

Abstract
Disulfide bond identification is important for a detailed understanding of protein structures, which directly affect their biological functions. Here we describe an integrated workflow for the fast and accurate identification of authentic protein disulfide bridges. This novel workflow incorporates acidic proteolytic digestion using pepsin to eliminate undesirable disulfide reshuffling during sample preparation and a novel search engine, SlinkS, to directly identify disulfide-bridged peptides isolated via electron transfer higher energy dissociation (EThcD). In EThcD fragmentation of disulfide-bridged peptides, electron transfer dissociation preferentially leads to the cleavage of the S–S bonds, generating two intense disulfide-cleaved peptides as primary fragment ions. Subsequently, higher energy collision dissociation primarily targets unreacted and charge-reduced precursor ions, inducing peptide backbone fragmentation. SlinkS is able to provide the accurate monoisotopic precursor masses of the two disulfide-cleaved peptides and the sequence of each linked peptide by matching the remaining EThcD product ions against a linear peptide database. The workflow was validated using a protein mixture containing six proteins rich in natural disulfide bridges. Using this pepsin-based workflow, we were able to efficiently and confidently identify a total of 31 unique Cys–Cys bonds (out of 43 disulfide bridges present), with no disulfide reshuffling products detected. Pepsin digestion not only outperformed trypsin digestion in terms of the number of detected authentic Cys–Cys bonds, but, more important, prevented the formation of artificially reshuffled disulfide bridges due to protein digestion under neutral pH. Our new workflow therefore provides a precise and generic approach for disulfide bridge mapping, which can be used to study protein folding, structure, and stability.
Disulfide bridges are one of the most common post-translational modifications in proteins (1). The formation of disulfide bonds between cysteine residues is a crucial component in the process of protein folding and plays an important role in stabilizing the tertiary and quaternary structures of proteins (2, 3). Therefore, detecting and characterizing the exact locations of disulfide bonds is an important aspect of proteomics, especially in the context of gaining a comprehensive understanding of protein folding and three-dimensional structures. Moreover, in the use of protein therapeutics (e.g. antibodies), it is also of interest to monitor the reshuffling of disulfide bonds during formulation, storage, and usage, which reflects the antibody structure, stability, and biological function (4).
Most knowledge about protein disulfide bridges comes from detailed molecular structures obtained via x-ray crystallography and NMR spectroscopy (5, 6), although regrettably such data are mostly obtained from overexpressed recombinant proteins. Mass spectrometry is gaining importance in the identification and characterization of protein disulfide bridges (7, 8). Some advantages of MS-based approaches include relatively easy sample preparation, short analysis time, and the capability to deal with more complex protein mixtures from endogenous sources. However, the detection of disulfide bridges remains challenging for a few reasons.
Firstly, the presence of free sulfhydryl groups can induce undesired sulfhydryl-disulfide reshuffling, especially under neutral and alkaline pH condition. As most standard proteomic strategies use enzymatic digestion in a pH range of 7.5–8.5, undesirable disulfide reshuffling can occur during sample handling (8). Secondly, most of the widely applied database searching programs, such as SEQUEST and Mascot, are not developed, and thus are not suitable, for analyzing fragmentation spectra originating from disulfide-bridged peptides (9).
Efforts have been directed at tackling these obstacles and facilitating the identification of authentic disulfide bridges. With respect to sample handling, it has been demonstrated by several groups that disulfide reshuffling can be reduced by (i) blocking free cysteines using alkylating reagents before denaturing the protein, (ii) lowering the pH to 6.0 to 7.0 during tryptic digestion (8, 10–13), and (iii) using the enzyme pepsin under acidic conditions for proteolytic digestion (13–17). Unfortunately, trypsin becomes less efficient and less specific at more acidic pH, and pepsin, which has an optimal pH range of 1–3, tremendously increases the complexity of both protein digests and data analysis (8). Regarding data analysis, one of the current approaches used for the identification of disulfide bridges involves chromatographic comparison between reduced and non-reduced protein digests, with disulfide-bridged peptides appearing only in non-reduced samples (8, 12). Alternatively, disulfide bonds can be identified directly from non-reduced protein digests using an electron transfer dissociation (ETD)1 MS2 and collision-induced dissociation (CID)/higher energy collision dissociation (HCD) MS3 fragmentation scheme (termed the ETD-MS2 CID/HCD-MS3 approach) (13, 18, 19). Thereby, ETD aids in the preferential cleavage of S–S linkages, generating two disulfide-cleaved peptides, which can be subsequently isolated and further fragmented via CID/HCD for sequence information. In addition, substantial efforts have been made to develop novel strategies specifically for interpreting spectra from disulfide-bridged peptides, including de novo sequencing approaches (20, 21) and database search engines such as MassMatrix and Dbond (9, 22).
A combined dual fragmentation scheme, referred to as electron-transfer and higher-energy collision dissociation (EThcD), was introduced by our group recently as implemented on an Orbitrap Elite (23–25) and will become available for the Orbitrap Fusion. In this approach, an initial ETD step is applied to fragment the isolated MS precursor, and subsequently all resulting ions are subjected to HCD fragmentation, generating a mixture of b/y and c/z ions. Here we explored the use of EThcD for disulfide bridge analysis. We reasoned that the previously reported ETD-MS2 CID/HCD-MS3 method could be integrated into a single EThcD experiment, with ETD applied first to preferentially break the disulfide bond and HCD employed next to enhance the number of peptide backbone fragments. Based on the fact that all the ions resulting from the ETD process are subjected to HCD simultaneously and thus no MS3 isolation is necessary, the sensitivity and duty cycle of the EThcD workflow should potentially be improved relative to the previous MS3 strategy.
In this work, we describe a fast and accurate framework for both intrapeptide and interpeptide disulfide bridge identification, including the acidic digestion procedure using pepsin, the usage of the dual-fragmentation scheme EThcD, and the development of a novel search engine, SlinkS. The workflow described herein diminishes issues induced by disulfide reshuffling during sample preparation and provides direct and efficient identification of intrapeptide and interpeptide disulfide bonds from LC/MS2 experiments. We evaluated the integrated workflow using a mixture of six standard proteins and confirmed that this approach enables reliable and robust identification of authentic disulfide bridges from protein mixtures. Furthermore, we assessed the capability of the workflow to quantitatively monitor the changes of disulfide bridges in stress-induced therapeutic antibodies.
EXPERIMENTAL PROCEDURES
Materials
Iodoacetamide, N-ethylmaleimide, urea, bovine serum albumin, ubiquitin, cytochrome C, lysozyme C, ribonuclease B, β-lactoglobulin, and pepsin were purchased from Sigma-Aldrich (Steinheim, Germany). Formic acid was purchased from Merck (Darmstadt, Germany). Acetonitrile was purchased from Biosolve (Valkenswaard, The Netherlands). Sequencing-grade trypsin was obtained from Promega (Madison, WI), and Lys-C was supplied by Wako Chemicals (Richmond, VA). Wild-type IgG1 antibody was obtained as previously described (26).
Proteolytic Digestion of Standard Protein Mixture
Five standard proteins, several of which are rich in disulfide bridges (namely, cytochrome C, bovine serum albumin, lysozyme C, ribonuclease B, and β-lactoglobulin) were reconstituted in 10 mm PBS (pH 7.4) at a concentration of 1 mg/ml and mixed in an equal ratio (w/w). The protein mixture was diluted 10 times with 0.04N HCl (pH 1.5) for pepsin digestion, 10 mm PBS (pH 7.8) for trypsin digestion at normal pH, and 10 mm PBS (pH 6.8) for trypsin digestion at low pH. Wild-type IgG1 (1 mg/ml in 20 mm Tris-HCl, pH 7.4) was added to each of the samples to reach a final concentration of 0.1 mg/ml. For pepsin digestion, 2 m urea was added to denature the protein, and pepsin was added in a 1:50 (w/w) ratio. The digestion was performed for 2 h at room temperature. For both normal and low-pH trypsin digestion, iodoacetamide (normal-pH digestion) or N-ethylmaleimide (low-pH digestion) was used to alkylate free Cys residues for 30 min at room temperature in the dark, and then 8 m urea was added to denature the protein. Lys-C was added in a 1:75 (w/w) ratio, and samples were incubated for 4 h at 37 °C. The sample was then diluted four times with 10 mm PBS (the same pH as the sample). Trypsin was added in a 1:100 (w/w) ratio, and incubation was carried out overnight at 37 °C.
Proteolytic Digestion of Heat-stressed Therapeutic Antibody
20 μl of wild-type IgG1 (1 mg/ml in 20 mm Tris-HCl, pH 7.4) was incubated at 37 °C, 60 °C, and 70 °C for 2 h. Then the non-heated and heated samples were diluted 10 times with 0.04N HCl (pH 1.5) prior to pepsin digestion. The digestion procedure was applied as described above.
LC/MS Analysis
Protein digests were analyzed by an ultra-HPLC Proxeon EASY-nLC 1000 (Thermo Fisher Scientific, Odense, Denmark) coupled on-line to an ETD-enabled LTQ Orbitrap Elite mass spectrometer (Thermo Fisher Scientific, Bremen, Germany). Reversed-phase separation was accomplished using a 100 μm inner diameter × 2 cm trap column (in-housed packed with ReproSil-Pur C18-AQ, 3 μm) (Dr. Maisch GmbH, Ammerbuch-Entringen, Germany) coupled to a 50 μm inner diameter × 50 cm analytical column (in-house packed with Poroshell 120 EC-C18, 2.7 μm) (Agilent Technologies, Amstelveen,The Netherlands). Mobile-phase solvent A consisted of 0.1% formic acid in water, and mobile-phase solvent B consisted of 0.1% formic acid in acetonitrile. The flow rate was set to 100 nL/min. A 50-min gradient was used (7% to 30% solvent B within 31 min, 30% to 100% solvent B within 3 min, 100% solvent B for 5 min, 100% to 7% solvent B within 1 min, and 7% solvent B for 10 min). The instrument firmware was modified to allow all-ion HCD fragmentation after the ETD step, as described earlier (23), allowing an EThcD fragmentation scheme. The five most abundant precursors were selected for EThcD data-dependent fragmentation. All data were acquired in the Orbitrap mass analyzer. For MS scans, the scan range was from 350 to 1500 m/z at a resolution of 60,000, and the automatic gain control target was set at 1 × 106. For MS2 scans, the resolution was 15,000, the automatic gain control target was set at 2 × 105, the precursor isolation width was 2 Da, and the maximum injection time was 500 ms. The ETD reaction time was set at 50 ms, the ETD automatic gain control target was 2 × 105, and the HCD normalized collision energy was 32% (calculation based on precursor m/z and charge).
Data Analysis
The raw data files were converted to .mgf files using Thermo Scientific Proteome Discoverer 1.4 software (Thermo Fisher Scientific) with the add-on node MS2-Spectrum Processor. The non-fragment filter node was used with the following parameters: remove precursors, 4-Da window offset; remove charge reduced precursors, 2-Da window offset; and remove neutral losses from charge reduced precursors, 2-Da window offset. The MS2-Spectrum processor was used to deconvolute and deisotope the original m/z values to singly charged masses. The in-house-developed algorithm SlinkS was used for the main search. SlinkS was developed in R (version 3.0.1). The source code and user manual are available via Sourceforge with the package name SlinkS. The singly charged precursors and product ions in the .mgf files were converted to neutral masses. The following settings were used: precursor ion mass tolerance, 10 ppm; product ion mass tolerance, 20 ppm; and variable modification, Met oxidation. For pepsin digestion, the setting “nonspecific enzymatic digestion” was used. For trypsin digestion, the setting “fully tryptic enzymatic specificity” and up to three miscleavages were used, and additional variable modification of carbamidomethyl (+57.021 Da) or N-ethylmaleimide (+125.0476 Da) was allowed to accommodate modifications induced by the alkylating reagents. The target database contained the sequences of six standard proteins, and the decoy database was generated by randomizing each sequence in the target database. SlinkS performed all the searches against a combined target and decoy database (12 protein entries as presented in supplemental Table S5). Of note, with the use of nonspecific in silico enzymatic digestion, this small protein database results in a peptide database containing 114,961 entries enabling proper false discovery rate (FDR) calculations. SlinkS searches both intrapeptide disulfide bridges (where the disulfide bridge is within one peptide) and interpeptide disulfide bridges (where the disulfide bridge is between two peptides). Intrapeptide disulfide bridges were searched against the peptide database containing at least two cysteines per peptide, whereas interpeptide disulfide bridges were searched against the peptide database containing at least one cysteine per peptide. All the identifications were filtered according to an estimated FDR of 1%, with a corresponding natural log based n-score cutoff of −21.5 for EThcD spectra and −20.5 for ETD spectra. All the fully annotated spectra are available in the supplemental material.
RESULTS AND DISCUSSION
Characterizing the Fragmentation Pattern of Disulfide-bridged Peptides under Different Activation Methods: HCD, ETD, and EThcD
To evaluate in detail the fragmentation patterns of interpeptide disulfide linkages under HCD/ETD/EThcD conditions, we selected five disulfide-containing peptides from a trypsin-digested standard protein mixture (see supplemental Table S1 for the list of selected peptides). Each peptide was fragmented by means of the three aforementioned fragmentation schemes, and all 15 MS2 spectra were manually annotated with the consideration of four possible fragment categories, which are summarized in Fig. 1. The fragment categories were chosen to contain (i) standard backbone fragments (annotated as b-, y-, c-, and z-ion series), (ii) disulfide-bond-containing backbone fragments (annotated as b-, y-, c-, and z- +A/B ion series), (iii) disulfide-bond-specific ions (cleaved at S–S or S–C bonds, annotated as S–S or S–C cleavage), and (iv) double-cleavage ions (referring to the specific situation in which one cleavage is at the peptide backbone and the other one is at the S–S/S–C bond; annotated as b-, y-, c-, or z- + S-S/S-C cleavage). A representative set of annotated HCD, ETD, and EThcD MS2 spectra is shown in Fig. 2 and supplemental Fig. S1.
Fig. 1.
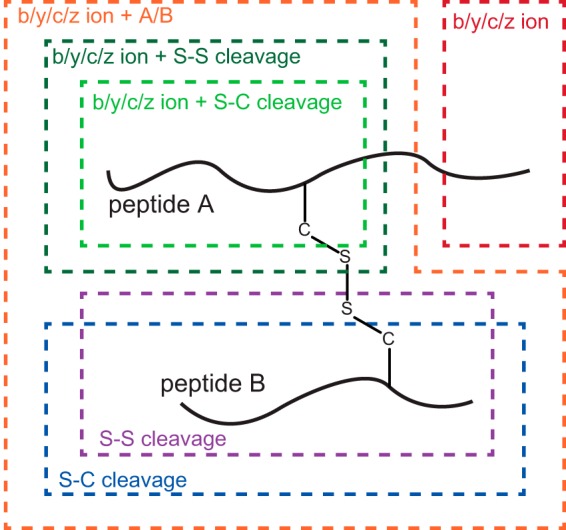
Scheme of generalized fragmentation mechanisms for interpeptide disulfide bridges. Four categories of MS2 fragment ions can be formed: (i) standard backbone fragments (b-, y-, c-, z-ion series) are shown in red, (ii) disulfide-bond-containing backbone fragments (b-, y-, c-, z- + A/B ions) are shown in orange, (iii) disulfide-bond-specific cleavages including S–S and S–C bond cleavage are shown in purple and blue, respectively, and (iv) double cleavage ions (one at the peptide backbone and the other at the S–S bond of disulfide bridges; b-, y-, c-, z- + S-S ions) are shown in dark and light green.
Fig. 2.

Illustrative HCD, ETD, and EThcD MS2 spectra of an interpeptide disulfide bridge. The MS2 product ion spectra are reconstructed based on deconvoluted neutral monoisotopic masses. The two disulfide-cleaved fragment ions are labeled as peptides A and B. Single-cleavage ions (including b-, y-, c-, z- and b-, y-, c-, z- + A/B ions) are labeled as b-, y-, c-, z-ion series. Double-cleavage ions (including b-, y-, c, z- + S-S ions) are labeled as b*-, y*-, c*-, z*-ion series.
The purpose of our SlinkS search algorithm is to determine the precursor masses of the two disulfide-cleaved peptides (e.g. those annotated as peptides A and B in Figs. 1 and 2) and search each of the linked peptides against a linear peptide database. The first critical challenge is to efficiently discriminate the two disulfide-cleaved peptides in the background of all the other MS2 fragments. To further evaluate the previous finding that the S–S bond is prone to cleavage under ETD-based conditions (13), we carefully examined the intensity ranking of each type of fragment ion presented in Fig. 1. As illustrated in Fig. 3A, in each of the five representative spectrum sets, the peptides derived from S–S bond cleavage were among the top three and top six most intense fragments in ETD and EThcD spectra, respectively, whereas such ions were nearly absent in HCD spectra. This observation suggests that both ETD and EThcD generate predominately disulfide-cleaved ions, although the relative intensities of these ions are slightly lower under EThcD conditions. Frese et al. showed that during EThcD fragmentation, HCD energy primarily targets the unreacted and charge-reduced precursors without inducing secondary fragmentation of c- or z-ions originating from the ETD reaction (23). In the case of disulfide-bridged peptide fragmentation, we need to preserve not only the c- and z-ions, but also, and more importantly, the two disulfide-cleaved peptides generated by the initial ETD reaction. Our finding indicates that, similarly to ETD, EThcD is also capable of generating highly abundant disulfide-cleaved peptides, although a small percentage of secondary fragmentation may also occur as a result of the subsequent HCD fragmentation step.
Fig. 3.

Quantitative characterization of fragmentation patterns of disulfide-bridged peptides under HCD, ETD, and EThcD conditions. A, a density plot of the intensity ranking of MS2 fragment ions indicates that the disulfide-cleaved peptides (labeled as S–S cleavage in purple) are among the most intense peaks in both ETD and EThcD spectra. Insets are the enlarged views of intensity rankings from 1 to 10. The y-axis is normalized to 0 to 1 scale. B, a bar plot illustrates the overall ion abundance contributed by each type of fragment ion. HCD ions, ETD ions, and ions derived from disulfide-bond-specific cleavages are labeled in orange, cyan, and purple. The y-axis is normalized to 0 to 1 scale.
The second interesting question is how much each type of fragment ion contributes to the total ion abundance during different fragmentation schemes. As shown in Fig. 3B, HCD and ETD fragmentation primarily generate, respectively, b-, y- and c-, z-ion series, whereas EThcD gives rise to all four types of fragment ions, with a slightly greater abundance of b-, y-ions over c-, z-ions. In all three types of fragmentation schemes, including EThcD, double cleavages (b-, y-, c-, z-, and S-S/S-C cleavage ions) were observed, although they were significantly less abundant than disulfide-bond-containing backbone fragments (b-, y-, c-, z-, and A/B ions). Based on the aforementioned observations of fragmentation patterns, the search engine SlinkS was developed to automatically identify disulfide bridges from a protein database, and this is detailed in the following section.
Software Design
SlinkS is a disulfide bridge search engine that is able to analyze LC/MS2 data derived from ETD and EThcD fragmentation schemes. Here ETD-based fragmentation is essential in order to generate high-abundant disulfide-cleaved peptides. SlinkS uses deconvoluted peak list files such as .mgf files as input. For this, Thermo Raw files need to be deconvoluted, deisotoped, and exported as .mgf files by existing software packages such as Proteome Discover and ProSightPC (Thermo Fisher Scientific) prior to the SlinkS search.
SlinkS searches both intrapeptide and interpeptide disulfide bridges. For intrapeptide disulfide identification, the software performs in a manner similar to that of linear peptide search engines, where a mass modification of −2H is applied during searching against an in silico digested peptide database. Peptide matching is accomplished by comparing the in silico generated theoretical peptide backbone fragments (c-, z-ion series for ETD and b-, y-, c-, z-ion series for EThcD) to all recorded MS2 fragments in the spectrum. Additionally, intrapeptide-specific internal ions (where peptide fragments are generated from double backbone cleavages between the two linked cysteines) are also considered in the algorithm. Notably, traditional internal fragment ions, such as water or ammonium loss, are not taken into account during the search. A set of annotated example intrapeptide disulfide bond spectra is shown in supplemental Fig. S2.
For interpeptide disulfide bond identification, as illustrated in Fig. 4, SlinkS first performs precursor mass selection to obtain the monoisotopic mass of each disulfide-cleaved peptide. This part is composed of two steps: (i) choosing the top n most abundant peaks from all the MS2 ions (n is a user-defined parameter in the search engine), and (ii) considering the sum of the masses from all possible peptide pairs and selecting the ones that lie within the user-defined error tolerance around the MS precursor ion mass. Previous studies showed that the dissociation of S–S bonds yields similar relative intensities of thiyl (–S·) and thiol (–SH) fragment ions (27); therefore, each of the disulfide-cleaved peptide masses (mp) obtained from precursor mass selection is extended to three masses (mp, mp + mH, and mp − mH, where mH is the mass of hydrogen) in order to include all the potential candidates of linked peptides.
Fig. 4.
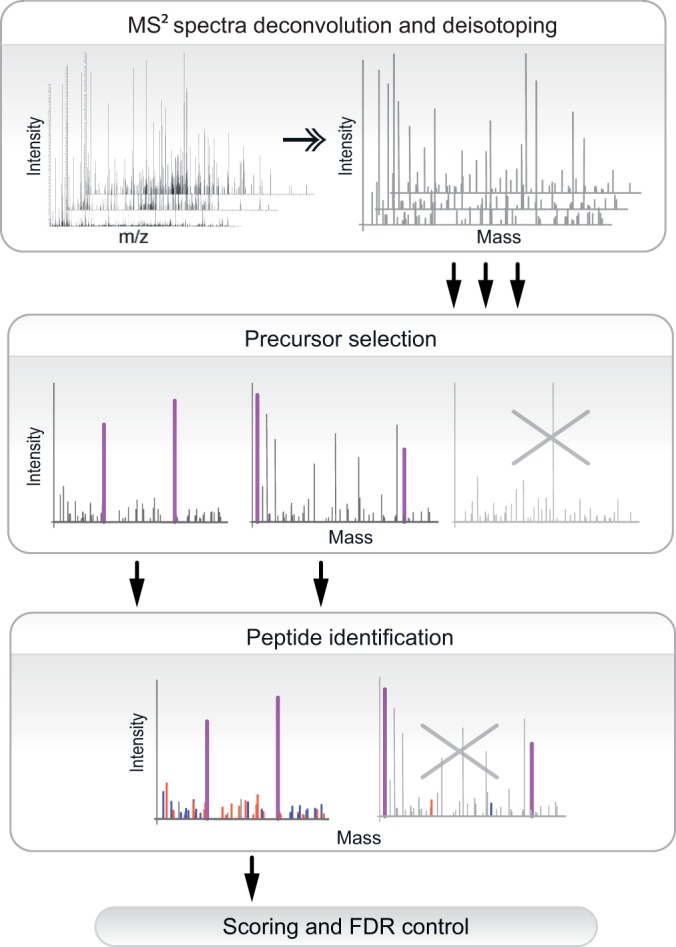
Schematic outline of SlinkS workflow for the identification of interpeptide disulfide bridges. Raw files are deconvoluted, deisotoped, converted to peak list files, and subjected to SlinkS analysis. SlinkS first performs precursor mass selection to obtain the monoisotopic mass of each disulfide-cleaved peptide, and spectra that do not contain any candidate peptide pairs are discarded. Next, all MS2 ions are matched against in silico fragments of each candidate peptide, and spectra that do not have sufficient matched fragment ions are omitted. Finally, the n-score and FDR are calculated, and identification results are filtered based on the user-defined n-score and FDR cutoff value.
The peptide database is created by in silico digestion of the protein database, where only peptides containing at least one cysteine are retained. SlinkS searches each recorded MS2 spectrum based on the determined precursor masses of disulfide-cleaved peptides and the remaining MS2 ions as backbone fragment ions for sequence information. In this study, only single cleavage ions (c-, z- and c-, z- + A/B ion series for ETD; b-, y-, c-, z-, and b-, y-, c-, z- + A/B ion series for EThcD) were considered for product ion matching. Because double cleavage ions (c-, z- + S-S/S-C cleavage ions for ETD; b-, y-, c-, z- + S-S/S-C cleavage ions for EThcD) are less frequent and contribute only marginally to the total ion current of the MS2 spectra, they are labeled in the spectra but not considered in SlinkS scoring.
SlinkS uses a probability score (termed the n-score) to calculate the confidence of each candidate sequence. The n-score has been introduced (28) and is used in ProSightPC (Thermo Fisher Scientific) for top-down/middle-down experiments. We adapted the principle of n-score calculation and modified it to match our disulfide bridge identification. Because interpeptide disulfide bridges are composed of two linked linear peptides, they are assigned when the n-scores from both of the peptides are less than the threshold. The use of an individual peptide score for each linked peptide has been described previously (29). It effectively avoids the frequently occurring scenario in which most of the matched fragments arise from only one of the linked peptides.
SlinkS uses a target-decoy strategy for peptide–spectrum match validation in which the FDR is calculated as the number of false positive hits divided by the total number of identifications (true and false positive hits). In contrast to the linear peptide FDR evaluation, the false positive hits of disulfide-bridged peptides refer to either or both of the linked peptides being matched in the decoy database (target-decoy + decoy-decoy), and the true positive hits refer to both of the linked peptides being matched in the target database (target-target).
Evaluating the Performance of SlinkS in the Identification of Disulfide Bridges under ETD and EThcD Conditions
To evaluate the reliability and specificity of the SlinkS algorithm in the identification of disulfide-bridged peptides, a defined mixture consisting of six well-characterized proteins was digested under acidic conditions using pepsin, and the resulting peptides were subjected to ETD and EThcD LC/MS2 experiments. Firstly we evaluated the n-score distributions of true and false positive hits by plotting the n-scores against the number of identifications in each category. For both ETD and EThcD data, the true positive matches (target-target) were clearly separated from the false positive hits (target-decoy and decoy-decoy) in the low n-score region (Fig. 5A). We also noticed that the hybrid false positive hits (target-decoy) contributed significantly more than the double false positive hits (decoy-decoy). This finding suggests that hybrid false positive hits, which are sometimes inappropriately estimated by other algorithms, have to be taken into account. As expected, when comparing to ETD data, the average n-score under EThcD is lower for true positive hits, which directly reflects the higher spectral quality due to the generation of all b-, y-, c-, z-ion series. However, the average n-score for false positive hits is also lower. One of the possible explanations is that more random matches are generated under EThcD because, in theory, EThcD spectra contain twice as many ions as ETD spectra. In addition, we plotted the estimated FDRs against different n-score thresholds used in the search (Fig. 5B). Under ETD and EThcD, respectively, a natural log-based n-score cutoff of −20.5 or −21.5 gave rise to an estimated FDR of ∼1%. This indicates that in order to reach the same desired FDR, a slightly lower n-score cutoff should be applied in EThcD than in ETD.
Fig. 5.

Validation of SlinkS performance in comparison of ETD and EThcD data. A, a density plot of n-score values versus the number of identified disulfide bridges in each category. For each of the disulfide-bridged peptides, the lower n-score value of the two linked peptides was used for plotting. B, a bar diagram of different n-score thresholds applied in the search against the corresponding FDRs. The actual dataset (shown as gray bars) is fitted to a power function (shown as blue lines). The red triangle indicates n-score cutoffs of −20.5 and −21.5 for ETD and EThcD spectra, respectively, to allow for an FDR of ∼1%. These n-score cutoff values are calculated based on the fitted curve.
Next, we assessed the number of identified disulfide bridges in ETD and EThcD experiments. Based on the results from n-score and FDR evaluation, we chose a peptide natural log n-score cutoff of −21.5 for EThcD and −20.5 for ETD data, both of which provided an estimated FDR of 1%. As a result, 13 unique Cys–Cys bridges were confidently identified via ETD, and an additional 11 (a total of 24) unique Cys–Cys bonds were identified via EThcD (supplemental Table S2). Together, these results suggest that SlinkS performs well in unambiguously and sensitively identifying disulfide-bridged peptides from both fragmentation schemes, but EThcD outperforms ETD substantially in terms of the number of identifications and the average n-score of all true positive matches.
Evaluation of Pepsin- and Trypsin-based Sample Preparation Strategies
In addition to the difficulties in spectra interpretation and database searching, sample preparation for disulfide bond identification is also quite challenging. The two major obstacles are (i) disulfide reshuffling caused by disulfide exchange or the formation of new artificial disulfide bridges, and (ii) low protein sequence coverage due to the presence of intertwined disulfide bridges.
To tackle these issues, pepsin was used for proteolytic digestion in this study. We chose pepsin over other frequently used enzymes because of the following advantages: (i) it is compatible with highly acidic conditions (pH 1–3), thus eliminating disulfide reshuffling, which typically occurs under neutral and alkaline pH, and (ii) it is a nonspecific enzyme and therefore works very well in generating small peptides and separating most cysteines into different peptides to facilitate spectra interpretation. However, pepsin is not widely applied to disulfide bond studies with the use of mass spectrometry (8), likely because of the complexity of the peptide mixture derived from nonspecific enzymatic digestion and the difficulties of spectra interpretation. During the database searching of disulfide bridges, because the precursor mass obtained from MS acquisition is the sum of the two linked peptides, all possible peptide pairs (the summed mass of which matches the precursor mass) in the peptide library have to be considered, which increases the size of the database quadratically. For example, an average-sized protein (50 kDa) generates ∼80 cysteine-containing tryptic peptides but ∼4000 peptic peptides. If we include all the possible peptide-pair combinations, the actual search space for a single pepsin-digested protein is 8 × 106, which is twice the size of a tryptic human proteome database. In our approach, because we were able to obtain the precursor mass of each linked peptide, we directly used the linear peptide database during the search and thus overcame the computational obstacle. In Fig. 6 we plot the number of theoretical peptides to be considered using different search strategies. It clearly shows the quadratic expansion of the search space when the combination of peptide pairs is taken into consideration. Therefore, the main feature of SlinkS, which is the ability to search each disulfide-linked peptide, is essential for reducing the search space.
Fig. 6.
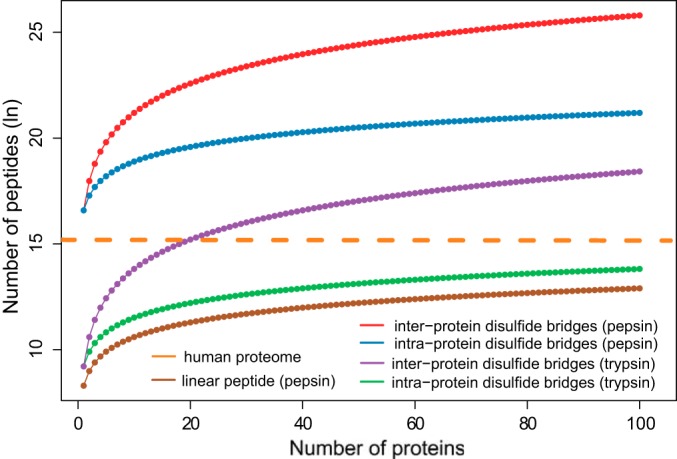
The size of the algorithm search space when different enzymatic digestion and searching strategies are employed. The number of peptides (natural log scaled) is plotted against the number of proteins in the database. The orange line indicates the number of tryptic peptides of human proteome. The dotted lines annotated as intraprotein disulfide bridges refer to the situation where only disulfide bonds within proteins are taken into account, and the dotted lines labeled as interprotein disulfide bridges imply that disulfide bonds between different proteins are also being considered by the search engine. Linear peptide corresponds to the actual database size used in SlinkS.
To further characterize the performance of pepsin digestion relative to that of the frequently used low-pH (pH 6–7) and normal-pH (pH 7–8) Lys-C plus trypsin digestion procedures, we used a protein mixture containing six well-characterized proteins with known disulfide bridges. In this case, we were able to evaluate both the efficacy of identifying authentic disulfide bonds and the level of reshuffling. We compared three different sample preparation conditions: pepsin digestion at pH 1.5 and Lys-C plus trypsin digestion at pH 6.8 and 7.8, with the two trypsin-digested samples alkylated by iodoacetamide or N-ethylmaleimide, respectively, prior to protein denaturation. Subsequently, peptide mixtures were subjected to EThcD LC/MS2 analysis. The data analysis was accomplished by SlinkS with the use of a natural log-based n-score cutoff value of −21.5 and an estimated FDR of ∼1%. The results are summarized in Table I, and all identifications are listed in supplemental Table S3. Based on crystal structures, out of 43 well-described disulfide bridges from all six proteins, 31,16, and 22 unique authentic disulfide bonds were identified in pepsin, low-pH trypsin, and normal-pH trypsin digestion workflows. Interestingly, in the low-pH trypsin-digested sample, we observed one abnormal disulfide bond, an intrapeptide linkage between the two cysteines in Cytochrome C. Because these two cysteines have been well characterized as free cysteines, our observation was very likely due to undesirable disulfide bond formation during sample preparation. This observation is probably promoted by the fact that these two cysteines are only two amino acid residues apart. Additionally, and more problematically, we identified eight intraprotein and six interprotein disulfide bridges in the pH 7.8 trypsin-digested sample. Because our starting material was a mixture of individually purified proteins, these interprotein disulfides were clearly an artifact from disulfide reshuffling during proteolytic digestion. Our results suggest that lowering the digestion pH to 6.8 can effectively decrease the undesirable disulfide reshuffling. However, because the entire sample preparation procedure is still performed under oxidative conditions (for example, the existence of oxygen in the air), the possibility of disulfide bond formation and exchange has to be taken into consideration. Convincingly, among all three tested digestion procedures, pepsin digestion resulted in the greatest number of identifications (outperforming trypsin digestion) and the most reliable results (providing no evidence of disulfide bond reshuffling).
Table I. Number of unambiguously identified disulfide-bridged peptides from a mixture of six standard proteins.
| Protein name | Number of free cysteinesa | Number of disulfide bridgesa | Trypsin (pH 7.8) | Trypsin (pH 6.8) | Pepsin (pH 1.5) |
|---|---|---|---|---|---|
| Cytochrome C | 2 | 0 | 0 | 0 | 0 |
| IgG | 0 | 16 | 8 | 6 | 12 |
| Bovine serum albumin | 1 | 17 | 11 | 7 | 13 |
| Lysozyme C | 1 | 4 | 1 | 1 | 4 |
| Ribonuclease B | 0 | 4 | 1 | 0 | 2 |
| β-lactoglobulin | 3 | 2 | 1 | 2 | 0 |
| Total | 7 | 43 | 22 | 16 | 31 |
| Observed artificial intraprotein disulfide bonds | - | - | 8 | 1 | 0 |
| Observed artificial interprotein disulfide bonds | - | - | 6 | 0 | 0 |
a As extracted from crystal structures.
Moreover, in our search results, many disulfide-bridged peptides containing two disulfide bonds were identified (supplemental Table S3), and an example EThcD spectrum of these double disulfide-bridged peptides is shown in Fig. 7. This observation is clearly an additional strength of SlinkS. As discussed in the previous section, during the precursor selection step in a SlinkS search, a ±1 Da window was included to allow the possibly formed thiyl (–S·) and thiol (–SH) ions after S–S bond cleavage. Beneficially, a disulfide-cleaved peptide containing an internal disulfide bridge, which is 2 Da smaller than its reduced form, is also included in the disulfide-cleaved peptide candidate list. The number of disulfide bonds in each disulfide-bridged peptide can be verified by calculating the mass difference between the sum of the two peptides and the observed precursor mass in the MS acquisition. In the example spectrum shown in Fig. 7, a 4-Da mass difference between the calculated and measured disulfide-bridged peptide precursor further proves the existence of two disulfide bonds in this identification.
Fig. 7.

An example EThcD spectrum of a disulfide-bridged peptide containing two disulfide bonds, illustrating that such spectra can also be identified by SlinkS. The color and label schemes are the same as in Fig. 2.
Applying Pepsin Digestion, EThcD, and SlinkS to Identify Disulfide Bridges in Therapeutic Antibodies
After the evaluation of a well-characterized standard protein mixture, we further applied our new workflow to an unknown system, namely, stress-induced therapeutic antibodies. As for many other therapeutic proteins, the correct formation of disulfide bridges is critical during antibody production, and undesirable disulfide reshuffling may cause antibody instability, unfolding, aggregation, and hence malfunction. It has been reported that therapeutic antibodies may become less effective under stress conditions, such as pH changes, temperature variations, and agitations (30–32). Heat stress may induce both intramolecule and intermolecule disulfide bridge reshuffling, which can be one of the causes of antibody unfolding and aggregation. We argue that assessing the changes in disulfide connections present in therapeutic antibodies under stress conditions, mimicking formulation, storage, and usage, provides useful information on their stability and long-term quality.
Here, we evaluated the disulfide bridge reshuffling of non-heated and heat-stressed IgG1 antibodies. The non-heated and heated (at 37 °C, 60 °C, or 70 °C) antibody samples were pepsin digested and EThcD LC/MS2 analyzed, and the data were searched by SlinkS. The results were filtered with a 1% FDR, and all unambiguously identified disulfide-bridged peptides are summarized in Fig. 8 and listed in supplemental Table S4. In total, out of 16 authentic disulfide-bridged peptides (including 12 intrachains and 4 interchains), all 12 intrachain disulfide bonds (6 unique ones in the monomer) were identified in both non-heated and heated samples. We did not detect any interchain disulfide bridges, which might have been due to the presence of intertwined disulfide bridges at the hinge region (Fig. 8A). In addition, we identified one and six unique reshuffled Cys–Cys bonds in the 60 °C and 70 °C heated samples, respectively. The relative abundances of these stress-induced, newly formed disulfide bridges were also quantified via spectra counting (Fig. 8B). Our observations suggest that with more elevated heat stress, both the number and the abundance of the reshuffled disulfide-bridged peptides increase. We also noticed that in heat-stressed antibody samples, disulfide reshuffling frequently happened at specific cysteine residues (i.e. it was not evenly spread among all cysteine residues), which strengthens our hypothesis that disulfide reshuffling is not a pure random process and correlates with the protein structure. Our results clearly show that heating as one of the environmental stresses can alter the structure and stability of therapeutic antibodies, which can be conveniently and efficiently detected via our pepsin-based disulfide bridge mapping approach.
Fig. 8.

A, structural view of a classical IgG1 antibody. Intrachain and interchain disulfide bridges are labeled as black and blue lines. Cysteines that are found to be involved in reshuffled disulfide bonds are shown in red. B, quantitative characterization of unambiguously identified disulfide-bridged peptides in non-heated and heated therapeutic antibodies. The authentic and reshuffled disulfide bonds are labeled in black and red, respectively. The inset shows an enlarged view of reshuffled disulfide bridges that were only observed in the heated samples.
CONCLUSIONS
We developed a novel integrated workflow to efficiently and reliably identify disulfide-bridged peptides from complex mixtures. The three integrated components of our workflow are the use of pepsin at highly acidic conditions for proteolytic digestion, EThcD for the generation of fragment ion spectra, and a novel search engine, SlinkS, for the automated interpretation of the data. SlinkS possesses a unique feature: for each MS2 spectrum, it first determines the precursor masses of the two disulfide-cleaved peptides and subsequently sequences each of them from a linear peptide database. This approach not only tremendously reduces the algorithm search space, but also provides a simple and efficient scoring scheme that employs individual peptide n-scores to evaluate the confidence of disulfide bond identification. Moreover, SlinkS allows one to define the FDR through a target-decoy strategy. In this study, we compared the performance of ETD and EThcD fragmentation schemes for disulfide bridge identification, and our data strongly suggest that EThcD outperforms ETD in terms of both the number of identifications and the average n-score of each linked peptide. We also assessed three different digestion strategies: pepsin digestion at pH 1.5, low-pH trypsin digestion at pH 6.8, and normal-pH trypsin digestion at pH 7.8. Our data highlight the benefits of using pepsin over trypsin digestion on the basis of much higher identification numbers and more reliable results (no artificial disulfide-bridged peptides observed). We conclude that our three-pronged integrated workflow is a powerful tool for the analysis of protein folding and three-dimensional structure in the native state, as well as conformational changes under stress-induced conditions.
Supplementary Material
Acknowledgments
We thank Dr. Christian Frese at the European Molecular Biology Laboratory for assistance in using EThcD fragmentation methods and Tao Chen from the Netherlands Cancer Institute for assistance in programming for SlinkS. We also thank Dr. Lucrèce Matheron for critical reading of the manuscript.
Footnotes
Author contributions: F.L. and A.J.H. designed research; F.L. performed research; B.v.B. and A.J.H. contributed new reagents or analytic tools; F.L. and B.v.B. analyzed data; F.L. and A.J.H. wrote the paper.
* Part of this research was performed within the framework of the PRIME-XS project (Grant No. 262067), funded by the European Union 7th Framework Program and the Netherlands Organization for Scientific Research (NWO)-supported large-scale proteomics facility Proteins@Work (project 184.032.201).
 This article contains supplemental material.
This article contains supplemental material.
1 The abbreviations used are:
- ETD
- electron transfer dissociation
- HCD
- higher energy collision dissociation
- CID
- collision-induced dissociation
- EThcD
- electron-transfer and higher-energy collision dissociation
- FDR
- false discovery rate.
REFERENCES
- 1. Thornton J. M. (1981) Disulphide bridges in globular proteins. J. Mol. Biol. 151, 261–287 [DOI] [PubMed] [Google Scholar]
- 2. Mamathambika B. S., Bardwell J. C. (2008) Disulfide-linked protein folding pathways. Annu. Rev. Cell Dev. Biol. 24, 211–235 [DOI] [PubMed] [Google Scholar]
- 3. Braakman I., Bulleid N. J. (2011) Protein folding and modification in the mammalian endoplasmic reticulum. Annu. Rev. Biochem. 80, 71–99 [DOI] [PubMed] [Google Scholar]
- 4. Liu H., May K. (2012) Disulfide bond structures of IgG molecules: structural variations, chemical modifications and possible impacts to stability and biological function. MAbs 4, 17–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Klaus W., Broger C., Gerber P., Senn H. (1993) Determination of the disulphide bonding pattern in proteins by local and global analysis of nuclear magnetic resonance data. Application to flavoridin. J. Mol. Biol. 232, 897–906 [DOI] [PubMed] [Google Scholar]
- 6. Jones T. A., Kjeldgaard M. (1997) Electron-density map interpretation. Methods Enzymol. 277, 173–208 [DOI] [PubMed] [Google Scholar]
- 7. Annan R. S., Carr S. A. (1997) The essential role of mass spectrometry in characterizing protein structure: mapping posttranslational modifications. J. Protein Chem. 16, 391–402 [DOI] [PubMed] [Google Scholar]
- 8. Gorman J. J., Wallis T. P., Pitt J. J. (2002) Protein disulfide bond determination by mass spectrometry. Mass Spectrom. Rev. 21, 183–216 [DOI] [PubMed] [Google Scholar]
- 9. Choi S., Jeong J., Na S., Lee H. S., Kim H. Y., Lee K. J., Paek E. (2010) New algorithm for the identification of intact disulfide linkages based on fragmentation characteristics in tandem mass spectra. J. Proteome Res. 9, 626–635 [DOI] [PubMed] [Google Scholar]
- 10. Yen T. Y., Joshi R. K., Yan H., Seto N. O., Palcic M. M., Macher B. A. (2000) Characterization of cysteine residues and disulfide bonds in proteins by liquid chromatography/electrospray ionization tandem mass spectrometry. J. Mass Spectrom. 35, 990–1002 [DOI] [PubMed] [Google Scholar]
- 11. Wu S. L., Jardine I., Hancock W. S., Karger B. L. (2004) A new and sensitive on-line liquid chromatography/mass spectrometric approach for top-down protein analysis: the comprehensive analysis of human growth hormone in an E. coli lysate using a hybrid linear ion trap/Fourier transform ion cyclotron resonance mass spectrometer. Rapid Commun. Mass Spectrom. 18, 2201–2207 [DOI] [PubMed] [Google Scholar]
- 12. Mo J., Tymiak A. A., Chen G. (2013) Characterization of disulfide linkages in recombinant human granulocyte-colony stimulating factor. Rapid Commun. Mass Spectrom. 27, 940–946 [DOI] [PubMed] [Google Scholar]
- 13. Ni W., Lin M., Salinas P., Savickas P., Wu S. L., Karger B. L. (2013) Complete mapping of a cystine knot and nested disulfides of recombinant human arylsulfatase A by multi-enzyme digestion and LC-MS analysis using CID and ETD. J. Am. Soc. Mass Spectrom. 24, 125–133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Haniu M., Horan T., Arakawa T., Le J., Katta V., Hara S., Rohde M. F. (1996) Disulfide structure and N-glycosylation sites of an extracellular domain of granulocyte-colony stimulating factor receptor. Biochemistry 35, 13040–13046 [DOI] [PubMed] [Google Scholar]
- 15. Haniu M., Arakawa T., Bures E. J., Young Y., Hui J. O., Rohde M. F., Welcher A. A., Horan T. (1998) Human leptin receptor. Determination of disulfide structure and N-glycosylation sites of the extracellular domain. J. Biol. Chem. 273, 28691–28699 [DOI] [PubMed] [Google Scholar]
- 16. Wallis T. P., Pitt J. J., Gorman J. J. (2001) Identification of disulfide-linked peptides by isotope profiles produced by peptic digestion of proteins in 50% (18)O water. Protein Sci. 10, 2251–2271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Moulaei T., Stuchlik O., Reed M., Yuan W., Pohl J., Lu W., Haugh-Krumpe L., O'Keefe B. R., Wlodawer A. (2010) Topology of the disulfide bonds in the antiviral lectin scytovirin. Protein Sci. 19, 1649–1661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Wu S. L., Jiang H., Hancock W. S., Karger B. L. (2010) Identification of the unpaired cysteine status and complete mapping of the 17 disulfides of recombinant tissue plasminogen activator using LC-MS with electron transfer dissociation/collision induced dissociation. Anal. Chem. 82, 5296–5303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Wang Y., Lu Q., Wu S. L., Karger B. L., Hancock W. S. (2011) Characterization and comparison of disulfide linkages and scrambling patterns in therapeutic monoclonal antibodies: using LC-MS with electron transfer dissociation. Anal. Chem. 83, 3133–3140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Nair S. S., Nilsson C. L., Emmett M. R., Schaub T. M., Gowd K. H., Thakur S. S., Krishnan K. S., Balaram P., Marshall A. G. (2006) De novo sequencing and disulfide mapping of a bromotryptophan-containing conotoxin by Fourier transform ion cyclotron resonance mass spectrometry. Anal. Chem. 78, 8082–8088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Shen Y., Tolic N., Purvine S. O., Smith R. D. (2010) Identification of disulfide bonds in protein proteolytic degradation products using de novo-protein unique sequence tags approach. J. Proteome Res. 9, 4053–4060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Xu H., Zhang L., Freitas M. A. (2008) Identification and characterization of disulfide bonds in proteins and peptides from tandem MS data by use of the MassMatrix MS/MS search engine. J. Proteome Res. 7, 138–144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Frese C. K., Altelaar A. F., van den Toorn H., Nolting D., Griep-Raming J., Heck A. J., Mohammed S. (2012) Toward full peptide sequence coverage by dual fragmentation combining electron-transfer and higher-energy collision dissociation tandem mass spectrometry. Anal. Chem. 84, 9668–9673 [DOI] [PubMed] [Google Scholar]
- 24. Frese C. K., Zhou H., Taus T., Altelaar A. F., Mechtler K., Heck A. J., Mohammed S. (2013) Unambiguous phosphosite localization using electron-transfer/higher-energy collision dissociation (EThcD). J. Proteome Res. 12, 1520–1525 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Mommen G. P., Frese C. K., Meiring H. D., van Gaans-van den Brink J., de Jong A. P., van Els C. A., Heck A. J. (2014) Expanding the detectable HLA peptide repertoire using electron-transfer/higher-energy collision dissociation (EThcD). Proc Natl Acad Sci U S A 111, 4507–4512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Rosati S., Yang Y., Barendregt A., Heck A. J. (2014) Detailed mass analysis of structural heterogeneity in monoclonal antibodies using native mass spectrometry. Nature protocols 9, 967–976 [DOI] [PubMed] [Google Scholar]
- 27. Turecek F., Julian R. R. (2013) Peptide radicals and cation radicals in the gas phase. Chem Rev 113, 6691–6733 [DOI] [PubMed] [Google Scholar]
- 28. Meng F., Cargile B. J., Miller L. M., Forbes A. J., Johnson J. R., Kelleher N. L. (2001) Informatics and multiplexing of intact protein identification in bacteria and the archaea. Nat. Biotechnol. 19, 952–957 [DOI] [PubMed] [Google Scholar]
- 29. Trnka M. J., Baker P. R., Robinson P. J., Burlingame A. L., Chalkley R. J. (2014) Matching cross-linked peptide spectra: only as good as the worse identification. Mol. Cell. Proteomics 13, 420–434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Vermeer A. W., Norde W. (2000) The thermal stability of immunoglobulin: unfolding and aggregation of a multi-domain protein. Biophys. J. 78, 394–404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Wang W., Singh S., Zeng D. L., King K., Nema S. (2007) Antibody structure, instability, and formulation. J. Pharm. Sci. 96, 1–26 [DOI] [PubMed] [Google Scholar]
- 32. Kukrer B., Filipe V., van Duijn E., Kasper P. T., Vreeken R. J., Heck A. J., Jiskoot W. (2010) Mass spectrometric analysis of intact human monoclonal antibody aggregates fractionated by size-exclusion chromatography. Pharm. Res. 27, 2197–2204 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.