and is working on the development of the exchange formats and terminological artifacts needed to describe, exchange, and query both metabolomics data and experimental metadata. In addition, mzQuantML has been designed to capture such data in a detailed manner, but its formalization is ongoing. We therefore added explicit structures to mzTab that can hold MS-based metabolomics identifications and quantification results. Small molecules are specified through an identifier in mzTab. These identifiers should generally be entries in compound databases used in the respective field—for example, Human Metabolome Database entries (29) or identifiers from Chemical Entities of Biological Interest (30), PubChem (31), LipidHome (32), or LIPID MAPS (33). Apart from these unique identifiers, small molecules can be assigned a chemical formula; Simplified Molecular-input Line-entry System (SMILES) and/or IUPAC International Chemical Identifier identifiers; or a human-readable description together with MS-related information such as m/z value, charge state, retention time, source database, and search engine, including, potentially, several scores. Furthermore, every small-molecule identification can also be linked to a source spectrum and given a reliability score based on an agreement from the Metabolomics Standards Initiative (24, 34) (see below). Below is an example from the “small molecule” section.
Use of Controlled Vocabulary
Because the use of CVs and ontologies has proved to be effective in other PSI formats, the same principle was applied for mzTab. CV terms help to make the stored data machine-comprehensible and comparable. Therefore, mzTab also makes use of CV terms to flexibly report several pieces of information and uses the same CVs that are being constantly enhanced for use with mzIdentML and mzQuantML (e.g. PSI-Mass Spectrometry).
In mzTab, CV parameters are reported using the simple structure [CV label, accession, name, value]—for example, [MS, MS:1001364, pep:global FDR, 0.1]. If a value cannot be represented using a CV term, the data producers must contact PSI to have the terms added to the CV in order to guarantee the integrity of the reported data. However, an alternative mechanism exists: so-called user parameters can be reported, and in this case reporting of the CV label and accession is not mandatory. This is useful, for example, when a new technology is introduced that is not yet represented in the respective CV or ontology. These parameters are not machine-comprehensible but at least provide the possibility of storing the data in human-readable form. The names of user parameters should be chosen by the user to describe the semantics of the reported value.

Identification Reliabilities
All protein, peptide, PSM, and small-molecule identifications reported in an mzTab file can be assigned a reliability score (“reliability” column in all tables).
The reliability is reported as an integer between 1 and 3 for proteomics results and should be interpreted as follows:
1: high reliability
2: medium reliability
3: poor reliability
For metabolomics (“small molecule” section), according to the current Metabolomics Standards Initiative agreement, the reliability should be reported as an integer between 1 and 4 and should be interpreted as follows:
1: identified metabolites
2: putatively annotated compounds
3: putatively characterized compound classes
4: unknown compounds
These abstract quality metrics were chosen over others to highlight the fact that scores from different sources and/or pipelines are not directly comparable, even if they are represented as p or e values. Thus, this score makes it apparent that any scoring approach is completely dependent on the data producer.
The idea behind this score was to mimic the general concept of “resource-based trust” and complement the rather complex search engine scores with a simple score accessible to non-proteomics experts. If resources/researchers now report their reliabilities using these metrics and document how their metric is generated, users can base their own interpretation of the results on their trust in the resource or research group.
Reporting Modifications
Modifications are modeled using a specific modification object with the following format: {position}{Parameter}-{Modification or Substitution identifier} {neutral loss}.
{position} is optional depending on the section where the modification is reported, as it might not be applicable to all types of small molecules. N- and C-terminal modifications in proteins and peptides are reported with the position set at zero or the amino acid length plus one, respectively. This element also allows modifications to be assigned to ambiguous locations and can be used to report reliabilities for every potential location using CV parameters. mzTab is thus the first PSI file format that explicitly models ambiguous modification positions as well as position-based scores. Neutral losses can also be reported using CV parameters. They may be either reported as separate modifications or associated with an existing one by appending it to the modification object.
For proteins and peptides, modifications should be reported using either Unimod (35) or the PSI-MOD (36) identifiers as {Modification identifier}. Because these two ontologies are not applicable to small molecules, so-called CHEMMODs were suggested for mzTab. Two types of CHEMMODs are allowed: specifying a chemical formula and specifying a mass shift. CHEMMODs must not be used for protein/peptide modifications if the respective entry is present in either the PSI-MOD or the Unimod ontology. Furthermore, mass differences must not be reported if the given difference can be expressed through a known and unambiguous chemical formula.
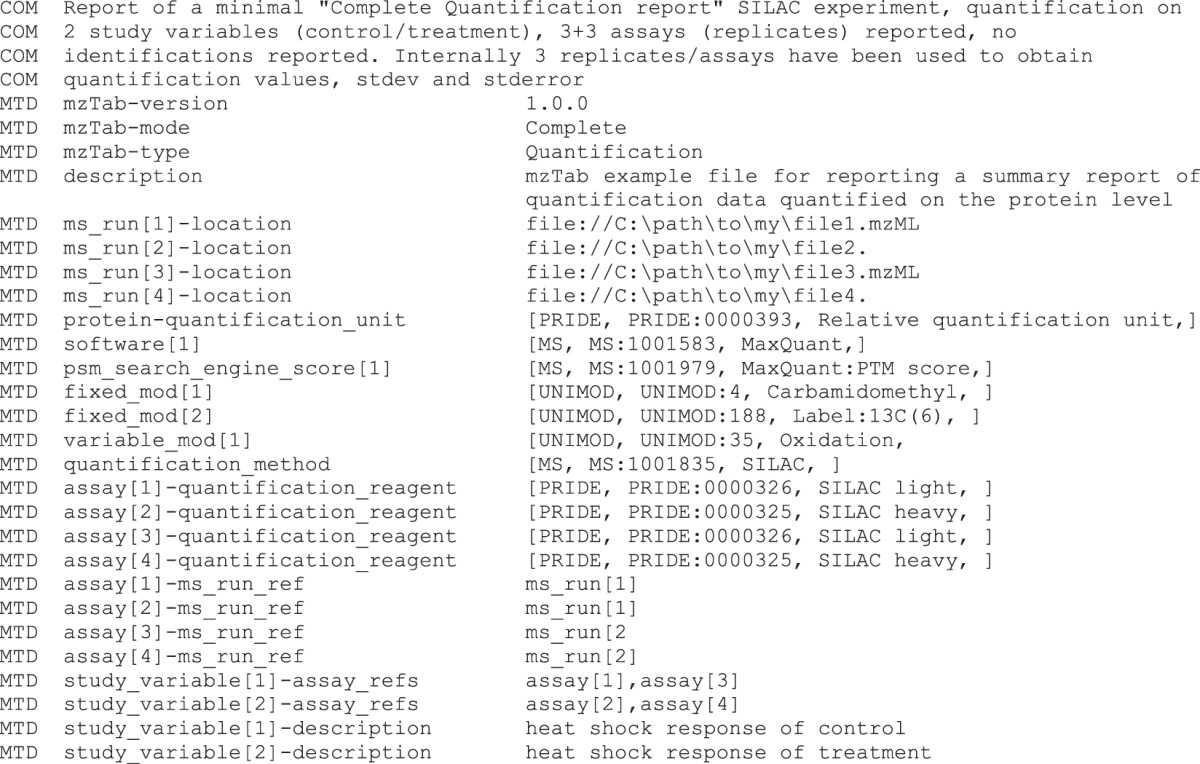
Quantitative Data
There are multiple quantification techniques available for MS-based experiments that often result in different types of data. The goal of mzTab was to provide a generic view on quantitative MS-based identification data. This is achieved using quantitation mzTab files. Additionally, the “complete” files allow reporting on the experimental design, a key point in every quantification study. The “peptide” section is used to aggregate quantification data on a per-peptide level.
Quantification techniques generally result in a numerical abundance measurement of an analyte. Some techniques allow or require multiple samples to be multiplexed and analyzed in a single MS run. When several biological samples are multiplexed, these samples are referred to as “assays” in mzTab. For every assay, an abundance value must be reported for each peptide, protein, or small-molecule identification (with nulls or zeros allowed if an entity is not measured or measured with a zero value in any given replicate). The used quantification method, metric units, and, for example, label reagent for a specific assay should be reported in the “metadata” section. Study variables can be included that encompass multiple assays and for which standard deviation and standard error should be reported.
The results of quantitative measurements are reported in additional columns that are appended after the mandatory columns in the respective section. The assay and/or study variables' I.D.s are part of the column name (for example, protein_abundance_assay[1] or protein_abundance_study_variable[1]).
It is not expected that these numbers will be comparable across multiple mzTab files, as different quantitative methods can generally not be compared directly. This method of reporting quantification data is used in exactly the same way for proteins, peptides, and small molecules. There are several example files online (http://mztab.googlecode.com) that exemplify how quantitative data can be reported in mzTab files including different approaches such as SILAC, iTRAQ, or label-free quantification. There is also an example from a quantitative lipidomics experiment. Below is an example corresponding to one SILAC experiment.
Comments on Specific Use Cases
Additional Columns to Report Additional Custom Information
It is desirable to be able to report information not included in the “core model” of any data format. For this reason, additional columns can be added to mzTab files to hold such custom data. Additional columns can be added to the end of the existing columns in all the table-based sections (“protein,” “peptide,” “PSM,” and “small molecule”). These column headers must start with the prefix “opt_” or “opt_global” (see details in the specification document) and can be used to store resource-dependent information. It is recommended that one use CV parameter accessions as optional column names and thereby explicitly define their contents. These column names follow the format opt_cv_{accession}_{parameter name} (for example, opt_cv_MS:1001975_delta_m/z to report the “delta m/z” for each peptide identification, or opt_cv_MS:1002217_decoy_peptide to identify decoy peptide identifications).
Referencing External Resources
All identifications reported in an mzTab file can reference external resources that contain detailed evidence supporting the identification. This link is stored in the “URI” column present in each of the table-based sections. The target that these universal resource identifiers point to is dependent on the resource that generated the mzTab file. If, for example, PeptideAtlas was exporting data in the mzTab format, the universal resource identifier would be expected to point to the identification's entry within the respective PeptideAtlas build. mzTab files originating from an mzIdentML file, for example, can reference the mzIdentML file using the “URI” column. When quantitative values are reported coming from an mzQuantML file, the mzQuantML file should be referenced, as it contains the reference to the underlying mzIdentML file. Thus, the simplified data represented in an mzTab file can easily be connected to the complete underlying evidence.
Referencing External Spectra
In mzTab, peptides and small molecules can be linked to source spectra in external files. This is accomplished through the “ms-run[1-n]-location” field in the “metadata” section, which holds all characteristics of a specific source file. The “spectra_ref” column in the appropriate tables is then used to unambiguously link to specific spectra within these source files.
Software Implementations
Several software implementations already exist for mzTab that can be used by developers and end-users (Table I). The mzTab schema was constantly tested during its development through various implementations. The jmzTab Java API is the current reference implementation for mzTab (37). It supports reading and writing of mzTab files and automatically checks their structural validity. The API provides functionality to convert PRIDE XML files to mzTab.
Table I. Software implementations of mzTab (by June 2014).
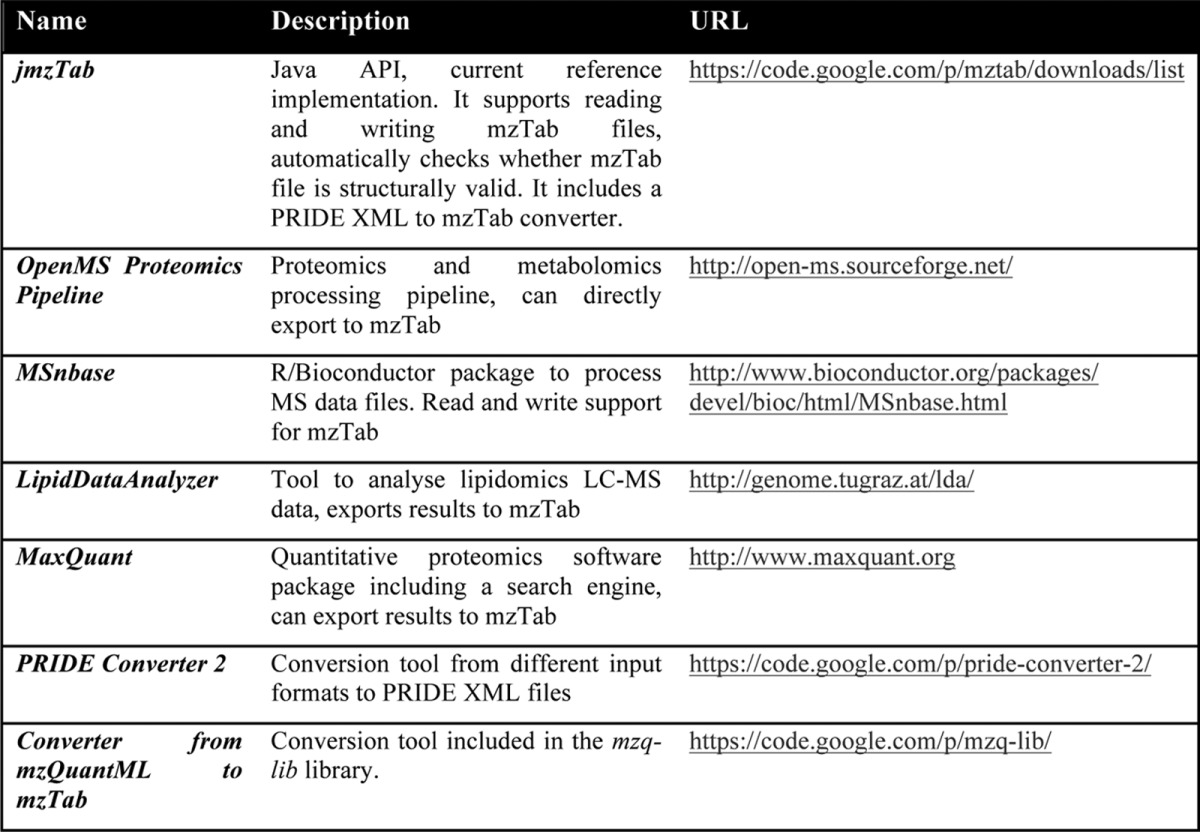
In addition, several popular proteomics software tools can already use mzTab files. OpenMS (19) offers an infrastructure for the development of MS-related software. Together with the OpenMS Proteomics Pipeline (18), it provides a full-featured proteomics and metabolomics data processing pipeline that can directly write mzTab files from version 1.11 on. In the MaxQuant software (17), a converter to mzTab format will be available from version 1.4.2 onward. The converter is implemented as a plugin for the Perseus software package.
mzTab read and write support has also been added to the R/Bioconductor package MSnbase (38), from version 1.5.6. It can seamlessly import data stored in mzTab files, including quantitative data and metadata, into flexible object classes to subsequently apply contemporary state-of-the-art statistical procedures readily available through the R statistical programming environment. The package provides ample documentation and examples on how to import from and export to mzTab.
The LipidDataAnalyzer (39), a tool for analyzing lipidomics LC-MS data developed at the Graz University of Technology, is the first non-proteomics tool to fully support mzTab export using the jmzTab API (from version 1.6.0). The LipidDataAnalyzer is able to export identified molecules from related experiments in one mzTab file including quantitative information extracted from MS data.
Apart from these tools, the new PRIDE submission tool, PRIDE Converter 2 (40), can convert multiple search engine output files to mzTab files and can also read quantitative and gel-based data from mzTab files to include them in the generated PRIDE XML files. PRIDE Converter 2 uses the jmzTab API to process mzTab files, allowing the inclusion of quantification information into PRIDE XML. Finally, a library of Java routines has been created for mzQuantML that contains a converter from mzQuantML to mzTab files.
To make the development of applications supporting mzTab as easy as possible, we created an additional document specifically targeted at developers: The 20 Minute Guide to mzTab. This is a quick introduction to the basics of mzTab and explains the format using examples rather than words. Additionally, it includes a comprehensive list of all fields and columns found in mzTab. This document, as well as the comprehensive specification document, can be found online (http://mztab.googlecode.com). We also created a comparison of the main features of mzIdentML, mzQuantML, and mzTab (supplemental Table S1) and a size comparison between different mzTab and PRIDE XML files (supplemental Table S2).
Additional Use Cases
One of the main goals of mzTab is to share and communicate MS-based proteomics and metabolomics results with researchers from other biological sciences. mzTab can provide a standardized summary of data submissions to proteomics repositories. mzTab is used in the context of the ProteomeXchange consortium data workflow. The ProteomeXchange consortium aims to promote standard submission and data-sharing policies among the main MS-based proteomics data repositories, including PRIDE and PeptideAtlas. At present, summary mzTab files are generated for each MS/MS “complete” submission to ProteomeXchange via PRIDE and are made available for download.
Laboratory information management systems often do not need to and cannot store the complete information acquired during a proteomics experiment. Still, researchers require a quick overview of previously performed experiments in their laboratory information management systems. mzTab might thus be an ideal basis for the development of laboratory information management systems that need to store basic proteomics/metabolomics results. The flexibility of the format and the possibility of adding custom fields make it suitable for storing highly heterogeneous data. Through the addition of optional columns after the mandatory fields, mzTab can be tailored for the specific needs of each research group.
For metabolomics, the development of mzTab might be even more helpful because, to the best of our knowledge, there is no widely accepted data format for MS-based results yet. mzIdentML was not developed with this use case in mind and cannot be easily adapted to support non-proteomics data. mzQuantML aims to store quantification information for small molecules, but the mechanism still needs to be formalized. At present, mzTab can report results from lipidomics approaches, as demonstrated by the implementation in the LipidDataAnalyzer (39). The MetaboLights repository (41) at the European Bioinformatics Institute for metabolomics experiments has so far used a version of mzTab that contains the “small molecule” section but not a fully compliant “metadata” section. However, for streamlining data submissions, it is planned to fully comply with the final version of the format specification.
We also envisage that the format will become a way to standardize the supplementary information provided by authors to scientific journals. In the case of proteomics, there are different reporting policies depending on the journal. Molecular & Cellular Proteomics, an early adopter in terms of guidelines, developed the so-called Paris guidelines for reporting proteomics data (42), which include a requirement to provide different metrics related to the published data. The reliability score could be used by the authors or the journal to report which proportion of the reported data in a manuscript met the quality criteria (if any) for each identification. Level 1 (high reliability) would mean “criteria passed,” whereas 3 (poor reliability) would mean the opposite, and 2 (medium reliability) would be reserved for borderline cases that are not straightforward to assess. Also, as mentioned before, different reporting requirements can be supported thanks to the flexibility of the format.
CONCLUSIONS
mzTab was developed to bridge the gap between the high level of detail found in existing XML-based standards formats, required in order to model complete proteomics data, and the need to easily report proteomics and metabolomics final results. The format was developed by members of the PSI Proteomics Informatics working group and other collaborators representing a wide array of academic research groups and projects. The complete development process was based around the mzTab Google Code page, which also hosts the complete format specification and the official software implementation jmzTab. The concept behind mzTab has previously been successfully applied in other bioinformatics fields (for instance, MAGE-TAB and MITAB).
After the standardization process is finished, mzTab will be a stable format. However, if needed, extensions to the proposed structure can be made in a flexible way. For instance, new optional sections in addition to the existing ones (“metadata,” “protein,” “peptide,” “PSM,” and “small molecule”) could be added without breaking existing software. However, alterations to the schema that would break existing parsing code will not be made without the format being reentered into the standardization process. We expect that the release of mzTab will increase data sharing for MS-based proteomics and metabolomics. We also envisage that the format could be extended to support other uses cases, such as peptide or small-molecule spectral clusters (43), or adapted to report selected reaction monitoring transitions, including the related quantification results.
We encourage readers to provide further input on the standard by contacting the authors, joining the PSI mailing list via the Google Code page, or attending a PSI meeting.
Supplementary Material
Acknowledgments
We acknowledge our colleagues in the Proteomics Standards Initiative for helpful discussions and feedback. We want to particularly thank Dr. Robert J. Chalkley for his input on the format.
Footnotes
Author contributions: A.R.J., O.K., J.V., and H.H. designed research; J.G., A.R.J., T.S., M.W., L.G., J.H., G.G.T., R.M.S., C.S., N.N., J.C., S.N., J.F., F.R., Q.X., N.d., Y.P., F.G., N.B., I.X., and J.V. performed research; J.G., J.V., and H.H. wrote the paper.
* J.G., F.R., N.d.T., and J.A.V. are supported by the Wellcome Trust (Grant Nos. WT085949MA and WT101477MA). J.G. is also funded by a grant from the Vienna Science and Technology Fund (WWTF) (Project LS11-045). J.A.V., A.R.J., and Q.W.X. acknowledge EU FP7 grant ProteomeXchange (Grant No. 260558). J.A.V. was also supported by EU FP7 grant LipidomicNet (Grant No. 202272). H.H. wants to acknowledge BBSRC (BB/I00095X/1). J.H. was supported by FFG, bmvit, mvwfi, ZIT, Zukunftsstiftung Tirol, and Land Steiermark within the Austrian COMET program (FFG Grant 824186). G.G.T. was supported by the Austrian Ministry of Science and Research GEN-AU Project BIN (FFG Grant 820962). A.R.J. and Y.P.-R. acknowledge support from BBSRC (Grant No. BB/K01997X/1). A.R.J. is also supported by grants from BBSRC (BB/H024654/1, BB/I00095X/1). O.K. is supported by grants from EU FP7 MARINA (Grant No. 236215), Deutsche Forschungsgemeinschaft (SFB685/B1 and SPP1335), and BMBF (SARA - FKZ 0315395F and BIOMARKERS - FKZ 01GI1104A). L.G. is supported by EU FP7 grant PRIME-XS (Grant No. 262067), also acknowledged by J.A.V. and O.K. I.X. was supported in part by the Swiss Federal Government through the State Secretariat for Education, Research and Innovation (SERI). R.M.S. is supported by EU FP7 grant COSMOS (Grant No. EC312941), also acknowledged by C.S. and S.N. N.B. is an Alfred P. Sloan Research Fellow and was supported by NIH/NIGMS (8P41GM103485-05).
 This article contains supplemental material.
This article contains supplemental material.
1 The abbreviations used are:
- MS
- mass spectrometry
- API
- application programming interface
- CV
- controlled vocabulary
- PSI
- Proteomics Standards Initiative
- PSM
- peptide spectrum match.
REFERENCES
- 1. Editors (2007) Mind the technology gap. Nat. Methods 4, 765. [DOI] [PubMed] [Google Scholar]
- 2. Vizcaino J. A., Deutsch E. W., Wang R., Csordas A., Reisinger F., Rios D., Dianes J. A., Sun Z., Farrah T., Bandeira N., Binz P. A., Xenarios I., Eisenacher M., Mayer G., Gatto L., Campos A., Chalkley R. J., Kraus H. J., Albar J. P., Martinez-Bartolome S., Apweiler R., Omenn G. S., Martens L., Jones A. R., Hermjakob H. (2014) ProteomeXchange provides globally coordinated proteomics data submission and dissemination. Nat. Biotechnol. 32, 223–226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Steinbeck C., Conesa P., Haug K., Mahendraker T., Williams M., Maguire E., Rocca-Serra P., Sansone S. A., Salek R. M., Griffin J. L. (2012) MetaboLights: towards a new COSMOS of metabolomics data management. Metabolomics 8, 757–760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Martens L., Chambers M., Sturm M., Kessner D., Levander F., Shofstahl J., Tang W. H., Rompp A., Neumann S., Pizarro A. D., Montecchi-Palazzi L., Tasman N., Coleman M., Reisinger F., Souda P., Hermjakob H., Binz P. A., Deutsch E. W. (2011) mzML—a community standard for mass spectrometry data. Mol. Cell. Proteomics 10, R110.000133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Jones A. R., Eisenacher M., Mayer G., Kohlbacher O., Siepen J., Hubbard S. J., Selley J. N., Searle B. C., Shofstahl J., Seymour S. L., Julian R., Binz P. A., Deutsch E. W., Hermjakob H., Reisinger F., Griss J., Vizcaino J. A., Chambers M., Pizarro A., Creasy D. (2012) The mzIdentML data standard for mass spectrometry-based proteomics results. Mol. Cell. Proteomics 11, M111.014381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Walzer M., Qi D., Mayer G., Uszkoreit J., Eisenacher M., Sachsenberg T., Gonzalez-Galarza F. F., Fan J., Bessant C., Deutsch E. W., Reisinger F., Vizcaino J. A., Medina-Aunon J. A., Albar J. P., Kohlbacher O., Jones A. R. (2013) The mzQuantML data standard for mass spectrometry-based quantitative studies in proteomics. Mol. Cell. Proteomics 12, 2332–2340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Cote R. G., Reisinger F., Martens L. (2010) jmzML, an open-source Java API for mzML, the PSI standard for MS data. Proteomics 10, 1332–1335 [DOI] [PubMed] [Google Scholar]
- 8. Reisinger F., Krishna R., Ghali F., Rios D., Hermjakob H., Vizcaino J. A., Jones A. R. (2012) jmzIdentML API: a Java interface to the mzIdentML standard for peptide and protein identification data. Proteomics 12, 790–794 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Qi D., Krishna R., Jones A. R. (2014) The jmzQuantML programming interface and validator for the mzQuantML data standard. Proteomics 14, 685–688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. R Development Core Team (2013) R: A Language and Environment for Statistical Computing [Google Scholar]
- 11. Orchard S., Albar J. P., Deutsch E. W., Eisenacher M., Vizcaino J. A., Hermjakob H. (2011) Enabling BioSharing—a report on the Annual Spring Workshop of the HUPO-PSI April 11–13, 2011, EMBL-Heidelberg, Germany. Proteomics 11, 4284–4290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kerrien S., Orchard S., Montecchi-Palazzi L., Aranda B., Quinn A. F., Vinod N., Bader G. D., Xenarios I., Wojcik J., Sherman D., Tyers M., Salama J. J., Moore S., Ceol A., Chatr-Aryamontri A., Oesterheld M., Stumpflen V., Salwinski L., Nerothin J., Cerami E., Cusick M. E., Vidal M., Gilson M., Armstrong J., Woollard P., Hogue C., Eisenberg D., Cesareni G., Apweiler R., Hermjakob H. (2007) Broadening the horizon—level 2.5 of the HUPO-PSI format for molecular interactions. BMC Biol. 5, 44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Rayner T. F., Rocca-Serra P., Spellman P. T., Causton H. C., Farne A., Holloway E., Irizarry R. A., Liu J., Maier D. S., Miller M., Petersen K., Quackenbush J., Sherlock G., Stoeckert C. J., Jr., White J., Whetzel P. L., Wymore F., Parkinson H., Sarkans U., Ball C. A., Brazma A. (2006) A simple spreadsheet-based, MIAME-supportive format for microarray data: MAGE-TAB. BMC Bioinformatics 7, 489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Spellman P. T., Miller M., Stewart J., Troup C., Sarkans U., Chervitz S., Bernhart D., Sherlock G., Ball C., Lepage M., Swiatek M., Marks W. L., Goncalves J., Markel S., Iordan D., Shojatalab M., Pizarro A., White J., Hubley R., Deutsch E., Senger M., Aronow B. J., Robinson A., Bassett D., Stoeckert C. J., Jr., Brazma A. (2002) Design and implementation of microarray gene expression markup language (MAGE-ML). Genome Biol. 3, RESEARCH0046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Perkins D. N., Pappin D. J., Creasy D. M., Cottrell J. S. (1999) Probability-based protein identification by searching sequence databases using mass spectrometry data. Electrophoresis 20, 3551–3567 [DOI] [PubMed] [Google Scholar]
- 16. Geer L. Y., Markey S. P., Kowalak J. A., Wagner L., Xu M., Maynard D. M., Yang X., Shi W., Bryant S. H. (2004) Open mass spectrometry search algorithm. J. Proteome Res. 3, 958–964 [DOI] [PubMed] [Google Scholar]
- 17. Cox J., Mann M. (2008) MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 26, 1367–1372 [DOI] [PubMed] [Google Scholar]
- 18. Kohlbacher O., Reinert K., Gröpl C., Lange E., Pfeifer N., Schulz-Trieglaff O., Sturm M. (2007) TOPP—the OpenMS proteomics pipeline. Bioinformatics 23, e191–e197 [DOI] [PubMed] [Google Scholar]
- 19. Sturm M., Bertsch A., Gropl C., Hildebrandt A., Hussong R., Lange E., Pfeifer N., Schulz-Trieglaff O., Zerck A., Reinert K., Kohlbacher O. (2008) OpenMS—an open-source software framework for mass spectrometry. BMC Bioinformatics 9, 163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Lam H., Deutsch E. W., Eddes J. S., Eng J. K., King N., Stein S. E., Aebersold R. (2007) Development and validation of a spectral library searching method for peptide identification from MS/MS. Proteomics 7, 655–667 [DOI] [PubMed] [Google Scholar]
- 21. Pedrioli P. G., Eng J. K., Hubley R., Vogelzang M., Deutsch E. W., Raught B., Pratt B., Nilsson E., Angeletti R. H., Apweiler R., Cheung K., Costello C. E., Hermjakob H., Huang S., Julian R. K., Kapp E., McComb M. E., Oliver S. G., Omenn G., Paton N. W., Simpson R., Smith R., Taylor C. F., Zhu W., Aebersold R. (2004) A common open representation of mass spectrometry data and its application to proteomics research. Nat. Biotechnol. 22, 1459–1466 [DOI] [PubMed] [Google Scholar]
- 22. Mayer G., Montecchi-Palazzi L., Ovelleiro D., Jones A. R., Binz P. A., Deutsch E. W., Chambers M., Kallhardt M., Levander F., Shofstahl J., Orchard S., Vizcaino J. A., Hermjakob H., Stephan C., Meyer H. E., Eisenacher M. (2013) The HUPO proteomics standards initiative—mass spectrometry controlled vocabulary. Database 2013, bat009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Taylor C. F., Paton N. W., Lilley K. S., Binz P. A., Julian R. K., Jr., Jones A. R., Zhu W., Apweiler R., Aebersold R., Deutsch E. W., Dunn M. J., Heck A. J., Leitner A., Macht M., Mann M., Martens L., Neubert T. A., Patterson S. D., Ping P., Seymour S. L., Souda P., Tsugita A., Vandekerckhove J., Vondriska T. M., Whitelegge J. P., Wilkins M. R., Xenarios I., Yates J. R., 3rd, Hermjakob H. (2007) The minimum information about a proteomics experiment (MIAPE). Nat. Biotechnol. 25, 887–893 [DOI] [PubMed] [Google Scholar]
- 24. Sumner L. W., Amberg A., Barrett D., Beale M. H., Beger R., Daykin C. A., Fan T. W., Fiehn O., Goodacre R., Griffin J. L., Hankemeier T., Hardy N., Harnly J., Higashi R., Kopka J., Lane A. N., Lindon J. C., Marriott P., Nicholls A. W., Reily M. D., Thaden J. J., Viant M. R. (2007) Proposed minimum reporting standards for chemical analysis Chemical Analysis Working Group (CAWG) Metabolomics Standards Initiative (MSI). Metabolomics 3, 211–221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Ashburner M., Ball C. A., Blake J. A., Botstein D., Butler H., Cherry J. M., Davis A. P., Dolinski K., Dwight S. S., Eppig J. T., Harris M. A., Hill D. P., Issel-Tarver L., Kasarskis A., Lewis S., Matese J. C., Richardson J. E., Ringwald M., Rubin G. M., Sherlock G. (2000) Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat. Genet. 25, 25–29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Jones A. R., Siepen J. A., Hubbard S. J., Paton N. W. (2009) Improving sensitivity in proteome studies by analysis of false discovery rates for multiple search engines. Proteomics 9, 1220–1229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Nahnsen S., Bertsch A., Rahnenfuhrer J., Nordheim A., Kohlbacher O. (2011) Probabilistic consensus scoring improves tandem mass spectrometry peptide identification. J. Proteome Res. 10, 3332–3343 [DOI] [PubMed] [Google Scholar]
- 28. Nesvizhskii A. I., Aebersold R. (2005) Interpretation of shotgun proteomic data: the protein inference problem. Mol. Cell. Proteomics 4, 1419–1440 [DOI] [PubMed] [Google Scholar]
- 29. Wishart D. S., Knox C., Guo A. C., Eisner R., Young N., Gautam B., Hau D. D., Psychogios N., Dong E., Bouatra S., Mandal R., Sinelnikov I., Xia J., Jia L., Cruz J. A., Lim E., Sobsey C. A., Shrivastava S., Huang P., Liu P., Fang L., Peng J., Fradette R., Cheng D., Tzur D., Clements M., Lewis A., De Souza A., Zuniga A., Dawe M., Xiong Y., Clive D., Greiner R., Nazyrova A., Shaykhutdinov R., Li L., Vogel H. J., Forsythe I. (2009) HMDB: a knowledgebase for the human metabolome. Nucleic Acids Res. 37, D603–D610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. de Matos P., Adams N., Hastings J., Moreno P., Steinbeck C. (2012) A database for chemical proteomics: ChEBI. Methods Mol. Biol. 803, 273–296 [DOI] [PubMed] [Google Scholar]
- 31. Sayers E. W., Barrett T., Benson D. A., Bolton E., Bryant S. H., Canese K., Chetvernin V., Church D. M., Dicuccio M., Federhen S., Feolo M., Fingerman I. M., Geer L. Y., Helmberg W., Kapustin Y., Krasnov S., Landsman D., Lipman D. J., Lu Z., Madden T. L., Madej T., Maglott D. R., Marchler-Bauer A., Miller V., Karsch-Mizrachi I., Ostell J., Panchenko A., Phan L., Pruitt K. D., Schuler G. D., Sequeira E., Sherry S. T., Shumway M., Sirotkin K., Slotta D., Souvorov A., Starchenko G., Tatusova T. A., Wagner L., Wang Y., Wilbur W. J., Yaschenko E., Ye J. (2012) Database resources of the National Center for Biotechnology Information. Nucleic Acids Res. 40, D13–D25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Foster J. M., Moreno P., Fabregat A., Hermjakob H., Steinbeck C., Apweiler R., Wakelam M. J., Vizcaino J. A. (2013) LipidHome: a database of theoretical lipids optimized for high throughput mass spectrometry lipidomics. PLoS One 8, e61951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Fahy E., Subramaniam S., Murphy R. C., Nishijima M., Raetz C. R., Shimizu T., Spener F., van Meer G., Wakelam M. J., Dennis E. A. (2009) Update of the LIPID MAPS comprehensive classification system for lipids. J. Lipid Res. 50 Suppl, S9–S14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Salek R. M., Steinbeck C., Viant M. R., Goodacre R., Dunn W. B. (2013) The role of reporting standards for metabolite annotation and identification in metabolomic studies. GigaScience 2, 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Creasy D. M., Cottrell J. S. (2004) Unimod: protein modifications for mass spectrometry. Proteomics 4, 1534–1536 [DOI] [PubMed] [Google Scholar]
- 36. Montecchi-Palazzi L., Beavis R., Binz P. A., Chalkley R. J., Cottrell J., Creasy D., Shofstahl J., Seymour S. L., Garavelli J. S. (2008) The PSI-MOD community standard for representation of protein modification data. Nat. Biotechnol. 26, 864–866 [DOI] [PubMed] [Google Scholar]
- 37. Xu Q. W., Griss J., Wang R., Jones A. R., Hermjakob H., Vizcaino J. A. (2014) jmzTab: a Java interface to the mzTab data standard. Proteomics 14, 1328–1332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Gatto L., Lilley K. S. (2012) MSnbase—an R/Bioconductor package for isobaric tagged mass spectrometry data visualization, processing and quantitation. Bioinformatics 28, 288–289 [DOI] [PubMed] [Google Scholar]
- 39. Hartler J., Trötzmüller M., Chitraju C., Spener F., Kofeler H. C., Thallinger G. G. (2011) Lipid Data Analyzer: unattended identification and quantitation of lipids in LC-MS data. Bioinformatics 27, 572–577 [DOI] [PubMed] [Google Scholar]
- 40. Cote R. G., Griss J., Dianes J. A., Wang R., Wright J. C., van den Toorn H. W., van Breukelen B., Heck A. J., Hulstaert N., Martens L., Reisinger F., Csordas A., Ovelleiro D., Perez-Rivevol Y., Barsnes H., Hermjakob H., Vizcaino J. A. (2012) The PRoteomics IDEntification (PRIDE) Converter 2 framework: an improved suite of tools to facilitate data submission to the PRIDE database and the ProteomeXchange consortium. Mol. Cell. Proteomics 11, 1682–1689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Haug K., Salek R. M., Conesa P., Hastings J., de Matos P., Rijnbeek M., Mahendraker T., Williams M., Neumann S., Rocca-Serra P., Maguire E., Gonzalez-Beltran A., Sansone S. A., Griffin J. L., Steinbeck C. (2013) MetaboLights—an open-access general-purpose repository for metabolomics studies and associated meta-data. Nucleic Acids Res. 41, D781–D786 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Bradshaw R. A., Burlingame A. L., Carr S., Aebersold R. (2006) Reporting protein identification data: the next generation of guidelines. Mol. Cell. Proteomics 5, 787–788 [DOI] [PubMed] [Google Scholar]
- 43. Griss J., Foster J. M., Hermjakob H., Vizcaino J. A. (2013) PRIDE Cluster: building a consensus of proteomics data. Nat. Methods 10, 95–96 1 1 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.