

Abstract
Fluorescent in situ hybridization (FISH) is a molecular technique which enables the detection of nucleic acids in cells. DNA FISH is often used in cytogenetics and cancer diagnostics, and can detect aberrations of the genome, which often has important clinical implications. RNA FISH can be used to detect RNA molecules in cells and has provided important insights in regulation of gene expression. Combining DNA and RNA FISH within the same cell is technically challenging, as conditions suitable for DNA FISH might be too harsh for fragile, single stranded RNA molecules. We here present an easily applicable protocol which enables the combined, simultaneous detection of Xist RNA and DNA encoded by the X chromosomes. This combined DNA-RNA FISH protocol can likely be applied to other systems where both RNA and DNA need to be detected.
Keywords: Biochemistry, Issue 88, Fluorescent in situ hybridization (FISH), combined DNA-RNA FISH, ES cell, cytogenetics, single cell analysis, X chromosome inactivation (XCI), Xist, Bacterial artificial chromosome (BAC), DNA-probe, Rnf12
Introduction
Studying cells and tissues by means of fluorescent in situ hybridization (FISH) has, since its introduction in the late 1970s1, allowed researchers to study genes, chromatin organization and gene expression at the subcellular level. DNA FISH is frequently used in cytogenetics, karyotyping2, cancer diagnostics3 and pre-implantation genetic screening4, and has an important role in molecular research5-6 since it allows the detection of nucleic acids in their native environment. Single cell expression analysis by RNA FISH can detect native primary transcripts and noncoding RNAs being transcribed from chromosomes, and offers advantages to other techniques which assess gene expression at a population level, including for example quantitative RT-PCR, genome wide expression analysis or Northern blotting. By visualizing RNA transcripts originating directly from their sites of origin, it has for example been noticed that gene expression is stochastic7, and can sometimes be allele specific8. Improvements in the technique have even allowed the detection and quantification of single mRNA molecules within cells9-11.
The basic principle of FISH consists of hybridization of nucleic acids within the cell to a nucleic acid probe by means of highly specific Watson and Crick base-pairing. The probe can be either directly or indirectly detected, resulting in a signal which can be microscopically visualized. Initial attempts consisted of radioactively labeled probes, which had drawbacks based on safety issues, limited spatial resolution and the ability to detect only one target at a time12-14. The subsequent development of nonradioactive labels, including fluorochromes, haptens, and enzymes, has allowed the wide spread use of FISH as a routine molecular biology technique. The FISH probe can either be directly labeled with fluorochromes by chemical linking of fluorescent molecules to nucleic acid sequences15 and integration of fluorescently labeled nucleotides16-19, or the probe can be indirectly visualized after integration of haptens (including biotin and digoxigenin) and immunological detection of haptens by hapten specific antibodies conjugated to fluorescent reporter molecules20-21. The latter approach allows signal amplification by using several layers of fluorescently labeled antibodies which are used to enhance the original signal, and enables the detection of RNA species which are expressed at low levels. By combining direct and indirect labeling techniques and various haptens, several targets can be simultaneously visualized within the same cell.
One of the most important steps in FISH protocols is the hybridization of the probe to its target. Although in theory, a probe will specifically bind only to its target, in practice, this specificity is not always achieved, as probes may bind to homologous regions, and hybridization conditions will not always be ideal for a certain DNA region or RNA species. Post-hybridization washes are therefore of particular importance, as they can increase the stringency of the FISH procedure, and can prevent nonspecific binding of FISH probes, which would result in a high level of background noise. As RNA molecules are single stranded they can easily be hybridized to a FISH probe. In contrast, the double stranded DNA molecule first needs a denaturation step, after which a probe can hybridize. This is usually achieved by heating of the specimen, which results in denaturation of the DNA. However, under these harsh conditions, fragile, single stranded RNA molecules might be lost. Therefore, combined DNA-RNA FISH requires significant optimization of the conditions, and is more technically challenging compared to the separate detection of only RNA or DNA.
Here we present a detailed protocol of combined, simultaneous DNA-RNA FISH which has allowed us to study X chromosome inactivation (XCI) in differentiating female mouse embryonic stem cells22-24. XCI is a crucial epigenetic mechanism for female embryonic development25, and results in heterochromatinization and hence silencing of one of the two X chromosomes in female individuals26-27. Essential to this process is the noncoding RNA Xist28-30, which is regulated by the RNF1222-23 and REX124 proteins. Xist expression becomes upregulated on the future inactive X chromosome (Xi) during embryonic development or upon ES cell differentiation in vitro, and can spread along the X chromosome and thereby attract chromatin remodeling enzymes which result in the transcriptional shutdown of the X chromosome31. This spreading of Xist RNA can be visualized by RNA FISH as a coating of the X chromosome, which is also referred to as an Xist cloud. Since female ES cells can lose one of their X chromosomes due to genomic instability, we and others have employed combined DNA-RNA FISH to study XCI, to make sure that only karyotypically stable cells are assessed in the analysis of this important process22,32-34. As with every molecular biology technique, several different excellent protocols have been published35-38. Here we present our method, starting from the induction of differentiation in mouse ES cells, fixation of cells, labeling of FISH probes by Nick-translation, pre-treatment of fixed cells to allow permeabilization and subsequent probe uptake, hybridization of the probe to the target, and finally detection of the probe by fluorescently labeled antibodies. The herein presented protocol allows the faithful detection of Xist RNA and the X chromosome within a period of two days, and the basics of this technique can likely be adapted to other systems and areas of research.
Protocol
1. Induction of Differentiation in Female Embryonic Stem Cells to Induce X Chromosome Inactivation
NOTE: Female mouse embryonic stem cells (available upon request) are grown under standard ES cell conditions on gelatinized culture dishes coated with mouse embryonic fibroblasts (MEFs). We here assume that the reader is familiar with standard cell culture techniques39-41. To induce differentiation, ES cells grown in T25 dishes will be separated from the MEFs, and will be plated in differentiation medium.
Remove ES medium from the ES cell culture, and wash twice with cell culture PBS.
Add 1.5 ml trypsin-EDTA (pre-warmed to 37 °C), and incubate cells at 37 °C for 7 min. After 3.5 min, shake the dish gently to break up the colonies.
Inactivate the trypsin-EDTA by adding 3.5 ml of differentiation medium to the cells. Pipette cells up and down to obtain a single cell solution, and collect in a 15 ml tube. Spin for 5 min at 200 x g, and collect cells in 10 ml of differentiation medium.
Add cell suspension to a non-gelatinized culture dish, and incubate for 1 hr in a cell culture incubator. NOTE: MEFs will be able to adhere to the non-gelatinized culture dish, whereas ES cells will remain in suspension.
In the meantime, add sterilized glass coverslips (24 x 24 mm) to 6-well plates, add 0.2% gelatin, and incubate for minimal 5 min at room temperature.
After 1 hr, collect the supernatant of the pre-plated ES cell culture, which will mainly consist of non-adhering ES cells. Plate ES cells to 6-well plates containing coverslips. NOTE: Use ⅙th of a confluent T25 dish for 6 wells of a 6-well plate. This will allow a confluent differentiation well after 3 days of differentiation. Change medium on a daily basis. For longer differentiation time courses, plate pre-plated ES cells in differentiation medium in bacterial dishes, and incubate on a cell shaker integrated into the cell culture incubator. This will allow the cells to form embryoid bodies, which can be plated to coverslips 1-2 days prior to fixation.
2. Fixation of Differentiated ES Cells for Subsequent DNA-RNA FISH Analysis
Remove differentiation medium from differentiation cultures, and wash twice with cell culture PBS. NOTE: Add PBS carefully to prevent the cells being washed away.
Remove cell culture PBS and fix cells by adding 4% paraformaldehyde (PFA)/PBS, and incubate for 10 min at room temperature. CAUTION: PFA is toxic and can cause significant health risk when being inhaled or when in contact to skin or eyes. Furthermore, PFA is carcinogenic.
Remove 4% PFA/PBS, and wash coverslips 3x with 70% ethanol. NOTE: Correct washing is important, as any trace of PFA will interfere with subsequent FISH steps. Coverslips can be stored in 70% ethanol at -20 °C for several months prior to FISH analysis.
3. Labeling of DNA Probes for FISH Experiments
NOTE: Obtain a DNA fragment of the gene of interest (minimal length >2 kb for RNA FISH, or >20 kb for DNA FISH), or a BAC covering the gene of interest. For RNA detection, a smaller probe can be sufficient compared to DNA detection, since most likely transcription will result in multiple transcripts, which can be simultaneously detected. For a strong signal on the single copy DNA strand, a longer probe is required, to be able to detect a sufficient number of incorporated haptens. Preferably, a non-transcribed genomic region is chosen for designing the DNA probe. In addition, when using a smaller probe to detect the RNA, it will be prevented that the genomic loci from which the RNA is transcribed is being detected in the combined DNA-RNA FISH approach, although this cannot be fully excluded. To detect mouse Xist, a 5.5 kb cDNA probe was used. For detection of the X chromosome, several BAC probes can be used. Good results have been obtained with a mixture of BAC RP23-100E1 and BAC CT7-474E4, which are located in close proximity to the Xist locus. Depending on the gene or chromosome of interest, several different probes might be required to obtain optimal results. Whenever working with materials which will be used for RNA-FISH, use sterile filter tips, RNAse free H2O, and work in a clean environment.
Take 1 µg of DNA and dilute in H2O in a total volume of 16 µl. Add 4 µl of digoxigenin or biotin labeling solution (see reagents), mix by vortexing and incubate at 16 °C for 90 min. NOTE: The biotin or digoxigenin-labeled nucleotides will now be incorporated by nick-translation.
In the meantime, prepare G50 columns to remove non-integrated nucleotides. Take a 2 ml syringe, remove the stamper, and add sterilized cotton into the syringe. Add G50 balls in solution to fill the syringe, place syringe in a 15 ml tube and centrifuge at 642 x g for 1 min. NOTE: Around 80% of the syringe should now be filled with G50. Repeat when less G50 is present.
After 90 min, add 40 µl of H2O to the nick-translation reaction mixture, and add the reaction to the G50 column. Add an empty tube under the column, and centrifuge at 642 x g for 2 min. Collect the flow through in a reaction tube.
Measure the volume of the flow through with a pipette, and add H2O to reach a total volume of 200 µl. Precipitate the flow through by adding 13.3 µl salmon sperm DNA (10 mg/ml), 13.3 µl yeast tRNA (10 mg/ml), 26.7 µl mouse Cot-1 DNA (1 mg/ml) and 28 µl 2 M NaAc, pH 5.6. Mix by vortexing, and add 533 µl ice cold 100% ethanol. Shake the tube, and centrifuge at 16,200 x g for 30 min at 4 °C.
Remove the supernatant, and wash pellet twice with 70% ethanol. Centrifuge at 16,200 x g for 5 min at 4 °C, and air dry the pellet. Add 50 µl of 50+ hybridization solution, and incubate at 37 °C for 30 min to facilitate dissolving. NOTE: Labeled probe can be stored for several years at -20 °C. CAUTION: 50+ hybridization solution contains formamide, which is toxic, can irritate skin, eyes and the respiratory system, and is also a teratogen.
4. Permeabilization, Pre-treatment and Hybridization of Fixed Cells for Combined DNA-RNA FISH
Rehydrate fixed cells on coverslips, by removing 70% ethanol and the addition of cell culture PBS to the 6-well plates. Incubate for 5 min. Repeat in total three times.
Remove cell culture PBS, and add 0.2% pepsin to the 6-well plates, to permeabilize the cells. Incubate 6-well plates in a water bath at 37 °C for 4 min. Remove pepsin, and inactivate with RNAse free H2O.
Post-fix cells by incubating with 4% PFA/PBS for 5 min at room temperature. After fixation, wash twice with cell culture PBS for 5 min.
Dehydrate cells by incubating with 70% ethanol for 3 min, 90% ethanol for 3 min and 100% ethanol for 3 min. Transfer coverslips out of 6-well plates, and air dry coverslips for several minutes.
Age cells by incubating coverslips at 65 °C for 1 hr on a heat plate.
Pre-hybridize FISH probe with mouse Cot-1 DNA by adding together for every coverslip to be analyzed, 25 µl of 50+ hybridization solution, 0.5 µl mouse Cot-1 DNA, 1 µl of biotin-labeled Xist probe and 1 µl of digoxigenin labeled BAC probe detecting the X chromosome. Mix by vortexing, and denature at 99 °C for 5 min. Incubate immediately at 37 °C for 45 min, to allow Cot-1 DNA to hybridize to repeat sequences present in the probes.
After aging, spot 50 µl of denaturation buffer (consisting of 70% formamide/2x SSC/10 mM phosphate buffer) on a glass slide, and put coverslips on top with cells facing towards the denaturation buffer. Incubate at 65 °C for 2 min.
Add coverslips back to 6-well plates, and post-fix cells by incubating in ice cold 70% ethanol for 5 min.
Dehydrate cells by subsequent incubation in 70% ethanol for 3 min, 90% ethanol for 3 min and 100% ethanol for 3 min. Remove coverslips from 6-well plates, and let air dry for several minutes.
Spot pre-hybridized probe on a glass slide, and incubate coverslip on top. Place slides in a humidified chamber filled with 50 ml 50% formamide/2x SSC, and incubate overnight at 37 °C.
5. Post-hybridization Washes and Antibody Mediated Detection of Probe
Fill 6-well plates with 2x SSC, and pre-warm till 42 °C. Add coverslips after overnight incubation, and incubate at 42 °C for 5 min.
Remove 2x SSC, and incubate coverslips with 50% formamide/2x SSC for 10 min at 42 °C. Repeat in total 3 times (30 min of washing altogether).
Remove 50% formamide/2x SSC, and wash slides with Tris-saline-Tween (TST) for 2 x 5 min at room temperature.
Spot 50 µl Tris-saline-BSA (TSBSA) per coverslip on new glass slides, and incubate coverslips upside down for blocking during 30 min at room temperature in a TST-humidified dark chamber.
Transfer coverslips back to 6-well plates, and wash 2 x 5 min with TST.
Spot 50 µl of sheep-anti-dig antibody (1:500 in TSBSA) per coverslip on new glass slides, and incubate coverslips upside-down for the first incubation step during 30 min at room temperature in a TST-humidified dark chamber.
Transfer coverslips back to 6-well plates, and wash 2 x 5 min with TST.
Spot 50 µl of rabbit-anti-sheep antibody conjugated with FITC (1:250 in TSBSA) per coverslip on new glass slides, and incubate coverslips upside down for the second incubation step during 30 min at room temperature in a TST-humidified dark chamber.
Transfer coverslips back to 6-well plates, and wash 2 x 5 min with TST.
Spot 50 µl of goat-anti-rabbit antibody conjugated with FITC (1:250 in TSBSA) per coverslip on new glass slides, and incubate coverslips upside down for the third incubation step during 30 min at room temperature in a TST-humidified dark chamber.
Transfer coverslips back to 6-well plates, and wash 2 x 5 min with TST.
Spot 50 µl of mouse-anti-biotin antibody (1:200 in TSBSA) per coverslip on new glass slides, and incubate coverslips upside down for the fourth incubation step during 30 min at room temperature in a TST-humidified dark chamber.
Transfer coverslips back to 6-well plates, and wash 2 x 5 min with TST.
Spot 50 µl of donkey-anti-mouse antibody conjugated with Rhodamine (1:250 in TSBSA) per coverslip on new glass slides, and incubate coverslips upside down for the fifth incubation step during 30 min at room temperature in a TST-humidified dark chamber.
Transfer coverslips back to 6-well plates, and wash 2 x 5 min with TST.
Spot 50 µl of goat-anti-horse antibody conjugated with Rhodamine (1:250 in TSBSA) per coverslip on new glass slides, and incubate coverslips upside down for the sixth incubation step during 30 min at room temperature in a TST-humidified dark chamber. NOTE: the goat-anti-horse antibody will recognize the donkey-anti-mouse antibody; when available to the researcher, a goat-anti-donkey antibody will work as well.
Transfer coverslips back to 6-well plates, and wash 2 x 5 min with TST. Wash additional 5 min with Tris-saline (TS).
Dehydrate cells by subsequent incubation in 70% ethanol for 3 min, 90% ethanol for 3 min and 100% ethanol for 3 min. Remove coverslips from 6-well plates, and let air dry for several minutes.
Spot a drop of mounting medium with DAPI (see reagents) on a glass slide, and add coverslip upside down on top. Seal slides with nail varnish and assess FISH results by fluorescent microscopy (Figure 1).
Representative Results
Using the above mentioned protocol for combined DNA-RNA FISH, we have been able to visualize X chromosome inactivation in differentiating female embryonic stem cells. Figure 1 shows a representative example of an DNA-RNA FISH experiment, where we detected both Xist (which is visible as an so-called Xist RNA cloud on the inactive X chromosome and a basal transcription pinpoint on the active X chromosome), and a region of the X chromosome, which is visible as a pinpoint signal. Note that one of the pinpoint signals is located within the Xist cloud, thereby showing that the Xist cloud is indeed localizing along, and coating the X chromosome. In more than 99% of cells, we detect using our protocol a correct DNA signal (data not shown). Compared to RNA-FISH, Xist clouds visualized using the combined DNA-RNA FISH procedure look sometimes larger, as the denatured DNA seems to occupy a larger area. We have obtained similar results using human ES cells, human lymphocytes and various fibroblast cell lines of various species, and the same protocol has been applied to study gene expression of various X-linked loci, including Rnf12. In addition, when this protocol was applied to undifferentiated female ES cells, basal Xist expression from both X chromosomes could be detected, which shows that using this protocol also genes expressed at low levels can be visualized (data not shown).
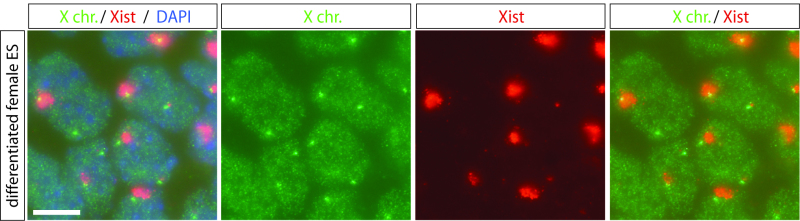 Figure 1. Combined DNA-RNA FISH in differentiated female ES cells. Representative results obtained after the herein described protocol for combined DNA-RNA FISH. The X chromosome (X chr.) is detected using a digoxigenin-labeled BAC probe, which is visualized using antibodies conjugated to FITC (green signal). In almost every cell, two X chromosomes are detected. Xist RNA is detected using a biotin labeled cDNA probe, which is visualized using antibodies conjugated to rhodamine (red signal). Both large Xist accumulations on the inactive X chromosome (Xist clouds) and basal Xist expression from the active X chromosome (Xist pinpoints) are detected (note: upon differentiation this basal Xist expression is ceased on the future active X chromosome, and therefore not detected in every nucleus). Nuclei are counterstained with DAPI (blue). Scale bar represents 10 µm. Click here to view larger image.
Figure 1. Combined DNA-RNA FISH in differentiated female ES cells. Representative results obtained after the herein described protocol for combined DNA-RNA FISH. The X chromosome (X chr.) is detected using a digoxigenin-labeled BAC probe, which is visualized using antibodies conjugated to FITC (green signal). In almost every cell, two X chromosomes are detected. Xist RNA is detected using a biotin labeled cDNA probe, which is visualized using antibodies conjugated to rhodamine (red signal). Both large Xist accumulations on the inactive X chromosome (Xist clouds) and basal Xist expression from the active X chromosome (Xist pinpoints) are detected (note: upon differentiation this basal Xist expression is ceased on the future active X chromosome, and therefore not detected in every nucleus). Nuclei are counterstained with DAPI (blue). Scale bar represents 10 µm. Click here to view larger image.
Discussion
Combined DNA-RNA FISH can be technically challenging, as conditions suitable for DNA FISH can be too harsh for less stable RNA molecules. Several approaches have been applied to study both DNA and RNA in the same cell, circumventing the need for simultaneous incubation with probes detecting both the DNA and RNA. For example, in a superimposition approach, first RNA FISH is performed, and cells are imaged, and coordinates are taken. Subsequently, the same slides are used for DNA FISH, during which the signal of RNA FISH is lost. After DNA FISH, the cells are again imaged, and the pictures obtained from both the RNA and DNA FISH are superimposed. Although this approach will work in almost all cases, as the DNA of fixed cells is stable, and is not affected by the treatment needed for initial RNA FISH, this is a very laborious approach which will cost at least four days. In another approach, first the RNA FISH is performed, the RNA FISH signal is fixed by addition of fixatives, after which DNA FISH is applied. Although also this approach can work, some of the fluorescent signal derived from the RNA FISH can be lost upon fixation. This might especially hinder the detection of transcripts which are only expressed at a low level. The herein presented approach is optimized for the simultaneous detection of both one RNA target and one DNA target in a single FISH experiment, and has the advantage that both results can be obtained in a single experiment within two days, with reliable results.
Several steps in the FISH protocol are crucial to obtain optimal results. First, whenever dealing with RNA molecules, one should be careful not to introduce RNAse in any buffer or solution used. Hence, a clean working environment should be established, and filter tips should be used when pipetting reagents being used for FISH. Care should be taken to use RNAse free compounds (for example RNAse free water, cell culture grade PBS, absolutely pure ethanol etc.). When these simple rules are followed, it is generally not necessary to use additional RNAse inhibitors supplemented to the reagents used. Second, the permeabilization procedure using pepsin is a gentle equilibrium between too little, or too much permeabilization. Depending on the particular cell type used, it might be required to optimize the time allowed for permeabilization, or the pepsin concentrations. The latter also depends on your particular batch of pepsin, as the activity might vary. As an alternative, permeabilization using 0.1% Triton-X100/PBS for 5 min results in good results under certain conditions, and has the advantage that FISH using this method of permeabilization can be combined with immunostaining to detect nuclear or cytoplasmic proteins. As a third important aspect of FISH, the aging, denaturation, hybridization and washing temperatures should be kept constant. Even small deviations from the target temperature can result in less efficient denaturation, hybridization, or less stringent washes, which will result in more background staining. As an alternative to the 1 hr aging step at 65 °C followed by denaturation at 65 °C for 2 min, also an initial denaturation at 80 °C for 5 min, without aging, can give good results. We prefer the aging procedure, as in general this procedure gives more reliable DNA FISH signals. Lastly, certain DNA probes will be so specific, that they will not require three layers of antibodies for sufficient detection. This should be empirically tested for every newly generated probe.
Although the current protocol works robustly in the study of X chromosome inactivation and detection of Xist, combined detection of RNA and DNA might be more complicated in other settings. This might especially be the case when the RNA species which is aimed to be detected is only expressed at very low levels, is full of repeat sequences or is a relatively short transcript. In these situations, it might be rather difficult to design an optimal probe, which will allow the efficient hybridization to a limited, not optimal available amount of target. In such situations, it might be worth optimizing first conditions for RNA FISH. Several alternative probes could be tried, and the duration of probe labeling by Nick-translation could be titrated, to optimize the amount of incorporated haptens. Importantly, it should be verified that the aimed RNA is expressed in the cells to be investigated, by for example verifying expression by RT-PCR. Also when analyzing a different type of cells, it is essential to include cells in which expression has previously been detected as positive controls, as this might allow the distinction between a technical, FISH related problem, or the simple absence of expression in the new type of cells analyzed. When, despite verified expression, the use of various probes and optimization of labeling conditions, RNA FISH does not result in a clear signal, it might be worth changing the hybridization conditions, by varying the amount of formamide used, or the duration and temperature of probe hybridization. Alternatively, directly labeled oligonucleotide probes might be used9-10. The same optimization steps can subsequently be applied to improve DNA FISH, and the simultaneous detection of DNA and RNA.
Using the combined DNA-RNA FISH protocol, we have been able to study X chromosome inactivation in differentiating female ES cells. Similar results have been obtained in our studies using different cell types and embryos, and we are currently further optimizing conditions to apply combined DNA-RNA FISH on tissue sections. As the important role of non-coding RNAs in many biological processes becomes more and more appreciated, we foresee that the same protocol can likely be used to study other non-coding RNAs in the context of their specific chromatin.
Disclosures
The authors have nothing to disclose.
Acknowledgments
The authors thank all previous and present members of the department of Reproduction and Development of the Erasmus MC for helpful and stimulating discussions during the past years. We also want to thank the three anonymous reviewers for providing helpful feedback. Work in the Gribnau lab is supported by funding from the Dutch research council (NWO-VICI) and an ERC starting grant.
References
- Rudkin GT, Stollar BD. High resolution detection of DNA-RNA hybrids in situ by indirect immunofluorescence. Nature. 1977;265:472–473. doi: 10.1038/265472a0. [DOI] [PubMed] [Google Scholar]
- Imataka G, Arisaka O. Chromosome analysis using spectral karyotyping (SKY) Cell Biochem Biophys. 2012;62:13–17. doi: 10.1007/s12013-011-9285-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross FM, et al. Report from the European Myeloma Network on interphase FISH in multiple myeloma and related disorders. Haematologica. 2012;97:1272–1277. doi: 10.3324/haematol.2011.056176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mastenbroek S, et al. Preimplantation genetic screening: a systematic review and meta-analysis of RCTs. Hum Reprod Update. 2011;17:454–466. doi: 10.1093/humupd/dmr003. [DOI] [PubMed] [Google Scholar]
- Martins-Taylor K, Xu RH. Concise review: Genomic stability of human induced pluripotent stem cells. Stem Cells. 2012;30:22–27. doi: 10.1002/stem.705. [DOI] [PubMed] [Google Scholar]
- Rens W, et al. Resolution and evolution of the duck-billed platypus karyotype with an X1Y1X2Y2X3Y3X4Y4X5Y5 male sex chromosome constitution. Proc Natl Acad Sci U S A. 2004;101:16257–16261. doi: 10.1073/pnas.0405702101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munsky B, et al. Using gene expression noise to understand gene regulation. Science. 2012;336:183–187. doi: 10.1126/science.1216379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyanari Y, Torres-Padilla ME. Control of ground-state pluripotency by allelic regulation of Nanog. Nature. 2012;483:470–473. doi: 10.1038/nature10807. [DOI] [PubMed] [Google Scholar]
- Kwon S. Single-molecule fluorescence in situ hybridization: quantitative imaging of single RNA molecules. BMB Rep. 2013;46:65–72. doi: 10.5483/BMBRep.2013.46.2.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji N, van Oudenaarden A. Single molecule fluorescent in situ hybridization (smFISH) of C. elegans worms and embryos. WormBook. 2012. pp. 1–16. [DOI] [PMC free article] [PubMed]
- Femino AM, et al. Visualization of single RNA transcripts in situ. Science. 1998;280:585–590. doi: 10.1126/science.280.5363.585. [DOI] [PubMed] [Google Scholar]
- Buongiorno-Nardelli M, Amaldi F. Autoradiographic detection of molecular hybrids between RNA and DNA in tissue sections. Nature. 1970;225:946–948. doi: 10.1038/225946a0. [DOI] [PubMed] [Google Scholar]
- Gall JG, Pardue ML. Formation and detection of RNA-DNA hybrid molecules in cytological preparations. Proc Natl Acad Sci U S A. 1969;63:378–383. doi: 10.1073/pnas.63.2.378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- John HA, et al. RNA-DNA hybrids at the cytological level. Nature. 1969;223:582–587. doi: 10.1038/223582a0. [DOI] [PubMed] [Google Scholar]
- Bauman JG, et al. A new method for fluorescence microscopical localization of specific DNA sequences by in situ hybridization of fluorochromelabelled RNA. Exp Cell Res. 1980;128:485–490. doi: 10.1016/0014-4827(80)90087-7. [DOI] [PubMed] [Google Scholar]
- Wiegant J, et al. In situ hybridization with fluoresceinated DNA. Nucleic Acids Res. 1991;19:3237–3241. doi: 10.1093/nar/19.12.3237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langer PR, et al. Enzymatic synthesis of biotin-labeled polynucleotides: novel nucleic acid affinity probes. Proc Natl Acad Sci U S A. 1981;78:6633–6637. doi: 10.1073/pnas.78.11.6633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manuelidis L, et al. High-resolution mapping of satellite DNA using biotin-labeled DNA probes. J Cell Biol. 1982;95:619–625. doi: 10.1083/jcb.95.2.619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kislauskis EH, et al. Isoform-specific 3'-untranslated sequences sort alpha-cardiac and beta-cytoplasmic actin messenger RNAs to different cytoplasmic compartments. J Cell Biol. 1993;123:165–172. doi: 10.1083/jcb.123.1.165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singer RH, Ward DC. Actin gene expression visualized in chicken muscle tissue culture by using in situ hybridization with a biotinated nucleotide analog. Proc Natl Acad Sci U S A. 1982;79:7331–7335. doi: 10.1073/pnas.79.23.7331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corput MP, et al. Fluorescence in situ hybridization using horseradish peroxidase-labeled oligodeoxynucleotides and tyramide signal amplification for sensitive DNA and mRNA detection. Histochem Cell Biol. 1998;110:431–437. doi: 10.1007/s004180050304. [DOI] [PubMed] [Google Scholar]
- Jonkers I, et al. RNF12 is an X-Encoded dose-dependent activator of X chromosome inactivation. Cell. 2009;139:999–101. doi: 10.1016/j.cell.2009.10.034. [DOI] [PubMed] [Google Scholar]
- Barakat TS, et al. RNF12 activates Xist and is essential for X chromosome inactivation. PLoS Genet. 2011;7 doi: 10.1371/journal.pgen.1002001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gontan C, et al. RNF12 initiates X-chromosome inactivation by targeting REX1 for degradation. Nature. 2012;485:386–390. doi: 10.1038/nature11070. [DOI] [PubMed] [Google Scholar]
- Lyon MF. Gene action in the X-chromosome of the mouse (Mus musculus L) Nature. 1961;190:372–373. doi: 10.1038/190372a0. [DOI] [PubMed] [Google Scholar]
- Barakat TS, Gribnau JX. chromosome inactivation in the cycle of life. Development. 2012;139:2085–2089. doi: 10.1242/dev.069328. [DOI] [PubMed] [Google Scholar]
- Augui S, et al. Regulation of X-chromosome inactivation by the X-inactivation centre. Nat Rev Genet. 2011;12:429–442. doi: 10.1038/nrg2987. [DOI] [PubMed] [Google Scholar]
- Brown CJ, et al. A gene from the region of the human X inactivation centre is expressed exclusively from the inactive X chromosome. Nature. 1991;349:38–44. doi: 10.1038/349038a0. [DOI] [PubMed] [Google Scholar]
- Brockdorff N, et al. Conservation of position and exclusive expression of mouse Xist from the inactive X chromosome. Nature. 1991;351:329–331. doi: 10.1038/351329a0. [DOI] [PubMed] [Google Scholar]
- Borsani G, et al. Characterization of a murine gene expressed from the inactive X chromosome. Nature. 1991;351:325–329. doi: 10.1038/351325a0. [DOI] [PubMed] [Google Scholar]
- Barakat TS, Gribnau J. X chromosome inactivation and embryonic stem cells. Adv Exp Med Biol. 2010;695:132–154. doi: 10.1007/978-1-4419-7037-4_10. [DOI] [PubMed] [Google Scholar]
- Chaumeil J, et al. A novel role for Xist RNA in the formation of a repressive nuclear compartment into which genes are recruited when silenced. Genes Dev. 2006;20:2223–2237. doi: 10.1101/gad.380906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okamoto I, et al. Eutherian mammals use diverse strategies to initiate X-chromosome inactivation during development. Nature. 2011;472:370–374. doi: 10.1038/nature09872. [DOI] [PubMed] [Google Scholar]
- Gribnau J, et al. X chromosome choice occurs independently of asynchronous replication timing. J Cell Biol. 2005;168:365–373. doi: 10.1083/jcb.200405117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Namekawa SH, Lee JT. Detection of nascent RNA, single-copy DNA and protein localization by immunoFISH in mouse germ cells and preimplantation embryos. Nat Protoc. 2011;6:270–284. doi: 10.1038/nprot.2010.195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Tine BA, et al. Simultaneous in situ detection of RNA, DNA, and protein using tyramide-coupled immunofluorescence. Methods Mol Biol. 2005;292:215–230. doi: 10.1385/1-59259-848-x:215. [DOI] [PubMed] [Google Scholar]
- Pollex T, et al. Live-Cell Imaging Combined with Immunofluorescence, RNA, or DNA FISH to Study the Nuclear Dynamics and Expression of the X-Inactivation Center. Methods Mol Biol. 2013;1042:13–31. doi: 10.1007/978-1-62703-526-2_2. [DOI] [PubMed] [Google Scholar]
- Chaumeil J, et al. Combined immunofluorescence, RNA fluorescent in situ hybridization, and DNA fluorescent in situ hybridization to study chromatin changes, transcriptional activity, nuclear organization, and X-chromosome inactivation. Methods Mol Biol. 2008;463:297–308. doi: 10.1007/978-1-59745-406-3_18. [DOI] [PubMed] [Google Scholar]
- Lin S, Talbot P. Methods for culturing mouse and human embryonic stem cells. Methods Mol Biol. 2011;690:31–56. doi: 10.1007/978-1-60761-962-8_2. [DOI] [PubMed] [Google Scholar]
- Meissner A, et al. Derivation and manipulation of murine embryonic stem cells. Methods Mol Biol. 2009;482:3–19. doi: 10.1007/978-1-59745-060-7_1. [DOI] [PubMed] [Google Scholar]
- Nichols J, Ying QL. Derivation and propagation of embryonic stem cells in serum- and feeder-free culture. Methods Mol Biol. 2006;329:91–98. doi: 10.1385/1-59745-037-5:91. [DOI] [PubMed] [Google Scholar]