

Abstract
Human genomic DNA extracted from urine could be an interesting tool for large-scale public health studies involving characterization of genetic variations or DNA biomarkers as a result of the simple and noninvasive collection method. These studies, involving many samples, require a rapid, easy, and standardized extraction protocol. Moreover, for practicability, there is a necessity to collect urine at a moment different from the first void and to store it appropriately until analysis. The present study compared seven commercial kits to select the most appropriate urinary human DNA extraction procedure for epidemiological studies. DNA yield has been determined using different quantification methods: two classical, i.e., NanoDrop and PicoGreen, and two species-specific real-time quantitative (q)PCR assays, as DNA extracted from urine contains, besides human, microbial DNA also, which largely contributes to the total DNA yield. In addition, the kits giving a good yield were also tested for the presence of PCR inhibitors. Further comparisons were performed regarding the sampling time and the storage conditions. Finally, as a proof-of-concept, an important gene related to smoking has been genotyped using the developed tools. We could select one well-performing kit for the human DNA extraction from urine suitable for molecular diagnostic real-time qPCR-based assays targeting genetic variations, applicable to large-scale studies. In addition, successful genotyping was possible using DNA extracted from urine stored at −20°C for several months, and an acceptable yield could also be obtained from urine collected at different moments during the day, which is particularly important for public health studies.
Keywords: epidemiological studies, real-time polymerase chain reaction, species specificity, 16S
INTRODUCTION
Biomarkers play a key role in public health-related studies, as they are indicators of hazard, exposure, disease, and population risk. There are different types of biomarkers, i.e., proteins, metabolites, and nucleic acids, of which, proteins and metabolites are the most widely used. Biomarkers provide information for early detection, prediction, prevention, prognosis, diagnosis, and response to therapy of diseases. Therefore, they can be used to make group and individual risk assessments to support a proactive public health policy.1 The measurement of DNA biomarkers, which are genetic variations that contribute to disease susceptibility as well as to treatment response1 at a large population level, is key to the development of public health genomics, where genome-based knowledge is used to benefit public health, by its integration into public health policy and services for the benefit of population health.2
However, in the context of public health genomics, DNA biomarkers have to be identified at population level, often on healthy persons out of the hospital, and therefore, some assay parameters must be improved and tested to facilitate and allow these epidemiological studies. Firstly, up until now, blood remains the commonly used source of human nucleic acids for biomarkers assays. However, blood has several limitations, such as the requirement of a professional staff, equipment, and infrastructure, thereby hampering sampling in a more epidemiological setting. Furthermore, certain groups of people (e.g., small children) are reluctant to give a blood sample. Regarding the sample itself, blood also contains several interfering proteins and PCR inhibitors and represents an infectious risk for HIV and other pathogens.3 Therefore, other sources of nucleic acids could be more appropriate. Saliva has already been used as a valuable, alternative source for DNA biomarkers.4,5 A saliva sample collection (e.g., using the Oragene kit; DNA Genotek, Ontario, Canada) holds several advantages compared with blood sampling, including a noninvasive sampling method, with an increased patient convenience and lower infection risk. Another potential alternative source for DNA biomarkers is urine. Indeed, besides proteins, urine also contains nucleic acids derived from epithelial cells (renal tubular, transitional urothelial, and squamous cells), leukocytes, and also malignant cells, which are liberated spontaneously into urine. Moreover, cell-free circulating nucleic acids from blood can pass through the kidney barrier into urine.6,7 Like saliva, it is readily available and can be obtained by a noninvasive collection method, which is an advantage for large-scale population studies. Compared with blood, urine also contains fewer interfering proteins and PCR inhibitors and is noninfectious for HIV and less infectious for many other pathogens.8,9 Although it holds great promise as an alternative source of DNA biomarkers, especially for specific cases, such as retrospective studies of banked urine when other sources of DNA were not available or in studies in which epigenetic DNA profiles in urine are different than in saliva, until now, urine has been less studied in the context of public health genomics. This also becomes clear when looking at the already reported human DNA extraction protocols for urine samples, which are a second important assay parameter to consider in epidemiological studies.
Indeed, several human DNA extraction protocols have been reported for urine samples, e.g., phenol-chloroform-based methods,9–11 involving highly toxic reagents, and in-house protocols.12,13 These methods are, however, usually time consuming and are more difficult to standardize. In this context, commercials kits14–17 are convenient, as they have the advantage of being easy to use and having standardized and time-saving protocols. To our knowledge, a study comparing commercial DNA extraction kits currently on the market for the extraction of human DNA from urine samples suited for large-scale population studies has not been reported yet.
Additionally, an optimal extraction procedure should allow an efficient target recovery in terms of yield of human DNA, together with a removal of amplification inhibitors. Indeed, one of the major limitations of PCR-based assays, which are predominantly used to measure DNA biomarkers, is the inhibition of the amplification process by substances remaining in the DNA extract. In urine, for example, urea at a concentration of 50 mM and above is inhibitory for PCR.18–21 Urea may cause inhibition by denaturing polymerases.19,22 Other examples of inhibitors are nucleases (DNAse and RNase), which can degrade target nucleic acids and/or oligonucleotide primers and can lead to PCR failure.19–21 Therefore, to perform PCR-based assays to measure DNA biomarkers in urine samples, PCR inhibitors must be inactivated or removed during the extraction procedure. Once the DNA is extracted, it should be verified that no PCR inhibitors are remaining in the DNA extract.
Thirdly, besides the extraction protocol used to obtain a satisfactory yield of human DNA, also, the sampling time is an important aspect for public health research. In literature, most of the studies are performed on the first morning urine, as it is reported that it is more concentrated in cells.23 However, in practice, for public health-related studies, first morning urine is not always easily available.
Finally, another practical issue is the storage of urine. Although urine can be obtained in large volumes, these precious samples must be handled and stored carefully for further use.24 Furthermore, to prevent bacterial growth and DNA degradation, it is important to store the urine samples under specific conditions before extraction, which might not be done immediately after sampling. Several storage conditions have been reported for urine, with and without additives,10,15,25–28 but until now, no best practices have been discussed for urine storage for public health-related studies.
The goal of this study was to select the most appropriate urinary human DNA extraction procedure for epidemiological studies to be able to measure genetic variations (DNA biomarkers) using a real-time qPCR assay. An additional aim of this study was to test different parameters of practical importance for epidemiological studies that can affect the efficiency of the analysis.
First, we have compared the relative efficacy of seven commercial DNA extraction kits to isolate human genomic DNA (gDNA) from urine samples with the goal of generating DNA suitable for PCR-based analysis. The DNA extraction kits were evaluated based on the following criteria: comparison of the yield of human DNA extracted from 1 ml urine, assessment of the presence of remaining PCR inhibitors (based on a real-time qPCR assay), evaluation of coextracted bacterial DNA, the cost, and the processing time.
The selected kit was then used to compare the yield of human DNA extracted from urine, collected at three different moments of the day, and from urine stored under different conditions (−20°C and −80°C), which are important issues to consider for routine and large-scale studies.
As a proof-of-concept, DNA extracted from urine has been used in a genotyping assay targeting a single nucleotide polymorphism (SNP) located in an important gene in public health, i.e., the neuronal cholinergic receptor nicotinic α-3 gene (CHRNA3) SNP rs1051730. This SNP has been classified as a tag SNP related to smoking and lung cancer by genome-wide association studies.29
MATERIALS AND METHODS
Urine Samples
Urine samples from six healthy volunteers [three women (samples 1–3) and three men (samples 4–6)] have been collected and processed immediately or immediately aliquoted and stored (−20°C or −80°C) for further use. This study was approved in the scope of a Ph.D. project by the Université Catholique de Louvain (UCL; Woluwe, Brussels) and the Scientific Institute of Public Health (WIV-ISP; Brussels) in Belgium. Urine was collected from volunteers among the scientific staff of WIV-ISP, where the research was carried out, and an informed consent has been signed by all of the participants.
DNA Extraction from Urine Samples
DNA from second morning urine was extracted in quadruplicate from each urine sample using the seven commercial kits (Table 1), according to their respective manufacturer's instructions. For each individual, the same second urine has been used for DNA extraction using the seven different kits. Therefore, variations in DNA yield will be attributable to the kit and not to a difference in cellular counts. The following kits have been used: QIAamp DNA Micro Kit (Qm; Qiagen); QIAamp Viral RNA Mini Kit (Qv; Qiagen); i-genomic Urine DNA Extraction Mini Kit (iG; Intron Biotechnology); ZR Urine DNA Isolation Kit (ZR; Zymo Research); Norgen RNA/DNA/Protein Purification Kit (N; Norgen Biotek); ReliaPrep Blood gDNA Miniprep System (P; Promega); and Abcam Urine Isolation Kit (Ab; Abcam). All of these kits are using the solid-phase extraction (or adsorption-based) methodology, which involves the use of silica or resin as the solid phase to which the DNA binds in the presence of chaotropic salts, followed by elution using low-salt concentrations in the elution buffer or water.
TABLE 1.
Commercial DNA Extraction and Purification Kits Used in This Study
| Full name of kit | Manufacturer | Kit name abbreviation | Recommended urine starting amount (ml) | Elution volume (μl) | Price per extraction (€) | Processing time (min) |
|---|---|---|---|---|---|---|
| QIAamp DNA Micro Kit | Qiagen | Qm | 1 | 40 | 5.08 | 110 |
| QIAamp Viral RNA Mini Kit | Qiagen | Qv | 1 | 40 | 4.54 | 60 |
| i-genomic Urine DNA Extraction Mini Kit | Intron Biotechnology | iG | 1 | 50 | 4.60 | 60 |
| ZR Urine DNA Isolation Kit | Zymo Research | ZR | 30 | 30 | 4.11 | 60 |
| Norgen RNA/DNA/Protein Purification Kit | Norgen Biotek | N | 1 | 30 | 10.25 | 90 |
| ReliaPrep Blood gDNA Miniprep System | Promega | P | 1 | 30 | 2.03 | 60 |
| Abcam Urine Isolation Kit | Abcam | Ab | 5 | 18 | 2.70 | 60 |
Qiagen, Valencia, CA, USA; Intron Biotechnology, Korea; Zymo Research, Irvine, CA, USA; Norgen Biotek, Ontario, Canada; Promega, Madison, WI, USA; Abcam, Cambridge, UK.
Yield and Purity of Total DNA Extracted from Urine Sample
The DNA yield (ng/ml urine) and purity [absorbance ratio at 260/280 (A260/A280)] were first determined by spectrophotometry using the NanoDrop 2000 (Thermo Fisher Scientific, Waltham, MA, USA),30 where pure DNA has an A260/A280 ratios, ranging between 1.8 and 2.0. A ratio <1.8 is indicative of residual protein, phenol, or other reagent associated with the extraction protocol, where a ratio >2.0 indicates RNA contamination.
The yield of total DNA was also determined using the Quant-iT PicoGreen (Invitrogen, Life Technologies, Grand Island, NY, USA) assay, according to the manufacturer's instructions (the PicoGreen dye is a fluorescent nucleic acid stain for quantitating dsDNA).
DNA Integrity
The integrity of gDNA was determined by the Agilent 2200 TapeStation Automated Electrophoresis System (Agilent Technologies, Waldbronn, Germany), according to the manufacturer's instructions. The DNA sample (1 μl) has been measured using the gDNA ScreenTape (#5067-5365) for a sizing range of 200 to >60,000 bp (Agilent Technologies). Results have been analyzed using the TapeStation Analysis Software, A.01.04.
Yield of Human DNA
The yield of human DNA was determined using a SYBR Green real-time qPCR assay, amplifying a 164-bp fragment of the human β-globin gene (Accession Number EF450778.1). The PCR oligonucleotides and amplification conditions used are listed in Table 2. The reaction was performed in a final volume of 25 μl, containing 5 μl of the DNA template, 1× SYBR Green Master Mix (Diagenode, Belgium), used as supplied in the kit, 0.25 μM forward primer (Eurogentec, Belgium) and 0.25 μM reverse primer (Eurogentec, Belgium). A standard curve was made using a known concentration of human gDNA [TaqMan control gDNA (human); Applied Biosystems, Life Technologies, Grand Island, NY, USA]. Four serial dilutions were performed, starting from 5000 copies until 10 copies of the human genome (i.e., copy numbers of the β-globin gene). In parallel, real-time qPCR was performed in duplicate for each of the quadruplicate DNA extracts using MicroAmp Fast 96-Well Reaction Plates (Life Technologies) with strips on the StepOnePlus Real-Time PCR System (Applied Biosystems, Life Technologies). Melt curves were used to verify the specificity of the primers. The melting temperature (Tm) of the expected amplicon was ±82°C, as determined by using the human gDNA control as a DNA template in the real-time qPCR assay.
TABLE 2.
Primers and Cycling Conditions
| Target gene | Amplicon size | Primers | Ref. | Cycling conditions | |||||
|---|---|---|---|---|---|---|---|---|---|
| Human β-globin gene | 164 bp | F: 5′-GGTTCTTTGAGTCCTTTGGGGATC-3′ | 6 | 40 cycles |
Melt curve |
||||
| R: 5′-GTCACAGTGCAGCTCACTCAGTGTG-3′ | 95°C | 95°C | 62°C | 95°C | 60°C | 95°C | |||
| 10 min | 15 s | 1 min | 15 s | 1 min | 15 s | ||||
| Bacterial 16S rRNA gene | 217 bp | F: 5′-GAGGAAGGTGGGGATGACGT-3′ | 13 | 40 cycles |
Melt curve |
||||
| R: 5′-AGGCCCGAACGTATTCAC-3′ | 95°C | 95°C | 62°C | 95°C | 60°C | 95°C | |||
| 10 min | 15 s | 1 min | 15 s | 1min | 15 s | ||||
F, Forward; R, reverse.
Determination of the Presence of PCR Inhibitors
The DNA extracts were also tested for amplification inhibition (inhibitors present in the DNA extract, i.e., coextracted compounds impairing the efficiency of the PCR reaction) by real-time qPCR, using the same conditions as for the standard curve for the human β-globin gene. Undiluted DNA (5 μl) and 10-fold diluted DNA were run in duplicate. The expected (theoretical) quantification cycle difference (ΔCq) between the Cq value of the 10-fold-diluted and the undiluted sample is 3.3 when the PCR efficiency is 100%. The experimental ΔCq is calculated based on the obtained Cq for both samples (difference between the obtained Cq for the 10-fold diluted and the undiluted sample). By taking into account the PCR efficiency of each run, we considered that there are no PCR inhibitors in the DNA extract if the experimental ΔCq value equals 3.3 ± 0.5.26,31
Evaluation of Coextracted Bacterial DNA
We used a universal 16S rRNA assay to determine the quantity of 16S rRNA gene copy numbers in the DNA extracts. A standard curve was made using a known concentration of bacterial gDNA of a Lactobacillus acidophilus strain, isolated from meat (at the National Reference Laboratory for Food-Borne Pathogens at the WIV-ISP),32 taking into account that L. acidophilus contains four 16S rRNA operons (copies).33 DNA was extracted from a 2-ml overnight culture of L. acidophilus using the DNeasy Blood & Tissue Kit (Qiagen). Five serial dilutions were performed, starting from 80,000 copies until 40 copies of the 16S rRNA gene (=10 bacterial genomes), calculated based on the DNA concentration and the genome size with following formula:
where N = copy of bacterial genomes; m = quantity of bacterial gDNA (grams); Na = Avogadro's constant (6.0221415×1023 mol-1); Mw = base pair mean MW (649 Da); and L = bacterial genome size (bp; for L. acidophilus = 1,993,564 bp).34
A 217-bp fragment of the 16S rRNA gene was amplified using universal bacterial primers.13 The PCR oligonucleotides and amplification conditions used are listed in Table 2. The assays were performed on the StepOnePlus Real-Time PCR System (Applied Biosystems, Life Technologies). We tested the specificity of the reactions by verifying that the universal 16S rRNA primers did not amplify the human DNA samples and that the human β-globin primers did not amplify the bacterial DNA. Melt curves were also used to verify the specificity of the primers. With the use of L. acidophilus DNA as template, an amplicon with a Tm of 81.5°C was obtained. In parallel to the standard curve, a real-time qPCR was performed in duplicate for each of the quadruplicate (undiluted) DNA extracts obtained with the Qm, Qv, and iG kits.
Sampling Time
Urine samples from three women and three men were collected at three different moments; i.e., first morning, second morning, and a urine sample from the afternoon (±15 h). Each urine sample (1 ml) was extracted immediately in quadruplicate using the Qv kit (Qiagen). The human DNA yield was determined using the real-time qPCR assay with human β-globin primers.
Storage Conditions
The second morning urine sample from three men was collected and aliquoted immediately. Urine has been handled differently: fresh urine (4×1 ml) has been processed immediately; 4 × 1 ml fresh urine has been stored at −20°C; 4 × 1 ml fresh urine has been stored at −80°C; 4 × 1 ml fresh urine pellet (after urine centrifugation at 8000 g during 10 min and supernatant removal) was stored at −20°C; 4 × 1 ml fresh urine pellet was stored at −80°C. DNA has been extracted 15 days after storage using the Qv kit (Qiagen). The human DNA yield was determined using the real-time qPCR assay with human β-globin primers.
CHRNA3 SNP rs1051730 Genotyping Assay
DNA (100 ng), extracted from urine samples (measured on the NanoDrop), has been used for an allelic discrimination assay performed on the StepOnePlus Real-Time PCR System (Applied Biosystems, Life Technologies). The commercially available kit (Life Technologies) TaqMan SNP Genotyping Assays (ID C___9510307_20), targeting the SNP rs1501730, has been used following the manufacturer's instructions. Allele 1 corresponds to the VIC dye (mutated allele), and allele 2 corresponds to the FAM dye (wild-type allele).
Sequences were determined using the ABI 3130xl Genetic Analyzer (Applied Biosystems, Life Technologies) and visualized with the Sequence Scanner V.1.0 software (Applied Biosystems, Life Technologies).
Statistical Analysis
The variances were statistically analyzed using ANOVA Tukey's multiple comparison test using the SPSS statistical package. The differences were considered significant if P < 0.05.
RESULTS
Total Yield
First, we have evaluated the yield of total extracted DNA, as for practicality reasons, it is important to obtain the highest DNA quantity of as little as possible volume to be able to do multiple tests with sometimes precious samples, even if large urine volumes are available. The yield was first quantified by two different methods: the NanoDrop 2000 spectrophotometer and Quant-iT PicoGreen assay (Fig. 1) Samples have been grouped by sex, as it is known that female urine contains more cells and higher amounts of DNA than male urine.10,35 The DNA yield/ml urine, obtained with NanoDrop, ranged from 19 to 7182 ng/ml for the female urine and from 6 to 7128 ng/ml for the male urine. Here, it must be taken into account that Qiagen kits are using carrier RNA to improve the recovery of DNA from urine samples. If carrier RNA is added, then the NanoDrop quantification leads to an overestimation of the DNA yield, thereby masking the difference in DNA yield between genders. These issues are reduced with the PicoGreen quantification, as it is specific to dsDNA. The DNA yield obtained with the PicoGreen for the different kits was lower than that measured with the NanoDrop and ranged from 12 to 439 ng/ml for the female urine and from 2 to 274 ng/ml for the male urine (Fig. 1).
Figure 1.

Yield of DNA extracted from fresh urine samples using seven different kits and grouped by gender. Yield was determined using: NanoDrop 2000 (Nano) spectrophotometer and Quant-iT PicoGreen assay (Pico). The same urine of six individuals was extracted each time (in quadruplicate) with the different kits. We performed a PicoGreen quantification on a DNA extract of a negative control sample (1 ml water) and found that the carrier RNA corresponds to 0.9 ng of the measured DNA amount when starting from a 1-ml vol for extraction. The impact of carrier RNA in the yield of DNA, extracted using the two kits, containing carrier RNA (i.e., Qv and Qm), was therefore found to be negligible (0.3–3.8%). Error bars represent the sd of the mean for 12 extractions (n=12). *Kit lysis buffer contains carrier RNA.
At this stage, three kits (ZR, N, and P) were no longer retained in the next steps of this study for the following reasons: when using the same urine sample for all kits tested, these three kits give a lower yield compared with the other kits; the ZR kit requires a high sample volume (30 ml urine); and N is rather expensive to be used in large-scale studies, where only DNA is required. The remaining kits giving an acceptable DNA yield (Qv, Qm, iG, and Ab) were selected for further analysis.
DNA Purity
Based on the ratio of A260/A280, as measured by spectrophotometry, the purity of the DNA extracts can be evaluated. The A260/A280 ratios for the DNA extracts obtained with the different kits have been summarized per gender and per kit in Table 3. It should be taken into account that several factors can interfere with spectrophotometric absorbance readings between 220 and 280 nm,36 e.g., the carrier RNA or other substances, such as sodium azide, present in the washing buffers. Therefore, the DNA purity is difficult to be determined by spectrophotometry only. These confounding factors have also been indicated in Table 3 for each of the extraction kits used. A260/A280 ratios are acceptable for Qv and Qm kits, as a high A260/A280 purity ratio is not necessarily indicative of a problem.37 However, DNA extracted with the iG and Ab kits corresponds to low A260/A280 ratios, therefore, suggesting that contaminants are not removed completely during the extraction procedure or indicating a very low concentration of nucleic acids.
TABLE 3.
A260/A280 Ratios of Four Kits (by gender)
| Kit/gender | Mean ratio A260/A280 (±SD) | Possible confounding factors |
|---|---|---|
| Qv Woman | 3.33 ± 0.12 | carrier RNA, sodium azide |
| Qv Man | 3.39 ± 0.06 | |
| Qm Woman | 2.53 ± 0.34 | carrier RNA, sodium azide |
| Qm Man | 2.48 ± 0.64 | |
| iG Woman | 1.19 ± 0.60 | residual chemicals (phenol, guanidine) or other reagent used in the extraction protocol |
| iG Man | 1.64 ± 0.19 | |
| Ab Woman | 0.93 ± 0.39 | residual chemicals (phenol, guanidine) or other reagent used in the extraction protocol and/or a very low DNA concentration |
| Ab Man | 0.96 ± 0.16 |
Pure DNA has a A260/A280 ratio of 1.8.
Determination of the Presence of PCR Inhibitors
The assessment of the presence of PCR inhibitors is very important for downstream molecular applications, as inhibitors can give false-negative results. A 10-fold dilution of the DNA extract has been used previously to detect the presence of PCR inhibitors.26,38
Our real-time qPCR results (Table 4) revealed that DNA extracted with Qm, iG, and Ab contains PCR inhibitors in most or all of the tested samples, whereas only one of the tested DNA samples extracted with Qv contains PCR inhibitors. Real-time qPCR amplifications of DNA extracted with Ab resulted in the largest amount of negative values obtained when making the subtraction of the Cq of the diluted and the undiluted sample, thereby suggesting that extracts obtained with the Ab kit contain more remaining PCR inhibitors than the DNA extracts obtained with the other kits.
TABLE 4.
Presence of PCR Inhibitors Assessed by qPCR
| Samples | Qv |
Qm |
iG |
Ab |
||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Cq mean undil | Cq mean 10-fold dil | ΔCq mean (10-fold dil-undil) | Cq mean undil | Cq mean 10-fold dil | ΔCq mean (10-fold dil-undil) | Cq mean undil | Cq mean 10-fold dil | ΔCq mean (10-fold dil-undil) | Cq mean undil | Cq mean 10-fold dil | ΔCq mean (10-fold dil-undil) | |
| 1 | 23.4 | 26.4 | 3.1 | 24.2 | 24.1 | −0.1 | 22.5 | 24.2 | 1.7 | 29.3 | 25.0 | −4.3 |
| 2 | 25.5 | 28.2 | 2.8 | 32.2 | 27.3 | −4.9 | 24.5 | 25.6 | 1.1 | 31.0 | 31.0 | 0.0 |
| 3 | 23.8 | 26.8 | 3.0 | 24.1 | 25.4 | 1.3 | 27.3 | 27.9 | 0.6 | 30.5 | 30.3 | −0.2 |
| 4 | 27.5 | 30.2 | 2.8 | 30.2 | 29.9 | −0.4 | 29.9 | 28.7 | −1.1 | 37.3 | 36.9 | −0.5 |
| 5 | 28.6 | 32.3 | 3.8 | 27.6 | 30.9 | 3.2 | 28.4 | 30.9 | 2.5 | 37.2 | 37.0 | −0.2 |
| 6 | 28.2 | 30.2 | 2.0 | 26.9 | 29.6 | 2.8 | 29.0 | 29.4 | 0.4 | 35.4 | 35.1 | −0.3 |
| No. of inhibited samples | 1/6 | 4/6 | 6/6 | 6/6 | ||||||||
Theoretical change in comparative threshold (ΔCq; Cq 10-fold diluted sample − Cq undiluted sample) = 3.3. There is no inhibition if ΔCq experimental (Cq 10-fold diluted sample − Cq undiluted sample) = 3.3 (±0.5). Inhibited qPCR reactions are indicated in bold italic. Negative ΔCq indicates a strong inhibition. undil, Undiluted DNA sample; dil, diluted sample.
Yield of Amplifiable Human DNA
Fragmented DNA not amplifiable in PCR is also measured using NanoDrop and PicoGreen methods. Moreover, besides epithelial cells, urine may also contain bacteria from epidermal and/or mucous membranes and viruses. Therefore, bacterial and viral DNA can be coextracted with human DNA, leading to an overestimation of the human DNA yield measured by classic, spectrophotometric, and fluorometric, nonspecific methods. To overcome this overestimation, a real-time qPCR assay using human-specific β-globin primers was used to determine more specifically the amount of human intact (amplifiable) DNA in the total DNA extracted from 1 ml urine (Fig. 2) The DNA yield/ml urine determined with real-time qPCR ranged from 1 to 215 ng/ml for the female urine and from 0.1 to 25 ng/ml for the male urine. More than a twofold difference was observed between the yield obtained with the PicoGreen and the real-time qPCR for female samples, and more than a 10-fold difference was observed for male samples. The yield determined with the PicoGreen method was overestimated. This can be explained by the presence of bacterial DNA and also by the presence of highly fragmented DNA, which cannot be amplified by real-time qPCR (see also below).
Figure 2.
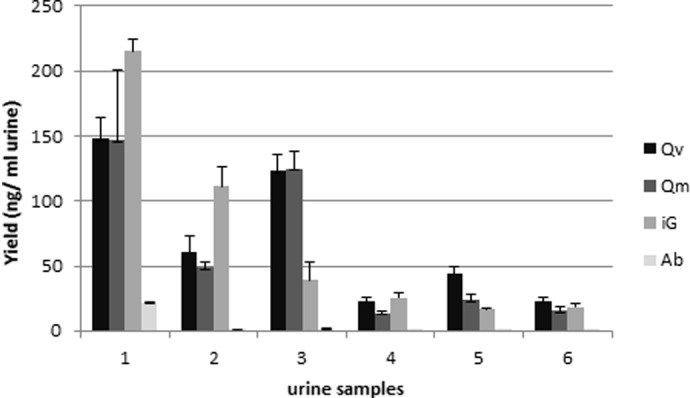
Yield of amplifiable human DNA. DNA quantification by real-time qPCR using human-specific β-globin primers on 10-fold-diluted DNA, which was extracted from fresh urine samples using different kits. Female urine samples: 1–3; male urine samples: 4–6. Error bars represent the sd of the mean for four extractions (n=4).
DNA Integrity
As shown in Fig. 3, gDNA extracted from urine is highly fragmented compared with human control gDNA. However, when comparing gDNA obtained by three different extraction kits, it was observed that gDNA extracted using Qm and Qv contains a higher proportion of longer DNA fragments compared with gDNA extracted with iG.
Figure 3.

Comparison of electropherogram of gDNA using the Agilent 2200 TapeStation. gDNA has been extracted from fresh urine samples of two female individuals using the Qv (blue, A, B), Qm (red, D, E) and iG (green, F, G) kits. TaqMan human gDNA control (C). L, Ladder: 100–48,500 bp.
Evaluation of Coextracted Bacterial DNA
As mentioned above, as extraction methods for DNA are not selective, they will extract all kind of gDNA present in the sample; i.e., human and microbial DNA. Real-time qPCR assays, using universal 16S rRNA gene primers, are used increasingly to estimate the quantity of bacteria in a DNA sample. We developed an assay to estimate the proportion of bacterial DNA in our urinary DNA extracts, based on a specific real-time qPCR assay with universal primers targeting a conserved region in the 16S rRNA gene and a standard curve. However, as a result of the diversity of the urine microbiota in healthy men39,40 and women41 and the large variance in genome size, genome number, and organization of ribosomal RNA (rrn) operons among bacterial species (the number of genes coding for 16S rRNA can vary from 142 to 1443), it is difficult to quantify absolutely the total load of bacteria in a DNA extract based on a standard curve, using universal 16S rRNA primers amplifying DNA of all bacteria present in the sample. This is because the standard curve should be different for each urine sample, and the bacterial composition of each sample for making this standard curve is a priori unknown. Therefore, we determined and compared the total number of 16S rRNA gene copies present in the urine DNA extracts, as an indication of the total load of bacteria. For this, we based our assay on a human urine-dominant bacterial species, i.e., Lactobacillus sp., which although being more abundant in female urine, is present in female and male urine.41 We used L. acidophilus, which contains four rrn operons,34 to make the standard curve for quantification in combination with universal 16S rRNA primers that will amplify all 16S rRNA gene copies (of all bacteria) present in the sample.13 Figure 4 shows an estimation of the bacterial DNA extracted from a 1-ml urine sample using the different extraction kits. Our results indicate that the quantity of extracted bacterial DNA is variable between individuals, with the iG kit, extracting in four out of six urine samples (individuals), a higher amount of bacterial DNA than Qm and Qv.
Figure 4.
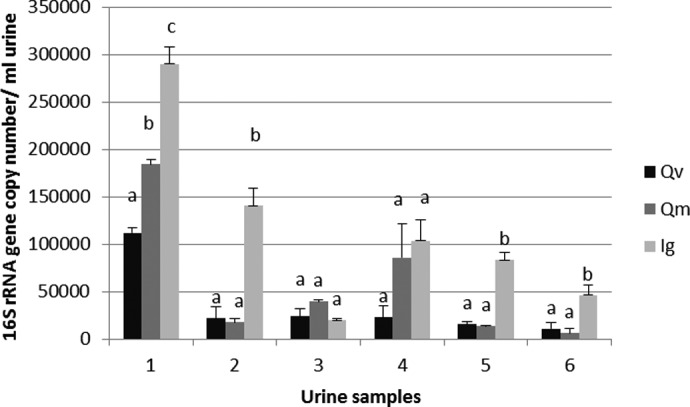
Bacterial DNA estimation by real-time qPCR using bacterial universal 16S rRNA primers. Results are reported as 16S rRNA gene copy numbers in DNA extracted from 1 ml urine sample. Female urine samples: 1–3; male urine samples: 4–6. Different letters indicate statistically different results (α=0.05), analyzed/individual. Error bars represent the sd of the mean for four extractions (n=4).
Selection of Kit Most Suited to Our Needs
Based on the results obtained at this stage of the study, we selected Qv as the kit the most suited to our needs in terms of yield of human DNA extracted and removal of PCR inhibitors. We also evaluated Qv regarding the extraction costs and the processing time and found them to be acceptable (see also Table 1). Only Qv will be used in the next step of our study.
Sampling Time
With the use of the Qv kit, we have compared the human DNA yield from urine collected at three different moments: first morning, second morning, and from the afternoon. When comparing the yield of human DNA from second morning urine with the first morning and the afternoon urine, our results revealed a significant difference between female and male samples. For female, the first morning urine resulted in the lowest human DNA yield, with two out of three samples from the afternoon urine giving the highest yield (Fig. 5). Furthermore, the DNA extracted from female afternoon urine contained longer DNA fragments than from the two other urines (Fig. 6) For male samples, the first morning urine gave the highest yield in human DNA (Fig. 5).
Figure 5.
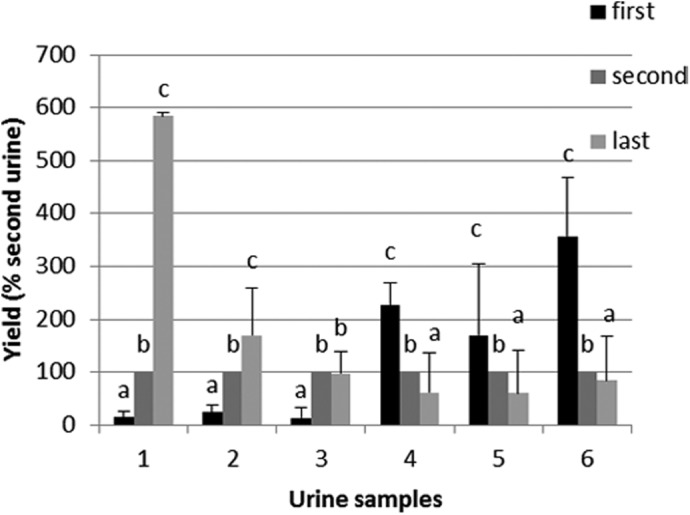
Sampling time: comparison of yield of human DNA obtained from 1 ml second morning urine with the first morning urine and the afternoon urine (“last”). Female urine samples: 1–3; male urine samples: 4–6. Different letters indicate statistically different results (α=0.05), analyzed/individual. Error bars represent the sd to the mean for four extractions (n=4).
Figure 6.
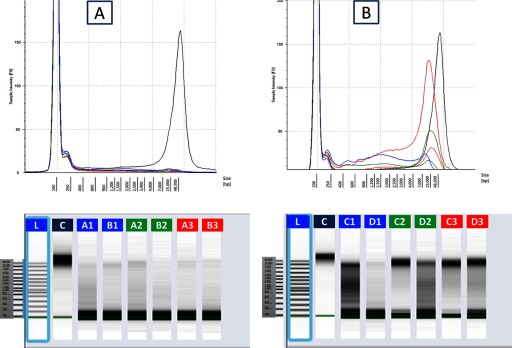
Sampling time: comparison of electropherogram of gDNA using the Agilent 2200 TapeStation. gDNA has been extracted from two male (A) and 2 female (B) urine samples, collected at three different moments of the day: first morning urine (blue, A1, B1, C1, D1), second morning urine (green, A2, B2, C2, D2), and afternoon urine (red, A3, B3, C3, D3). TaqMan human gDNA control (black, C). L, Ladder: 100–48,500 bp.
Storage Conditions
We have also compared the yield of human DNA obtained from 1 ml urine stored under different conditions. To study the worst case scenario, only male urine has been used. Results of the multiple comparisons test are displayed in Fig. 7. In general, no differences were seen among the fresh, whole urine at −80°C and urine sediment at −80°C. Likewise, no differences were demonstrated among the whole urine at −20°C and the urine sediment at −20°C. However, compared with fresh urine, storage at −80°C resulted in a higher yield of human DNA than storage at −20°C, which induced a major DNA loss. However, the yield after storage at −20°C remains acceptable for downstream analysis.
Figure 7.
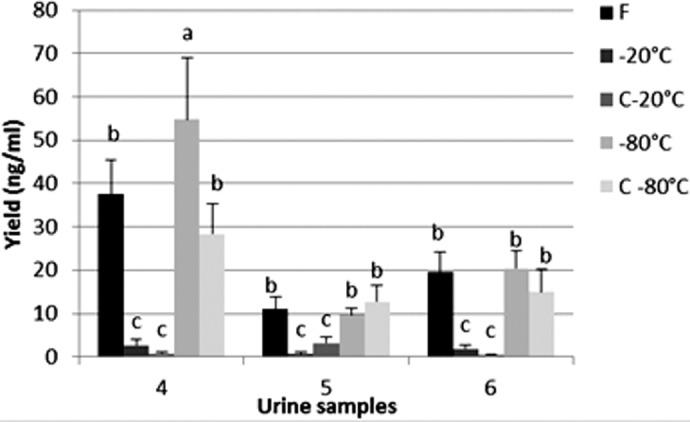
Storage conditions: comparison of yield of human DNA obtained from 1 ml fresh urine or urine stored at different conditions. DNA quantification was performed by real-time qPCR using human-specific β-globin primers on 10-fold-diluted DNA. The same urine has been used for the fresh and the frozen conditions. F, Fresh urine; −20°C, urine stored at −20°C; C−20°C, urine sediment stored at −20°C; −80°C, urine stored at −80°C; C−80°C, urine sediment stored at −80°C. Male urine samples: 4–6. Different letters indicate statistically different results (α=0.05), analyzed/individual. Error bars represent the sd of the mean for four extractions (n=4).
Proof-of-Concept
As a proof-of-concept for public health-related studies, the DNA extracted from urine stored at −20°C for 6 months using the Qv kit was genotyped for SNP rs1051730. The allelic discrimination plot displayed in Fig. 8 shows a clear discrimination between the homozygote and the heterozygote genotypes. These results were confirmed by sequencing.
Figure 8.

Genotyping results for the CHRNA3 SNP of six DNA samples. Allelic discrimination assay using DNA extracted from whole urine stored at −20°C for 6 months. The assay was performed in duplicate. Fluorescence for the wild-type allele is plotted along the y-axis (the dye is “FAM”) and for the mutated allele, labeled with the dye “VIC”, along the x-axis. WT, Samples homozygous for the wild-type allele; MUT, samples homozygous for the mutated allele; HET, samples heterozygous (wild-type alleles+mutated allele); ×, “no template” controls.
DISCUSSION
DNA biomarkers (genetic variations) are an important tool for public health, as they contribute to disease susceptibility and treatment response. They have the advantage to be stable over the entire lifetime and to be reproducible. Population-based studies of these biomarkers are needed to quantify the impact of gene variants on the risk of disease and to quantify the effect of modifiable factors that interact with gene variants, thereby translating such DNA biomarkers into opportunities for a proactive public health policy based on public health genomics.2,44 Such studies require easy-accessible, good-quality human gDNA for molecular downstream applications.
Regarding the source of gDNA, blood remains, up to now, the commonly used source of human gDNA. However, urine, compared with blood, is a very interesting alternative source of gDNA for epidemiologic and public health-related studies, as it can be collected by a noninvasive method; requires no special staff, equipment, nor infrastructure; and can be obtained in large volumes several times/day. As already mentioned, saliva could also be a valuable alternative as a source for DNA and other biomarkers.4,5 For some prospective collections, it might be a more patient-friendly method than urine. However, it is not yet known completely whether all biomarkers in urine also occur in saliva and vice versa. For DNA biomarkers, such as SNPs, this should give no difference, but for others, such as epigenetic biomarkers or RNA biomarkers, this might be more important. Indeed, biofluid samples are generally linked to anomalies in particular tissues. For instance, urine is more likely to reflect renal disease, whereas saliva may more accurately reflect lung disease. Furthermore, for applications where other biomarkers (proteins, metabolites) are to be measured simultaneously with the DNA biomarkers, it should be evaluated which source is the most appropriate, or both sources might be complementary.
Different extraction methods have been reported in the literature to obtain DNA from urine ready to use for amplification by PCR, enzyme restriction, and genotyping.13,14 Conventional methods, such as phenol-chloroform methods, are time consuming and might be more difficult to standardize. Epidemiological studies, which involve a high number of samples, require a rapid, easy, and standardized protocol. Therefore, commercial kits are highly convenient. However, to our knowledge, there are no studies yet comparing commercial human DNA extraction kits currently available on the market with the focus on large-scale epidemiologic and public health-related studies.
In the present study, we have compared seven commercial kits for DNA extraction based on critical parameters for large-scale population studies, such as a high yield of extracted human DNA of high quality and purity, sufficient removal of PCR inhibitors, sampled volume, processing time, and the cost. Initially, we measured the yield of total DNA using classical methods, i.e., spectrophotometry and fluorometry. These methods are largely used but have some disadvantages. For instance, spectrophotometric methods cannot distinguish RNA from DNA, which is a big problem for DNA extraction protocols requiring the addition of carrier RNA. This issue was largely resolved by using the PicoGreen method, only measuring dsDNA. Three kits were not retained for further analysis, as a result of the low DNA total yield (ZR, N, and P), the large-sampled volume (ZR), and/or the expensive price (N), regarding the need in our study. The four remaining kits were subsequently assessed regarding the remaining PCR inhibitors in the DNA extracts. PCR inhibitor removal is crucial, as PCR inhibitors can lead to PCR failure and false-negative results. A260/A280 ratios can already give an indication of the presence of remaining contaminants. For both kits (iG and Ab), we obtained low A260/A280 ratios, which could be indicative of remaining contaminants (possible PCR inhibitors) in the extract. However, there exist more specific assays, based on PCR, to determine the presence of PCR inhibitors. They are sometimes assessed by dilution (e.g., 10-fold) of the DNA sample, reducing the concentration of inhibitors related to target DNA.26,45,46 Especially in large-scale epidemiological studies, where cost and time are important, and where only little sample might be available, this approach can already give an indication of the presence of PCR inhibitors. We found that based on a 10-fold-diluted DNA extract, using a human β-globin real-time qPCR assay, amplification of DNA extracted with the Qv kit contains less PCR inhibitors compared with DNA extracted using the Qm, iG, and Ab kits, which contain more PCR inhibitors. These results confirm the conclusions drawn from the A260/A280 ratios. The results for the Qv kit correspond to those obtained with the same kit by Siddiqui et al.,17 who used a commercial human DNA quantification kit to assess PCR inhibitors. For DNA extracted with the other kits, no data on the presence of remaining PCR inhibitors are available yet in the literature. There have been other, more elaborated, methods reported in literature for the determination of PCR inhibitors,19–21 which should be used to perform a more detailed analysis of the PCR inhibitors remaining in the DNA extracts of some tested kit. As it has been shown in this study that even with specific washing steps in the extraction protocol or with the use of specific kits for urine samples, some PCR inhibitors might remain in the extract, the presence of PCR inhibitors should be verified before performing downstream PCR analyses, and measures should be taken (e.g., by diluting the DNA).
The human β-globin real-time qPCR assay was not only interesting to evaluate remaining PCR inhibitors but also to compare the true yield of amplifiable human DNA obtained with the different kits. Indeed, DNA extracted from urine also contains coextracted DNA from microorganisms, leading to quantification errors (i.e., overestimation) of the human DNA yield when measured with classical DNA quantification methods, such as NanoDrop and PicoGreen. This is another disadvantage of these quantification methods. As mentioned above, these methods are not specific for quantifying human DNA but are largely used in routine and epidemiological studies, as they have the advantage to be rapid and easy to use. They have also been used in several other studies reporting on DNA extraction methods for urine samples.8,9,26,28 For those methods, there is the contribution of coextracted DNA from microorganisms and from fragmented DNA, which cannot be amplified by PCR and hence, might be unusable in downstream molecular assays. Indeed, when assessing the DNA integrity of the extracted DNA, a large proportion of smaller DNA fragments was observed. For urine, it is well known that the DNA is fragmented as a result of the activity of endogenous nucleases present in urine.47 It has been reported that the predominant fraction of DNA fragments found in human urine ranges from 150 to 250 bp.8,48 Therefore, with the use of urine as a source for DNA extraction, large DNA fragments (1–7 kbp) will be difficult to obtain. If they are present in the urine, they can be extracted, but this will depend on the kit used; e.g., in our study, DNA fragments >10 kb were obtained using the Qm kit. The presence of smaller DNA fragments might be an issue to be taken into account when determining the yield of DNA based on PCR analyses. DNA fragments smaller than the target to be amplified in the PCR assay (in our case, smaller than 164 bp, which is the size of the fragment of the human β-globin gene to be amplified in the real-time qPCR assay) will not be included, leading to an underestimation of the DNA yield. Nevertheless, given the fact that bacterial DNA is coextracted, a true comparison of human DNA extraction kits for urine can only be made when using specific human DNA quantification assays, such as the human β-globin gene real-time qPCR assay used in our study. With the use of this method, we obtained a human DNA yield from 1 ml urine, using the Qv, Qm, and iG kits, ranging from 39 to 215 ng for female urine and from 13 to 44 ng for male urine. A similar DNA yield has been reported for Qv and other extraction methods, based on commercial human DNA quantification assays.10,15,17,27 Disappointingly, in our hands, the Ab kit did not result in the yield promised by the manufacturer and was not further included in our study. The human β-globin method that we used has the advantage to be very simple and cheaper than commercially available assays. For some applications, the exact quantity of human DNA might not be important to know. However, for some genome-wide molecular technologies, such as SNP arrays and exome or whole genome sequencing [so-called next-generation sequencing (NGS) approaches], the use of specific methods for human DNA quantification is highly recommended to avoid an overestimation of the human DNA quantity.
Although it is known that urine DNA extracts contain bacterial DNA, as is also the case for saliva,5 not many others have reported specific data on this. Therefore, we sought to quantify the contribution of bacterial DNA in urinary total DNA extracts obtained with the tested kits. A real-time qPCR method, using universal 16S rRNA primers13 and a standard curve based on DNA of an abundant bacterial species in human urine, i.e., L. acidophilus, has been developed to assess the presence of bacterial DNA in the urine DNA extracts, based on the quantity of 16S rRNA gene copies. An absolute quantification of bacterial load based on a single standard curve is impossible as a result of the diversity of bacterial species present in urine. This assay revealed that all of the tested kits extract a high quantity of bacterial DNA, individual dependent, and that this DNA largely contributes to the DNA yield measured by classical methods. iG extracted more bacterial DNA than the other tested kits. Therefore, this kit might be more advantageous for other purposes, e.g., for urine microbiome studies. If specific human DNA primers/probes are used in the downstream molecular assays, then the presence of bacterial DNA might not be a problem. However, when using NGS technologies, the presence of bacterial DNA will lead to the generation of a high number of expensive, unusable sequence reads. Therefore, in these cases, it should be considered to remove microorganisms from the urine before DNA extraction, e.g., by centrifugation of the urine sample at 2500 g during 15 min.49
Other parameters that we have considered for efficient DNA extraction during epidemiological studies are the processing time and the price per extraction, which should be acceptable when performing large-scale population studies involving many samples. By taking all of these criteria into account, i.e., a good yield of human DNA, the sufficient removal of PCR inhibitors, the processing time, and the costs, we finally selected Qv as the kit the best suited to our needs among the seven tested kits.
Another important aspect when thinking of using urine as an alternative source of human DNA for large-scale population studies is the time of sampling. Most studies on urine samples used the first morning urine, as it is considered to contain more cells,23 but in epidemiological studies, first morning urine is not always easy to obtain and can be restrictive for volunteers' participation. Additionally, first morning urine has some disadvantages. DNA extracted from first morning urine is more degraded than DNA extracted from urine at other moments during the day, as it contains more degenerated cells and a higher nuclease activity.8,50 Moreover, first voided female urine also contains more bacteria than the midstream urine.51,52 A question raised during this study was whether urine collected at other moments of the day could also give an acceptable yield of good-quality human DNA. Our results showed that the second morning urine and also the urine of the afternoon give a good yield of DNA. However, we observed a difference between female and male urine. With females, the afternoon urine gives the highest human yield, but with males, the first urine gives the highest yield. This issue would need further investigations. However, we can conclude that urine from other moments of the day gives an acceptable yield and can be used in routine and epidemiological studies. Additionally, we found that gDNA extracted from female afternoon urine samples is less fragmented and gives longer fragments than morning urines. This is of high importance when PCR methods are targeting longer DNA fragments, although in general, gDNA from urine is suitable for real-time qPCR assays, as these are usually targeting smaller DNA fragments (<200 bp).
In epidemiologic studies, the assays are rarely performed on fresh urine samples. Storage conditions are very important to keep the integrity of the sample and to avoid DNA degradation. Several studies on the storage of urine, with and without additive, have been reported.10,13,15,36 We were interested in the storage of the whole urine samples but also of only the urinary sediments (without additives). This might give a difference, as freezing and thawing of urine might lead to bursting of cells and thereby, to the loss of DNA.16,53 However, urine centrifugation before storage did not lead to a higher DNA yield in our hands. This is an advantage in our set-up, as this would require an additional handling that might be difficult to perform in epidemiological studies as a result of the high number of samples. As reported in literature,26,28 we could confirm that storage of urine samples (whole or sediment) at −80°C is the best method compared with storage at −20°C, which leads to a big loss of DNA. However, −80°C freezers are expensive equipment and are not always available in clinical laboratories. For that reason, −20°C freezers are more used for sample storage. Our study case showed that genotyping, using DNA extracted with the Qv kit from urine stored at −20°C during 6 months without additives, leads to a good discrimination between homozygote and heterozygote genotypes. Furthermore, in other studies, human molecular data have been obtained from urine stored without additives at −20°C for up to 7 (DNA extracted by the alcohol-precipitation method)13 or even 25 (DNA isolated by a filtration technique) years.25 In clinical research, a likely workflow for biomarker measurements would include the urine collection, followed by an initial refrigeration at 4°C before transportation to a −20°C or −80°C freezer as archival temperature. Therefore, it would be interesting to study in the future the impact of the combination of different parameters of the initial conditions of collection, temporary storage, and time between collection and freezing on the measurement of different types of (DNA) biomarkers.
In summary, compared with blood, urine is a valuable source of high-quality DNA for molecular assays in epidemiological studies as a result of the simple collection method that could also promote the volunteers' participation. For the processing of a large number of samples, the commercial kits are very convenient for their standardized and time-saving protocol. In our study, three of the seven tested kits performed very well and equally in terms of DNA quantity, but caution must be taken regarding the PCR inhibitors remaining in the DNA extracts. Compared with blood, urine contains different species of DNA. Therefore, the DNA quantification methods must be chosen depending on the downstream applications. Another advantage of urine for epidemiological studies is the possibility to use urine collected at different moments of the day when the first urine is not available. Regarding storage conditions, storage of urine at −80°C is highly recommended to prevent DNA loss, although storage at −20°C remains acceptable for PCR-based assays, as these techniques are highly sensitive.
For our proof-of-concept, gDNA extracted from urine stored at −20°C was used in a genotyping assay targeting a SNP in the CHRNA3 gene that has been related to smoking and lung cancer by genome-wide association studies.29 Tobacco smoking is the single largest cause of preventable mortality worldwide.54 Smoking behavior is influenced by genetic (60–70%) and nongenetic (environmental) factors.55 Until now, urine samples are largely used to assess the nicotine metabolites.56,57 However, to our knowledge, CHRNA3 genotyping has not been performed on DNA extracted from urine but only from blood samples. Therefore, urine would be an interesting source of different types of biomarkers, including several DNA biomarkers, to be included in a large-scale population study, for example, on smoking behavior.
ACKNOWLEDGMENTS
This project was funded by an Ylieff grant from the Belgian Federal Public Service: Public Health, Food Chain Safety and Environment. Sequencing reactions were run at the Platform Biotechnology and Molecular Biology at the WIV-ISP. The authors thank all of the volunteers who provided urine samples and Wim Coucke for his help in the statistical analysis. We also gratefully acknowledge the two anonymous reviewers for their valuable suggestions and remarks. There are no financial supports or associations that may pose a conflict of interest.
REFERENCES
- 1. Ziegler A, Koch A, Krockenberger K, Grosshennig A. Personalized medicine using DNA biomarkers: a review. Hum Genet 2012;131:1627–1638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Cleeren E, Van der Heyden J, Brand A, Van Oyen H. Public health in the genomic era: will Public Health Genomics contribute to major changes in the prevention of common diseases? Arch Public Health 2011;69:8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Kendall TL, Byerley DJ, Dean R. Isolation of DNA from blood. Anal Biochem 1991;195:74–76. [DOI] [PubMed] [Google Scholar]
- 4. Yoshizawa JM, Schafer CA, Schafer JJ, Farrell JJ, Paster BJ, Wong DT. Salivary biomarkers: toward future clinical and diagnostic utilities. Clin Microbiol Rev 2013;26:781–791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Abraham JE, Maranian MJ, Spiteri I, et al. Saliva samples are a viable alternative to blood samples as a source of DNA for high throughput genotyping. BMC Med Genomics 2012;5:19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Bryzgunova OE, Skvortsova TE, Kolesnikova EV, et al. Isolation and comparative study of cell-free nucleic acids from human urine. Ann N Y Acad Sci 2006;1075:334–340. [DOI] [PubMed] [Google Scholar]
- 7. Anker P, Mulcahy H, Stroun M. Circulating nucleic acids in plasma and serum as a noninvasive investigation for cancer: time for large-scale clinical studies? Int J Cancer 2003;103:149–152. [DOI] [PubMed] [Google Scholar]
- 8. Botezatu I, Serdyuk O, Potapova G, et al. Genetic analysis of DNA excreted in urine: a new approach for detecting specific genomic DNA sequences from cells dying in an organism. Clin Chem 2000;46:1078–1084. [PubMed] [Google Scholar]
- 9. Yokota M, Tatsumi N, Tsuda I, Takubo T, Hiyoshi M. DNA extraction from human urinary sediment. J Clin Lab Anal 1998;12:88–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Vu NT, Chaturvedi AK, Canfield DV. Genotyping for DQA1 and PM loci in urine using PCR-based amplification: effects of sample volume, storage temperature, preservatives, and aging on DNA extraction and typing. Forensic Sci Int 1999;102:23–34. [DOI] [PubMed] [Google Scholar]
- 11. Johnson DJ, Calderaro AC, Roberts KA. Variation in nuclear DNA concentrations during urination. J Forensic Sci 2007;52:110–113. [DOI] [PubMed] [Google Scholar]
- 12. Bergallo M, Costa C, Gribaudo G, et al. Evaluation of six methods for extraction and purification of viral DNA from urine and serum samples. New Microbiol 2006;29:111–119. [PubMed] [Google Scholar]
- 13. Van der Hel OL, van der Luijt RB, Bueno de Mesquita HB, et al. Quality and quantity of DNA isolated from frozen urine in population-based research. Anal Biochem 2002;304:206–211. [DOI] [PubMed] [Google Scholar]
- 14. Haufroid V, Clippe A, Knoops B, Bernard A, Lison D. Genotyping in urine: an interesting tool for epidemiological studies. Clin Chem 1998;44:2210–2211. [PubMed] [Google Scholar]
- 15. Milde A, Haas-Rochholz H, Kaatsch HJ. Improved DNA typing of human urine by adding EDTA. Int J Legal Med 1999;112:209–210. [DOI] [PubMed] [Google Scholar]
- 16. Yasuda T, Iida R, Takeshita H, et al. A simple method of DNA extraction and STR typing from urine samples using a commercially available DNA/RNA extraction kit. J Forensic Sci 2003;48:108–110. [PubMed] [Google Scholar]
- 17. Siddiqui H, Nederbragt AJ, Jakobsen KS. A solid-phase method for preparing human DNA from urine for diagnostic purposes. Clin Biochem 2009;42:1128–1135. [DOI] [PubMed] [Google Scholar]
- 18. Khan G, Kangro HO, Coates PJ, Heath RB. Inhibitory effects of urine on the polymerase chain reaction for cytomegalovirus DNA. J Clin Pathol 1991;44:360–365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Hedman J, Radstrom P. Overcoming inhibition in real-time diagnostic PCR. Methods Mol Biol 2013;943:17–48. [DOI] [PubMed] [Google Scholar]
- 20. Hedman J, Knutsson R, Ansell R, Radstrom P, Rasmusson B. Pre-PCR processing in bioterrorism preparedness: improved diagnostic capabilities for laboratory response networks. Biosecur Bioterror 2013;11(Suppl 1):S87–S101. [DOI] [PubMed] [Google Scholar]
- 21. Alaeddini R. Forensic implications of PCR inhibition—a review. Forensic Sci Int Genet 2012;6:297–305. [DOI] [PubMed] [Google Scholar]
- 22. Saulnier P, Andremont A. Detection of genes in feces by booster polymerase chain reaction. J Clin Microbiol 1992;30:2080–2083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kavaler E, Landman J, Chang Y, Droller MJ, Liu BC. Detecting human bladder carcinoma cells in voided urine samples by assaying for the presence of telomerase activity. Cancer 1998;82:708–714. [DOI] [PubMed] [Google Scholar]
- 24. Holland NT, Smith MT, Eskenazi B, Bastaki M. Biological sample collection and processing for molecular epidemiological studies. Mutat Res 2003;543:217–234. [DOI] [PubMed] [Google Scholar]
- 25. Prinz M, Grellner W, Schmitt C. DNA typing of urine samples following several years of storage. Int J Legal Med 1993;106:75–79. [DOI] [PubMed] [Google Scholar]
- 26. Cannas A, Kalunga G, Green C, et al. Implications of storing urinary DNA from different populations for molecular analyses. PLoS One 2009;4:e6985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Zhang SH, Zhao SM, Zhao ZM, Li CT. Genotyping of urinary samples stored with EDTA for forensic applications. Genet Mol Res 2012;11:3007–3012. [DOI] [PubMed] [Google Scholar]
- 28. Hilhorst M, Theunissen R, van Rie H., van Paassen P, Tervaert JW. DNA extraction from long-term stored urine. BMC Nephrol 2013;14:238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Amos CI, Wu X, Broderick P, et al. Genome-wide association scan of tag SNPs identifies a susceptibility locus for lung cancer at 15q25.1. Nat Genet 2008;40:616–622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Sambrook J, Fritsch EF, Maniatis T, Maniatis T. Molecular Cloning: A Laboratory Manual. New York: Cold Spring Harbor Laboratory Press, 1989. [Google Scholar]
- 31. European Network of Genetically Modified Organism Laboratories. Definition of Minimum Performance Requirements for Analytical Methods of GMO Testing, European Commission, Brussels, Belgium, 2008;8. [Google Scholar]
- 32. Argentinian Meat (A19), Labo National Reference Laboratory Food Born Pathogen, Scientific Institute of Public Health (WIV-ISP), Brussels, 2010. [Google Scholar]
- 33. El-Osta YG, Hillier AJ, Dobos M. Construction of a combined physical and genetic map of the chromosome of Lactobacillus acidophilus ATCC 4356 and characterization of the rRNA operons. Microbiology 2005;151:875–892. [DOI] [PubMed] [Google Scholar]
- 34. Altermann E, Russell WM, Azcarate-Peril MA, et al. Complete genome sequence of the probiotic lactic acid bacterium Lactobacillus acidophilus NCFM. Proc Natl Acad Sci USA 2005;102:3906–3912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Nakazono T, Kashimura S, Hayashiba Y, Hara K, Miyoshi A. Successful DNA typing of urine stains using a DNA purification kit following dialfiltration. J Forensic Sci 2005;50:860–864. [PubMed] [Google Scholar]
- 36. QIAamp DNA Micro Kit protocol, QIAamp Viral RNA Mini Kit protocol. Valencia, CA, USA: Qiagen, 2014. [Google Scholar]
- 37. Thermo Scientific. Assessment of Nucleic Acid Purity, T042 Technical Bulletin, NanoDrop Spectrophotometers. Wilmington, DE, USA: Thermo Fisher Scientific, 2011. [Google Scholar]
- 38. Huggett JF, Novak T, Garson JA, et al. Differential susceptibility of PCR reactions to inhibitors: an important and unrecognised phenomenon. BMC Res Notes 2008;1:70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Nelson DE, Van Der Pol B, Dong Q, et al. Characteristic male urine microbiomes associate with asymptomatic sexually transmitted infection. PLoS One 2010;5:e14116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Dong Q, Nelson DE, Toh E, et al. The microbial communities in male first catch urine are highly similar to those in paired urethral swab specimens. PLoS One 2011;6:e19709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Siddiqui H, Nederbragt AJ, Lagesen K, Jeansson SL, Jakobsen KS. Assessing diversity of the female urine microbiota by high throughput sequencing of 16S rDNA amplicons. BMC Microbiol 2011;11:244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Cole ST, Saint Girons I. Bacterial genomics. FEMS Microbiol Rev 1994;14:139–160. [DOI] [PubMed] [Google Scholar]
- 43. Young M, Cole ST. Clostridium. In Sonenshein AL, Hoch JA, Losick R. (eds): Bacillus subtilis and Other Gram-Positive Organisms. Washington, DC, USA: American Society for Microbiology, 1993;35–52. [Google Scholar]
- 44. Beskow LM, Khoury MJ, Baker TG, Thrasher JF. The integration of genomics into public health research, policy and practice in the United States. Community Genet 2001;4:2–11. [DOI] [PubMed] [Google Scholar]
- 45. Wilson IG. Inhibition and facilitation of nucleic acid amplification. Appl Environ Microbiol 1997;63:3741–3751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Wiedbrauk DL, Werner JC, Drevon AM. Inhibition of PCR by aqueous and vitreous fluids. J Clin Microbiol 1995;33:2643–2646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Ghatak S, Muthukumaran RB, Nachimuthu SK. A simple method of genomic DNA extraction from human samples for PCR-RFLP analysis. J Biomol Tech 2013;24:224–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Rauter C, Mueller M, Diterich I, et al. Critical evaluation of urine-based PCR assay for diagnosis of Lyme borreliosis. Clin Diagn Lab Immunol 2005;12:910–917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Merchant ML, Powell DW, Wilkey DW, et al. Microfiltration isolation of human urinary exosomes for characterization by MS. Proteomics Clin Appl 2010;4:84–96. [DOI] [PubMed] [Google Scholar]
- 50. Sullivan PS, Chan JB, Levin MR, Rao J. Urine cytology and adjunct markers for detection and surveillance of bladder cancer. Am J Transl Res 2010;2:412–440. [PMC free article] [PubMed] [Google Scholar]
- 51. Manoni F, Gessoni G, Alessio MG, et al. Mid-stream vs. first-voided urine collection by using automated analyzers for particle examination in healthy subjects: an Italian multicenter study. Clin Chem Lab Med 2012;50:679–684. [DOI] [PubMed] [Google Scholar]
- 52. Schaub S, Wilkins J, Weiler T, Sangster K, Rush D, Nickerson P. Urine protein profiling with surface-enhanced laser-desorption/ionization time-of-flight mass spectrometry. Kidney Int 2004;65:323–332. [DOI] [PubMed] [Google Scholar]
- 53. Kishi K, Yasuda T, Takeshita H. DNase I: structure, function, and use in medicine and forensic science. Leg Med (Tokyo) 2001;3:69–83. [DOI] [PubMed] [Google Scholar]
- 54. Tobacco Free Initiative, World Health Organization. WHO Report on the Global Tobacco Epidemic, 2009: Implementing Smoke-Free Environments. Geneva, Switzerland: World Health Organization, 2009. [Google Scholar]
- 55. De Viron S, Malats N, Van der Heyden J, Van Oyen H, Brand A. Environmental and genomic factors as well as interventions influencing smoking cessation: a systematic review of reviews and a proposed working model. Public Health Genomics 2013;16:159–173. [DOI] [PubMed] [Google Scholar]
- 56. Le Marchand L, Derby KS, Murphy SE, et al. Smokers with the CHRNA lung cancer-associated variants are exposed to higher levels of nicotine equivalents and a carcinogenic tobacco-specific nitrosamine. Cancer Res 2008;68:9137–9140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Wassenaar CA, Dong Q, Wei Q, Amos CI, Spitz MR, Tyndale RF. Relationship between CYP2A6 and CHRNA5-CHRNA3-CHRNB4 variation and smoking behaviors and lung cancer risk. J Natl Cancer Inst 2011;103:1342–1346. [DOI] [PMC free article] [PubMed] [Google Scholar]