

Abstract
We report the application of peptide-embedded imidazoles as catalysts for the site-selective delivery of the phenyl thionoformate unit as a prelude to deoxygenation reactions of polyols. Methodology was developed that allows for the synthesis of thiocarbonyl derivatives based on a combination of additives that include N-alkylimidazoles and FeCl3 as co-catalysts. The use of this reagent combination leads to increased reaction rates and efficient yields relative to simple base-mediated reactions. In terms of controlling regioselectivity during the course of polyol modification, we found that histidine-containing peptides, in combination with FeCl3, could lead to modulation of the product distribution. Through screening of peptides and control of reaction conditions, products could be observed that reflected both the inherent preference of substrates, and also reversal of inherent selectivity.
Keywords: peptide-based catalysis, deoxygenation, polyol, regioselectivity, thionoformate
Introduction
The deoxygenation of alcohols is a fundamental transformation in organic synthesis.1 The development of catalysts that could selectively remove a hydroxyl group from a polyol structure would be a particularly advantageous method in medicinal chemistry settings since one might use the technique to obtain deoxyanalogs of polyol agents without elaborate syntheses, or complicated protecting group schemes (Scheme 1).2 In order to develop such methodology, we began an investigation of site-selective transfer of the thiocarbonyl group as a prelude to substrate deoxygenation, utilizing the venerable Barton-McCombie reaction.3 Any successful approach to this problem requires catalysts that are particularly effective for mediating regioselective reactions, a current challenge in stereoselective synthesis.4
Scheme 1.

Our overall strategy was to employ the emerging paradigm of asymmetric nucleophilic catalysis5 for the projected group transfer reactions. While this field has exploded with a myriad of exciting catalyst types, including DMAP analogs,6 phosphines,7 amidines,8 and other nucleophilic functional groups,9 we sought to capitalize on developments in our own laboratory around peptide-embedded nucleophiles (e.g., 1).10 In this spirit, we have demonstrated that short peptide derivatives of histidine allow for highly enantioselective and site-selective alcohol derivatizations,11 among them acylations (i.e., to deliver 2),12 phosphorylations (3)13 and transfer of sulfur-based electrophiles (4) (Scheme 2).14,15
Scheme 2.

The portability of the catalysis strategy to thiocarbonyl transfer is not straightforward. Although the literature contains numerous examples of thiocarbonylation as a prelude to deoxygenation,16 the details of catalytic protocols are often variable. There are cases in which nucleophilic catalysts such as DMAP are employed,17 but often pyridine is used simultaneously;18 there are also reports where thiocarbonylation is carried out at high temperature in the absence of a catalyst.19 Nonetheless, we began our studies with the goal of exploring whether the catalytic cycle described in Scheme 3 might be viable. Our plan was to examine the potential of catalysts like 1 to capture phenyl chlorothionoformate (5) with the expectation that intermediates related to 6 would be formed. Transfer of thiocarbonyl to substrate would then deliver product 7, while regenerating catalyst 1. Described below are our findings in connection to our exploration of this potential catalytic cycle.
Scheme 3.

Results and Discussion
Preliminary experiments
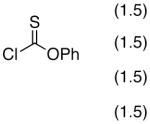
To clarify the basics of catalytic thiocarbonyl transfer, we carried out several simple experiments that show that neither DMAP nor NMI is generally effective as a substoichiometric catalyst for the thiocarbonylation of simple substrates to give 8 from cyclohexanol. Notably, as shown in equation 1, when reactions are conducted with DMAP in the presence of Et3N, thiocarbonate 8 is not formed. Rather, the addition product 9, derived from triethylamine addition to phenyl chlorothionoformate followed by dealkylation, is formed preferably as the major product (eq 1).20 On the other hand, thiocarbonate 8 is readily obtained when the reaction is performed with a variety of pyridines, suggesting that a general base mechanism may result in efficient reactions at room temperature (eq 2).
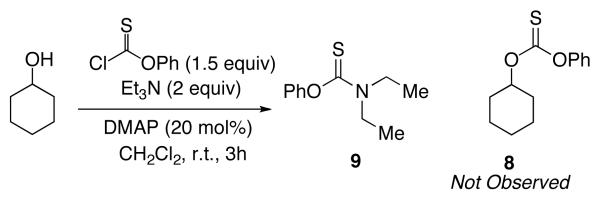 |
(1) |
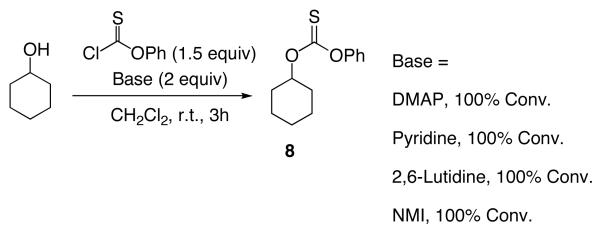 |
(2) |
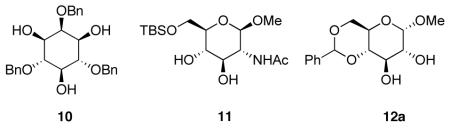
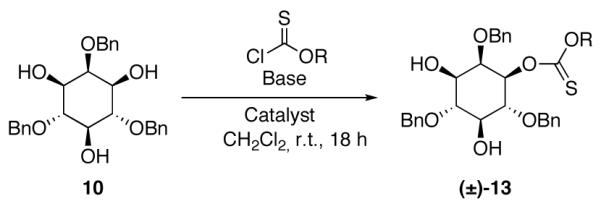
In order to explore a possible catalytic reaction in which an N-alkylimidazole might function by a nucleophilic mechanism,21 we sought to identify conditions that would employ a base (to scavenge the full equivalent of HCl generated in these reactions) that was not itself a promoter of the reaction. During the course of these studies, we found an intriguing difference in reactivity between simple substrates like cyclohexanol, and oxygenated substrates such as carbohydrate derivatives like inositide 10,22 glucosamine derivative 1123 and glucose derivative 12a.24
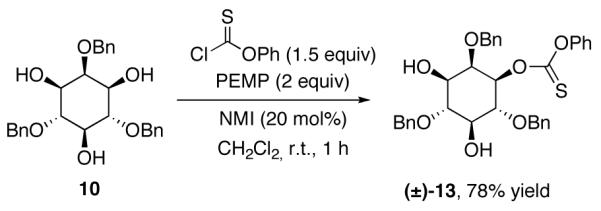
As shown in Table 1, with myo-inositol derivative 10 as a substrate, we observed generally slower base-promoted thiocarbonylation reactions. Thus, as shown in entry 1, use of pyridine as a promoter allowed observation of (±)−13 in only 20% conversion, as measured by 1H NMR. Inclusion of DMAP (20 mol%) in the reaction mixture lead to essentially identical results (entry 2), while the combination of 2,6-lutidine with DMAP actually led to no reaction (entry 3). On the other hand, robust catalytic conditions were identified when 1,2,2,6,6-pentamethylpiperidine (PEMP) was employed as a base in combination with NMI as a catalyst. PEMP alone was found to afford no background rate under the conditions we explored (entry 4); use of PEMP, in combination with 20 mol% NMI on the other hand, led to 90% conversion to (±)−13 within 18 h (entry 5); a combination of PEMP and 20 mol% DMAP led to a slightly less efficient reaction (76% conversion; entry 6). As shown in entries 7-10, PEMP was found to be superior to a range of other bases in combination with NMI as a catalyst. The thionoformate was also varied (entries 11-13), which leads to comparable results.
Table 1.
Thiocarbonylation reaction of 2,4,6-tribenzyl myo-inositol
| Entry | Thionoformate (equiv) | Base (equiv) | Catalyst (20 mol%) | Conversion (%)a |
|---|---|---|---|---|
| 1 |

|
Pyridine (1.2) | - | 20 |
| 2 | Pyridine (1.2) | DMAP | 25 | |
| 3 | 2,6-Lutidine (1.2) | DMAP | <5 | |
|
| ||||
| 4 |
|
PEMP (2) | - | N.R. |
| 5 | PEMP (2) | NMI | 90 | |
| 6 | PEMP (2) | DMAP | 76 | |
|
| ||||
| 7 |
|
K2CO3 (2) | NMI | N.R. |
| 8 | Proton Sponge (2) | NMI | 13 | |
| 9 | DABCO (2) | NMI | N.R. | |
| 10 | DIPEA (2) | NMI | 45 | |
|
| ||||
| 11 | ClC(S)O-p-tolyl (1.5) | PEMP (2) | NMI | 91 |
| 12 | ClC(S)O-p-C6H4Cl (1.5) | PEMP (2) | NMI | 78 |
| 13 | ClC(S)O-p-C6H4F (1.5) | PEMP (2) | NMI | 76 |
Conversions determined by 1H-NMR integration
PEMP = 1,2,2,6,6-pentamethyl piperidine: 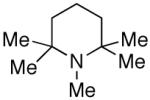
 |
(3) |
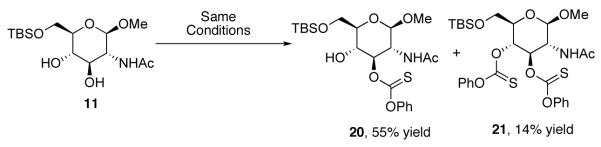
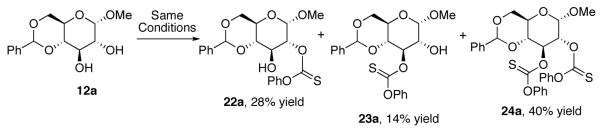
With seemingly robust conditions identified for NMI-catalyzed thionoformate transfer in hand (Table 1, entry 5; eq 3), we sought to examine the scope for a wide variety of substrates. Surprisingly, as shown in Chart 1, the conditions were strikingly specific for carbohydrate derivatives. Examination of seven “simple” substrates revealed that compounds like cyclohexanol and 14-19 underwent no reaction under the identical conditions that led to efficient formation of (±)−13. Indeed, inositol derivative (±)−13 could be obtained in 78% isolated yield, within 1 h of reaction time, under the preparative, catalytic conditions (eq 4). Glucosamine derivative 11 is efficiently converted to a 4:1 mixture of the corresponding mono- and bis(thionoformates) 20 and 21 in 69% combined yield (eq 5). Glucose derivative 12a is converted to an 82% combined, isolated yield of thionoformates 22a, 23a, and 24a, within 1 h, under the identical conditions (eq 6).
Chart 1.

 |
(4) |
 |
(5) |
 |
(6) |
Identification of an FeCl3 effect
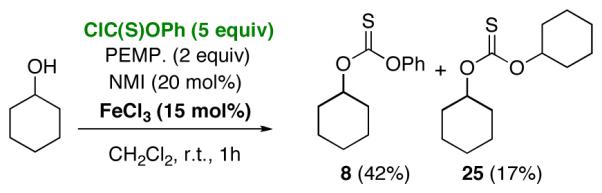
In order to address the lack of generality of the PEMP/NMI conditions, we began to explore additives that might accelerate the reactions of recalcitrant substrates. Among the hypotheses we explored was the idea that phenyl chlorothionoformate could be activated through the use of a co-catalytic amount of a Lewis acid. Indeed, we found that incorporation of a substoichiometic amount of FeCl3 (15 mol%) led to a dramatic increase in the rate of the reactions in the presence of PEMP (2 equiv) and NMI (20 mol%). Whereas no reaction was observed with cyclohexanol in the presence of phenyl chlorothionoformate and PEMP/NMI (Scheme 4, Reaction A), rapid consumption of cyclohexanol was observed when FeCl3 (15 mol%) was introduced (Scheme 4, Reaction B). In terms of control experiments, no reaction was observed in the corresponding reaction when cyclohexanol was exposed to phenyl chlorothionoformate, PEMP, and FeCl3, in the absence of catalytic NMI (Scheme 4, Reaction C). Of further note, in the rapid reaction containing each of the catalytic additives, monoaddition product 8 is not the sole significant product. In fact, over-addition product 25 is also observed (70% combined yield), with compound 8 and 25 being formed in a 1:1 ratio. However, the amount of the overaddition product may be decreased using a larger excess of phenyl chlorothionoformate, as is exemplified in equation 7. Similar results were obtained with cyclooctanol, while other “simple” alcohols such as 15 and 16 turned out to be less reactive, requiring 50 mol% FeCl3 and refluxing dichloromethane for 5 hours to give the desired thiocarbonylation products.
Scheme 4.

 |
(7) |
At this stage, it is too preliminary to state definitively what the role of the FeCl3 co-catalyst is for these more rapid thionoformylation reactions. Among the potential roles we have considered are: (a) that FeCl3 activates phenyl chlorothionoformate in analogy to its role in Friedel-Crafts chemistry (e.g., Figure 1, complex A or complex B);25 (b) that NMI-FeCl3 complexes are formed that in turn activate the reagent (e.g., Figure 1, complex C); (c) that substrate-metal alkoxides are formed that perturb reactivity (e.g., Figure 1, complex D).26 These mechanistic possibilities are not easily resolved and will be the subject of future studies in our laboratory.
Figure 1.
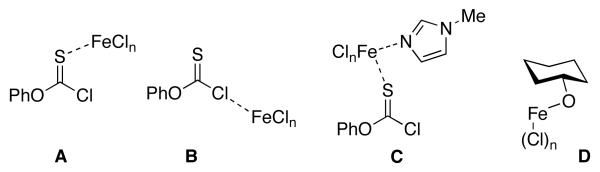
Some potential modes of FeCl3 activation of thionoformylation reactions.
Case studies in regioselective polyol thiocarbonylation and deoxygenation
Given the role of deoxysugars in numerous natural products,27 we chose several simple carbohydrate derivatives as a testing ground for the application of these catalytic thiocarbonylation conditions to site-selective deoxygenations. Our studies began with an examination of α-methyl glucoside 12a (eq 8), and the goal of identifying two sets of catalytic conditions – one that would favor the formation of the mono-functionalized 2-thiocarbonate 22a, and an orthogonal catalytic reaction that would favor the regioisomer, 3-thiocarbonate 23a. Of course, we were keenly aware of the possibility of overfunctionalization to give bis(thiocarbonate) 24a, and also the possible formation of cyclic thiocarbonate 26a.
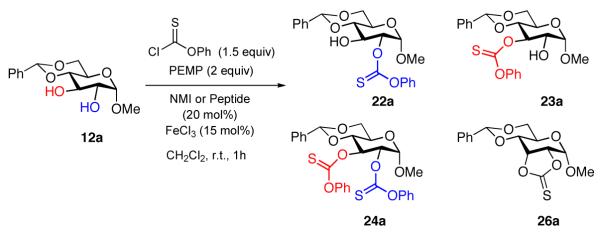 |
(8) |
Any study of regiochemical functionalization also requires a set of minimal control experiments that document (a) the inherent preferences in the hierarchical reactivity within the substrate, and (b) the potential for thermodynamic equilibration of products such as 22a-24a and 26a under the reaction conditions. In the studies we describe below, each case includes a delineation of a hierarchical reactivity of the regiotopic hydroxyl groups in the presence of a given set of reaction conditions. Typically, these involve the use of NMI and FeCl3, each in substoichiometric quantity, in the presence of a stoichiometric quantity of PEMP. Subsequently, we present the perturbation of the inherent product distribution (defined by the outcome with NMI/FeCl3-PEMP), to give different product ratios as a function of the NMI-embedded peptide structure (e.g., 27 versus 28; Chart 2). In the following cases, isolation of the pure mono(thiocarbonates), and resubmission to the reaction conditions revealed them to be non-interconverting under the reaction conditions. As a result, the data presented seem to reflect kinetic selectivities for the site-selective derivatizations we studied. Gratifyingly, in each of the cases we examined, we were able to identify catalyst-dependent reversals of inherent site-selectivity, a manifestation of the ability of these reaction conditions to exhibit kinetic selectivity.
Chart 2.

Thus, our investigation of regioselective thiocarbonylation of 12a began with a documentation of the “inherent” regioselectivity under the optimized conditions, and under conditions of various control experiments. As shown in Table 2, entries 1-2, the inherent NMI-catalyzed ratio of products appears to be ~2:1, either when the reaction is monitored by 1H NMR (entry 1) or when ratios are assessed by quantitative product isolation by careful silica gel chromatography (entry 2). As in our earlier studies, FeCl3 alone does not promote the reaction (entry 3). The inherent selectivity under the NMI/FeCl3 conditions is somewhat different. As illustrated in entries 4 and 5, under these conditions 22a and 23a appear in essentially a 1:1 ratio. In each of the reactions containing NMI, a substantial amount of the over-addition product 24a is also observed. Nonetheless, the equivalence in reactivity of the 2- and 3-positions of 12a under these conditions sets the stage for diverting the product distribution in either direction, as a function of the NMI-embedded peptide structure.28 Our choice of peptides for screening derives from our historical catalyst libraries that we have amassed over several years of study. In particular, catalyst libraries have been assembled from both designed libraries, and random libraries that we prepared for various projects involving alcohol derivatization.29 Notably, catalyst 27 contributes to an enhancement of the 2-substituted product 22a, whether FeCl3 is excluded as a co-catalyst (22a: 23a = 11.7:1, entry 6), or whether it is included (7.6:1, entry 7). Furthermore, when this reaction is conducted at −40 °C, the 2-substituted product 22a is formed in a 22:1 ratio, and may be isolated in 67% yield (entry 8).
Table 2.
| Entry | Catalyst | 12a (%) | 22a (%) | 23a (%) | 22a : 23a | 24a (%) | 26a (%) |
|---|---|---|---|---|---|---|---|
| 1b | NMI (20 mol%) | 4 | 32 | 14 | 2.3 : 1 | 50 | - |
| 2c | NMI (20 mol%) | 28 | 14 | 2 : 1 | 40 | 6 | |
|
| |||||||
| 3 | FeCl3 (15 mol%) | No Reaction | |||||
|
| |||||||
| 4b | NMI (20 mol%) + FeCl3 (15 mol%) | 10 | 30 | 30 | 1 : 1 | 28 | 2 |
| 5c | NMI (20 mol%) + FeCl3 (15 mol%) | 26 | 26 | 1 : 1 | 20 | 16 | |
|
| |||||||
| 6b | 27 (20 mol%) | 14 | 70 | 6 | 11.7 : 1 | 10 | - |
| 7b | 27 (20 mol%) + FeCl3 (15 mol%) | 40 | 53 | 7 | 7.6 : 1 | - | - |
| 8c,d | 27 (20 mol%) | 67 | 3 | 22 : 1 | 10 | 7 | |
|
| |||||||
| 9b | 28 (20 mol%) | 17 | 42 | 25 | 1.7 : 1 | 16 | - |
| 10c | 28 (20 mol%) + FeCl3 (15 mol%) | 40 | 11 | 47 | 1 : 4 | - | 2 |
| 11c,e | 28 (20 mol%) + FeCl3 (15 mol%) | 8 | 53 | 1 : 6.6 | 5 | 8 | |
All reactions conducted in the presence of PEMP (2 equiv), and run according to the standard conditions described in the Experimental Section.
Conversion determined by 1H NMR.
Isolated yield after flash chromatography.
The reaction was conducted at −40 °C, 5 h.
The reaction was conducted at −25 °C, 5 h.
A peptide-dependent reversal in regioselectivity was observed when peptide 28 was used as a co-catalyst in place of 27. Interestingly, the reversal was only observed when the peptide-FeCl3 co-catalyst combination was employed. When catalyst 28 is used exclusively, a 22a:23a ratio of 1.7:1 is observed (entry 9). In the presence of FeCl3, however, the ratio reverses to 1:4 (entry 10), and is amplified to 1:6.6 when the reaction is conducted at low temperature, enabling isolation of 23a in 53% yield under these conditions (entry 11). These results represented an encouraging illustration of the possibility of controlling the regiochemical outcome of these thiocarbonylations as a function of the specific catalytic conditions. Moreover, contrasting entries 7 and 11 illustrates the potential for different regioselectivity outcomes that are fully attributable to the chiral, functionality-rich environment provided by the peptide structure.
We note parenthetically, that each of the compounds prepared with the site-selective thiocarbonylations is readily converted to the deoxygenated species using a common set of standard Barton-McCombie reaction conditions. Thus, deoxysugars 29-31 are readily obtained by this method (eq 9-11).
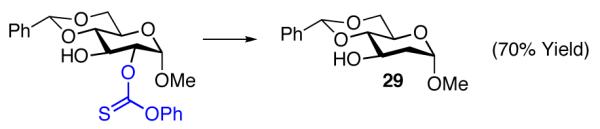 |
(9) |
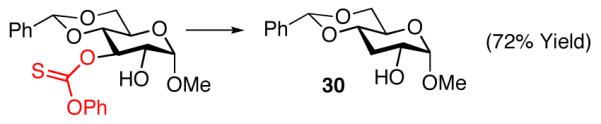 |
(10) |
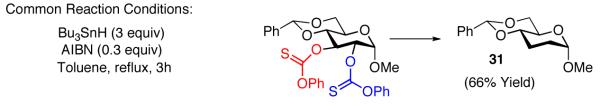 |
(11) |
In an effort to establish the generality of this first-generation set of conditions, we explored several other simple derivatives of glucose. Thus, the thiocarbonylation on the α-isopropyl glucoside 12b gave essentially the same product distribution as the corresponding α-methyl derivative 12a: peptide 27 enhanced the reactivity for the 2 position to give 22b preferentially, while peptide 28, in combination with FeCl3, reversed the product distribution to favor the 3-thiocarbonyl derivative 23b (eq 12).
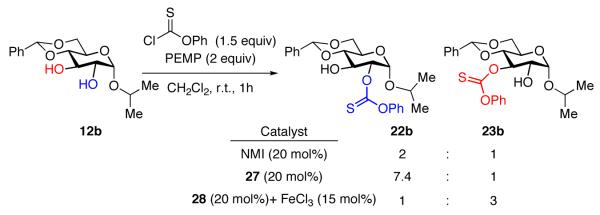 |
(12) |
Clearly, in the absence of a detailed role of the peptide structure in translating stereochemical information to the substrate, screening of diverse peptide catalyst libraries is one way forward in finding analogous reversals of kinetic regioselectivity. One such study of interest includes the simple β-methyl glucoside 32, which provides a direct comparison to our study of α-anomer 12a. As shown in equation 13, the issues are similar in terms of the potential for a complex product distribution, with derivatives 33-36 possible. With the β-anomer, the inherent product distribution for the formation of 2 -versus 3-mono(thiocarbonates) (i.e., 33:34) is 1:2 under catalysis by NMI in the absence of FeCl3, and a similar ratio (1:1.6) is observed in the presence of both NMI and FeCl3 (Table 3, entries 1 and 2). In this case, our “hit” peptides for the α-glucoside were less successful in terms of regioselectivity. Entry 3 shows that peptide catalyst 27, in the absence of FeCl3, provides a modest preference for 3-thiocarbonate 34 (1:1.5). Alternatively, peptide 28 provides a modest preference for 2-thiocarbonate 33 (1.5:1; entry 5). In both cases, the inclusion of FeCl3 in the reaction mixture provides subtle modulation of ratios, but the effects overall with β-glucoside 32 are small. It is possible that a more comprehensive screen of peptide co-catalysts could provide a more dramatic result. In the context of the present study, it may suffice to say that the differentiation of β-glucosides of glucose, with the 4-position protected, may provide a particular challenge to regioselective catalysis in this “all equatorial” array.
Table 3.
| Entry | Catalyst | 32 (%) | 33 (%) | 34 (%) | 33 : 34 | 35 (%) | 36 (%) |
|---|---|---|---|---|---|---|---|
| 1 | NMI (20 mol%) | - | 7 | 13 | 1 : 2 | 76 | 4 |
| 2 | NMI (20 mol%) + FeCl3(15 mol%) | 5 | 11 | 18 | 1 : 1.6 | 38 | 28 |
|
| |||||||
| 3 | 27 (20 mol%) | - | 18 | 27 | 1 : 1.5 | 25 | 30 |
| 4 | 27 (20 mol%) + FeCl3(15 mol%) | 9 | 33 | 23 | 1.4 : 1 | 7 | 29 |
|
| |||||||
| 5 | 28 (20 mol%) | 7 | 14 | 10 | 1.5 : 1 | 36 | 33 |
| 6 | 28 (20 mol%) + FeCl3(15 mol%) | 15 | 10 | 18 | 1 : 2 | 4 | 54 |
All reactions conducted in the presence of PEMP, and run according to the standard conditions described in the Experimental Section.
Conversion determined by 1H NMR.
27: BOC-Pmh-DPro-Aib-DTrp(Boc)-DPhe-OMe
28: BOC-Pmh-Thr(tBu)-DVal-His(trt)-DPhe-DVal-Thr(tBu)-Ile-OMe
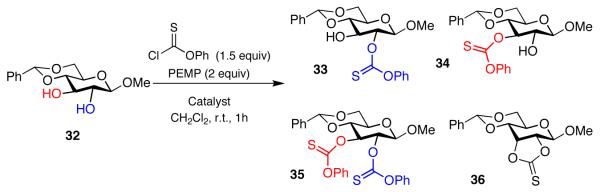 |
(13) |
Finally, we decided to investigate the reactivity in a more complex setting. In order to compare with the glucose derivatives previously studied, we synthesized the 4,6-O-benzylidene −1′, 6′-bis-O-t-butyldiphenylsilyl protected sucrose derivative 37 (eq 14).30 As shown in Table 4, with NMI as catalyst, the 3′-position is slightly more reactive than the 2-position on the hexose ring (entry 1). Notably, the 3-position of the glucose ring is substantially less reactive still. A brief screen of peptide catalysts and conditions revealed that the potential exists for modulating the site selectivity of these thiocarbonylations. For example, when peptide 27 is employed as a catalyst, modest 2:1 preference for reaction of the 2-position of the glucoside is favored to deliver 38 as the major product (entry 2). In contrast, peptide 28 reverses the site-selectivity to the 3-position of the fructoside to favor 39 in a ratio of 1:2 (entry 3). While the reversal is modest in these cases, we suspect that a more extensive screening of catalysts and conditions could lead to more dramatic reversals in terms of magnitude.
Table 4.
Regioselectivity in the thiocarbonylation of sucrose derivative 37.a
| Entry | Catalyst | 38 : 39 | Yield (38 + 39)b |
|---|---|---|---|
| 1 | NMI (20 mol%) | 1 : 1.6 | 39 |
| 2 | 27 (20 mol%) | 2 : 1 | 66 |
| 3 | 28 (20 mol%) + FeCl3(15 mol%) | 1 : 2 | 40 |
All reactions conducted in the presence of PEMP, and run according to the standard conditions described in the Experimental Section.
Isolated yield after flash chromatography
27: BOC-Pmh-DPro-Aib-DTrp(Boc)-DPhe-OMe
28: BOC-Pmh-Thr(t-Bu)-DVal-His(trt)-DPhe-DVal-Thr(t-Bu)-Ile-OMe
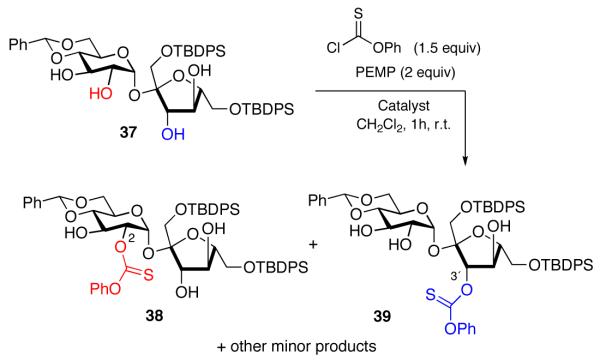 |
(14) |
Notably, sucrose derivatives 38 and 39 were readily deoxygenated to give deoxy-sucrose derivatives 40 and 41 (eq 15 and 16). Such sequences may provide straightforward access to deoxygenated disaccharide derivatives with enhanced efficiencies in comparison to the corresponding reactions wherein a single catalyst is employed to obtain a given product distribution.
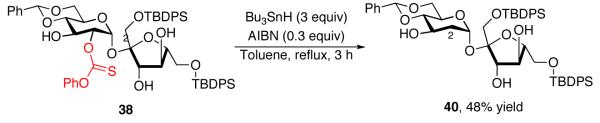 |
(15) |
 |
(16) |
Conclusions
In these studies of site-selective thiocarbonylation, we have established a number of important milestones. Initially, we identified a reliable reagent combination that leads to consistent and efficient thiocarbonylation reactions using phenyl chlorothionoformate as a reagent, and a combination of NMI and FeCl3 (each in substoichiometric quantities) as co-catalysts. Importantly, we then showed that site-selectivity of thiocarbonylation may be modulated in a catalyst-dependent fashion, and, in particular, peptidic structures containing the N-alkylimidazole substructure are critical variables in screening and optimization. Furthermore, in selected cases, we showed that one could observe reversals of inherent selectivity such that catalyst control, rather than substrate control only, could be utilized to deliver alternative major products in the polyol settings. Such reactions hold the potential to reduce the chemist’s dependence on protecting groups by designing direct thiocarbonylation-deoxygenation sequences that may be of interest in synthetic and medicinal chemistry applications.
The mechanistic basis of these reversals is a frontier activity in the developing area of asymmetric catalysts that modulate regioselectivity. We have taken important first steps in our studies of chiral, peptide-based asymmetric catalysts.31 That these new protocols include a significant, metal-based co-catalyst increases the complexity of these catalytic reactions in significant ways. The elucidation of how peptide-based systems function in the presence of additives like FeCl3 is an exciting challenge that we are now addressing in our laboratory.
Experimental Section
General Procedures for the Thiocarbonylation Reaction
General Procedure A
Phenyl chlorothionoformate (1.5 equiv), 1,2,2,6,6-pentamethyl piperidine (2.0 equiv), and N-methylimidazole or a peptide catalyst (0.20 equiv) were added sequentially to a flame dried 10 mL round bottom flask containing a solution of alcohol, diol, or polyol (1.0 equiv) in anhydrous dichloromethane (1 mL). The reaction (yellow or brown) solution was stirred at room temperature for 1 hour, then quenched with 1 mL of methanol and concentrated under reduced pressure. The mixture of reaction products was isolated by silica gel flash chromatography.
General Procedure B
Phenyl chlorothionoformate (1.5 equiv), 1,2,2,6,6-pentamethyl piperidine (2.0 equiv), N-methylimidazole or a peptide catalyst (0.20 equiv), and FeCl3 (0.15 equiv) were added sequentially to a flame dried 10 mL round bottom flask containing a solution of alcohol, diol, or polyol (1.0 equiv) in anhydrous dichloromethane (1 mL). The reaction (yellow or brown) mixture was stirred at room temperature for 1 hour, then quenched with 1 mL of methanol and concentrated under reduced pressure. The mixture of reaction products was isolated by silica gel flash chromatography.
NOTE: Optimized reaction conditions are described for each respective substrate. Unless otherwise noted, reactions were run according to conditions (General Procedure A or B) described above.
4, 6-O-Benzylidene 2-mono(thiocarbonate) α-methyl glucoside 22a
General procedure A for thiocarbonylation was followed, glucose derivative 12a (40 mg, 0.14 mmol) was employed as starting material with peptide 27 (25 mg, 0.028 mmol), phenyl chlorothionoformate (28 μL, 0.21 mmol), and 1,2,2,6,6-pentamethyl piperidine (51 μL, 0.28 mmol) in CH2Cl2 (1.5 mL) and the reaction was allowed to stir for 5 h at −40°C. Purification through silica gel flash chromatography (7:1 to 2:1 hexanes-EtOAc) afforded a 22:1 product ratio of 22a:23a. Both 22a (39 mg, 67%) and 23a (2.0 mg, 3%) were isolated as white solids. In addition, minor amounts of 24a (8 mg, 10%) and 26a (3 mg, 7%) were also isolated, each as a white solid.
Data for 22a: The spectral data for compound 22a matched that which had been previously reported.32
4, 6-O-Benzylidene 3-mono(thiocarbonate) α-methyl glucoside 23a
General procedure B for thiocarbonylation was followed, glucose derivative 12a (30 mg, 0.11 mmol) was employed as starting material with peptide 28 (32 mg, 0.022 mmol), phenyl chlorothionoformate (22 μL, 0.165 mmol), 1,2,2,6,6-pentamethyl piperidine (40 μL, 0.22 mmol), and FeCl3 (2.7 mg, 0.0165 mmol) in CH2Cl2 (1.0 mL) and the reaction was allowed to stir for 5 h at −25°C. Purification through silica gel flash chromatography (7:1 to 2:1 hexanes-EtOAc) afforded a 1:6.6 product ratio of 22a:23a. Both 22a (4.0 mg, 8%) and 23a (24 mg, 53%) were isolated as white solids. In addition, minor amounts of 24a (3 mg, 5%) and 26a (3 mg, 8%) were also isolated, each as a white solid.
Data for 23a: mp: 97-101 °C; 1H NMR (CDCl3, 500 MHz) Δ 7.51-7.49 (m, 2H), 7.41-7.27 (m, 6H), 7.12-7.10 (m, 2H), 5.98 (t, J = 9.5 Hz, 1H), 5.56 (s, 1H), 4.89 (d, J = 3.5 Hz, 1H), 4.35 (dd, J = 4.7, 10.4 Hz, 1H), 3.98-3.91 (m, 2H), 3.83-3.77 (m, 2H), 3.52 (s, 3H), 2.31 (d, J = 9.5 Hz, 1H); 13C NMR (CDCl3, 125 MHz) Δ 195.8, 153.5, 136.9, 129.4, 129.1, 128.2, 126.5, 126.2, 101.5, 100.1, 82.1, 78.7, 71.9, 68.9, 62.7, 55.7, 53.4; IR (film, cm−1) 3457, 2917, 1483, 1283, 1201, 1070, 1050; TLC Rf0.22 (2:1 hexanes-EtOAc); [α]20.0D +84.1 (c 1.0, CHCl3); HRMS calcd for [C21H23O7S H]+ requires m/z 419.1165; found 419.1167 (ESI+).
4, 6-O-Benzylidene 2,3-bis(thiocarbonate) α-methyl glucoside 24a
Data for 24a: mp: 58-60 °C; 1H NMR (CDCl3, 500 MHz) Δ 7.41-7.39 (m, 2H), 7.33-7.13 (m, 9H), 7.03-7.01 (m, 2H), 6.95-6.93 (m, 2H), 6.22 (t, J = 9.6 Hz, 1H), 5.57 (dd, J = 3.8, 9.5 Hz, 1H), 5.48 (s, 1H), 5.17 (d, J = 3.8 Hz, 1H), 4.27 (dd, J = 4.7, 10.3 Hz, 1H), 3.97 (dt, J = 4.7, 10.0 Hz, 1H), 3.83 (t, J = 9.6 Hz, 1H), 3.74 (t, J = 10.2 Hz, 1H), 3.40 (s, 3H); 13C NMR (CDCl3, 125 MHz) Δ 194.4, 194.3, 153.5, 153.4, 136.7, 129.6, 129.4, 129.1, 128.2, 126.8, 126.6, 126.2, 121.8, 121.7, 101.7, 96.9, 79.6, 79.1, 78.5, 68.8, 62.4, 55.7; IR (film, cm−1) 2918, 1487, 1454, 1272, 1225, 1192, 1148, 1123; TLC Rf0.56 (2:1 hexanes-EtOAc); [α]20.0D −30.5 (c 1.0, CHCl3); HRMS calcd for [C28H27O8S2 H]+ requires m/z 555.1147; found 555.1145 (ESI+).
Compound 26a
Data for 26a: The spectral data for compound 26a matched that which had been previously reported.33
General Procedure for the Deoxygenation Reaction
To a solution of the corresponding mono or bis thiocarbonate (1 equiv) in toluene (4 mL), tributyltin hydride (3 equiv) and AIBN (0.3 equiv) were added and the mixture was heated at reflux for 3 h. Solvent was then removed under reduced pressure and the resulting residue was purified by silica gel flash chromatography (3:1 to 2:1, hexanes-EtOAc).
Compound 29
General procedure for deoxygenation was followed, glucose derivative 22a (54 mg, 0.13 mmol) was employed as starting material with AIBN (6.4 mg, 0.039 mmol) and tributyltin hydride (100 μL, 0.39 mmol). Compound 29 was isolated as a white solid (24 mg, 70%).
Data for 29: The spectral data for compound 29 matched that which had been previously reported.34
Compound 30
General procedure for deoxygenation was followed, glucose derivative 23a (50 mg, 0.12 mmol) was employed as starting material with AIBN (6.0 mg, 0.036 mmol) and tributyltin hydride (95.0 μL, 0.36 mmol). Compound 30 was isolated as a white solid (23 mg, 72%).
Data for 30: The spectral data for compound 30 matched that which had been previously reported.35
Compound 31
General procedure for deoxygenation was followed, glucose derivative 24a (79 mg, 0.14 mmol) was employed as starting material with AIBN (7.0 mg, 0.042 mmol) and tributyltin hydride (110 μL, 0.42 mmol). Compound 31 was isolated as a white solid (23 mg, 66%).
Data for 31: mp: 199-202 °C; 1H NMR (CDCl3, 400 MHz) Δ 7.50-7.48 (m, 2H), 7.39-7.34 (m, 3H), 5.57 (s, 1H), 4.70 (d, J = 3.3 Hz, 1H), 4.22 (dd, J = 4.5, 10.1 Hz, 1H), 3.83 (dt, J = 4.8, 9.7 Hz, 1H), 3.72 (t, J = 10.2 Hz, 1H), 3.63-3.57 (m, 1H), 3.38 (s, 3H), 2.02-1.84 (m, 4H); 13C NMR (CDCl3, 125 MHz) Δ 137.7, 129.0, 128.3, 126.2, 101.9, 97.9, 78.3, 69.6, 64.8, 54.6, 29.5, 24.0; IR (film, cm−1) 2946, 2901, 2868, 1377, 1123, 1095, 1054, 997, 919, 694; TLC Rf0.44 (2:1 hexanes-EtOAc); [α]20.0D +93.5 (c 0.62, CHCl3); HRMS calcd for [C14H18O4 Na]+ requires m/z 273.1103; found 273.1112 (ESI+).
Supplementary Material
Acknowledgement
This work is supported by the NIH (GM-068649). M. S.-R. would like to thank the Ministerio de Educacion y Ciencia of Spain for a postdoctoral fellowship.
Footnotes
Supporting Information Available: Experimental procedures, characterization data, 1H and 13C NMR spectra for compounds 8, (±)−13, 20, 21, 22b-24b, 26b, 33, 34, 35, 36, 38, 39. This material is provided free of charge via the Internet at http://pubs.acs.org.
REFERENCES
- 1.(a) Hartwig W. Tetrahedron. 1983;39:2609–2645. [Google Scholar]; (b) Crich D, Quintero L. Chem. Rev. 1989;89:1413–1432. [Google Scholar]; (c) Lopez RM, Hays DS, Fu GC. J. Am. Chem. Soc. 1997;119:6949–6950. [Google Scholar]; (d) Yasuda M, Onisi Y, Ueba M, Miyai T, Baba A. J. Org. Chem. 2001;66:7741–7744. doi: 10.1021/jo0158534. [DOI] [PubMed] [Google Scholar]; (e) Schlaf M, Ghosh P, Fagan PJ, Hauptman E, Bullock R. M. Angew. Chem., Int. Ed. 2001;40:3887–3890. [PubMed] [Google Scholar]
- 2.(a) Graham AE, Thomas AV, Yang R. J. Org. Chem. 2000;65:2583–2585. doi: 10.1021/jo991645o. [DOI] [PubMed] [Google Scholar]; (b) Morgan AJ, Wang YK, Roberts MF, Miller SJ. J. Am. Chem. Soc. 2004;126:15370–15371. doi: 10.1021/ja047360x. [DOI] [PubMed] [Google Scholar]; (c) Gómez AM, Moreno E, Valverde S, López JC. Eur. J. Org. Chem. 2004:1830–1840. [Google Scholar]; (d) Wang YK, Morgan AJ, Stieglitz K, Stec B, Thompson B, Miller SJ, Roberts MF. Biochemistry. 2006;45:3307–3314. doi: 10.1021/bi052467y. [DOI] [PubMed] [Google Scholar]
- 3.(a) Barton DHR, McCombie SW. J. Chem. Soc., Perkin Trans. 1975;1:1574–1585. [Google Scholar]; (b) Barton DHR, Blundell P, Dorchak J, Jang DO, Jaszberenyi J. Cs. Tetrahedron. 1991;47:8969–8984. [Google Scholar]; (c) Barton DHR, Dorchak J, Jaszberenyi J. Cs. Tetrahedron. 1992;48:7435–7446. [Google Scholar]
- 4.(a) Kurahashi T, Mizutani T, Yoshida J. J. Chem. Soc., Perkin Trans. 1999;1:465–473. [Google Scholar]; (b) Gani D. Nature. 2001;414:703–705. doi: 10.1038/414703a. [DOI] [PubMed] [Google Scholar]; (c) Kurahashi T, Mizutani T, Yoshida J. Tetrahedron. 2002;58:8669–8677. [Google Scholar]; (d) Hu G, Vasella A. Helv. Chim. Acta. 2002;85:4369–4391. [Google Scholar]; (e) Kattnig E, Albert M. Org. Lett. 2004;6:945–948. doi: 10.1021/ol0364935. [DOI] [PubMed] [Google Scholar]; (f) Moitessier N, Englebienne P, Chapleur Y. Tetrahedron. 2005;61:6839–6853. [Google Scholar]; (g) Wang C-C, Lee J-C, Luo S-Y, Kulkarni SS, Huang Y-W, Lee C-C, Chang K-L, Hung S-C. Nature. 2007;446:896–899. doi: 10.1038/nature05730. [DOI] [PubMed] [Google Scholar]
- 5.(a) Fu GC. Acc. Chem. Res. 2000;33:412–420. doi: 10.1021/ar990077w. [DOI] [PubMed] [Google Scholar]; (b) Vedejs E, Daugulis O, MacKay JA, Rozners E. Synlett. 2001:1499–1505. [Google Scholar]; (c) France S, Guerin DJ, Miller SJ, Lectka T. Chem. Rev. 2003;103:2985–3012. doi: 10.1021/cr020061a. [DOI] [PubMed] [Google Scholar]; (d) Denmark SE, Fu J. Chem. Commun. 2003:167–170. doi: 10.1039/b208065f. [DOI] [PubMed] [Google Scholar]; (e) Spivey AC, Arseniyadis S, Fekner T, Maddaford A, Leese DP. Tetrahedron. 2006;62:295–301. [Google Scholar]
- 6.(a) Vedejs E, Chen X. J. Am. Chem. Soc. 1996;118:1809–1810. [Google Scholar]; (b) Kawabata T, Nagato M, Takasu K, Fuji K. J. Am. Chem. Soc. 1997;119:3169–3170. [Google Scholar]; (c) Arai S, Bellemin-Laponnaz S, Fu GC. Angew. Chem., Int. Ed. 2001;40:234–236. [PubMed] [Google Scholar]; (d) Shaw SA, Aleman P, Vedejs E. J. Am. Chem. Soc. 2003;125:13368–13369. doi: 10.1021/ja037223k. [DOI] [PubMed] [Google Scholar]; (e) Spivey AC, Zhu F, Mitchell MB, Davey SG, Jarvest LR. J. Org. Chem. 2003;68:7379–7385. doi: 10.1021/jo034603f. [DOI] [PubMed] [Google Scholar]; (f) Fu GC. Acc. Chem. Res. 2004;37:542–547. doi: 10.1021/ar030051b. [DOI] [PubMed] [Google Scholar]; (g) Spivey AC, Arseniyadis S. Angew. Chem., Int. Ed. 2004;43:5436–5441. doi: 10.1002/anie.200460373. [DOI] [PubMed] [Google Scholar]
- 7.(a) Denmark SE, Fu J. J. Am. Chem. Soc. 2001;123:9488–9489. doi: 10.1021/ja016552e. [DOI] [PubMed] [Google Scholar]; (b) Vedejs E, Daugulis O, Harper LA, MacKay JA, Powell DR. J. Org. Chem. 2003;68:5020–5027. doi: 10.1021/jo030007+. [DOI] [PubMed] [Google Scholar]; (c) Methot JL, Roush WR. Adv. Synth. Catal. 2004;346:1035–1050. and references therein. [Google Scholar]
- 8.(a) Birman VB, Uffman EW, Jiang H, Li X, Kilbane CJ. J. Am. Chem. Soc. 2004;126:12226–12227. doi: 10.1021/ja0491477. [DOI] [PubMed] [Google Scholar]; (b) Birman VB, Jiang H. Org. Lett. 2005;7:3445–3447. doi: 10.1021/ol051063v. [DOI] [PubMed] [Google Scholar]; (c) Birman VB, Li X, Jiang H, Uffman EW. Tetrahedron. 2006;62:285–294. [Google Scholar]; (d) Birman VB, Li X, Han Z. Org. Lett. 2007;9:37–40. doi: 10.1021/ol0623419. [DOI] [PubMed] [Google Scholar]
- 9.Chiral diamines: Oriyama T, Imai K, Hosoya T, Sano T. Tetrahedron Lett. 1998;39:397400. Oriyama T, Imai K, Sano T, Hosoya T. Tetrahedron Lett. 1998;39:3529–3532. Nucleophilic carbenes: Enders D, Balensiefer B. Acc. Chem. Res. 2004;37:534–541. doi: 10.1021/ar030050j.
- 10.Miller S. J. Acc. Chem. Res. 2004;37:601–610. doi: 10.1021/ar030061c. [DOI] [PubMed] [Google Scholar]
- 11.(a) Griswold KS, Miller SJ. Tetrahedron. 2003;59:8869–8875. [Google Scholar]; (b) Lewis CA, Miller S. J. Angew. Chem. Int. Ed. 2006;45:5616–5619. doi: 10.1002/anie.200601490. [DOI] [PubMed] [Google Scholar]
- 12.(a) Jarvo ER, Copeland GT, Papaioannou N, Bonitatebus PJ, Jr., Miller SJ. J. Am. Chem. Soc. 1999;121:11638–11643. [Google Scholar]; (b) Copeland GT, Miller SJ. J. Am. Chem. Soc. 2001;123:6496–6502. doi: 10.1021/ja0108584. [DOI] [PubMed] [Google Scholar]; (c) Fierman MB, O’Leary DJ, Steinmetz WE, Miller SJ. J. Am. Chem. Soc. 2004;126:6967–6971. doi: 10.1021/ja049661c. [DOI] [PubMed] [Google Scholar]; (d) Lewis CA, Chiu A, Kubryk M, Balsells J, Pollard D, Esser CK, Murry J, Reamer RA, Hansen KB, Miller SJ. J. Am. Chem. Soc. 2006;128:16454–16455. doi: 10.1021/ja067840j. [DOI] [PubMed] [Google Scholar]
- 13.(a) Sculimbrene BR, Miller SJ. J. Am. Chem. Soc. 2001;123:10125–10126. doi: 10.1021/ja016779+. [DOI] [PubMed] [Google Scholar]; (b) Sculimbrene BR, Morgan AJ, Miller SJ. J. Am. Chem. Soc. 2002;124:11653–11656. doi: 10.1021/ja027402m. [DOI] [PubMed] [Google Scholar]; (c) Sculimbrene BR, Morgan AJ, Miller S. J. Chem. Commun. 2003;1781:1785. doi: 10.1039/b304015c. [DOI] [PubMed] [Google Scholar]; (d) Sculimbrene BR, Xu Y, Miller SJ. J. Am. Chem. Soc. 2004;126:13182–13183. doi: 10.1021/ja0466098. [DOI] [PubMed] [Google Scholar]
- 14.Evans JW, Fierman MB, Miller SJ, Ellman JA. J. Am. Chem. Soc. 2004;126:8134–8135. doi: 10.1021/ja047845l. [DOI] [PubMed] [Google Scholar]
- 15.For a related approach involving alcohol silylation, see: Isobe T, Fukuda K, Araki Y, Ishikawa T. Chem. Commun. 2001:243–244. Zhao Y, Rodrigo J, Hoveyda AH, Snapper ML. Nature. 2006;443:67–70. doi: 10.1038/nature05102.
- 16.Barton DHR, Ferreira JA, Jaszberenyi JC. Preparative Carbohydrate Chemistry. 1997:151-172. [Google Scholar]
- 17.Rawal VH, Newton RC, Krishnamurthy V. J. Org. Chem. 1990;55:5181–5183. [Google Scholar]
- 18.For three examples of syntheses where both DMAP and pyridine are used in combination to effect thiocarbonylation, see: Denmark SE, Thorarensen A. J. Org. Chem. 1994;59:5672–5680. Rigby JH, Mateo ME. Tetrahedron. 1996;52:10569–10582. Shimokawa J, Shirai K, Tanatani A, Hashimoto Y, Nagasawa K. Angew. Chem., Int. Ed. 2004;43:1559–1562. doi: 10.1002/anie.200353200.
- 19.Gaudino JJ, Wilcox CS. J. Am. Chem. Soc. 1990;112:4374–4380. [Google Scholar]
- 20.(a) Hsu FL, Zhang X, Hong SS, Berg FJ, Miller DD. Heterocycles. 1994;39:801–809. [Google Scholar]; (b) Millan DS, Prager RH. Aust. J. Chem. 1999:841–849. [Google Scholar]
- 21.(a) Guibe-Jampel E, Bram G, Vilkas M. Bull. Soc. Chim. Fr. 1973:1021–1027. [Google Scholar]; (b) Pandit NK, Connors KA. J. Pharm. Sci. 1982;71:485–491. doi: 10.1002/jps.2600710503. [DOI] [PubMed] [Google Scholar]; (c) Breslow R. J. Mol. Catal. 1994;91:161–174. [Google Scholar]
- 22.(a) Lee HW, Kishi Y. J. Org. Chem. 1985;50:4402–4404. [Google Scholar]; (b) Billington DC, Baker R, Kulagowski JJ, Mawer IM, Vacca JP, Jane deSolms S, Huff JR. J. Chem. Soc., Perkin Trans. 1989;1:1423–1429. [Google Scholar]
- 23.RajanBabu TV, Ayers TA, Halliday GA, You KK, Calabrese JC. J. Org. Chem. 1997;62:6012–6028. [Google Scholar]
- 24.Ferro V, Mocerino M, Stick RV, Tilbrook DM. G. Aust. J. Chem. 1988;41:813–815. [Google Scholar]
- 25.Diaz DD, Miranda PO, Padron JI, Martin VS. Curr. Org. Chem. 2006;10:457–476. [Google Scholar]
- 26.David S, Hanessian S. Tetrahedron. 1985;41:643–663. [Google Scholar]
- 27.See ref. 2.
- 28.After screening about 100 peptides of different length and structure, we found pentapeptide 27 and octapeptide 28 showed the most regiodivergent behavior in the thiocarbonylation of α-methyl glucoside 12.
- 29.A representative comparison of such libraries may be found in the following article: Jarvo ER, Evans CA, Copeland GT, Miller SJ. J. Org. Chem. 2001;66:5522–5527. doi: 10.1021/jo015803z.
- 30.(a) Karl H, Lee CK, Khan R. Carbohydrate Research. 1982;101:31–38. [Google Scholar]; (b) Adinolfi M, De Napoli L, Di Fabio G, Iadonisi A, Montesarchio D. Org. Biomol. Chem. 2004;2:1879–1886. doi: 10.1039/b401819b. [DOI] [PubMed] [Google Scholar]
- 31.Vasbinder MM, Jarvo ER, Miller S. J. Angew. Chem., Int. Ed. 2001;40:2824–2827. doi: 10.1002/1521-3773(20010803)40:15<2824::aid-anie2824>3.3.co;2-a. [DOI] [PubMed] [Google Scholar]; (b) See ref. 12.
- 32.Petra̧ková E, Glaudemans CP. J. Glycoconjugate Journal. 1994;11:17–22. doi: 10.1007/BF00732428. [DOI] [PubMed] [Google Scholar]
- 33.Tsuda Y, Noguchi S, Kanemitsu K, Sato Y, Kakimoto K, Iwakura Y, Hosoi S. Chem. Pharm. Bull. 1997;45:971–980. [Google Scholar]
- 34.Minami A, Eguchi T. J. Amer. Chem. Soc. 2007;129:5102–5107. doi: 10.1021/ja0685250. [DOI] [PubMed] [Google Scholar]
- 35.Lee E, McArdle P, Browne P, Gavin N. Carbohydrate Research. 1989;191:351–356. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.