

Abstract
Objectives:
Recently, 2 independent studies reported that a rare missense variant, rs75932628 (R47H), in exon 2 of the gene encoding the “triggering receptor expressed on myeloid cells 2” (TREM2) significantly increases the risk of Alzheimer disease (AD) with an effect size comparable to that of the APOE ε4 allele.
Methods:
In this study, we attempted to replicate the association between rs75932628 and AD risk by directly genotyping rs75932628 in 2 independent Caucasian family cohorts consisting of 927 families (with 1,777 affected and 1,235 unaffected) and in 2 Caucasian case-control cohorts composed of 1,314 cases and 1,609 controls. In addition, we imputed genotypes in 3 independent Caucasian case-control cohorts containing 1,906 cases and 1,503 controls.
Results:
Meta-analysis of the 2 family-based and the 5 case-control cohorts yielded a p value of 0.0029, while the overall summary estimate (using case-control data only) resulted in an odds ratio of 1.67 (95% confidence interval 0.95–2.92) for the association between the TREM2 R47H and increased AD risk.
Conclusions:
While our results serve to confirm the association between R47H and risk of AD, the observed effect on risk was substantially smaller than that previously reported.
Alzheimer disease (AD) is the most common form of dementia in the elderly. AD is a genetically heterogeneous disorder characterized by the coexistence of monogenic vs genetically complex forms. Given the disease's established high heritability, the quest for identifying novel AD genes has been pursued for more than 3 decades.1 Late-onset AD (LOAD) genome-wide association studies (GWAS) in case-control datasets have yielded several novel AD candidate genes beyond APOE, including CD33,2–4 CLU, CR1, PICALM,5,6 BIN1,7 ABCA7,3 MS4A6A/MS4A4E, EPHA1, and CD2AP,3,4 all of which have subsequently been validated in independent samples (www.alzgene.org).
Recent reports from case-control studies in AD8,9 have implicated a rare missense variant (rs75932628, R47H substitution) in TREM2 (encoding the triggering receptor expressed on myeloid cells 2 protein) that significantly increases the risk of LOAD. This association between the minor T allele at rs75932628 and increased risk of AD was subsequently confirmed9–11 in several independent study samples. In this study, we attempt to independently replicate the previously reported rare-variant association (TREM2, R47H) with a specific direction of effect (i.e., an increase in AD risk conferred by the minor T allele) association with AD. We also performed conditional logistic regression to report the odds ratio (OR) as an informative parameter for comparing our results with those from previous studies.
METHODS
AD study cohort details and genotype generation.
For the purpose of this study, we utilized several large sample collections with more than 9,300 individuals originating from both family-based and case-control datasets. We first genotyped rs75932628 in 2 family-based study samples: National Institute of Mental Health (NIMH) AD Genetics Initiative Study Family Sample,12,13 and the National Institute on Aging Genetics Initiative for Late Onset Alzheimer's Disease Family Study Sample14 (NIA-LOAD) datasets. In addition, rs75935628 was also genotyped in the German Study on Ageing, Cognition and Dementia in Primary Care Patients (AgeCoDe) case-control samples.15–17 Furthermore, rs75932628 genotypes were imputed in 3 publicly available GWAS datasets, GlaxoSmithKline (GSK), the Alzheimer's Disease Neuroimaging Initiative (ADNI), and the Translational Genomics Research Institute series 2 (TGEN2), which were not analyzed in the previous studies.
Family-based replication assessment by direct genotyping.
The portion of the NIMH AD family sample analyzed in this study consisted of 410 Caucasian families containing 917 affected (average age at onset 72 ± 8.3 years, range 41–93) and 199 unaffected subjects (average age at last cognitive examination 78 ± 9.4 years, range 49–100). All NIMH families were prescreened for mutations in the known early-onset familial AD (i.e., APP, PSEN1, or PSEN2) or frontotemporal dementia (i.e., MAPT, GRN, TARDBP, VCP, or CHMP2B) genes, resulting in an exclusion of 11 families from analysis. The NIA-LOAD familial cohort contained 517 Caucasian families with 860 affected (average age at onset 73 ± 7.8 years, range 43–99) and 1,036 unaffected subjects (average age at last cognitive examination 65 ± 9.9 years, range 50–100). To reduce the potential of diagnostic misclassification, we excluded unaffected subjects who were younger than 49 years of age at last cognitive examination from the family-based analyses. Both family cohorts included primarily sibling pairs. The NIMH contained 105 families with at least one sibling with an age at onset younger than 65 years, whereas the NIA-LOAD contained 92 families with one unaffected subject younger than 65 years. In addition, we genotyped the unrelated case-control portion of NIA-LOAD that included 378 cases and 686 controls. Note that this dataset was included in the original study,8 but limited to imputed genotypes and not by direct genotyping.
Case-control replication assessment by direct genotyping or imputation.
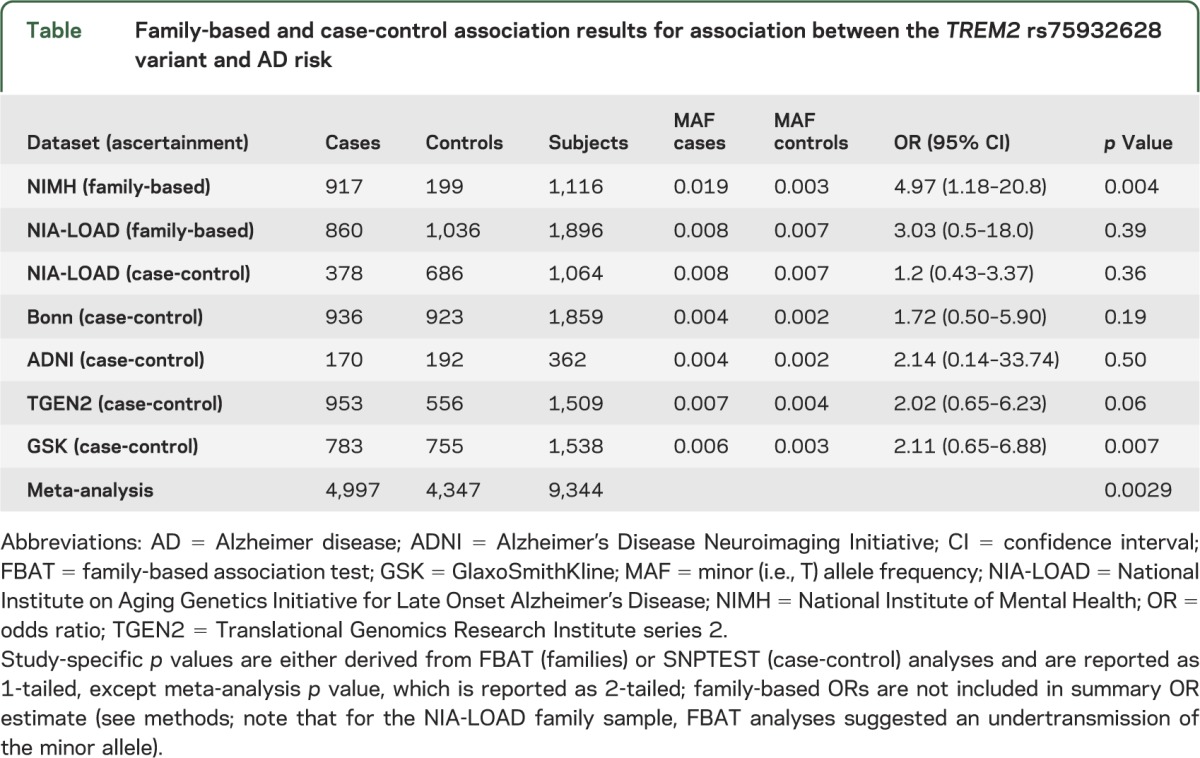
In addition to direct genotyping in the family-based studies, we imputed rs75932628 in 3 case-control cohorts: the GSK, ADNI, and TGEN2 utilizing IMPUTE218,19 and the latest 1000 Genome project (phase I) reference panel. The imputation information quality scores for the GSK, ADNI, and TGEN2 cohorts were 0.30, 0.37, and 0.71, respectively. In addition, rs75932628 was determined by direct genotyping (using the MassARRAY system on a Sequenom Compact matrix-assisted laser desorption/ionization time-of-flight instrument [Sequenom Inc., San Diego, CA]) in 936 patients with AD and 923 healthy controls from Germany (here collectively summarized as “Bonn cohort”).15–17 See the table for a summary of demographic characteristics of all datasets included in this study.
Table.
Family-based and case-control association results for association between the TREM2 rs75932628 variant and AD risk
Standard protocol approvals, registrations, and patient consents.
Informed consent was provided by all participants, and research approval was established by the relevant institutional review boards for each study cohort.
Statistical analyses.
A family-based association test (FBAT) approach was used to test for over- or undertransmission (i.e., risk or protection) of the minor allele in affected offspring as implemented in the PBAT20,21 (version 3.6) software. Conditional logistic regression stratified by family was used to estimate the OR of the underlying risk effects in the family-based datasets. Because OR estimates in the context of family studies are inherently difficult to estimate precisely, the ORs calculated here are displayed merely for informational purposes but are not included in the fixed-effect meta-analyses described below. To assess population substructure within the case-control studies, principal component analysis was performed using EIGENSTRAT.22 Allelic association analysis was performed as implemented in SNPTEST v223 for the case-control datasets with a logistic regression model adjusting for sex, age, and the first 3 principal components determined by EIGENSTRAT. Similarly, logistic regression adjusting for sex, age, and APO ε4 status was used to estimate the OR and standard error in the case-control sample from Bonn. To aggregate data across the datasets analyzed here, we performed 2 types of meta-analyses: first, to estimate the overall level of statistical significance of the association evidence in our study, we performed a p value–based meta-analysis (using the METAL software24) across all family-based and case-control datasets. For this purpose, we used the 2-tailed p values for each dataset weighted by their standard error. Second, to estimate the size of the underlying risk effect, we performed fixed-effect meta-analyses limited to the case-control samples only, given the above-mentioned uncertainties inherent in OR estimations derived from family data. Because the aim of this study was to specifically assess whether or not the minor T allele at rs75932628 increases the risk of AD, all reported study-specific p values are 1-tailed (2-tailed values are used for the meta-analyses; see above). We estimated the power of our study by utilizing the “Genetic Power Calculator.”25 Combining all datasets, power to detect an allelic OR of 1.7 (i.e., the lowest 95% confidence bound suggested in the original study8) at minor allele frequency (MAF) 0.004 (i.e., the median MAF observed in unaffected individuals here) at α = 0.05 and a disease prevalence of 20% exceeds 80%.
RESULTS
Family-based association analyses.
Family-based association analysis of experimentally determined genotypes in both the NIMH and NIA-LOAD cohorts provided independent support of an increase in AD risk conferred by the minor T allele at rs75932628, albeit at differing significance levels. In the NIMH cohort, there were 12 FBAT informative families (defined as those with at least one heterozygous parent) yielding a 1-tailed p value of 0.004 (table), with an OR of 4.9 (95% confidence interval [CI] 1.18–20.83), which was comparable to previous studies.8,9 With an OR of 3.03 (95% CI 0.51–18.0), based on 7 FBAT informative families, the NIA-LOAD family cohort's effect estimate pointed in the same direction, but did not reach statistical significance (table), likely because of insufficient sample size.
Rare variant (burden) family-based association analysis in NIMH.
We also assessed the potential effects of other rare variants in exon 2 of TREM2 on AD as suggested in the original report.8 For this purpose, 410 probands from the NIMH family sample collection were resequenced in the TREM2 exon 2 region. Twenty-five nuclear families whose probands carried rare functional variants (a total of 65 affected and 17 unaffected subjects) were resequenced in the follow-up step. To perform “burden” analysis on 4 missense single nucleotide polymorphisms (SNPs) (rs75932628, rs143332484, rs2234253, and rs200392967) in exon 2 of TREM2 identified by our sequencing efforts, we based our statistical analysis on the rare variant extension of FBAT26,27 (version 2.0.4 beta). Based on the MAF of the subset of unrelated probands (n = 25) in our follow-up sample, SNPs with MAF <0.08 (arbitrarily chosen) were considered as rare variants; rs75932628-T was the most common of the 4 SNPs with a MAF = 0.19. As expected, this higher MAF (0.19) in rs75932628-T frequency across all members of this subsample enriched for the minor allele is much higher than in the datasets overall (or the general population). Two options are available in FBAT's rare variant approach, such that each variant can contribute (1) equally (unweighted) or (2) unequally (weighted) to the rare variant statistic. In the latter option, each contributing SNP was inversely weighted, by the square root of (n × f × [1 − f]), were n is the number of informative families and f is the MAF of the SNP. Thus, when weighting a set of SNPs, the contribution from the most common variant (here: rs75932628) was downweighted, while the contributions from other rare variants were upweighted. Setting the 4 SNPs to contribute equally, the contribution from rs75932628 dominated (1-tailed p value for the burden statistic = 4.8 × 10−4). However, using the weighted option, the signal from the rare variants did not improve the strength of the association: 1-tailed p = 1.8 × 10−3. Overall, burden analysis of rare variants in exon 2 of TREM2 in a subset of the NIMH cohort corroborated the rs75932628 association with AD risk observed in the entire Caucasian portion of the NIMH cohort.
Case-control association analyses.
The association results for rs75932628 using experimentally derived (NIA-LOAD and Bonn) or imputed (TGEN2, GSK, and ADNI) genotypes all indicated a risk effect for the minor T allele with ORs ranging from approximately 1.2 to 2.1 (table). Combining the case-control results by meta-analysis yielded an OR of 1.67 (95% CI 0.95–2.92, p [1-tailed] = 0.034). The case-control portion of the NIA-LOAD sample included here was directly genotyped, whereas the genotypes for the NIA-LOAD case-control replication sample (included in the original study8; supplementary table S2) were imputed. The difference in NIA-LOAD case-control OR (1.2) estimated here vs the OR (1.9) reported previously8 is most likely attributable to the difference in the number of cases included in each analysis (378 vs 1,026, respectively). Fixed-effect meta-analysis using risk estimates from the case-control studies only resulted in a summary OR of 1.67 (95% CI 0.95–2.92; figure). A p value–based meta-analysis combining all available data generated here, i.e., from the 2 family-based and 5 case-control datasets using METAL, suggests that the association between the minor T allele at rs75932628 and AD risk is statistically significant overall (z score 2.978, p value = 0.0029; table).
Figure. Forest plot of meta-analysis for TREM2 (rs75932628): T vs C; combining all the case-control datasets analyzed in this study.
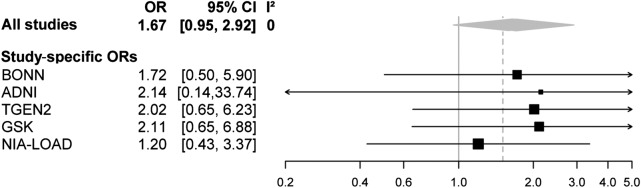
ADNI = Alzheimer's Disease Neuroimaging Initiative; CI = confidence interval; GSK = GlaxoSmithKline; NIA-LOAD = National Institute on Aging Genetics Initiative for Late Onset Alzheimer's Disease; OR = odds ratio; TGEN2 = Translational Genomics Research Institute series 2; TREM2 = triggering receptor expressed on myeloid cells 2.
DISCUSSION
The results from our extensive analyses of 9,344 subjects using both family-based and case-control designs provide additional independent evidence supporting the previously reported association between the T allele at SNP rs75932628 in TREM2 with increased AD risk. However, the risk effect estimated here (OR = 1.67) is considerably smaller than those reported in the previous studies: OR = 2.9,8 OR = 4.5,9 and OR = 4.0510 (specific effect size is not reported in one previous study11). The relatively low quality of the genotype imputation (information score <0.40) in the GSK and ADNI studies could contribute to this difference, although imputation uncertainty is accounted for in the association statistics. We also note that 4 of the 7 datasets (64% of the total subjects) included in our meta-analysis were directly genotyped. Thus, we believe the OR calculated in our meta-analysis remains largely reliable. This is also supported by analyses excluding the GSK and ADNI datasets, which reduced the OR estimates even further, although not statistically so (OR = 1.43, 95% CI 0.90–2.28). To date, the NIMH and NIA-LOAD cohorts are the first family-based datasets to independently show evidence for the association between the minor T allele at rs75932628 in TREM2 and increased risk of AD, although the penetrance in the NIMH and NIA-LOAD cohort differed substantially (97% vs 61%, respectively). Note that although all controls in our study were deemed as “cognitively healthy” at the time of last examination, this status could possibly change over time, potentially leading to a reduction in power (and possibly underestimation of OR and statistical significance) in our analyses.
While our data suggest that the association between rs75932628 and AD risk is most likely genuine, the combination of a modest effect size and very low MAF posits that this association finding bears very little epidemiologic relevance for the population at large (unlike the other known common AD risk alleles). At the same time, the penetrance of rs75932628 on AD risk is too small to be clinically meaningful to the individual (unlike any of the known early-onset AD-causing mutations in APP, PSEN1, and PSEN2). TREM2 shares its tiny population impact with the effects of the even lower prevalence, early-onset AD mutations in APP, PSEN1, and PSEN2. However, these latter mutations in APP, PSEN1, and PSEN2 are disease-causing and strike essentially with complete penetrance, making them reliable prognostic and diagnostic markers for individual patients or families. This characteristic is not shared with the R47H variant reported for TREM2.
TREM2 serves as a receptor in the innate immune system and is upregulated in microglia surrounding senile plaques in older APP23 mice28 and CRND8 transgenic mice (expressing mutant KM670/671NL and V717F APP).9 TREM2 also promotes the phagocytosis and clearance of β-amyloid protein.29 Similarly, another LOAD gene, CD33,2 is expressed in microglia and has been shown to inhibit microglial uptake and clearance of β-amyloid.30 The CD33 variant, rs3865444, which protects against AD, is associated with reduced CD33 microglial expression, and inactivation of CD33 in mice leads to reduced β-amyloid levels.30 Together, the genetic and transgenic mouse findings support both that TREM2, similar to CD33, is a critical player in AD pathogenesis and a promising target for therapeutic treatment based on enhancement of β-amyloid uptake and clearance.
Based on our analysis, TREM2 harbors a low-frequency variant associated with AD risk that bears very little epidemiologic relevance for the population at large. Given the low frequency of R47H and overall modest effects on risk for AD (OR = 1.67), this variant would also possess minimal clinical utility as a predictor or diagnostic for AD. Nonetheless, further elucidation of how TREM2 and CD33 regulate β-amyloid uptake and clearance will be important for further development of these targets in the quest to effectively treat and prevent AD.
ACKNOWLEDGMENT
The authors thank all patients and their families whose help and participation made this research possible. The authors thank contributors, including the Alzheimer's Disease Centers, who collected samples used in this study. Thanks to Kelly Faber at NCRAD for her insight of the NIA-LOAD/NCRAD data and for coordinating data transfer. For the ascertainment of the case-control sample from Bonn, the authors thank members of the AgeCoDe Study Group: Wolfgang Maier, Martin Scherer, Heinz-Harald Abholz, Cadja Bachmann, Hendrik van den Bussche, Sandra Eifflaender-Gorfer, Marion Eisele, Annette Ernst, Kathrin Heser, Teresa Kaufeler, Mirjam Köhler, Alexander Koppara, Melanie Luppa, Manfred Mayer, Anna Schumacher, Janine Stein, Franziska Tebarth, Michael Wagner, Klaus Weckbecker, Dagmar Weeg, Thomas Zimmermann, Steffen Wolfsgruber, Carolin Lange, Hanna Kaduszkiewicz, Siegfried Weyerer, Jochen Werle, Michael Pentzek, Angela Fuchs, Steffi G. Riedel-Heller, Tobias Luck, Edelgard Mösch, Horst Bickel, Birgitt Wiese, Jana Prokein, Hans-Helmut König, Christian Brettschneider, and Frank Jessen. In addition, the authors thank Mrs. Stefanie Heilmann and Dr. Markus Nöthen for their technical and scientific support. T.B. is an associate member of the ImmunoSensation Cluster of Excellence.
GLOSSARY
- AD
Alzheimer disease
- ADNI
Alzheimer's Disease Neuroimaging Initiative
- AgeCoDe
German Study on Ageing, Cognition and Dementia in Primary Care Patients
- CI
confidence interval
- FBAT
family-based association test
- GSK
GlaxoSmithKline
- GWAS
genome-wide association study
- LOAD
late-onset Alzheimer disease
- MAF
minor allele frequency
- NIA
National Institute on Aging
- NIA-LOAD
National Institute on Aging Genetics Initiative for Late Onset Alzheimer's Disease
- NIMH
National Institute of Mental Health
- OR
odds ratio
- SNP
single nucleotide polymorphism
- TGEN2
Translational Genomics Research Institute series 2
- TREM2
triggering receptor expressed on myeloid cells 2
AUTHOR CONTRIBUTIONS
Dr. Hooli processed DNA samples on human microarray, SNP genotype data generation and quality control, and drafted the manuscript. Dr. Parrado performed statistical analysis of family-based cohorts and drafted the manuscript. Ms. Mullin performed genotyping, data generation, and quality assessment. Mr. Yip performed statistical rare variant test. Dr. Liu performed the imputation of the GWAS datasets and statistical analysis of case-control datasets. Mr. Roehr performed the imputation of the various GWAS datasets. Dr. Qiao performed family-based association testing and burden analysis. Dr. Jessen, Dr. Peters, Dr. Becker, and Dr. Ramirez designed the study in Bonn and revised the manuscript for intellectual content. Dr. Lange helped design and plan the study. Dr. Bertram helped in the design, conceptualization, and planning of the study, performed and plotted the meta-analysis graphs, and helped in writing and revising the manuscript. Dr. Tanzi designed and conceptualized the study, and helped in revising the manuscript.
STUDY FUNDING
Supported by the Cure Alzheimer Fund (to L.B. and R.E.T.) as part of the “Alzheimer Genome Project,” and by federal grants from the NIA (5R01AG23667 to L.B.), the NIMH (5R37MH60009 to R.E.T.), and the German Federal Ministry of Education and Research (BMBF grant 16SV5538 to L.B.). Data and biomaterials were collected in 3 projects that participated in the NIMH Alzheimer Disease Genetics Initiative. From 1991 to 1998, the principal investigators and coinvestigators included the following: Massachusetts General Hospital, Boston, U01 MH46281, Marilyn S. Albert, PhD, and Deborah Blacker, MD, ScD; Johns Hopkins University, Baltimore, MD, U01 MH46290, Susan S. Bassett, PhD, Gary A. Chase, PhD, and Marshal F. Folstein, MD; and University of Alabama, Birmingham, U01 MH46373, Rodney C.P. Go, PhD, and Lindy E. Harrell, MD. Samples from the National Cell Repository for Alzheimer's Disease (NCRAD), which receives government support under a cooperative agreement grant (U24 AG21886) awarded by the NIA, were used in this study. This publication is part of the German Research Network on Dementia (KND) and the German Research Network on Degenerative Dementia (KNDD) and was funded by the German Federal Ministry of Education and Research (grants KND: 01GI0102, 01GI0420, 01GI0422, 01GI0423, 01GI0429, 01GI0431, 01GI0433, 01GI0434; grants KNDD: 01GI0710, 01GI0711, 01GI0712, 01GI0713, 01GI0714, 01GI0715, 01GI0716, 01 ET1006B). This study was not industry-sponsored.
DISCLOSURE
The authors report no disclosures relevant to the manuscript. Go to Neurology.org for full disclosures.
REFERENCES
- 1.Bertram L, Lill CM, Tanzi RE. The genetics of Alzheimer disease: back to the future. Neuron 2010;68:270–281 [DOI] [PubMed] [Google Scholar]
- 2.Bertram L, Lange C, Mullin K, et al. Genome-wide association analysis reveals putative Alzheimer's disease susceptibility loci in addition to APOE. Am J Hum Genet 2008;83:623–632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hollingworth P, Harold D, Sims R, et al. Common variants at ABCA7, MS4A6A/MS4A4E, EPHA1, CD33 and CD2AP are associated with Alzheimer's disease. Nat Genet 2011;43:429–435 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Naj AC, Jun G, Beecham GW, et al. Common variants at MS4A4/MS4A6E, CD2AP, CD33 and EPHA1 are associated with late-onset Alzheimer's disease. Nat Genet 2011;43:436–441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Harold D, Abraham R, Hollingworth P, et al. Genome-wide association study identifies variants at CLU and PICALM associated with Alzheimer's disease. Nat Genet 2009;41:1088–1093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lambert JC, Heath S, Even G, et al. Genome-wide association study identifies variants at CLU and CR1 associated with Alzheimer's disease. Nat Genet 2009;41:1094–1099 [DOI] [PubMed] [Google Scholar]
- 7.Seshadri S, Fitzpatrick AL, Ikram MA, et al. Genome-wide analysis of genetic loci associated with Alzheimer disease. JAMA 2010;303:1832–1840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jonsson T, Stefansson H, Steinberg S, et al. Variant of TREM2 associated with the risk of Alzheimer's disease. N Engl J Med 2013;368:107–116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Guerreiro R, Wojtas A, Bras J, et al. TREM2 variants in Alzheimer's disease. N Engl J Med 2013;368:117–127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pottier C, Wallon D, Rousseau S, et al. TREM2 R47H variant as a risk factor for early-onset Alzheimer's disease. J Alzheimers Dis 2013;35:45–49 [DOI] [PubMed] [Google Scholar]
- 11.Benitez BA, Cooper B, Pastor P, et al. TREM2 is associated with the risk of Alzheimer's disease in Spanish population. Neurobiol Aging 2013;34:1711.e15–1711.e17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Blacker D, Bertram L, Saunders AJ, et al. Results of a high-resolution genome screen of 437 Alzheimer's disease families. Hum Mol Genet 2003;12:23–32 [DOI] [PubMed] [Google Scholar]
- 13.Blacker D, Haines JL, Rodes L, et al. ApoE-4 and age at onset of Alzheimer's disease: the NIMH genetics initiative. Neurology 1997;48:139–147 [DOI] [PubMed] [Google Scholar]
- 14.Wijsman EM, Pankratz ND, Choi Y, et al. Genome-wide association of familial late-onset Alzheimer's disease replicates BIN1 and CLU and nominates CUGBP2 in interaction with APOE. PLoS Genet 2011;7:e1001308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jessen F, Wiese B, Bickel H, et al. Prediction of dementia in primary care patients. PloS One 2011;6:e16852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Luck T, Riedel-Heller SG, Kaduszkiewicz H, et al. Mild cognitive impairment in general practice: age-specific prevalence and correlate results from the German Study on Ageing, Cognition and Dementia in Primary Care Patients (AgeCoDe). Dement Geriatr Cogn Disord 2007;24:307–316 [DOI] [PubMed] [Google Scholar]
- 17.Kornhuber J, Schmidtke K, Frolich L, et al. Early and differential diagnosis of dementia and mild cognitive impairment: design and cohort baseline characteristics of the German Dementia Competence Network. Dement Geriatr Cogn Disord 2009;27:404–417 [DOI] [PubMed] [Google Scholar]
- 18.Howie B, Marchini J, Stephens M. Genotype imputation with thousands of genomes. G3 2011;1:457–470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Marchini J, Howie B, Myers S, McVean G, Donnelly P. A new multipoint method for genome-wide association studies by imputation of genotypes. Nat Genet 2007;39:906–913 [DOI] [PubMed] [Google Scholar]
- 20.Won S, Wilk JB, Mathias RA, et al. On the analysis of genome-wide association studies in family-based designs: a universal, robust analysis approach and an application to four genome-wide association studies. PLoS Genet 2009;5:e1000741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lange C, DeMeo D, Silverman EK, Weiss ST, Laird NM. PBAT: tools for family-based association studies. Am J Hum Genet 2004;74:367–369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Price AL, Patterson NJ, Plenge RM, Weinblatt ME, Shadick NA, Reich D. Principal components analysis corrects for stratification in genome-wide association studies. Nat Genet 2006;38:904–909 [DOI] [PubMed] [Google Scholar]
- 23.Marchini J, Howie B. Genotype imputation for genome-wide association studies. Nat Rev Genet 2010;11:499–511 [DOI] [PubMed] [Google Scholar]
- 24.Willer CJ, Li Y, Abecasis GR. METAL: fast and efficient meta-analysis of genomewide association scans. Bioinformatics 2010;26:2190–2191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Purcell S, Cherny SS, Sham PC. Genetic Power Calculator: design of linkage and association genetic mapping studies of complex traits. Bioinformatics 2003;19:149–150 [DOI] [PubMed] [Google Scholar]
- 26.De G, Yip WK, Ionita-Laza I, Laird N. Rare variant analysis for family-based design. PloS One 2013;8:e48495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yip WK, De G, Raby BA, Laird N. Identifying causal rare variants of disease through family-based analysis of Genetics Analysis Workshop 17 data set. BMC Proc 2011;5(suppl 9):S21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Frank S, Burbach GJ, Bonin M, et al. TREM2 is upregulated in amyloid plaque-associated microglia in aged APP23 transgenic mice. Glia 2008;56:1438–1447 [DOI] [PubMed] [Google Scholar]
- 29.Melchior B, Garcia AE, Hsiung BK, et al. Dual induction of TREM2 and tolerance-related transcript, Tmem176b, in amyloid transgenic mice: implications for vaccine-based therapies for Alzheimer's disease. ASN Neuro 2010;2:e00037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Griciuc A, Serrano-Pozo A, Parrado AR, et al. Alzheimer's disease risk gene CD33 inhibits microglial uptake of amyloid beta. Neuron 2013;78:631–643 [DOI] [PMC free article] [PubMed] [Google Scholar]