

Abstract
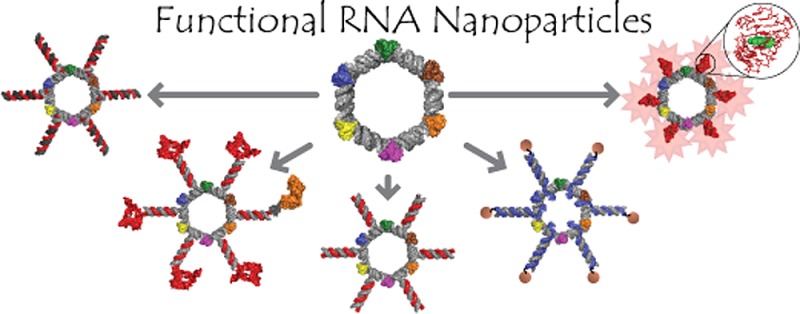
Our recent advancements in RNA nanotechnology introduced novel nanoscaffolds (nanorings); however, the potential of their use for biomedical applications was never fully revealed. As presented here, besides functionalization with multiple different short interfering RNAs for combinatorial RNA interference (e.g., against multiple HIV-1 genes), nanorings also allow simultaneous embedment of assorted RNA aptamers, fluorescent dyes, proteins, as well as recently developed RNA–DNA hybrids aimed to conditionally activate multiple split functionalities inside cells.
Keywords: RNA nanotechnology, RNA nanoparticles, RNA interference, aptamers, RNA−DNA hybrid reassociation
Improving the quality of life in modern society promotes longer life expectancies of the population. Consequently, the chance of contracting a serious infection or illness increases. Lately, there is considerable hope that nanotechnologies will provide new, revolutionary approaches for the detection and therapy of different life-threatening diseases. Nanotechnology promises to completely change, for example, the way we diagnose and treat cancers by substantially increasing the concentrations of drugs delivered to the targets while minimizing their toxicity.1,2
The use of inorganic or synthetic materials to produce nanoparticles (NPs) for diagnostics and treatment is often accompanied by high levels of endotoxin content and sterility issues coming from commercial starting materials or residual manufacturing components.3,4 Therefore, these NPs require additional purification or remanufacturing even before initiating preclinical studies. Another problem with some synthetic and inorganic compounds is their bioincompatibility and accumulation in the human body which may cause some health complications later in a patient’s life.2,4 The use of biological materials (such as RNA or DNA) for drug formulation may become the next big step in NP therapy development. Also, over the past few years, the total number of RNA interference (RNAi)-based preclinical and clinical trials has increased significantly.5 RNAi is a naturally occurring cellular post-transcriptional gene regulation process employing small double-stranded RNAs to direct and trigger homology-dependent gene silencing.6 The RNAi machinery is increasingly being harnessed for therapeutic gene modulation and treatment of various diseases through the exogenous introduction of short synthetic RNA duplexes called small-interfering RNAs (siRNAs).7 Currently, more than 20 different therapeutic siRNAs are in clinical trials.8 Besides specific siRNAs (or micro-RNAs), several other promising therapeutically potent RNA classes such as antisenses, aptamers, and ribozymes are worthy of consideration. Simultaneous use of multiple different RNA therapeutics is expected to have significant synergistic effects. One of the well-known examples is combinatorial RNAi used for highly effective simultaneous multiple gene suppression preventing the possibility of mutation-assisted escape from RNAi (e.g., in the case of HIV).9
Using natural or artificially selected RNA motifs and modules, RNA molecules can be programmed to form a wide variety of compact and stable artificial three-dimensional nanostructures (called RNA NPs)10−101 suitable for the broad range of clinical and nanotechnological applications.10,15−26 Therapeutic nucleic acids, proteins, or small molecules can be individually attached using different techniques20 to programmable RNA monomers entering the composition of RNA NP. The assembly of the monomers will bring the desired functionalities together, thus providing precise control over their topology, composition, and modularity. The use of functional RNA NP in vivo will guarantee a higher concentration and desired stoichiometry of therapeutic moieties locally.
Herein, we report the multifunctional RNA NPs built based on the previously designed RNA nanorings13,27 to illustrate how this system can be used to address several present challenges associated with RNA NPs including functionalization with different classes of molecules such as multiple siRNAs (Figure 1a), aptamers (Figure 1b), proteins (Figure 1c), and small molecules (Figure 1e). Detailed characterization of the resulting functional RNA NPs in vitro (by native-PAGE, DLS, cryo-EM, and fluorescent studies), in various cell cultures and in vivo, is demonstrated.
Figure 1.

Schematic representation of assemblies leading to the formation of RNA nanorings functionalized with (a) Dicer substrate RNAs, (b) malachite green (MG) aptamers for in vitro visualization, (c) J18 aptamers for cell targeting and phycoerythrin for visualization in vivo, (d) Dicer substrate RNAs introduced via the toehold interactions, and (e) RNA–DNA hybrids with split functionalities (RNAi and FRET). Functional siRNAs can be released by Dicer nuclease. KLs stand for kissing loops.
We also demonstrate how the nanoring design can achieve cell-targeting properties through incorporation of RNA aptamers specific for the human epidermal growth factor receptor, EGFR (Figure 1c). EGFR is highly overexpressed on the surface of a number of cancer cell types, which has made it an ideal candidate for targeting through aptamer-mediated delivery of cancer therapeutics.28 DNA nanostructures29 were previously targeted to cancer cell lines and specifically attached through antibodies to EGF receptors to bridge multiple cells and create cellular assemblies.30
The incorporation of RNA functionalities such as Dicer substrate (DS) RNAs31 into the nanoscaffolds presents difficulties in terms of solid state chemical synthesis as RNA components generally cannot exceed ∼60 nucleotides in length. We address this problem by annealing DS RNAs to the nanoscaffolds using single-stranded toehold recognition sites (Figure 1d).
Lastly, we have shown how the therapeutic functionality of the nanoring can be triggered through the incorporation of RNA–DNA hybrids (Figure 1e). This newly developed technique32,33 involves splitting the different functionalities between a RNA–DNA nanoring and cognate RNA–DNA hybrids with further conditional intracellular activation of these functionalities.
Functional Nanoring Assembly and Characterization
The assembly process depicted in Figure 1 requires several incubation steps and certain buffer conditions detailed elsewhere.13,15 In vitro assembled nanorings functionalized with different numbers of elongated DS RNAs31 were characterized structurally by native PAGE and dynamic light scattering (Supporting Figure S1a). The release of functional moieties (siRNAs) through the process of dicing was confirmed by in vitro assays with human recombinant Dicer (Supporting Figure S1b).13
To demonstrate the combinatorial nature of the scaffolds, nanorings were functionalized with up to six RNA aptamers (Figure 1b and Supporting Figure S1c) selected to bind the malachite green (MG) dye and significantly increase its emission, which is otherwise undetectable in aqueous solutions.34−36 This aptamer37 was previously used for the laser-mediated inactivation of RNA transcripts,35 biosensing of native RNAs,34 DNAs,36 and small molecules,38 real-time visualization of cotranscriptional assemblies,11 RNA–DNA hybrid reassociation,32 as well as the formation of RNA nanoparticles.11,39 Current fluorescence studies indicate that the sequential increase in fluorescence of MG is directly proportional to the number of aptamers introduced to nanoring scaffolds. Moreover, the functional scaffolds can be produced cotranscriptionally,40 and the assembly of nanorings carrying six aptamers can be tracked in real time by fluorescence or using native-PAGE experiments (Supporting Figure S1d).
The DS RNA containing nanorings were visualized using single-particle cryo-electron microscopy (cryo-EM) (Figure 2). The three-dimensional structure of the RNA particles was obtained using EMAN2 single-particle reconstruction (Methods). The cryo-EM images show that the RNA particles have the expected size and uniform distribution throughout the imaging field (Figure 2a). The computed projections from these three-dimensional reconstructions match well with the class averages of observed particles with similar views (Figure 2a). The final 16 Å cryo-EM map with imposed 6-fold symmetry showed that the arms in the siRNA ring do not point straight out (Figure 2b, Supporting Figure S2). Looking from the side, siRNA arms point about 25 degrees upward, thus creating a crown shape in the hexagonal molecule. Also, looking from the top, the DS RNA arms are positioned in a pinwheel fashion around the ring. The six DS RNA arms point about 53 degrees clockwise compared to the arms in Figure 1 model. Computational modeling of the DS RNA ring generated a cluster of crown-shaped models, as well as alternatives varying the up or down orientation of the DS arms, and most suggesting the top-view pinwheel positioning. The model yielding the best fit into the cryo-EM density map is illustrated in Figure 2b.
Figure 2.

Structural characterization by cryo-EM of RNA nanorings functionalized with six DS RNAs. (a) A typical cryo-EM image of the DS RNA nanoring particles (left panel). Class averages for each DS RNA nanoring as observed by cryo-EM (central panel), with corresponding projections of the reconstructed three-dimensional structure (right panel). (b) Single particle reconstruction of functionalized RNA nanorings. Different views of the model fit with the electron density volume are shown. The volume map is thresholded at the minimum level at which all the atoms of the model can be fit inside the volume. The resolution is 16 Å.
Nanoring-Mediated Gene Silencing and Cell Targeting in Vitro
To study the potential use of nanorings as scaffolds for simultaneous delivery of multiple siRNAs, nanorings functionalized with six fluorescently tagged DS RNAs were transfected into human breast cancer cells (Figure 3a, Supporting Figure S3). One day later, transfection efficiencies were determined by confocal fluorescence microscopy and analyzed using fluorescence-activated cell sorting (FACS). The results presented in Figure 3a revealed a higher intracellular uptake through endocytosis (endocytic uptake was confirmed by the colocalization experiments shown in Figure 3b) for functionalized nanorings compared to the uptake of fluorescently labeled individual DS RNAs transfected at a six times higher concentration. This can be attributed to the tighter binding of the RNA NPs (due to their size and total charge) to the polycationic carriers (Lipofectamine2000 or L2K) compared to the free siRNA duplexes.41
Figure 3.

Cell uptake, endosomal colocalization, silencing, and RNA aptamer mediated binding efficiencies of functional nanorings. (a) Transfection efficiencies using human breast cancer cells (MDA-MB-231). DS RNAs (60 nM final) covalently labeled with one Alexa 546 per duplex were compared to the functionalized nanorings (10 nM final) labeled with six Alexa 546 dyes. One day after the transfection, the efficiencies were analyzed by confocal fluorescence microscopy and flow cytometry experiments. (b) Studying the localization of nanorings with commonly used markers for endosomal compartments Early Endosome Antigen 1 (EEA1) and Rab7. (c) GFP knockdown assays using human breast cancer cells (MDA-MB-231/GFP) which stably express enhanced GFP (eGFP). Fluorescence microscopy (left panel) and statistical analysis (30000 cells per sample) of flow cytometry experiments (right panel) of eGFP expression 3 days after the transfection of cells with DS RNA duplexes and nanorings functionalized with six DS RNAs against eGFP. The ratio of DS RNA duplexes to DS RNA functionalized nanorings was 6:1. (d) Nanorings labeled with phycoerythrin (PE) and containing different numbers of the EGFR-specific J18 aptamer selected to specifically bind EGFR expressed on A431 cells were tested for relative binding efficiencies using FACS. The J18 RNA aptamer model is a conceptual cartoon, based on the minimum free energy secondary structure (MFE). Image numbers in (b) correspond to differential interference contrast (DIC) images (1), Alexa546 emission (2), EAA1 antibody staining (3), and Rab7 antibody staining (4). Images (1 + 2 + 3) and (1 + 2 + 4) are superpositions of three different images.
The use of nanoparticles functionalized with siRNAs provides a precise control over the formulation and higher local concentration of siRNAs, which in turn may improve the loading of RISC, presented only in specific cytoplasmic locations.42,43 To assess the release of siRNAs from the functionalized nanorings upon dicing inside the cells, experiments with human breast cancer cells stably expressing enhanced green fluorescent protein (GFP) were carried out (Figure 3b and Supporting Figures S4–5). First, cells were transfected with different concentrations of nanorings carrying six DS RNAs against GFP and the individual DS RNAs. Due to the use of one-type of DS RNA (against GFP) attached to the nanorings in six copies, free DS RNAs or siRNA are always compared at a 6-fold higher concentration compared to the corresponding functionalized nanorings. After 3 days, the amounts of GFP production were examined (Supporting Figure S4). The visual analysis revealed significant and comparable silencing efficiencies both for the DS RNA decorated nanorings and DS RNA duplexes at concentrations as low as 0.75 nM and 4.5 nM, respectively. To statistically compare the extents of silencing, cells transfected with small amounts of functionalized nanorings (1 nM final) and siRNA or DS RNA duplexes (6 nM final) were analyzed by FACS (Figure 3c). The results demonstrate significant levels of silencing of GFP at low concentrations of functional RNA nanoparticles (1 nM). As a negative control, the nanorings without DS RNAs and nanorings functionalized with DS RNAs designed against a different gene were used (Supporting Figure S5a). The specific gene silencing was observed only in the case of nanorings designed to target GFP. The functional nanorings had a smaller effect on cell viability compared to DS RNA (Supporting Figure S5b). The effect of gene silencing persisted over a nine day period (Supporting Figure S5c) and was comparable for the functional nanorings and DS RNAs introduced at six times higher concentration. This phenomenon is consistent with previously published results for RNAi activation by branched RNA nanostructures.44
Targeting of nanorings using receptor-specific aptamers was also assessed. Specific targeting of NPs to cells of interest for biosensing and in vivo application has posed challenges. To demonstrate that NPs can be targeted to specific cells, we generated NPs containing up to five copies of the J18 RNA aptamer that is specific for the human epidermal growth factor receptor (EGFR).28,45 For visualization, a biotinylated oligonucleotide was coupled to phycoerythrin (PE) through a streptavidin linkage and used in the assembly of the nanorings (Figure 3d and Supporting Figure S6). This coupling system illustrates how protein moieties can be incorporated into the nanoscaffolds. We observed that nanorings can bind to target human epidermoid carcinoma cells (A431) that express high levels of EGFR. NPs with four and five aptamers revealed the strongest signal compared to the rings bearing fewer copies of the aptamers. For example, the fluorescence signal of cells treated with nanorings bearing one aptamer was more than 3-fold weaker compared to nanorings with four aptamers. This suggests that higher numbers of aptamers per NP provide a higher binding affinity to target cells. Our results indicate that binding of NPs to cells is mediated by the RNA aptamer molecule since cotreatment of cells with RNases led to a complete loss of fluorescence (Supporting Figure S6b). Loss of the signal was due to the enzymatic degradation of the RNA molecules and not their target, since the binding of anti-EGFR monoclonal antibodies to EGFR in the presence of RNases was unchanged (Supporting Figure S6c). Furthermore, addition of recombinant epidermal growth factor (rEGF), a ligand for EGFR, led to a decrease in the fluorescent signal (Supporting Figure S6d), suggesting that rEGF competed with the J18 aptamer in binding to the cellular EGFR. The decrease of the signal was not caused by nonspecific degradation of the aptamer by rEGF, since the presence of an unrelated recombinant protein (rIgG) had no negative effect on NP binding. A similar effect was also seen for cells treated using PE labeled J18 aptamers (data not shown).
Functionalization of Nanorings through Toehold Interactions
In addition to synthesizing the nanoring scaffold monomers concatenated with the DS RNA strands, it is possible to alternatively functionalize the nanoring scaffolds through toehold interactions. This system of attachment allows for the multifunctional use of a single nanoscaffold since different nucleic acid functionalities can be joined as long as they bear the cognate toehold complementary to the one found in the nanoscaffold. To demonstrate this, the six scaffold monomers were engineered to carry 10 nt single-stranded RNA toeholds on the 3′-end, which were designed to anneal to a complementary toehold sequence in the antisense component of the GFP DS RNAs (Supporting Figure S7). With this method of assembly, the same nanoring scaffold can be packaged with several different functionalities based upon toehold recognition. Additionally, the length of the scaffold strands can be reduced with this bipartite assembly process as the siRNA components are no longer concatenated, which increases the efficiency of synthesis. To confirm the formation of the nanoring construct with six GFP DS RNAs annealed at the 3′ ends, native-PAGE was performed using nanorings with and without toeholds as the controls. The release of siRNAs upon dicing of the annealed DS RNAs was confirmed by GFP knockdown assays.
Controlled Activation of Intracellular FRET and RNAi by Nanorings with RNA–DNA Hybrids
Additional control over activation of different functionalities can be achieved by using the recently developed technique based on RNA–DNA hybrids.32,33,46 In this scheme, we have split multiple functionalities—DS RNAs and a Förster resonance energy transfer (FRET) pair between the hybrid RNA–DNA nanoring and hybrid RNA–DNA duplexes, thus deactivating the functionalities (Figure 4a). Dicer is an RNaseIII-like enzyme, which is incapable of processing the RNA–DNA hybrids32,33,47 to make them loadable into the RISC. The strands of DS RNAs concatenated to the 3′-end of the nanoring monomers are annealed to the complementary DNAs, thus preventing Dicer from processing these duplexes and making the nanorings nonfunctional. These DNAs contain single-stranded 3′-end toeholds complementary to the toeholds situated at 5′-ends of the DNAs forming hybrids with the sense strands of the DS RNAs. In addition to splitting the DS RNA, we have separated a FRET pair (Alexa488 and Alexa546) between the nonfunctional RNA–DNA rings and hybrids through the conjugation of dyes to the DNA components. The ssDNA complementary toeholds, when in close proximity, can recognize each other and trigger reassociation. This results in the simultaneous formation of DS RNAs functionalized nanorings together with a FRET induction.
Figure 4.

Activation of different functionalities by RNA–DNA hybrids. (a) Scheme showing an activation of multiple functionalities (RNAi, FRET) upon reassociation of nonfunctional nanorings decorated with RNA–DNA hybrids and six nonfunctional cognate RNA–DNA hybrids. (b) FRET time traces during reassociation of hybrid nanorings labeled with Alexa546 and cognate hybrids labeled with Alexa488. (c) Intracellular FRET experiments: cells were cotransfected with hybrid nanorings and cognate hybrids labeled with Alexa546 and Alexa488, respectively. Images were taken the next day. (d) GFP knockdown assays. Three days after transfection of MDA-MB-231/GFP cells with hybrid nanorings and cognate hybrids programmed to release DS RNAs, eGFP expression was statistically analyzed with flow cytometry experiments. As the control, DS RNA duplexes against eGFP were used. Please note that individually neither hybid nanorings nor hybrids cause decrease in eGFP production. Image numbers in (c) correspond to differential interference contrast (DIC) images (1), Alexa488 emission (2), Alexa546 emission (3), bleed-through corrected FRET image (4), and 3D chart representation of zoomed fragment indicated by a yellow box of bleed-through corrected FRET image with the yellow dot indicating the correspondence (5).
To follow the reassociation in real time, FRET time-traces were performed. When the nonfunctional RNA–DNA ring is mixed with six RNA–DNA hybrids, the dsDNA formation brings Alexa488 within the Förster distance (R0 = 6.31 nm) of Alexa546. As a result, the emission of Alexa546 increases, while the signal of Alexa488 drops (Figure 4b). The results of FRET time-traces reveal a quick burst phase of partial reassociation followed by a more complete pairing of fluorescent tags. To visualize intracellular reassociation, nonfunctional RNA–DNA rings and hybrids labeled with Alexa546 and Alexa488 (Figure 4c) were cotransfected into MDA-MB-231 cells and examined by confocal microscopy the next day. The FRET signal after bleed-through correction was calculated as detailed previously32 and is presented in Figure 4c (1 + 4 and 5).
To gauge whether the cognate hybrid rings and duplexes can intracellularly recombine to form functional DS RNA nanorings, human breast cancer cells stably expressing GFP were cotransfected with the nonfunctional components (Figure 4d). Cells were also separately treated with the hybrid rings or just hybrids to determine whether the individual components could induce knockdown of GFP expression. Three days after transfection, the level of GFP expression was measured with flow cytometry. The results demonstrate no silencing of GFP production caused by the individual components. However, when cells were cotransfected with separately prepared complexes of L2K/hybrid rings and L2K/hybrids, the level of silencing measured after 3 days was comparable to the silencing resulting from the transfections with control, preformed GFP siRNAs.
Implementation of Functional Nanorings in Vivo
Additionally, we performed in vivo gene silencing experiments in athymic nude mice bearing xenograft tumors expressing GFP (Figure 5). Functionalized nanorings and control DS RNAs were administrated by intratumor injections into different mice. Five days later, the silencing efficiencies were analyzed ex vivo by measuring the fluorescent intensities of native GFP in treated tumors compared to the tumor of a control animal. Both injections resulted in a significant decrease in GFP fluorescence intensities of ∼90% for functionalized nanorings and ∼80% for control DS RNAs. These results are in a good agreement with multiple cell culture experiments, confirming an efficient delivery and further silencing of target genes by functionalized nanorings.
Figure 5.
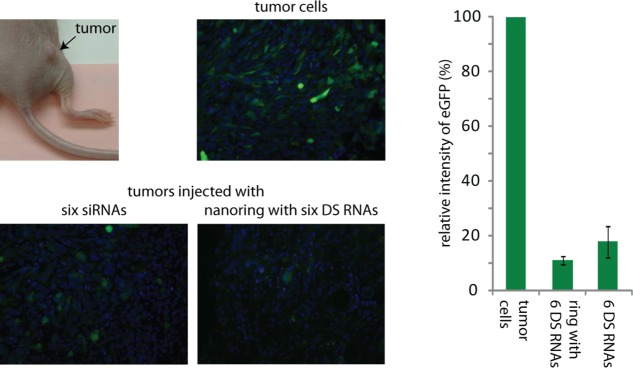
In vivo studies of nanorings functionalized with six DS RNAs in a tumor xenograft mouse model. Fluorescent imaging of tumors and corresponding quantification after 5 days postinjections in vivo demonstrate significant levels of eGFP silencing caused by nanorings functionalized with six DS RNAs compared to free siRNAs. Free DS RNAs were used at six times higher concentrations. Error bars denote ±SEM; N = 2.
Functional Nanorings against HIV-1
To show the feasibility of the nanorings, we developed a set of two nanorings constructs (designated as nanorings A and B) functionalized with different composition of DS RNAs specified in Methods. Each nanoring targets six different regions of HIV-1: PBS-matrix, capsid, protease, reverse transcriptase, envelope, Nef, and Rev-Tat.102 The experiments demonstrate the decrease in virus protein expression inside the transfected cells by 74–83%, for both nanorings A and B at 1 nM concentrations (Figure 6a). The levels of HIV-1 structural proteins (Gag) were quantified (55 kDa Gag precursor + matrix/capsid p41 + capsid, capsid/SP1 p24/p25) to evaluate the efficiency of proteins knockdown. Both nanorings were able to inhibit HIV-1 production in the supernatant. Virus inhibition reached levels of ∼100% at 1 nM concentrations of nanorings. Values were comparable to background levels detected by the assay (Figure 6b). These results were equivalent to the levels of inhibition achieved by the controls, a mixture of six corresponding DS RNAs. Under lower concentrations of the nanorings (0.1 nM) virus production was inhibited in 71–75%. Cytotoxicity was minimal for nanoring B at 1 nM concentration, highlighting the specificity of the knockdown (Supporting Figure S8).
Figure 6.
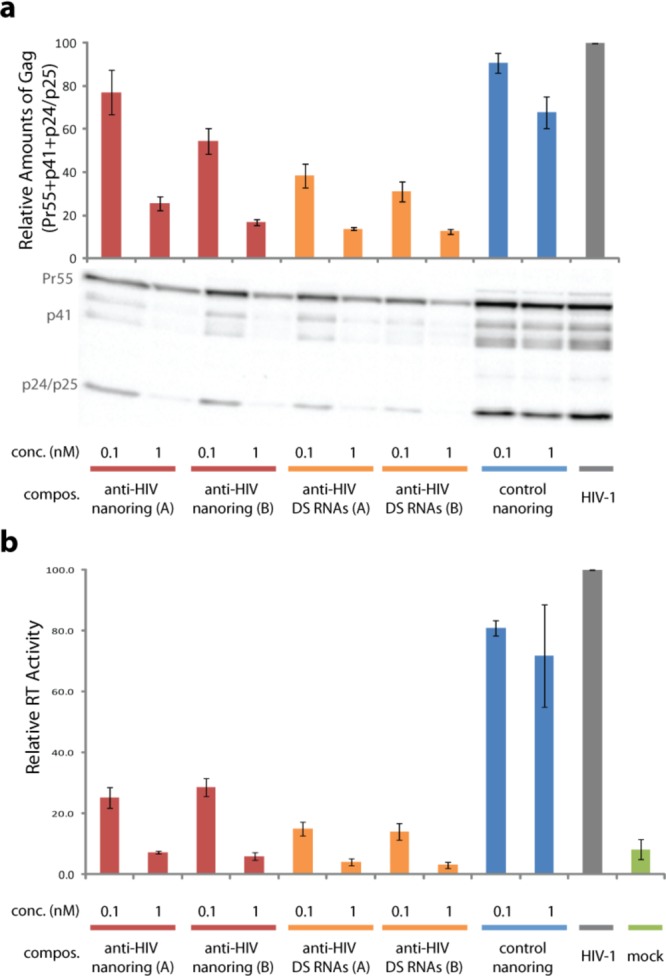
HIV-1 expression and production is inhibited by functional nanorings. (a) HIV-1 expression inside the cell was measured at 48 h post-transfection. HeLa cells were lysed and probed by Western blotting for HIV-1 proteins. Positions of Pr55Gag (Pr55), matrix-capsid (p41), and capsid/capsid-SP1 (p24/p25) are indicated. Quantification of total cell-associated Gag: Pr55 + p41 + p25 + p24. Total Gag in virus control (HIV-1) without nanorings or Dicer substrate (DS) RNAs set at 100. Error bars denote ±SEM; N = 4. (b) HeLa cells were transfected with pNL4-3 (full-length HIV-1 molecular clone), with and without nanorings or DS RNAs. Virus supernatant was harvested 48 h post-transfection, and the reverse transcriptase (RT) production was measured (this assay quantifies the amounts of virus produced by the cells); data are shown normalized to virus controls (HIV-1) without functional nanorings or DS RNAs. Mock represents untrasfected HeLa cells. Corresponding mixtures of six different anti-HIV DS RNAs (A and B) were used as positive controls. Nanoring control without any anti-HIV DS RNAs was used as a negative control. Error bars denote ±SEM; N = 4.
Methods Summary
RNA Nanoring Sequence Design Assemblies and Native PAGE
The detailed design and production of RNA strands entering the composition of nanorings functionalized with six siRNAs is comprehensively described in our previous work.15 The full list of RNA sequences used is available (Supporting Information). RNA molecules were purchased (in the case of short RNAs, from Integrated DNA Technologies, Inc.) or prepared by transcription of PCR amplified DNA templates; synthetic DNA molecules coding for the sequence of the designed RNA were purchased already amplified by PCR using primers containing the T7 RNA polymerase promoter. PCR products were purified using the QiaQuick PCR purification kit, and RNA molecules were prepared enzymatically by in vitro transcription using T7 RNA polymerase. Transcription was performed in 50 mM Tris-HCl, pH 7.5, 2 mM spermidine, 1 mM DTT, 0.4 unils/Al RNasine (Promega), 5 mM MgCl2, 0.5 mM MnCl2, 1 mM NTPs, 0.1 μM of DNA template (or mixture of DNA templates for cotranscriptional assemblies), and T7 RNA polymerase (Promega). For the visualization of assembled RNA NPs quality control experiments, [32P]Cp labeled RNA molecules were used (T4 RNA ligase is used to label the 3′-ends of RNA molecules by attaching [32P]Cp).48 In the case of the initial radiolabeled native-PAGE assays, radiolabeled RNA scaffold strands were mixed with concatenated strands individually followed by the assembly protocol.15 For dicing functional control experiments, RNA molecules were cotranscriptionally α[P32]-ATP body-labeled. Native PAGE experiments were performed as described.49 Typically, assembly experiments reported were analyzed at 10 °C on 7% (29:1) native polyacrylamide gels in the presence of 89 mM Tris-borate, pH 8.3, 2 mM Mg(OAc)2. A Hitachi FMBIO II Multi-View Imager was used to visualize SYBR gold staining.
Dynamic Light Scattering (DLS) Experiments
For DLS, 10 μL of sample solution containing preassembled nanorings with six DS RNAs were measured by DynaPro99 (Protein Solution/Wyatt) with a laser wavelength of 824 nm at 24 °C.11 The theoretical hydrodynamic radii (Rh) were calculated by measuring three-dimensional CPK models.
Recombinant Human Dicer Assay
Nanorings (having one of the strands radiolabeled) with six DS RNAs were prepared as described above to a final concentration of 3 μM. For dicing experiments, samples were incubated for 4 h at 37 °C with recombinant human turbo Dicer enzyme kit (Genlantis), containing an ultra-active form of human recombinant Dicer enzyme, according to the manufacturer’s suggested protocol.13 Dicing reactions were quenched by adding Dicer stop solution (provided by the manufacturer) prior to analysis on 2 mM Mg(OAc)2 native 7% PAGE (described above).
Malachite Green (MG) Aptamers Fluorescent Experiments
All fluorescent studies of MG aptamer functionalized nanorings (at 1 μM final) were carried out in assembly buffer during the incubation at 37 °C. For all samples, the excitation was set at 425 nm. For cotranscriptional assemblies of RNA nanorings functionalized with up to six MG aptamers, aliquots of transcription mixture were taken, MG was added (10 μM final) to each aliquot, and the emission was measured promptly. Some bleaching of MG by transcription mixture was observed over time.
Cryo-Electron Microscopy (cryo-EM) Experiments
Quantifoil Copper 200 mesh R 3.5/1 grids were washed overnight with acetone. To prepare a frozen, hydrated grid, 2.5 μL of sample was applied to the grid, blotted, and plunged into liquid ethane using Vitrobot III (FEI, Hillsboro, OR). 200 keV images were collected from frozen hydrated specimens kept at liquid nitrogen temperature (∼100 K) in a Gatan cryo-holder (model 626, Gatan Inc., Pleasanton, CA) on either JEM2200FS or JEM2010F electron microscope (JEOL, Tokyo) both equipped with a field emission gun and a 4k × 4k CCD camera (Gatan Inc., Pleasanton, CA). JEM-2200FS has an in-column energy filter. Samples were imaged at 83555× effective magnification targeted at 2–5 μm underfocus. We used a total specimen exposure for each image of 33 e–/Å2sec.
Cryo-EM Reconstruction
11067 RNA particles were boxed using EMAN2 boxer. 3D reconstruction was carried out with the EMAN2 software.50 6-fold symmetry was imposed for structure determination. The resolution of the map was assessed to be 16 Å using the gold-standard criterion of Fourier shell correlation (FSC) cutoff at 0.143 from two independent half-sets of data.51 The map was deposited to EMDB (EMD-2783).
Hexameric Nanoring Models
Models of hexmeric nanorings with six DS siRNA arms were created by merging the model of the hexameric ring scaffold, built with the aid of our program NanoTiler (http://www.lecb.ncifcrf.gov/bshapiro/software.html)52 with several alternative models of one monomer with the siRNA arm. Monomer models were built with the aid of programs RNA2D3D (http://www.lecb.ncifcrf.gov/bshapiro/software.html),53 MCSym (http://www.major.iric.ca/MC-Pipeline/)54 and RNAComposer (http://euterpe.man.poznan.pl/Home).55 All three programs take sequence and secondary structure descriptors as input and output 3D structures (PDB format files). From among multiple models generated by the programs, several representatives were selected based on the combination of the best (lowest) free energy and the best structural fit of the 3D structures to the hexameric ring, performed with the aid of the PyMOL Molecular Graphics System (using custom scripts) (Schrödinger, LLC, http://www.pymol.org/). Models were also selected to represent potential alternative orientations of the siRNA arms relative to the plane of the nanoring. All models were subjected to GBSA-based energy minimization (implicit solvent method) in Amber12 with the RNA force field ff10(56−58) and thus structurally refined.
Fitting Hexameric Nanoring Models to the Cryo-EM Density Map
Finally, given the cryo-EM reconstruction, the UCSF Chimera package (http://www.cgl.ucsf.edu/chimera)59 was used to best fit models in the density volume. The fit shown in Figure 2b has the volume map thresholded at the minimum level at which all the atoms of the model can be fit inside the volume (or maximum density level accommodating all of the atoms of the model).
Transfection Experiments
For assaying the delivery of functionalized nanorings, the human breast cancer cell line MDA-MB-231 (with and without GFP) was grown in D-MEM medium (Gibco BRL) supplemented with 10% FBS and penicillin–streptomycin (pen-strep) in a 5% CO2 incubator. All in vitro transfections in this project were performed using Lipofectamine 2000 (L2K) purchased from Invitrogen. 10× or 50× transfection solutions were preincubated at 30 °C with L2K. For all transfections (unless indicated otherwise), the concentration of DS RNAs was six times higher compared to nanorings functionalized with six DS RNAs. Prior to each transfection, the cell media was replaced with OPTI-MEM and previously prepared 10× or 50× RNA/L2K complexes were added at a final concentration of 1×. The cells were incubated for 4 h followed by the media change (D-MEM, 10% FCS, 1% pen-strep).
For targeting experiments, A431 cells were washed three times in DPBS/5 mM MgCl2, and 2 × 105 cells were incubated in the presence of ∼170 nM (final concentration) of nanoring RNA particles in the dark for 30 min at room temperature. Subsequently, cells were washed three times with DPBS 5 mM MgCl2, and 10 000 cells were analyzed using a BD FACSCanto II (BD Bioscience) flow cytometer. Data were analyzed using FlowJo_V10 software. The RiboShredder RNase blend (Epicenter, Madison, WI) was added at a final concentration of ∼0.03 U/μL, and cells were kept on ice to prevent endosomal uptake of bound NPs. The final concentrations of the rEGF (GenScript, Piscataway, NJ) and rIgG proteins (ACRObiosystems, Bethesda, MD) were 500 nM and 150 nM, respectively.
Microscopy
To assess the delivery of functionalized nanorings in cells, measurements were performed using a LSM 710 confocal microscope (Carl Zeiss) with a 63×, 1.4 NA magnification lens. MDA-MB-231 cells were plated in glass bottom Petri dishes (Ibidi, Germany) and subjected to transfection with nanorings as described above. Images of the cells were then taken to assess the appearance of FRET within the sample. For Alexa546 imaging, a DPSS 561 laser was used for excitation, and emission was collected between 566 and 680 nm. All images were taken with a pinhole adjusted to 1 airy unit.
Endosomal Colocalization Studies
To confirm the endosomal location of endocytosed fluorescently labeled functional RNA nanorings in cells, costaining experiments with endosomal markers (EEA1 and Rab7) were performed.32 Cells were transfected with RNA NPs labeled with six Alexa546 dyes. On the next day, transfected cells were fixed with 4% paraformaldehyde for 20 min at room temperature and handled at this temperature thereafter. Samples were washed three times with PBS and then permeabilized with 0.2% Triton X-100 for 20 min. Upon washing three times with PBS, samples were blocked for 1 h with 1% BSA and then exposed to primary antibodies against the early endosome associated protein EEA1 (cell signaling) or against the late endosome marker Rab7 (cell signaling). Upon washing three times with PBS, the samples were stained with a secondary Alexa 488 antibody (Molecular Probes). As the comparison, fluorescently labeled DS RNAs were used at 6-fold higher concentrations.
Reassociation of RNA–DNA Hybrids in Cells Assessed through FRET32
All measurements were performed using a LSM 710 confocal microscope (Carl Zeiss) with a 63×, 1.4 NA magnification lens. All images were taken with a pinhole adjusted to 1 airy unit. Fluorescently labeled hybrid NPs and cognate hybrids were individually preincubated with L2K and cotransfected into cells. On the next day, the samples were fixed by incubation in 4% paraformaldehyde for 20 min at room temperature. Images of the cells were then taken to assess the appearance of FRET within the sample. For Alexa 488 imaging, the 488 nm line of an argon laser was used as excitation, and the emission was collected between 493 and 557 nm. For Alexa 546 imaging, a DPSS 561 laser was used for excitation, and emission was collected between 566 and 680 nm. In order to evaluate the sensitized emission through FRET, images were taken by exciting the sample with the 488 nm line and collecting emission between 566 and 680 nm. Because of spectral overlap, the FRET signal is contaminated by donor emission into the acceptor channel and by the excitation of acceptor molecules by the donor excitation wavelength. This bleed-through was assessed through measurements performed with samples transfected with individual dyes and mathematically removed from the images of FRET.
Flow Cytometry Experiments
For analysis with flow cytometry experiments, MDA-MB-231 cells (with and without GFP) were grown in 12-well plates (10 × 104 cells per well), lifted with cell dissociation buffer, and washed twice with PBS. The level of expression of GFP was determined by fluorescence-activated cell sorting (FACS) analysis on a FACScalibur flow cytometer (BD Bioscience). At least 30 000 events were collected and analyzed using the Cell quest software.
Cell Viability Assay
Cells were seeded in 96 well plates at a density of 10 000 cells/well in serum containing media 24 h prior to experiments. Samples were added to the cells in triplicate in serum free media and incubated for 4 h at 37 °C. After incubation the serum-free media was replaced with serum-containing media. At different time points, according to the manufacturer’s protocol, cell titer blue reagent was added to each well, and the cells were further incubated for 3 h at 37 °C. The fluorescence of the resofurin (converted from resazurin by viable cells) was measured at λex 560 nm and λem 590 nm with an auto cutoff in a fluorescent ELISA plate reader (SpectraMAX, Molecular Devices, Sunnyvale, CA).
In Vivo Silencing Experiments
Animal studies were performed according to the Frederick National Laboratory for Cancer Research (Frederick, MD) Animal Care and Use Committee guidelines. Imaging studies were performed using MDA-MB-231 tumor-bearing athymic nude mice (Charles River Laboratories, Frederick, MD). For tumor induction, a single cancer cell suspension of MDA-MB-231/GFP human breast cancer cell line expressing GFP was prepared in Hank’s balanced salt solution (HBSS). 7–9 week old female athymic nude mice were subcutaneously implanted with 1 × 107 cancer cells in 100 μL of HBSS in the mouse flank. For in vivo delivery, DS RNAs and functional nanorings were associated with bolaamphiphilic (bolas) cationic carriers as described in Kim et al.60 After sufficient growth of soft tumors (∼1 week), two mice were injected intratumorally with DS RNAs (300 nM RNA and 10 μg/mL bola in 100 μL of the PBS injection mixture), and two mice were injected with nanorings functionalized with six DS RNAs (50 nM RNA and 10 μg/mL bola in 100 μL of the PBS injection mixture). One control mouse was injected with 100 μL 1× PBS buffer. After 5 days (120 h), mice were sacrificed. Tumors were removed from mice, fixed overnight at 4 °C in 4% PFA, and then transferred to 20% sucrose overnight at 4 °C. Excess sucrose was blotted from the tumor, and the tumor was embedded in OCT compound (Tissue-Tek). 10 μm cryosections were mounted on slides and stained with DAPI (Invitrogen) then coverslipped with Prolong Gold a/Fade reagent (Invitrogen). Images were captured using Nikon’s Eclipse 80i microscope, with a QImaging Retiga-2000R camera and Nikon’s NIS-Elements AR Imaging Software. The data were quantified and presented based on the total GFP signal normalized to the total number of cells in the given field.
HIV-1 Inhibition by Functional Nanorings
To test inhibition of HIV-1 gene expression mediated by nanorings functionalized with six Dicer substrates (DS) RNAs were selected against multiple regions of the HIV-1 genome.102 After cleavage by Dicer inside cells, these siRNAs are able to knock down HIV-1 gene expression and virus particle production. Nanoring A targets the HIV-1 genome at: primer-binding site (PBS)–matrix (PBS–MA), capsid (CA), protease (PR), reverse transcriptase (RT), surface envelope glycoprotein (gp120), and nef. nanoring construct B targets the HIV-1 genome at: PBS–matrix (PBS–MA), capsid (CA), protease (PR), reverse transcriptase (RT), Nef, and Rev-Tat. Rev stands for regulator of expression virion proteins. Tat stands for trans-activator of transcription, and Nef stands for negative factor. To validate the knockdown of the nanorings constructs A and B, a corresponding mixture of individual DS RNAs was used. As negative control, a nanoring containing six copies of DS RNAs against the cellular protein GSTP1 was used.32,33 Hela cells were cotransfected with the WT HIV-1 molecular clone, pNL4-3, psiCHECK-1 (Renilla Luciferase expression vector, Promega), and the functional nanorings or DS mixtures using Lipofectamine 2000 (Invitrogen). At 48 h post-transfection, the supernatants were harvested, and the reverse transcriptase (RT) activity was measured in an in vitro reaction.61 Levels of RT activity are directly proportional to the amount of released virus. Viral protein expression was analyzed by Western blotting. Cells were lysed using 1× renilla lysis buffer (Promega) according to manufacturer’s protocol. Protein samples were separated by SDS-PAGE and transferred to a polyvinylidene fluoride (PVDF) membrane (Immobilon, Millipore) by semidry electroblotting. Membranes were probed with primary antibody (pooled immunoglobulin from HIV-1-infected patients, HIV-Ig; NIH AIDS Research and Reference Reagent Program) overnight at 4 °C, washed, then incubated for 1 h with human specific horseradish peroxidase-conjugated secondary antibody. Membranes were then incubated with SuperSignal West Pico Chemiluminescent Substrate (Thermo Scientific). After incubation at room temperature, membranes were exposed to a charge-coupled device in a Universal Hood II (Biorad). Quantification was performed using ImageLab software (Biorad). Total HIV-1 Gag protein was measured (55 kDa Gag precursor + matrix/capsid p41 + capsid, capsid/SP1 p24/p25), and values were normalized to virus control (no siRNA cotransfected with pNL4-3). No signal was detected in untransfected cell lysates (data not shown). N = 4.
Acknowledgments
We thank Faye M. Walker for her assistance at early stages of this project. We thank the Pathology Histology Lab of Leidos Biomedical Research, Inc. at the Frederick National Laboratory for Cancer Research for help with ex vivo imaging. We thank Patricia S. Steeg at the National Cancer Institute for providing GFP expressing cells for the in vivo experiments. We thank Andrew Anderson for technical assistance during electron microscopy reconstruction. This research was supported [in part] by the Intramural Research Program of the NIH, Center for Cancer Research, NCI-Frederick. This work has been funded in whole or in part with Federal funds from the Frederick National Laboratory for Cancer Research, National Institutes of Health, under contract no. HHSN261200800001E. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the U.S. Government. This research was supported in part by the National Institutes of Health P41GM103832 and P50 GM1003297 (to W.C.) and the Postdoctoral Training Fellowship from the Keck Center Computational Cancer Biology Training Program of the Gulf Coast Consortia to A.Y.K. (CPRIT grant no. RP101489). This research was also supported by NIH grant no. R01GM-079604 (to L.J.). M.P. is supported in part by an appointment to the Research Participation Program at the Center for Biologics Evaluation and Research administered by the Oak Ridge Institute for Science and Education through an inter agency agreement between the U.S. Department of Energy and the U.S. Food and Drug Administration.
Supporting Information Available
Additional information as noted in text. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Farokhzad O. C.; Langer R. ACS Nano 2009, 3116–20. [DOI] [PubMed] [Google Scholar]
- Petros R. A.; DeSimone J. M. Nat. Rev. Drug Discovery 2010, 98615–27. [DOI] [PubMed] [Google Scholar]
- Crist R. M.; Grossman J. H.; Patri A. K.; Stern S. T.; Dobrovolskaia M. A.; Adiseshaiah P. P.; Clogston J. D.; McNeil S. E. Integr. Biol. (Cambridge) 2013, 5166–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moghimi S. M.; Hunter A. C.; Andresen T. L. Annu. Rev. Pharmacol. Toxicol. 2012, 52, 481–503. [DOI] [PubMed] [Google Scholar]
- Chen J.; Xie J. Int. J. Nanomed. 2012, 7, 3971–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fire A.; Xu S.; Montgomery M. K.; Kostas S. A.; Driver S. E.; Mello C. C. Nature 1998, 3916669806–11. [DOI] [PubMed] [Google Scholar]
- Bramsen J. B.; Kjems J. Front Genet. 2012, 3, 154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou J.; Shum K. T.; Burnett J. C.; Rossi J. J. Pharmaceuticals (Basel) 2013, 6185–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grimm D.; Kay M. A. Mol. Ther. 2007, 155878–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Afonin K. A.; Kasprzak W. K.; Bindewald E.; Kireeva M.; Viard M.; Kashlev M.; Shapiro B. A. Acc. Chem. Res. 2014, 47, 1731–1741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Afonin K. A.; Bindewald E.; Yaghoubian A. J.; Voss N.; Jacovetty E.; Shapiro B. A.; Jaeger L. Nat. Nanotechnol. 2010, 59676–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Severcan I.; Geary C.; Chworos A.; Voss N.; Jacovetty E.; Jaeger L. Nat. Chem. 2010, 29772–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grabow W. W.; Zakrevsky P.; Afonin K. A.; Chworos A.; Shapiro B. A.; Jaeger L. Nano Lett. 2011, 112878–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo P.; Zhang C.; Chen C.; Garver K.; Trottier M. Mol. Cell 1998, 21149–55. [DOI] [PubMed] [Google Scholar]
- Grabow W. W.; Jaeger L. Acc. Chem. Res. 2014, 47, 1871–80. [DOI] [PubMed] [Google Scholar]
- Chworos A.; Severcan I.; Koyfman A. Y.; Weinkam P.; Oroudjev E.; Hansma H. G.; Jaeger L. Science 2004, 306, 2068–72. [DOI] [PubMed] [Google Scholar]
- Afonin K. A.; Grabow W. W.; Walker F. M.; Bindewald E.; Dobrovolskaia M. A.; Shapiro B. A.; Jaeger L. Nat. Protocols 2011, 6122022–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo P. Nat. Nanotechnol. 2010, 512833–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shukla G. C.; Haque F.; Tor Y.; Wilhelmsson L. M.; Toulme J. J.; Isambert H.; Guo P.; Rossi J. J.; Tenenbaum S. A.; Shapiro B. A. ACS Nano 2011, 553405–3418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shu Y.; Haque F.; Shu D.; Li W.; Zhu Z.; Kotb M.; Lyubchenko Y.; Guo P. RNA 2013, 196767–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koyfman A. Y.; Braun G.; Magonov S.; Chworos A.; Reich N. O.; Jaeger L. J. Am. Chem. Soc. 2005, 1273411886–7. [DOI] [PubMed] [Google Scholar]
- Shu Y.; Pi F.; Sharma A.; Rajabi M.; Haque F.; Shu D.; Leggas M.; Evers B. M.; Guo P. Adv. Drug Delivery Rev. 2014, 66C, 74–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khisamutdinov E. F.; Jasinski D. L.; Guo P. ACS Nano 2014, 8, 4771–4781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hao C.; Li X.; Tian C.; Jiang W.; Wang G.; Mao C. Nat. Commun. 2014, 5, 3890. [DOI] [PubMed] [Google Scholar]
- Ohno H.; Kobayashi T.; Kabata R.; Endo K.; Iwasa T.; Yoshimura S. H.; Takeyasu K.; Inoue T.; Saito H. Nat. Nanotechnol. 2011, 62116–20. [DOI] [PubMed] [Google Scholar]
- Osada E.; Suzuki Y.; Hidaka K.; Ohno H.; Sugiyama H.; Endo M.; Saito H. ACS Nano 2014, 8, 8130–8140. [DOI] [PubMed] [Google Scholar]
- Haque F.; Shu D.; Shu Y.; Shlyakhtenko L. S.; Rychahou P. G.; Evers B. M.; Guo P. Nano Today 2012, 74245–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarapore P.; Shu Y.; Guo P.; Ho S. M. Mol. Ther. 2011, 192386–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yingling Y. G.; Shapiro B. A. Nano Lett. 2007, 782328–34. [DOI] [PubMed] [Google Scholar]
- Li N.; Ebright J. N.; Stovall G. M.; Chen X.; Nguyen H. H.; Singh A.; Syrett A.; Ellington A. D. J. Proteome Res. 2009, 852438–48. [DOI] [PubMed] [Google Scholar]
- Koyfman A. Y.; Magonov S. N.; Reich N. O. Langmuir 2009, 2521091–6. [DOI] [PubMed] [Google Scholar]
- Koyfman A. Y.; Braun G. B.; Reich N. O. J. Am. Chem. Soc. 2009, 1314014237–9. [DOI] [PubMed] [Google Scholar]
- Rose S. D.; Kim D. H.; Amarzguioui M.; Heidel J. D.; Collingwood M. A.; Davis M. E.; Rossi J. J.; Behlke M. A. Nucleic Acids Res. 2005, 33134140–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Afonin K. A.; Viard M.; Martins A. N.; Lockett S. J.; Maciag A. E.; Freed E. O.; Heldman E.; Jaeger L.; Blumenthal R.; Shapiro B. A. Nat. Nanotechnol. 2013, 84296–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Afonin K. A.; Desai R.; Viard M.; Kireeva M. L.; Bindewald E.; Case C. L.; Maciag A. E.; Kasprzak W. K.; Kim T.; Sappe A.; Stepler M.; KewalRamani V. N.; Kashlev M.; Blumenthal R.; Shapiro B. A. Nucl. Acids Res. 2014, 4232085–2097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Afonin K. A.; Danilov E. O.; Novikova I. V.; Leontis N. B. ChemBioChem 2008, 9121902–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grate D.; Wilson C. Proc. Natl. Acad. Sci. U.S.A. 1999, 96116131–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolpashchikov D. M. J. Am. Chem. Soc. 2005, 1273612442–3. [DOI] [PubMed] [Google Scholar]
- Baugh C.; Grate D.; Wilson C. J. Mol. Biol. 2000, 3011117–28. [DOI] [PubMed] [Google Scholar]
- Stojanovic M. N.; Kolpashchikov D. M. J. Am. Chem. Soc. 2004, 126309266–70. [DOI] [PubMed] [Google Scholar]
- Shu D.; Shu Y.; Haque F.; Abdelmawla S.; Guo P. Nat. Nanotechnol. 2011, 610658–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Afonin K. A.; Kireeva M.; Grabow W. W.; Kashlev M.; Jaeger L.; Shapiro B. A. Nano Lett. 2012, 12105192–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang C. I.; Lee T. Y.; Kim S.; Sun X.; Hong S. W.; Yoo J. W.; Dua P.; Kang H. S.; Kim S.; Li C. J.; Lee D. K. J. Gene Med. 2012, 142138–46. [DOI] [PubMed] [Google Scholar]
- Lee Y. S.; Pressman S.; Andress A. P.; Kim K.; White J. L.; Cassidy J. J.; Li X.; Lubell K.; Lim do H.; Cho I. S.; Nakahara K.; Preall J. B.; Bellare P.; Sontheimer E. J.; Carthew R. W. Nat. Cell Biol. 2009, 1191150–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sen G. L.; Blau H. M. Nat. Cell Biol. 2005, 76633–6. [DOI] [PubMed] [Google Scholar]
- Nakashima Y.; Abe H.; Abe N.; Aikawa K.; Ito Y. Chem. Commun. (Cambridge) 2011, 47298367–9. [DOI] [PubMed] [Google Scholar]
- Li N.; Larson T.; Nguyen H. H.; Sokolov K. V.; Ellington A. D. Chem. Commun. (Cambridge) 2010, 463392–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Afonin K. A.; Lindsay B.; Shapiro B. A. RNA Nanotechnology 2013, 1, 1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H.; Kolb F. A.; Brondani V.; Billy E.; Filipowicz W. EMBO J. 2002, 21215875–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Low J. T.; Knoepfel S. A.; Watts J. M.; ter Brake O.; Berkhout B.; Weeks K. M. S. Mol. Ther. 2012, 20, 820–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Afonin K. A.; Cieply D. J.; Leontis N. B. J. Am. Chem. Soc. 2008, 130193–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Afonin K. A.; Leontis N. B. J. Am. Chem. Soc. 2006, 1285016131–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang G.; Peng L.; Baldwin P. R.; Mann D. S.; Jiang W.; Rees I.; Ludtke S. J. J. Struct Biol. 2007, 157138–46. [DOI] [PubMed] [Google Scholar]
- Scheres S. H.; Chen S. Nat. Methods 2012, 99853–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bindewald E.; Grunewald C.; Boyle B.; O’Connor M.; Shapiro B. A. J. Mol. Graph. Model. 2008, 273299–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez H. M.; Maizel J. V. Jr.; Shapiro B. A. J. Biomol Struct Dyn 2008, 256669–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parisien M.; Major F. Nature 2008, 452718351–5. [DOI] [PubMed] [Google Scholar]
- Popenda M.; Szachniuk M.; Antczak M.; Purzycka K. J.; Lukasiak P.; Bartol N.; Blazewicz J.; Adamiak R. W. Nucleic Acids Res. 2012, 4014e112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Case D. A.; Darden T. A.; Cheatham T. E. 3rd; Simmerling C. L.; Wang J.; Duke R. E.; Luo R.; Walker R. C.; Zhang W.; Merz K. M.; Roberts B.; Hayik S.; Roitberg A.; Seabra G.; Swails J.; Goetz A. W.; Kolossváry I.; Wong K. F.; Paesani F.; Vanicek J.; Wolf R. M.; Liu J.; Wu X.; Brozell S. R.; Steinbrecher T.; Gohlke H.; Cai Q.; Ye X.; Wang J.; Hsieh M. J.; Cui G.; Roe D. R.; Mathews D. H.; Seetin M. G.; Salomon-Ferrer R.; Sagui C.; Babin V.; Luchko T.; Gusarov S.; Kovalenko A.; Kollman P. A.. AMBER12; University of California: San Francisco, 2012. [Google Scholar]
- Essmann U.; Perera L.; Berkowitz M. L.; Darden T. A.; Lee H.; Pedersen L. G. J. Chem. Phys. 1995, 103198577–93. [Google Scholar]
- Wang J.; Cieplak P.; Kollman P. A. J. Comput. Chem. 2000, 21121049–74. [Google Scholar]
- Pettersen E. F.; Goddard T. D.; Huang C. C.; Couch G. S.; Greenblatt D. M.; Meng E. C.; Ferrin T. E. J. Comput. Chem. 2004, 25131605–12. [DOI] [PubMed] [Google Scholar]
- Kim T.; Afonin K. A.; Viard M.; Koyfman A. Y.; Sparks S.; Heldman E.; Grinberg S.; Linder C.; Blumenthal R. P.; Shapiro B. A. Mol. Ther. Nucleic Acids 2013, 2, e80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freed E. O.; Martin M. A. J. Virol. 1994, 6842503–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.