

Abstract
Background:
Cleistanthins A and B are isolated compounds from the leaves of Cleistanthus collinus Roxb (Euphorbiaceae). This plant is poisonous in nature which causes cardiovascular abnormalities such as hypotension, nonspecific ST-T changes and QTc prolongation. The biological activity predictions spectra of the compounds show the presence of antihypertensive, diuretic and antitumor activities.
Objective:
Objective of the present study was to determine the in silico molecular interaction of cleistanthins A and B with Angiotensin I- Converting Enzyme (ACE-I) using Induced Fit Docking (IFD) protocols.
Materials and Methods:
All the molecular modeling calculations like IFD docking, binding free energy calculation and ADME/Tox were carried out using Glide software (Schrödinger LLC 2009, USA) in CentOS EL-5 workstation.
Results:
The IFD complexes showed favorable docking score, glide energy, glide emodel, hydrogen bond and hydrophobic interactions between the active site residues of ACE-I and the compounds. Binding free energy was calculated for the IFD complexes using Prime MM-GBSA method. The conformational changes induced by the inhibitor at the active site of ACE-I were observed based on changes of the back bone Cα atoms and side-chain chi (x) angles. The various physicochemical properties were calculated for these compounds. Both cleistanthins A and B showed better docking score, glide energy and glide emodel when compared to captopril inhibitor.
Conclusion:
These compounds have successively satisfied all the in silico parameters and seem to be potent inhibitors of ACE-I and potential candidates for hypertension.
Keywords: ADME/Tox, Chi angles, Cleistanthin A, Cleistanthin B, Induced fit docking, Prime MM-GBSA
INTRODUCTION
Plants are found to be the treasure chest of biological substances/drugs such as anti-cholinergic agents, skeletal muscle relaxants and salicylates. In some cases, compounds that exhibit medicinal properties have been obtained from plants which are considered to be toxic. Atropine, D-tubocurarine, hyoscine, digoxin and strychnine were isolated from toxic plants. Cleistanthus collinus Roxb., (Euphorbiaceae) is one such toxic plant, which exerts significant toxicity on cardiovascular, renal and respiratory system. The toxic effect also induces metabolic acidosis and alters liver and kidney functions. From the leaves of Cleistanthus collinus, more than 25 compounds were identified in which some compounds including cleistanthin A and cleistanthin B were reported as toxic.[1] Both cleistanthins A and B are glycosides commonly present in Euphorbiaceae family plants.[2]
Cleistanthin A is also present in the leaves of Phyllanthus taxodiifolius Beille., (Euphorbiaceae) and this plant is commonly used as a traditional diuretic agent among Thai people.[3] The predicted biological activity spectra of cleistanthins A and B showed the presence of hypotensive effect, diuretic and antitumor activities. Both compounds had significant antineoplastic and hypotensive effects in rodents and its cell lines.[4,5] The in vitro studies of cleistanthins A and B compounds showed a significant diuretic effect but the effect was not comparable with standard diuretic agents.[4] Hence; the present study aims to determine the possible interactions and binding free energy of cleistanthins A and B with target of ACE-I using Induced Fit Docking and Prime Molecular Mechanics Generalized Born Surface Area (MM-GBSA) analysis.
MATERIALS AND METHODS
Ligand preparation and biological activity prediction
The natural compounds of cleistanthins A and B were isolated and purified from the leaves of Cleistanthus collinus using column chromatographic method and the structures were determined.[4] These structures [Figure 1] were built using builder panel in Maestro and ligand preparation was carried out for these compounds by Ligprep 2.3 module (Schrödinger, USA, 2009). Ligprep performs addition of hydrogens, 2D to 3D conversion, realistic bond lengths and bond angles, low energy structure with correct chiralities, ionization states, tautomers, stereochemistries and ring conformations. The energy minimized compounds were subjected to biological activity prediction based on their structural orientation using PASS (Prediction of Activity Spectra for Substances) tool.[6]
Figure 1.
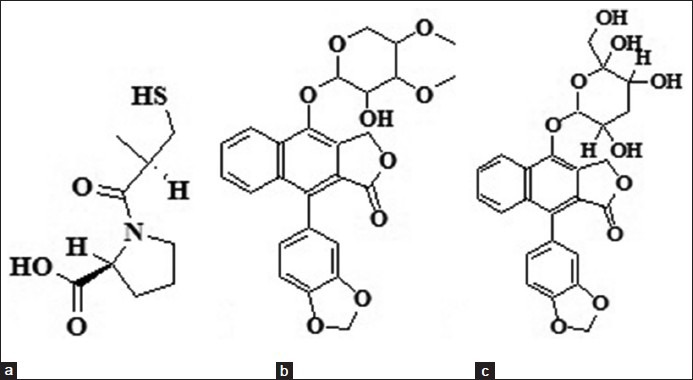
Chemical diagrams of (a) Captopril, (b) cleistanthin A and (c) cleistanthin B inhibitors used in the in silico study
Protein preparation
The PASS prediction results also showed that these compounds have anti-pulmonary hypertension property. Based on the results of in vitro, in vivo and in silico PASS studies,[1,3] the x-ray crystal structure of human testicular Angiotensin I-Converting Enzyme (tACE-I) with captopril complex was recovered from Protein Data Bank (1UZF). The ACE is a zinc metallopeptidase that plays an important role of catalyzing the proteolysis of angiotensin I to the vasopressor angiotensin II. ACE, angiotensin I and II are part of renin-angiotensin system which regulates the blood pressure, volume of fluids in the body. ACE catalyses the conversion of angiotensin I to II leading to vasoconstriction. ACE inhibitors block the conversion of angiotensin I to II thereby reducing the cardiac index and increasing natriuresis.[7] Selection of potent inhibitors to this enzyme, may lead to development of new drugs for the treatment of cardiovascular diseases. Captopril is the first approved drug as an orally active ACE inhibitor for treatment of human hypertension, which was accomplished in 1981 by Cushman and Ondetti.[8]
Induced fit docking
In the standard (rigid) mode of docking, as the protein is held rigid and the ligand is free to rotate, the simulation may provide misleading results. Also, many proteins undergo side-chain (χ angles) or backbone (Cα) conformational changes or both, while ligand binds at the active site of the target. These conformational changes allow the protein to generate close conformations to the shape of the ligand and lead to good binding affinity complex. In this study, the IFD (flexible docking) was carried out using Glide software (Schrödinger LLC 2009, USA) to predict accurate concomitant structural movements when ligand binds at the active site of the protein.[9] The energy minimized human tACE-I-captopril complex was subjected for IFD studies. At the active site of tACE-I complex, the captopril binding was used to generate the grid for IFD calculation. While docking, the default van der Waals radii (0.50) was used for non-polar atoms of the receptor and ligands (captopril, cleistanthin A and cleistanthin B). In IFD study, the active site residues of tACE-I conform more closely to the shape and binding mode of the compounds. The energetically favorable IFD complexes were obtained for all compounds and the best poses were selected by docking score, glide energy and glide emodel for further calculations. Ligplot[10] and PyMol[11] were used for analyzing the hydrogen bond and hydrophobic interactions.
Prime MM-GBSA
The ligand strain energies and ligand binding energies were calculated for the natural and captopril compounds using Prime MM-GBSA modules (Schrφdinger LLC 2009, USA). The best poses of IFD complexes were chosen to obtain the binding free energy calculation. Prime MM-GBSA is a method that combines Optimized Potential for Liquid Simulations-All Atoms (OPLSAA) force field, molecular mechanics energies (EMM), an SGB salvation model for polar solvation (GSGB), and a non-polar salvation term (GNP) composed of the non-polar solvent accessible surface area and van der Waals interactions.
The total binding free energy: ΔGbind = Gcomplex − (Gprotein + Gligand)
Cα and χ angles conformational changes
The best IFD complexes and the crystal structure of tACE-I were taken for analyzing the conformational changes of Cα atoms and χ angles at the active site. The IFD complexes were superimposed with the crystal structure of tACE-I for the structure based alignment matching using Chimera.[12] Needleman-Wunsch algorithm and BLOSUM-62 matrix were used in chimera for aligning the amino acid residues based on the alignment of the helix and strand (Cα atom) of the IFD complexes with the respective helix and strand (Cα atom) of the crystal structure. Then, the side-chain χ angles (χ1 (N-Cα-Cβ-Cγ), χ2 (Cα-Cβ-Cγ-Cδ), χ3 (Cβ-Cγ-Cδ-Cε), and χ4 (Cγ-Cδ-Cε-Cζ)) were analyzed to understand the conformational changes at the active site of tACE-I in the IFD complexes. These side-chain χ angles were calculated and analyzed using MolProbity[13] and Chimera software.
Absorption, Distribution, Metabolism and xcretion (ADME)/ Tox
The energy minimized natural compounds were subjected into ADME/Tox calculations using Qikprop 3.2 (Schrödinger 2009). Qikprop is quick and accurate; it predicts structurally significant descriptors and pharmaceutically relevant properties of organic molecules. QikProp provides ranges for comparing a particular molecule's physicochemical properties with those of 95% of known drugs.
RESULTS AND DISCUSSION
Docked complex analyses
Captopril was redocked at the active site of tACE-I [Figure 2a]. The docked complex has the docking score = -4.09, Glide energy = -32.19 Kcal/mol and Glide emodel = −45.66 Kcal/mol [Table 1]. The redocked complex was superimposed with the crystal structure of tACE-I and the RMSD was 0.237Å. Captopril has maintained four hydrogen bond interactions with the active site residues. In this compound, the O2 atom has trifurcated hydrogen bond acceptor interactions with Gln 281 (NE2), Lys 511 (NZ) and Tyr 520 (OH) and O3 atom has interacted with His 513 (NE2) residue. Zn (II) ion forms tetrahedral coordination geometry with Glu 384 (OE2), Glu 411 (OE1), Glu 411 (OE2) residues and sulfur atom in the ligand. This complex has the binding free energy of −16.41 Kcal/mol.
Figure 2.
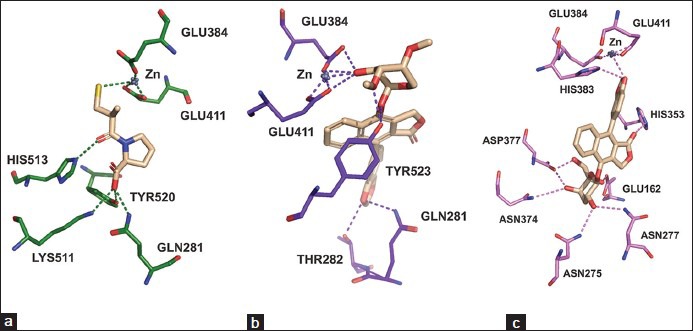
Hydrogen bond interactions (dotted lines) of (a) captopril, (b) cleistanthin A and (c) cleistanthin B with active site residues of tACE
Table 1.
Docking score, glide energy, glide emodel, hydrogen bond interactions and binding free energy of Induced fit docked complexes
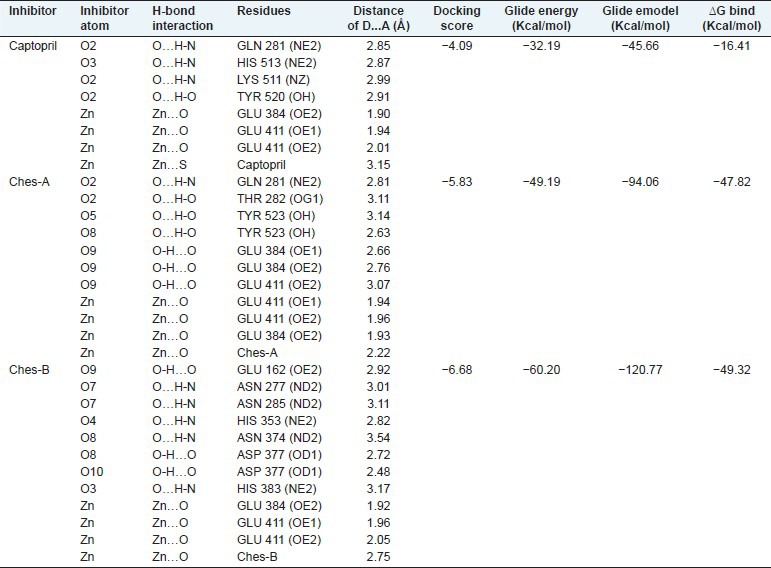
Seven hydrogen bond interactions were observed in the tACE-I-cleistanthin A complex and trifurcated hydrogen bond donor interactions were observed between ligand O9 atom and the active site residues of Glu 384 (OE2), Glu 384 (OE1) and Glu 411 (OE2). The residue Tyr 523 (OH) has maintained two hydrogen bond interactions with ligand atoms of O5 and O8 with distances 3.14 Å and 2.63 Å, respectively [Table 1 and Figure 2b]. Zn (II) ion has formed a tetrahedral coordination geometry which has three interactions with the active site residues (Glu 384 (OE2), Glu 411 (OE1) and Glu 411 (OE2)) and one interaction with O9 atom in the ligand. The ligand showed the docking score = −5.83, Glide energy = −49.19 Kcal/mol and Glide emodel = −94.06 Kcal/mol at the active site of tACE-I [Table 1]. This complex had binding free energy = -47.82 Kcal/mol.
Docked orientation of the cleistanthin B with the active site residues of tACE-I showed that the docking score = −6.68, Glide energy = −60.20Kcal/mol and Glide emodel = -120.77 Kcal/mol. The ligand has eight hydrogen bond interactions with the active site residues. The atom O7 has two hydrogen bond interactions with Asn 227 (ND2) and Asn 285 (ND2). The residues Asn 374 (ND2) and Asp 377 (OD1) interact with O8 atom of the ligand. Moreover, the O9 atom also has two hydrogen bond interactions with His 353 (NE2) and Glu 162 (OE2) residues. O3 and O10 atoms have one interaction with His 383 (NE2) and Asp 377 (OD2), respectively. The Zn (II) ion has tetrahedral coordination geometry with Glu 384 (OE2), Glu 411 (OE1), Glu 411 (OE2) residues and O3 atom of the ligand [Table 1 and Figure 2c]. This complex shows the binding free energy of −49.32 Kcal/mol.
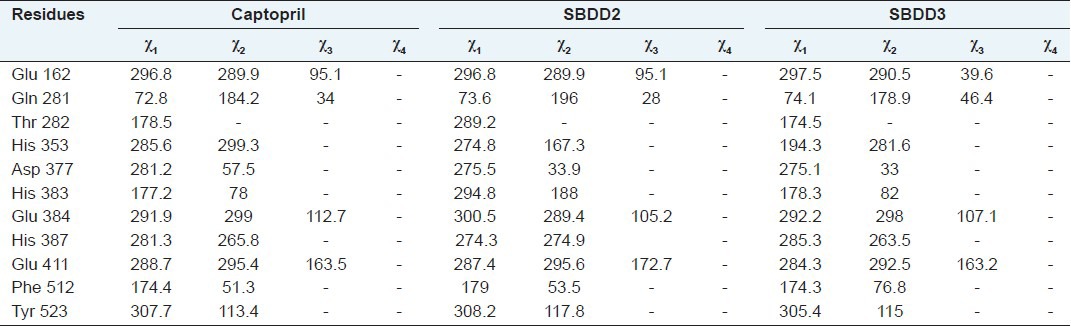
Conformation changes of Cα and side-chain X angles
The Cα conformational changes of Ches A and B complexes were compared with crystal structure of tACE-I and the RMSD values are 0.24 Å and 0.23 Å for Ches A and B compounds, respectively. The side-chain conformational changes were analyzed by measuring chi angles at the active site residues for all the complexes. Glu 162, Gln 281, Thr 282, His 353, Asp 377, His 383, Glu 384, His 387, Glu 411, Phe 512 and Tyr 523 residues showed side-chain conformational changes at the active site of the tACE-I. These results show that all the compounds induced conformational changes at the active site residues [Figure 3 and Table 2].
Figure 3.
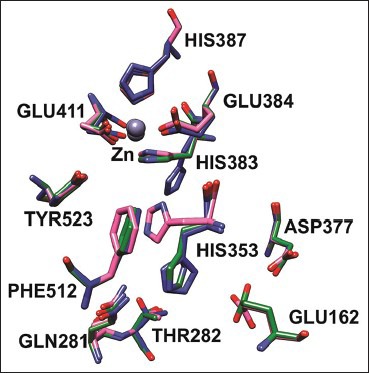
Side-chain conformational changes in the tACE complexes captopril, cleistanthin A and B bound complexes are colored in green, blue and pink, respectively
Table 2.
Conformational changes of the side.chain Chi angles (χ1, χ2, χ3, and χ4) of the docked complexes
Analysis of physicochemical properties
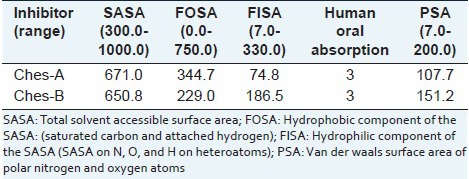
The natural compounds were taken for physicochemical properties analyzed using Qikprop simulation. The molecular weight of the compounds is less than 500Da and QPlogPo/w (octanol/water partition coefficient) for all the inhibitors is less than five. These compounds satisfy the partition coefficient of octanol/gas (QPlogPoct), water/gas (QPlogPw) and brain/blood (QPlogBB) values as per the rules. The aqueous solubility (QPlogS) and the skin permeability (QPlogKp) were predicted for the compounds. Total solvent accessible surface area (SASA), hydrophobic component of the SASA (FOSA) and hydrophilic component of the SASA (FISA) values abide the ranges specified in the Qikprop. The natural compounds have favorable oral absorption score of 3, when qualitative human oral absorption was predicted. Polar nitrogen and oxygen van der Waals surface area (PSA) of compounds satisfy the range in physicochemical calculation [Tables 3a and b].
Table 3a.
Calculation of Lipinski's rule of five, partition coefficient and skin permeability
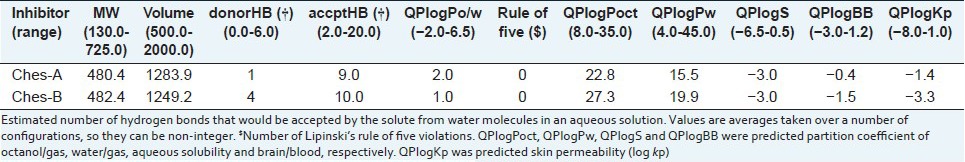
Table 3b.
Surface area calculation and human oral absorption
CONCLUSION
The natural compounds and captopril were subjected for IFD and Prime MM-GBSA studies to observe docking score, glide energy, glide emodel, hydrogen bond and hydrophobic interactions and binding free energy between these ligands and the active site residues of tACE-I. The values of these for natural compounds were compared with redocked captopril-tACE-I complex. The comparison shows that cleistanthins A and B have better docking score, glide energy, glide emodel and binding free energy compared to captopril. Moreover, hydrogen bond and hydrophobic interactions of the natural compounds have shown favorable interactions with active site residues.
The natural compounds have favorable results in physicochemical properties of partition coefficient of QPlogPoct, QplogPw and QplogBB and surface area calculations of SASA, FOSA, FISA, and PSA. According to molecular weight calculation and the human oral absorption properties, both the compounds are below 500 Da with high oral absorption value. Both compounds satisfy all the in silico parameters like docking score, glide energy, glide emodel, binding free energy, ADME/Tox and non-bonded interactions. Hence, cleistanthins A and B could exert potent inhibition of the tACE-I and they have favorable physicochemical properties. The inhibition of ACE by cleistanthins A and B could be revealed by in vivo and in vitro studies and if so, these compounds may be attempted to be used against cardiovascular diseases.
ACKNOWLEDGEMENTS
BV thanks the University Grant Commission (UGC), Government of India, New Delhi, for the award of a meritorious fellowship. BV and DV thank UGC (SAP-CAS) for the department facilities and Bioinformatics Infrastructure Facility (DBT-supported) for computing facilities.
Footnotes
Source of Support: Nil
Conflict of Interest: None declared.
REFERENCES
- 1.Parasuraman S, Raveendran R. Computer-aided prediction of biological activity spectra, pharmacological and toxicological property of Cleistanthin A and B. Int J Res Pharm Sci. 2010;1:333–7. [Google Scholar]
- 2.Parasuraman S, Raveendran R. Diuretic effects of cleistanthin A and cleistanthin B from the leaves of Cleistanthus collinus in wistar rats. J Young Pharm. 2012;4:73–7. doi: 10.4103/0975-1483.96616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tuchinda P, Kumkao A, Pohmakotr M, Sophasan S, Santisuk T, Reutrakul V. Cytotoxic arylnaphthalide lignan glycosides from the aerial parts of Phyllanthus taxodiifolius. Planta Med. 2006;72:60–2. doi: 10.1055/s-2005-873141. [DOI] [PubMed] [Google Scholar]
- 4.Parasuraman S, Raveendran R, Vijayakumar B, Velmurugan D, Balamurugan S. Molecular docking and ex vivo pharmacological evaluation of constituents of the leaves of Cleistanthus collinus (Roxb.) (Euphorbiaceae) Indian J Pharmacol. 2012;44:197–203. doi: 10.4103/0253-7613.93848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pradheepkumar CP, Shanmugam G. Anticancer potential of cleistanthin A isolated from the tropical plant Cleistanthus collinus. Oncol Res. 1999;11:225–32. [PubMed] [Google Scholar]
- 6.Poroikov VV, Filimonov DA, Ihlenfeldt WD, Gloriozova TA, Lagunin AA, Borodina YV, et al. PASS biological activity spectrum predictions in the enhanced open NCI database browser. J Chem Inf Comput Sci. 2003;43:228–36. doi: 10.1021/ci020048r. [DOI] [PubMed] [Google Scholar]
- 7.Natesh R, Schwager SL, Sturrock ED, Acharya KR. Crystal structure of the human angiotensin-converting enzyme-lisinopril complex. Nature. 2003;42:551–4. doi: 10.1038/nature01370. [DOI] [PubMed] [Google Scholar]
- 8.Cushman DW, Ondetti MA. Design of angiotensin converting enzyme inhibitors. Nat Med. 1999;5:1110–3. doi: 10.1038/13423. [DOI] [PubMed] [Google Scholar]
- 9.Vijayakumar B, Umamaheswari A, Puratchikody A, Velmurugan D. Selection of an improved HDAC8 inhibitor through structure-based drug design. Bioinformation. 2011;7:134–41. doi: 10.6026/97320630007134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wallace AC, Laskowski RA, Thornton JM. LIGPLOT: A program to generate schematic diagrams of protein-ligand interactions. Protein Eng. 1995;8:127–34. doi: 10.1093/protein/8.2.127. [DOI] [PubMed] [Google Scholar]
- 11.DeLano WL. San Carlos, CA, USA: DeLano Scientific; 2002. The PyMOL Molecular Graphics System. Available from: http://www.pymol.org . [Google Scholar]
- 12.Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, et al. UCSF Chimera: A visualization system for exploratory research and analysis. J Comput Chem. 2004;25:1605–12. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- 13.Davis IW, Leaver-Fay A, Chen VB, Block JN, Kapral GJ, Wang X, et al. MolProbity: All-atom contacts and structure validation for proteins and nucleic acids. Nucleic Acids Res. 2007;35:W375–83. doi: 10.1093/nar/gkm216. [DOI] [PMC free article] [PubMed] [Google Scholar]