

Abstract
Instability of targeting ligand is a roadblock towards successful development of folate targeted liposomes. Folate ligands have been linked to polyethylene glycol (PEG) and cholesterol by an amide bond to form folate-CONH-PEG-CONH-Cholesterol (F-CONH-PEG-CONH-Chol), which is subject to hydrolysis. To increase the stability of folate ligands and promote the long circulation and targeting effects, we synthesized a chemically stable lipophilic folate derivative, folate-CONH-PEG-NH-Cholesterol (F-CONH-PEG-NH-Chol), where the amide bond was replaced by a C-N bond, to deliver liposomal doxorubicin (Dox). Its physical stability, cellular uptake, cellular toxicity, pharmacokinetics, distribution, anti-tumor efficacy, and cardiac toxicity were investigated. Our results indicate that F-CONH-PEG-NH-Chol conjugated liposomes are taken up selectively by folate receptor-positive HeLa and KB cells. Compared with F-CONH-PEG-CONH-Chol with two carbonate linkages, F-CONH-PEG-NH-Chol better retained its drug entrapment efficiency and folate receptor-targeting activity during prolonged circulation. F-CONH-PEG-NH-Chol thus represents a physically stable and effective ligand for delivering folate receptor-targeted liposomes, with prolonged circulation time and efficient tissue distribution, as well as higher efficacy and less cardiac toxicity. Collectively, these results suggest that this novel conjugate can serve as a promising derivative for the delivery of anti-tumor therapeutic agents.
Keywords: liposome, doxorubicin, folate ligand, FR-targeting, stability
Introduction
Folate receptor (FR) is a glycosyl phosphatidylinositol (GPI)-anchored membrane protein, which is over-expressed in over 90% of ovarian carcinomas and in other epithelial cancers to varying degrees.1–5 Conversely, the expression levels of FR in normal tissues are much lower than in tumor tissues. The distinct expression pattern of FR between normal and malignant tissues makes it an ideal target for drug delivery. The natural ligand of FR, namely folic acid or folate, shows a highly selective affinity to FR. Folate has been extensively explored as the targeting ligand for chemotherapeutic nanoparticle delivery because of its inherent high affinity, small size, and non-toxicity.6–11
Among the various types of delivery systems, liposomes have attracted considerable attention. It is one of the only two families of therapeutic nanocarriers that have been approved in clinical practice.12 Compared with free molecules, liposomes can help prolong systemic circulation time, enhance the tumor localization, and overcome multi-drug resistance.13,14 Liposomes can also reduce the adverse effects of chemotherapeutic drugs, such as the cardiac toxicity of doxorubicin (Dox).15–18 For two decades, studies have been focused on the development of novel liposomal therapeutic agents with high encapsulation efficiency and advanced passive targeting efficiency.
Folate ligand has been incorporated into liposomes to enhance the targeting efficiency for tumors.6,19,20 Lee and Low synthesized folate-conjugated liposomes by incorporating 0.1 mol% of folate-polyethylene glycol-distearoylphatidylethanolamine (folate-PEG-DSPE) construct into the lipid bilayer. They found uptake of folate-PEG-liposomal Dox by KB cells was 45-fold higher than that of non-targeted liposomal Dox, and 1.6 times higher than that of free Dox, while the cytotoxicity was 86 and 2.7 times higher respectively.21 Since then, different lipophilic derivatives, such as folate-PEG-DSPE, folate-PEG-cholesterol (F-PEG-Chol), and folate-PEG-cholesteryl hemisuccinate (F-PEG-CHEMS) have been evaluated to improve the targeting efficiency and the stability of liposomes.22–24
Dox, a potent anticancer drug, is effective against a wide range of human neoplasms. It has been widely used as a chemical agent for treating solid tumors. However, the clinical uses of Dox have been restricted largely due to lack of tissue specificity and its serious cardiotoxic effects, resulting from the generation of free radicals and lipid peroxidation. To reduce the toxicity and improve the targeting efficiency of Dox, we have reported two novel liposome delivery ligands, the lipophilic folate derivatives F-PEG-Chol and F-PEG-CHEMS, which showed excellent colloidal stability and prolonged circulation properties.23
However, both the F-PEG-Chol and the F-PEG-CHEMS derivatives are subject to hydrolysis due to the presence of amide and ester linkages. To reduce the possible hydrolysis and improve its stability, here we report a stable folate derivative folate-CONH-PEG-NH-Cholesterol (F-CONH-PEG-NH-Chol), in which the cholesterol (Chol) is connected with PEG by a C-N bond instead of an amide bond. After the preparation of liposomes targeted with F-CONH-PEG-NH-Chol, the physical stability, FR dependent cellular uptake, cytotoxicity, pharmacokinetic properties, distribution and anti-tumor efficacy, as well as cardiac toxicity were investigated to determine its feasibility to deliver either calcein or Dox (Figure 1).
Figure 1.
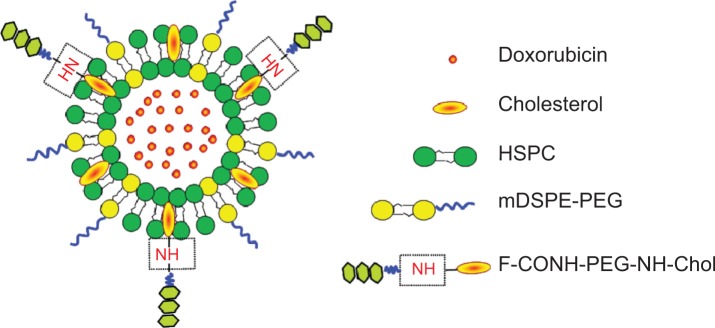
Schematic structure of the F-CONH-PEG-NH-Chol conjugated Dox liposomes.
Abbreviations: Dox, doxorubicin; HSPC, hydrogenated soybean phosphatidylcholine; F-CONH-PEG-NH-Chol, folate-CONH-PEG-NH-Cholesterol; mDSPE-PEG, poly(ethylene glycol)-distearoylphosphatidylethanolamine-maleimide.
Materials and methods
Chemicals for preparation of folate binding Dox liposomes
Doxorubicin hydrochloride (Dox·HCl), folic acid, dicyclohexylcarbodiimide (DCC), N-hydroxysuccinimide (NHS), Chol, L-glutamate, calcein, Sepharose CL-4B chromatography media and folate-free Roswell Park Memorial Institute (RPMI) 1640 cell culture medium were purchased from Sigma-Aldrich Chemical Co., (St Louis, MO, USA). High glucose Dulbecco’s Modified Eagle’s Medium (DMEM) was purchased from the Hyclone of Thermo Fisher Scientific (Waltham, MA, USA). Trypsin was purchased from Beyotime Institute of Biotechnology (Wuhan, People’s Republic of China). Hydrogenated soybean phosphatidylcholine (HSPC), monomethoxy polyethylene glycol 2000-distearoyl phosphatidylethanolamine (mPEG-DSPE) were purchased from Avanti Polar Lipid (Alabaster, AL, USA). PD-10 desalting columns were purchased from GE Healthcare Bio-Sciences Corp., (Piscataway, NJ, USA). All reagents and solvents were of analytical or high-performance liquid chromatography grade and were used without further purification.
Cell culture
HeLa and KB cells are from the American Type Culture Collection (ATCC), Manassas, VA, USA. SMMC-7721 cells are from the China Type Culture Collection at Wuhan University (Wuhan, People’s Republic of China). The cells were cultured with folate free RPMI 1640 or high glucose DMEM supplemented with penicillin, streptomycin, and 10% fetal bovine serum (FBS) in 37°C and 5% CO2 incubators. M-plasmocin (Invivogen, San Diego, CA, USA) at a concentration of 2.5 μg/mL was used to prevent possible mycoplasma infection.
Synthesis of F-CONH-PEG-NH-Chol
The synthesis procedure is shown in Figure 2. First, folate-PEG-bis-amine and Chol-p-toluenesulfonate (Chol-OTs) were synthesized by methods described in the literature.25,26 Briefly, for the synthesis of folate-PEG-bis-amine, folic acid of 26.5 mg (60 μmol) was dissolved in DMSO (1 mL), and then 167.5 mg of PEG-bis-amine (50 μmol), 15.5 mg of DCC (75 μmol), 8.63 mg of NHS (75 μmol) and triethylamine (34.8 μL, 7.18 M) were added into the solution and then reacted at 30°C overnight. The product was centrifuged at 7,000 rpm (Eppendorf 5415D; Eppendorf, Hamburg, Germany) for 7 minutes (min), and then the supernatant was transferred into a new centrifuge tube. Three mL of Na2CO3 (50 mM) was added and the mixture was centrifuged for 15 min at 4,000 rpm. The upper layer was purified by Sephadex G-25 gel-filtration chromatography and downy yellow solid was obtained by lyophilization. For the synthesis of Chol-OTs, pyridine (30 mL), 5 g of Chol (12.93 mmol) and 2.71 g of 4-toluene sulfonyl chloride (TsCl) (14.22 mmol) were mixed and stirred at 25°C for 12 hours (h). The reactant was mixed with 25 mL of purified water and extracted with ethyl ether 3 times. The organic phases were merged and dried with Na2SO4 for 1 h. The residue was then recrystallized with petroleum ether. Finally, for the synthesis of F-CONH-PEG-NH-Chol, folate-PEG-bis-amine (128 mg, 33.9 μmol), Chol-OTs (22.3 mg, 42.3 μmol), and triethylamine (11.8 μL, 7.18 M) were dissolved in CHCl3 (20 mL), and the reaction was allowed to proceed under stirring at 25°C for 48 h. The solvent (CHCl3) was then removed by rotary evaporation and the residue was treated with 8 mL Na2CO3 (50 mM), the mixture was sonicated for 10 min and further centrifuged at 4,000 rpm for 20 min. Supernatant was then dialyzed with dialysis membrane (molecular weight cut off 14 kD). The product F-CONH-PEG-NH-Chol was then dried by lyophilization, which yielded a yellow powder product (115 mg) with a yield of 81.6%. The molecular weight of the conjugate was confirmed by matrix-assisted laser desorption/ionization/time-of-flight mass spectroscopy (ultra flextreme MALDI-TOF-MS; Bruker Daltonics Inc., [Bruker Corporation, Billerica, MA, USA]).
Figure 2.

The synthesis of F-CONH-PEG-NH-Chol (folate-CONH-PEG-NH-Cholesterol).
Notes: Folate was linked to PEG-bis-amine by an amide bond first. Then cholesterol–OTs was reacted with folate-CONH-PEG-NH2 to form folate-CONH-PEG-NH-Chol by C-N bond connection.
Abbreviation: cholesterol-OTs, cholesterol-p-toluenesulfonate.
Liposome preparation
The liposomes were prepared by thin film hydration and polycarbonate membrane extrusion method according to the literature.8,27 The lipid compositions of the FR-targeted liposomes and non-targeted liposomes were HSPC/Chol/mPEG-DSPE/F-CONH-PEG-NH-Chol at a molar ratio of 55:40:4.5:0.5 and HSPC/Chol/mPEG-DSPE at a molar ratio of 55:40:5 respectively. Dox was remote loaded into the liposomes by a transmembrane pH gradient. Briefly, the total lipid (85 mg) was dissolved in CHCl3 and dried to a thin film by rotary evaporation followed by further drying under vacuum at 40°C. The lipid film was then hydrated with 2 mL (NH4)2SO4 (250 mM) for 30 min at 60°C. The resultant multilamellar vesicles were extruded three times through 0.2 μm pore-size polycarbonate membranes and three times through 0.1 μm pore-size polycarbonate membranes using a Lipex Extruder (Northern Lipids Inc., Burnaby, Canada) driven by pressurized nitrogen at 60°C to produce homogeneous unilamellar vesicles. The external (NH4)2SO4 was removed with phosphate-buffered saline (PBS, pH 7.4) by size-exclusion chromatography on a PD-10 column. Dox·HCl dissolved in deionized H2O (10 mg/mL) was added to the liposomes at a Dox-to-lipid ratio of 1:20 (weight/weight), followed by a 30 min incubation at 65°C. Free Dox was separated from liposomes by size exclusion chromatography on a Sepharose CL-4B column. Dox concentration in the liposomes and drug encapsulation efficiency of the collected fraction were determined by measuring absorption at 480 nm on an UV-Vis spectrophotometer (SP-756PC, Shanghai Spectrum Instruments, Shanghai, People’s Republic of China) following liposome lysis in ethanol. The calibration curve of Dox concentration for blank liposome lysate was obtained as y =0.0217x +0.0063 (r2=0.9995), in which “y” is the absorbance and “r” is the correlation coefficient. The particle size of the liposomes and zeta potential were measured by Zeta PALS (Zeta Potential Analyzer; Brookhaven Instruments Corporation, Long Island, NY, USA).
FR-targeted calcein liposomes (F-Calcein-L) and non-targeted calcein liposomes (Calcein-L) were prepared according to the procedures described above except that the lipid film was hydrated in calcein (50 mM) instead of the (NH4)2SO4 solution.
Cellular uptake of liposomes
FR positive HeLa, KB and FR negative SMMC-7721 cells grown in a monolayer were suspended by a brief treatment with trypsin and then washed once with fresh folate free RPMI 1640 medium. Aliquots of cell suspensions were incubated with Calcein-L or F-Calcein-L for 1 h at 37°C as previously reported.23 For free folate blocking group, 1 mM free folate was added to the incubation media previous to the addition of F-Calcein-L. After incubation, the cells were washed three times with cold PBS and were then taken to be examined either by fluorescence microscope or flow cytometry.
Pharmacokinetic and distribution analysis
Male Kunming mice (body weight ∼25 g, 6–8 weeks old) were obtained from Laboratory Animal Center of the Huazhong University of Science and Technology. Liposomes or free drug were injected as a single intravenous bolus via the lateral tail vein at a dose of 5 mg/kg Dox. At 5 min, 15 min, 30 min, 1 h, 2 h, 4 h, 8 h, 12 h, 24 h, and 48 h after the injection, 500 μL blood was collected in heparin-treated tubes and then centrifuged (5,000 rpm, 5 min) to obtain plasma. Plasma aliquots of 100 μL were added to 1 mL ethanol and 100 μL deionized water, followed by addition of 25 μL 20% sodium dodecyl sulfate. These were then vortexed for 30 seconds (s) and centrifuged to extract the Dox from the liposomes. Dox in plasma was measured by its fluorescence intensity at 495 nm by fluorescence spectrophotometer (Hitachi F-2700, software FL solution, Hitachi Ltd., Tokyo, Japan). The concentration of Dox in each sample was calculated by using a calibration curve, with Dox itself as the internal standard. Pharmacokinetic variables including area under the curve (AUC), maximal concentration in plasma (Cmax), half-life (t1/2) and clearance were calculated by using pharmacological software Drug And Statistics (version 2.0, Hefei, People’s Republic of China).
For tissue distribution experiments, liposomes or free Dox were injected intravenously at a dose of 5 mg/kg. Tissues from the heart, liver, spleen, and kidney were collected at 5 min, 15 min, 30 min, 1 h, 2 h, 4 h, 8 h, 12 h, 24 h, and 48 h respectively after injection. Tissues of 0.1 mg were homogenized with the buffer (300 μL ethanol, 15 μL 37% HCl, 185 μL H2O) and then centrifuged (12,000 rpm, 15 min) to collect the supernatant. The concentration of Dox was determined by fluorescence spectrophotometry.
Microscopy
Microscopic examination of cells or tissue section slides was performed with an Olympus SZX12 fluorescence microscope equipped with a digital camera and connected to a PC running MagnaFire 2.0 camera software (Optronics, Goleta, CA, USA). Pictures were taken at equal exposure times for each sample.
MTT assay
Cytotoxicity of F-CONH-PEG-NH-Chol Dox liposomes (F-D-L) and non-targeted PEGylated Dox liposomes (D-L) was determined in HeLa and KB cells respectively by an MTT assay as described in the literature.25,28 Simply put, HeLa, KB and SMMC-7721 cells cultured in folate free RPMI 1640 medium were digested by trypsin and seeded in 96-well culture plates at 5×103 cells per well. After 24 h growth, the medium was removed and 200 μL fresh medium containing serial dilutions of the formulations was added, including free Dox, D-L, F-D-L, and F-D-L plus 1 mM free folic acid. The cells were incubated for 2 h at 37°C, and then cultured in fresh folate-free RPMI 1640 medium for 72 h. Then, 20 μL MTT (5 mg/mL) was added into each well and the plates were incubated for an additional 4 h. After that, the medium was removed and 200 μL DMSO was added into each well to dissolve the blue formazan crystal. Cell viability was assessed on a microplate reader at 490 nm.
Anti-tumor efficacy in vivo
Female nude mice (6–8 weeks of age, 16–18 g) were obtained from Huafukang Technology Corporation (Beijing, People’s Republic of China), kept in filter-topped cages with standard rodent chow, water available ad libitum, and a 12 h light/dark cycle. The experiments were performed according to national regulations and approved by the Animal Experiments Ethical Committee of Huazhong University of Science and Technology. Subcutaneous KB tumors were seeded by inoculation of 2.5×106 KB cells in the front armpit of the mice. At the tumor volume of 100 mm3, mice received liposomes or free Dox by tail vein injection at the dose of 5 mg/kg. Tumor size was determined at every other day and calculated by the formula volume =0.52×a×b2, “a” represents the long diameter and “b” represents the short diameter.
Cardiac toxicity assay
Severe Dox toxicity was induced in mice by four intraperitoneal injections of Dox, D-L and F-D-L at 4 mg/kg body weight each time at an interval of 48 h, for a cumulative dose of 16 mg/kg body weight. Body weight and heart weight were measured at the end of the experiment. The level of glutathione peroxidase (GSH-Px) was measured after the administration of Dox. Death of any of the animals was recorded during the observation period.
Statistical analysis
Significance analysis of all the data was performed using the Student’s t-test (SPSS Software, version 12, SPSS Inc., Chicago, IL, USA). A P-value of <0.05 was considered significant and P<0.01 was considered highly significant when compared with the corresponding control.
Results
Synthesis of F-CONH-PEG-NH-Chol
F-CONH-PEG-NH-Chol was synthesized, as described in Materials and methods section and illustrated in Figure 2. The product F-CONH-PEG-NH-Chol was suspended in Na2CO3 (50 mM), and then the by-products and unreacted materials were removed by dialysis. Formation of the F-CONH-PEG-NH-Chol product was confirmed by thin layer chromatography (CH2Cl2/methanol at 80:20) and it had an Rf (rate of flow) value of 0.6, less than that of Chol-OTs. Then, the structure was validated by the mass spectrum (Figure S1). Matrix-assisted laser desorption/ionization time of flight mass spectrometry confirmed that the molecular mass of the conjugate presented a bell-shaped distribution with 44-Da spaced lines, indicating that the PEG conjugate is mono charged. The distribution curve was centered at 4,140 Da, in agreement with the calculated molecular weight of 4,158 Da. These results confirmed that the F-CONH-PEG-NH-Chol derivatives were correctly synthesized as we have expected and can be used in the preparation of targeted liposomes.
Characterization and physical stability test of liposomes incorporating F-CONH-PEG-NH-Chol
Liposomes incorporating F-CONH-PEG-NH-Chol were prepared as indicated in the Materials and methods section and were used to carry calcein (F-Calcein-L) or Dox (F-D-L) (Figure 1). We then characterized the liposomes extensively. We first determined their diameter and found that the F-D-L had a mean diameter of 137.0±0.9 nm and a narrow distribution (Table 1). The efficiency of remote loading of Dox into the liposomes was >90% at the drug/lipid ratio of 1:20 (weight/weight). The zeta potentials of F-D-L and D-L were −12.41±3.97 mV and −12.99±4.04 mV, respectively. The negative zeta potentials might be caused by the charges of mPEG-DSPE, because F-CONH-PEG-NH-Chol overall is a neutral molecule and does not contribute to the surface charge.29
Table 1.
Particle size, zeta potential, and entrapment efficiency of D-L and F-D-L
| Liposome | Particle size (nm) |
Zeta potential (mV) |
Entrapment (%) |
|---|---|---|---|
| D-L | 129.8±0.9 | −12.41±3.97 | 89.5±2.3 |
| F-D-L | 137.0±0.9* | −12.99±4.04 | 90.1±1.5 |
| F-Calcein-L | 131±0.8 | −12.69±3.04 | 92.1±3.6 |
Notes: The entrapment was calculated as the encapsulated Dox or calcein divided by the total used in the preparation. The encapsulated Dox or calcein was determined by the spectrometer after the demulsification of the liposomes. Data are presented as the mean ± SD (n=3).
P<0.05 compared with D-L.
Abbreviations: Dox, doxorubicin; D-L, PEGylated Dox liposomes; F-D-L, F-CONH-PEG-NH-Chol Dox liposomes; F-CONH-PEG-NH-Chol, folate-CONH-PEG-NH-Cholesterol; F-Calcein-L, folate receptor targeted calcein liposomes; SD, standard deviation.
Liposomes dispersed in aqueous solution are physically and chemically unstable in general under long-term storage.30 Hydrolysis and oxidation of phospholipids and liposome aggregation are the major causes of liposome instability. We believe that replacing the amide bond of folate-CONH-PEG-CONH-Cholesterol (F-CONH-PEG-CONH-Chol) with a C-N bond will increase the stability of this folate derivative. So the physical stability of F-CONH-PEG-NH-Chol-containing Dox liposomes and F-CONH-PEG-CONH-Chol-containing Dox liposomes was compared. After 3 months of storage at 4°C, the mean particle size of F-D-L showed a very slight increase, from 128.0±1.0 nm to 138.4±2.4 nm (Figure 3A). The particle size of F-CONH-PEG-CONH-Chol Dox liposomes increased to 165.3±6.8 nm, which might have resulted from the aggregation of liposomes due to loss of PEG molecules. Meanwhile, the entrapment of Dox in F-CONH-PEG-NH-Chol liposomes remained more than 90% (Figure 3B). In contrast, F-CONH-PEG-CONH-Chol liposomes retained less than 80% of the Dox after 3 months of storage (Figure 3B). These results indicate that the F-CONH-PEG-NH-Chol liposomes are stable at 4°C for at least 3 months and are more stable than the liposomes with F-CONH-PEG-CONH-Chol.
Figure 3.
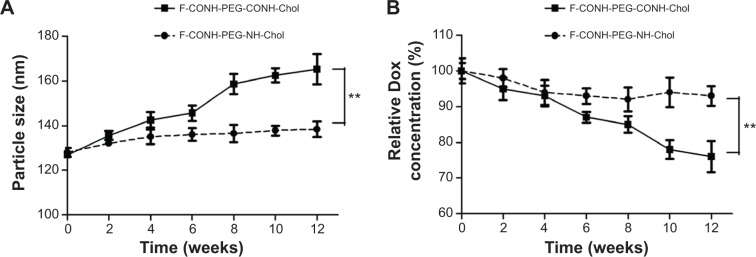
The physical stability test of F-CONH-PEG-NH-Chol and F-CONH-PEG-CONH-Chol liposomes.
Notes: Both liposomes were stored at 4°C for 3 months after preparation and particle size and drug encapsulation were determined every 2 weeks. (A) Particles size changes with time at 4°C. (B) Dox content changes with time at 4°C. Data are expressed as the mean ± SD (n=3). **P<0.01.
Abbreviations: Dox, doxorubicin; SD, standard deviation; F-CONH-PEG-NH-Chol, folate-CONH-PEG-NH-Cholesterol; F-CONH-PEG-CONH-Chol, folate-CONH-PEG-CONH-Cholesterol.
Increased targeted cellular uptake of F-CONH-PEG-NH-Chol liposomes in vitro
To evaluate whether the F-CONH-PEG-NH-Chol derivative can efficiently target cancer cells, we tested the uptake of liposomes with or without F-CONH-PEG-NH-Chol in HeLa, KB, and SMMC-7721 cells, respectively. The HeLa, KB, and SMMC-7721 cells were treated with liposomes for 24 h and then the cellular uptake was evaluated by both flow cytometry and fluorescence microscopy.
As shown in Figure 4 (left lane), fluorescence microscopy showed the uptake of calcein encapsulated by F-Calcein-L was much higher than non-targeting Calcein-L in both HeLa and KB cells, which express high level FRs, but not in SMMC-7721 cells, which express little FR. Moreover, the specific uptake of F-Calcein-L by HeLa and KB cells can be blocked by 1 μM folic acid pretreatment (Figure 4). We also confirmed this phenomenon by flow cytometry (Figure 4, right lane). Quantitative analysis showed that the uptake of F-Calcein-L was about ten times higher than that of Calcein-L and this increase could be blocked by free folic acid. These data demonstrated that the F-CONH-PEG-NH-Chol derivative can mediate the uptake of liposomes efficiently in vitro and this function is highly dependent on the existence of FRs and is sensitive to the presence of folic acid.
Figure 4.

Uptake of F-Calcein-L and Calcein-L by HeLa, KB and SMMC-7721 cells.
Notes: The cells were cultured with Calcein-L, F-Calcein-L and F-Calcein-L pretreated with 1 mM free folic acid (F-Cal-L + folate) for 1 hour and the uptake was assayed by both the fluorescence microscopy (×100) and the FACS. The images were taken in bright fields or under fluorescence for equal exposure time. The FACS measured 2×106 cells for each sample and the blank means no drug was added and was used as a control. Note when cells were pretreated with folic acid, the uptake of F-Calcein-L was inhibited.
Abbreviation: F-Calcein-L, folate receptor targeted calcein liposomes; FACS, fluorescence activated cell sorter.
Enhanced cytotoxicity of F-D-L compared with non-targeted Dox liposomes
The cytotoxicity of both F-D-L and D-L were evaluated in HeLa and KB cells by MTT assay. As shown in Table 2, the cytotoxicity of free Dox was the greatest. When Dox was encapsulated with liposome, the toxicity was reduced markedly. In HeLa cells, the half maximal inhibitory concentration (IC50) value of non-targeted D-L was 178.5 μM, 37-fold that of free Dox. In KB cells, the IC50 of D-L is 111.1 μM, 110-fold that of free Dox (Table 2). These data confirmed again that liposomal encapsulation blocked the penetration and release of Dox into cells. However, the F-D-L incorporating F-CONH-PEG-NH-Chol showed improved cytotoxicity. In HeLa cells, the IC50 value of F-D-L was 82.2 μM, only half that of the D-L. In KB cells, the IC50 of F-D-L was 39.5 μM, one third that of D-L (Table 2). Importantly, the presence of 1mM folic acid could diminish the difference in IC50 values between D-L and F-D-L in both KB and HeLa cells. In summary, by incorporating F-CONH-PEG-NH-Chol, Dox liposomes showed significantly active targeting effects and improved cytotoxicity in vitro. These data suggest that the F-CONH-PEG-NH-Chol ligand has the potential to improve the anti-tumor effect of D-L.
Table 2.
IC50 of Dox formulation in HeLa and KB cells
Notes: The IC50 of F-D-L, D-L and Dox were determined in HeLa and KB cells by incubating for 2 hours at 37°C. Data are presented as the mean ± SD (n=6).
P<0.01 compared with D-L in the same cell lines.
Abbreviations: Dox, doxorubicin; F-D-L, F-CONH-PEG-NH-Chol conjugated doxorubicin liposome; F-CONH-PEG-NH-Chol, folate-CONH-PEG-NH-Cholesterol; D-L, PEGylated doxorubicin liposome; SD, standard deviation; IC50, half maximal inhibitory concentration.
F-CONH-PEG-NH-Chol ligands maintain the long circulation time of liposomes in vivo
For nanoparticles, long circulation is the most prominent feature, which may improve the therapeutic efficacy and help reduce the toxicity to normal tissues. To evaluate the impact of F-CONH-PEG-NH-Chol on the circulation time of liposomes, we determined the pharmacokinetics of both F-D-L and D-L. F-D-L, D-L, and free Dox were injected in mice as a single intravenous bolus via the lateral tail vein at an equivalent dose of 5 mg/kg Dox. Plasma clearance kinetics of liposomal Dox is shown in Figure 5 and pharmacokinetic parameters obtained by using the DAS software (version 2.0, Drug and Statistics, Hefei, People’s Republic of China) with non-compartmental model are summarized in Table 3. As expected, free Dox showed rapid clearance from the plasma and its concentration was almost undetectable after 5 min (Figure 5A). Meanwhile, D-L showed a much longer circulation time, and its t1/2 increased to 15.49 h (Table 3). Although the t1/2 of F-D-L was slightly shorter than that of D-L, it showed a larger AUC than non-targeted D-L (Table 3). The larger AUC of F-D-L can be attributed to the lower clearance rate and higher maximal concentration (Figure 5A and Table 3). The lower clearance rate of F-D-L can be a result from the hydrolysis resistant C-N bond between PEG and Chol, which better maintained the connection between PEG and the liposomes and hence retained the long circulation of Dox liposomes. This is consistent with the data on F-PEG-DSPE and F-PEG-Chol-containing liposomes reported previously.20,31 Importantly, the larger AUC and Cmax suggest that F-CONH-PEG-NH-Chol can both maintain longer circulation and should not reduce the treatment efficacy.
Figure 5.
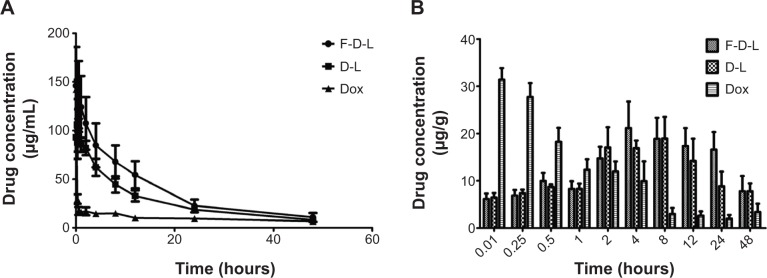
F-D-L, D-L and free Dox concentrations in mouse plasma and liver.
Notes: (A) Pharmacokinetics of F-D-L, D-L and free Dox in the mouse plasma. (B) Dox distribution in the mouse livers administered with F-D-L, D-L and free Dox (n=6). Data are expressed as the mean ± SD (n=6).
Abbreviations: F-D-L, F-CONH-PEG-NH-Chol conjugated doxorubicin liposome; F-CONH-PEG-NH-Chol, folate-CONH-PEG-NH-Cholesterol; D-L, PEGylated doxorubicin liposome; Dox, doxorubicin; SD, standard deviation.
Table 3.
Plasma pharmacokinetic analysis
| Parameter | Unit | D-L | F-D-L |
|---|---|---|---|
| AUC (0–∞) | μg/mL·h | 1,773.41 | 2,076.50 |
| t1/2 | H | 15.49 | 14.92 |
| CL | mL/h/kg | 2.819 | 2.408 |
| Cmax | μg/L | 105.287 | 136.661 |
Abbreviations: AUC, area under the curve; t1/2, half-life; CL, clearance rate; Cmax, maximal concentration in plasma; D-L, PEGylated doxorubicin liposome; F-D-L, F-CONH-PEG-NH-Chol conjugated doxorubicin liposome; F-CONH-PEG-NH-Chol, folate-CONH-PEG-NH-Cholesterol.
F-CONH-PEG-NH-Chol ligand does not affect the distribution of liposomal Dox in normal tissues
For nanoparticulate carriers, their distributions in the body following systemic administration is one of their critical characteristics.12 To determine whether the folate ligand affects the redistribution of Dox from plasma, we examined the distribution of Dox in the liver, heart, spleen, and kidney. We first examined the distribution of Dox encapsulated by D-L and F-D-L in the liver. In comparison with free Dox, encapsulated Dox by D-L and F-D-L showed a significant delay in reaching the Cmax. They took 4–8 h to reach the Cmax in the liver, while free Dox took only 5 min (Figure 5B). While both the D-L and F-D-L showed long circulation features, the F-D-L reached the Cmax faster than the D-L (4 h versus 8 h, Figure 5B). This phenomenon suggests that F-CONH-PEG-NH-Chol may accelerate the redistribution of Dox after administration whereas it does not affect the long circulation of liposomes. Besides the liver, we also evaluated the distribution of Dox in the heart, kidney, and spleen. The distribution kinetics of Dox in kidneys and spleens was similar to that in livers, in which F-D-L and D-L delayed the uptake of Dox. In addition, we noticed that after reaching the Cmax in the liver, spleen, and kidney, free Dox was rapidly cleared from these organs. However, D-L and F-D-L administration retained relatively higher Dox concentrations (>10 μg/mL) until 24 h (Figure 6A). In contrast, the Dox in hearts delivered by D-L and F-D-L both showed very low concentration from 5 min to 24 h, almost identical to that of the free Dox (Figure 6A). Besides the difference of distribution speed between these Dox formulations, the distribution mode of Dox in these organs was altered too. D-L did not alter the distribution of Dox in the liver, but it reduced the Dox distribution in the heart from 2.3% to 1.3% among the total amount of Dox in these four organs. F-D-L further reduced the Dox distribution in the heart to 1.1% although it elevated the Dox distribution in livers for some extent (Figure 6B). These data suggest that F-CONH-PEG-NH-Chol ligand can help liposomes maintain long circulation property and can reduce the distribution of Dox in the heart, a main toxic organ of Dox.
Figure 6.

Tissue distribution and percentage of Dox by F-D-L, D-L and free Dox administration in the four organs of mice.
Notes: (A) Tissue distribution of Dox in the heart, liver, spleen, and kidney at 5 minutes, 2 hours, 12 hours, and 24 hours. (B) The percentages of Dox distribution in these organs at their peak concentrations for each formulation. The percentages of Dox distribution in these organs were calculated by the respective Dox weight in each organ divided by the total amount of the Dox in these four organs. The Dox weight in each organ was obtained by multiplying their peak concentration (drug weight/gram tissue) and the weight of the organ. The data are expressed as the mean ± SD (n=6).
Abbreviations: F-D-L, F-CONH-PEG-NH-Chol conjugated doxorubicin liposome; F-CONH-PEG-NH-Chol, folate-CONH-PEG-NH-Cholesterol; D-L, PEGylated doxorubicin liposome; Dox, doxorubicin; SD, standard deviation.
F-CONH-PEG-NH-Chol liposomal Dox showed better efficacy than non-targeted liposomal Dox
As we showed before, the F-D-L showed higher cytotoxicity in vitro, longer circulation and improved distribution in vivo than non-targeted D-L. We then evaluated whether F-D-L showed better anti-tumor capability than D-L in mice. We inoculated 5×106 KB cells per mouse and then gave a dose of free Dox or liposomal Dox intravenously at a dose of 10 mg/kg body weight when the tumor grew to 100 mm3 (about 2 weeks after the inoculation).
After drug administration, the tumor sizes of F-D-L treated mice were significantly smaller than those of the D-L and free Dox treated mice (Figure 7A). The tumor growth rates of F-D-L treated mice were correspondingly lower than the D-L and free Dox treated mice (Figure 7B). The gap of tumor sizes between F-D-L and D-L groups was maintained during our 3 weeks observation (Figure 7B). Importantly, the tumor size difference between the F-D-L and D-L group increased along with the growth of the tumors (Figure 7B). Upon the final determination on day 20, the average tumor sizes for Dox, D-L, and F-D-L treated mice were 1,280, 1,060 and 750 mm3 respectively. There was a significant difference between the tumor sizes of F-D-L and Dox treated mice, while no significant difference between Dox treated mice and D-L treated mice. Besides inhibiting the tumor growth, F-D-L also contributed to the prolonged survival time of treated mice compared with D-L and Dox. The median survival time of mice treated with Dox, D-L, and F-D-L was 40 days, 43 days, and 48 days, respectively (Figure 7C). These results suggested that the F-D-L can improve the therapeutic efficacy of D-L in vivo although the effects were not as evident as shown in vitro.
Figure 7.

Anti-tumor efficacy of F-D-L, D-L and Dox in tumor bearing mice.
Notes: (A) Tumor bearing mice treated with drugs. The KB cells cultured with folate free medium were inoculated subcutaneously at the front limb and drug was administered by tail vein injection when the tumors grew to 100 mm3. (B) Tumor growth curve. Tumor sizes were determined every other day from the time of drug administration and the volumes were calculated by V =0.52×a×b2, “a” means the long diameters and “b” means the short diameters. (C) Survival curve of tumor bearing mice. The survival of mice was recorded since the mice were inoculated with cancer cells. The drugs were administered at 2 weeks when the tumor grew to 100 mm3. Data are expressed as the mean ± SD (n=6). *P<0.05.
Abbreviations: F-D-L, F-CONH-PEG-NH-Chol conjugated doxorubicin liposome; F-CONH-PEG-NH-Chol, folate-CONH-PEG-NH-Cholesterol; D-L, PEGylated doxorubicin liposome; Dox, doxorubicin; SD, standard deviation.
F-CONH-PEG-NH-Chol improved liposomal Dox biodistribution and release in tumor tissues
The next question is how the F-CONH-PEG-NH-Chol ligand contributes to the tumor targeting delivery of liposomal Dox. To address this question, we compared F-D-L and D-L for their ability to deliver the Dox to tumor tissues. We injected xenograft tumor bearing mice with saline, free Dox, D-L, and F-D-L respectively and then the mice were sacrificed at 4 h after the injections. The tumors were then removed and fixed for microscopic examination or homogenized for Dox concentration determination. Compared with saline injections, the free Dox injection resulted in strong tissue fluorescence (Figure 8A, a and b). However, when the Dox was delivered by D-L, it was restricted to the vicinity of blood capillary (Figure 8A, c). This phenomenon indicated that the liposomes either extravasated poorly or released the drug insufficiently in tumor tissues. Surprisingly, the Dox spread to a much broader area in tumor tissues when delivered by F-D-L (Figure 8A, d). This result suggests that the F-CONH-PEG-NH-Chol ligand may help liposomes penetrate the blood capillary or help the Dox release from liposomes. Then we determined Dox concentration in tumor tissues and the results showed that Dox concentration in F-D-L treated tumor was the highest, much higher than that in D-L treated and Dox treated tumor (Figure 8B). This quantitative analysis supports the view that F-CONH-PEG-NH-Chol can function as an active ligand to help overcome the barrier to drug release from the liposomes in tumor tissues.
Figure 8.
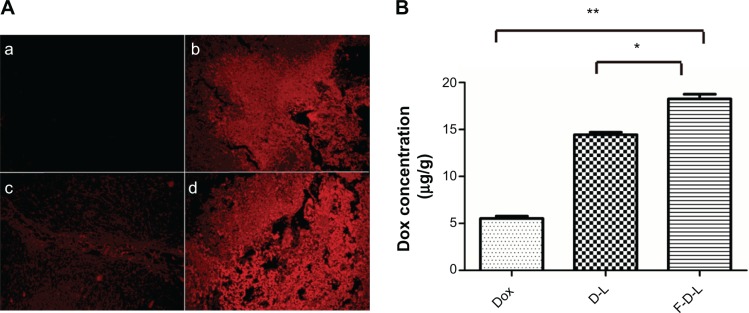
The distribution of Dox in the tumors of treated mice.
Notes: Two hours after injections, mice were sacrificed and tumors were resected and fixed for photography check. (A): (a) saline; (b) free Dox; (c) D-L; (d) F-D-L. (B) The concentration of Dox in the mice treated with Dox, D-L and F-D-L. The tumor tissue was homogenized and Dox concentration was determined on a microplate reader (n=6). *P<0.05; **P<0.01.
Abbreviations: F-D-L, F-CONH-PEG-NH-Chol conjugated doxorubicin liposome; F-CONH-PEG-NH-Chol, folate-CONH-PEG-NH-Cholesterol; D-L, PEGylated doxorubicin liposome; Dox, doxorubicin.
F-CONH-PEG-NH-Chol liposomal Dox maintains low cardiotoxicity
Application of Dox in the clinics was restricted largely because of its cardiac toxicity. In this study, we also observed that the mice treated with free Dox died at an early stage before the tumors reached very large sizes, which infers that the mice may have died from the cardiotoxicity of Dox (Figure 7C). Because liposomes are believed to be able to reduce the side effects of chemotherapeutic agents, we then evaluated whether the addition of F-CONH-PEG-NH-Chol will affect the cardiotoxicity of liposomal Dox. As shown in Figure 9A, when the mice were given free Dox, the heart index (heart weight/body weight) showed marked decrease, from 0.43 down to 0.37, which hints at the injury of cardiac muscles. However, administration of the D-L and F-D-L did not result in significant heart injury although the uptake of free Dox and liposomal formulations was at the same level (Figure 9A). The severe body weight loss (13%) caused by Dox was significantly attenuated by D-L or F-D-L capsulation and there was no significant difference between D-L and F-D-L treatment (Figure 9A). Moreover, the cardiac GSH-Px level, an anti-oxidative stress marker, was reduced following Dox injection, but not reduced after the D-L or F-D-L administration (Figure 9B). As the Dox concentrations in the hearts of D-L and F-D-L treated mice were lower than free Dox (Figure 6B), these data indicate that F-CONH-PEG-NH-Chol facilitated the cellular uptake of D-L in the tumors, but not in the hearts. A possible reason is that the FR in the heart is very little and there is almost no uptake of folate conjugated liposomes by the cardiac muscle cells.32,33 Taken together, both the F-D-L and D-L can reduce the exposure of Dox to cardiac myocytes and keep the heart from oxidation stress induced by Dox. These data collectively showed that the F-CONH-PEG-NH-Chol ligand does not affect the safety of Dox liposomes while improving their efficacy.
Figure 9.
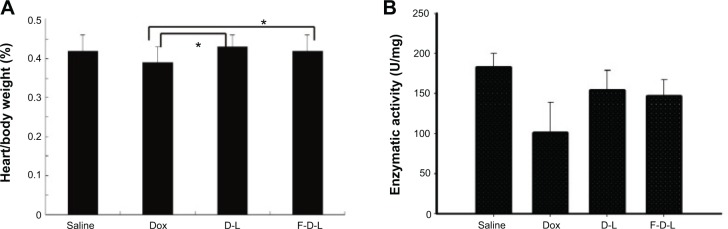
Folate conjugated liposomes have reduced cardiotoxicity.
Notes: (A) Heart/body weight index. (B) GSH-Px enzymatic activities. Data are expressed as the mean ± SD (n=6). *P<0.05.
Abbreviations: F-D-L, F-CONH-PEG-NH-Chol conjugated doxorubicin liposome; F-CONH-PEG-NH-Chol, folate-CONH-PEG-NH-Cholesterol; D-L, PEGylated doxorubicin liposome; Dox, doxorubicin; SD, standard deviation; GSH-Px, glutathione peroxidase.
Discussion
Since the first liposomal pharmaceutical product, Doxil® (Janssen Products, LP, Titusville, NJ, USA), received US Food and Drug Administration approval in 1995, liposomes have been intensively investigated for applications in clinics. Many types of liposomes, such as PEGylated liposomes (Doxil, Lipo-dox), temperature sensitive liposomes (ThermoDox), cationic liposomes (EndoTAG-1), and virosomes (Epaxal and Inflexal V) have been used in clinics or developed for clinical use at different stages.34 Collective evidence has been obtained over time that improved liposomal formulations, such as PEGylated liposomes, may prolong blood circulation time, alter drug distribution in the body, and hence reduce the possible side effects related to the drugs (eg, cardiotoxicity). However, significant challenges still impede expanded clinical applications of liposomal formulations. PEGylated liposomes (Doxil and Lipo-dox), for example, displayed significant incidence of stomatitis in clinical trials,35,36 which may have resulted from PEGylation. Moreover, some of the new generation liposomes showed only comparable or even poorer therapeutic efficiency compared with free drug or conventional vesicles in clinical trials. For instance, in comparison with Doxil, ThermoDox displayed significantly weaker Dox accumulation in mice tumors at 24 h after administration.37
Advanced approaches are explored to overcome these limitations, in order to fully utilize the drug delivery potential of liposomes. Some of these approaches are based on reducing the toxicity or side effects of drugs, instead of pursuing solely improved efficacy. Among many types of liposomes, targeted delivery by liposomes has long been explored to reduce the toxicity to non-targeted organs and promote the efficacy by enhancing the drug concentration in target organs, especially in tumors. We have explored various types of FR targeting ligands through chemical design and in vitro studies.23,38–40 Although most types of these folate ligands showed effectiveness in vitro, their effects in mouse models have not been determined. The current study represents our first effort in evaluating this novel F-CONH-PEG-NH-Chol ligand for targeting liposomes.
When delivering drugs by liposomes or other nanoparticles, reduced efficacy has been one major obstacle to successful translation of these therapeutic modalities into the clinic. Our data suggest again that even though some liposomes or other nanoparticles showed satisfactory targeting delivery effects in vitro, significant barriers still exist for clinical translation. This observation is consistent with other earlier reports from clinical studies. For example, SPI-77, the first liposomal formulation of cisplatin, had limited clinical efficacy in a phase II clinical trial of advanced non-small-cell lung cancer, even though SPI-77 demonstrated enhanced cisplatin tumor accumulation in preclinical models.42 Similar to SPI-77, a phase II study of liposomal annamycin in the treatment of Dox-resistant breast cancer failed because of diminished anti-tumor activity.43 Although new liposome-based drugs have been well studied in preclinical animal models, these agents may be unable to provide promising therapeutic effects in clinical trials. Better understanding of the pharmacokinetics of liposomes and other nano-formulations, and rational design based on these understandings are urgently needed to promote the clinical development of these advanced delivery systems.
In this study, we noticed the accumulation of Dox delivered by FR binding liposomes in tumors. Although the relative higher concentration gave rise to improved treatment effects, the result was not as evident as shown by in vitro study. Multiple factors could contribute to this unsatisfactory outcome. First, it was possible that F-D-L penetrated into the tumor tissue and attached to the tumor cells, but was not internalized by the cells. Second, the liposome vehicles were internalized by the tumor cells, but the encapsulated Dox molecules did not escape from the particles. Third, the Dox may be released from liposomes, but was restricted only to small parts of the tissues. And accompanying the fast growing of the tumors, the high concentration of Dox in local area can reduce and the drug can also be excreted. The special cancer microenvironment can explain many failed nano-chemotherapeutics, since it is totally different from the cell culture environment in vitro.
Besides increasing the concentration of drugs in tumors, the targeting liposomes enhance the treatment efficacy also by reducing the toxicity of chemotherapeutic agents to normal tissues or organs.44 In this study, we hope to see further reduced distribution of Dox in heart than D-L by which to further improve the safety of D-L. As expected, we observed the reduced cardiac toxicity of F-D-L, which was even better than D-L. This effect can be attributed to reduced distribution of Dox in heart tissues which is supported by distribution study in normal tissues. When administered by free Dox, hearts absorbed 2.3% Dox among the four organs we tested. When administered with D-L and F-D-L, the heart only absorbed 1.3% and 1.1% Dox, respectively (Figure 6B). This result suggests reduced Dox distribution helps lower the toxicity of Dox to hearts. On the other hand, F-D-L showed enhanced distribution of Dox in the liver, which is an reticular epithelial system-rich organ. This result suggests that the C-N bond between PEG and Chol molecules can avoid the disassociation of PEG from the liposomes and maintain the long circulation and improve the uptake of liposomal Dox to reticular epithelial system-rich organs including tumors.45
Of note, observations based on in vitro study only may give rise to incorrect conclusions. In our study, for example, increased transportation of Dox into the tumor cells via FR targeted uptake should result in greater delivery and therefore more cytotoxicity of Dox as compared with free Dox. But the IC50 value of Dox is lower than the F-D-L. This is because the free Dox has excellent membrane permeability and immediate cytotoxic effects in vitro. Conversely, F-D-L needs longer uptake time and hence showed slower and weaker killing effects. In vivo, however, F-D-L showed competent, even better treatment efficacy than Dox because of longer circulation and targeted uptake.
Considering the complex distribution, metabolism and excretion process of the nanoparticle drugs, the comparison of drug circulation time in blood, drug accumulation in tissues, and potential efficacy between conventional vesicles and new generations liposome-based drugs should be investigated more extensively in animal models in the future. The in vivo approach is recommended to avoid some of the pitfalls in nanomedicine development primarily based on in vitro studies that are not representative of in vivo conditions. In addition, the application of physiologically based pharmacokinetics models could be utilized to facilitate the interspecies extrapolation, which may potentially address the disconnection between the preclinical and clinical studies. Furthermore, clinical studies designed to investigate the toxicity and therapeutic effects unique to the nanoparticles of different compositions will eventually allow development of nanomedicine in therapeutic areas beyond oncology.
Conclusion
A chemically stable folate ligand, F-CONH-PEG-NH-Chol, was successfully synthesized and investigated for liposomal Dox delivery. The F-CONH-PEG-NH-Chol conjugated liposomes can successfully target tumor cells both in vitro and in vivo. In mouse models, F-CONH-PEG-NH-Chol liposomes maintained long circulation and efficient tissue distributions, including in tumor tissues. The F-CONH-PEG-NH-Chol liposomes loaded with Dox also showed better treatment benefits in comparison with non-conjugated liposomes, and successfully reduced the cardiotoxicity associated with Dox therapy. These results suggest that F-CONH-PEG-NH-Chol is a stable and effective folate ligand for liposomal drug delivery.
Supplementary material
Mass spectrum of F-CONH-PEG-NH-Chol (folate-CONH-PEG-NH-Cholesterol).
Notes: The calculated molecular weight of F-CONH-PEG-NH-Chol is 4,158 Da and the mass spectrum was determined by matrix-assisted laser desorption/ionization/time-of-flight mass spectroscopy. The fragment of F-CONH-PEG-NH-Chol was distributed in a bell shape centered at 4,140 Da, consistent with calculated molecular weight.
Acknowledgments
This work was financially supported by the National Natural Science Foundation of China (No 81370058, 81101824, 81301235), Wuhan Young Talents Program (No 2013070104010042 to CX) and by the Outstanding Youth Fund of Tongji Hospital (No XQN005 to XH).
Footnotes
Disclosure
The authors have no conflicts of interest to disclose.
References
- 1.Elwood PC. Molecular cloning and characterization of the human folate-binding protein cDNA from placenta and malignant tissue culture (KB) cells. J Biol Chem. 1989;264(25):14893–14901. [PubMed] [Google Scholar]
- 2.Lacey SW, Sanders JM, Rothberg KG, Anderson RG, Kamen BA. Complementary DNA for the folate binding protein correctly predicts anchoring to the membrane by glycosyl-phosphatidylinositol. J Clin Invest. 1989;84(2):715–720. doi: 10.1172/JCI114220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wu M, Gunning W, Ratnam M. Expression of folate receptor type alpha in relation to cell type, malignancy, and differentiation in ovary, uterus, and cervix. Cancer Epidemiol Biomarkers Prev. 1999;8(9):775–782. [PubMed] [Google Scholar]
- 4.Ross JF, Chaudhuri PK, Ratnam M. Differential regulation of folate receptor isoforms in normal and malignant tissues in vivo and in established cell lines. Physiologic and clinical implications. Cancer. 1994;73(9):2432–2443. doi: 10.1002/1097-0142(19940501)73:9<2432::aid-cncr2820730929>3.0.co;2-s. [DOI] [PubMed] [Google Scholar]
- 5.Parker N, Turk MJ, Westrick E, Lewis JD, Low PS, Leamon CP. Folate receptor expression in carcinomas and normal tissues determined by a quantitative radioligand binding assay. Anal Biochem. 2005;338(2):284–293. doi: 10.1016/j.ab.2004.12.026. [DOI] [PubMed] [Google Scholar]
- 6.Leamon CP. Folate-targeted drug strategies for the treatment of cancer. Curr Opin Investig Drugs. 2008;9(12):1277–1286. [PubMed] [Google Scholar]
- 7.Zhao XB, Lee RJ. Tumor-selective targeted delivery of genes and antisense oligodeoxyribonucleotides via the folate receptor. Adv Drug Deliv Rev. 2004;56(8):1193–1204. doi: 10.1016/j.addr.2004.01.005. [DOI] [PubMed] [Google Scholar]
- 8.Pan XQ, Zheng X, Shi G, Wang H, Ratnam M, Lee RJ. Strategy for the treatment of acute myelogenous leukemia based on folate receptor beta-targeted liposomal doxorubicin combined with receptor induction using all-trans retinoic acid. Blood. 2002;100(2):594–602. doi: 10.1182/blood.v100.2.594. [DOI] [PubMed] [Google Scholar]
- 9.Wang S, Low PS. Folate-mediated targeting of antineoplastic drugs, imaging agents, and nucleic acids to cancer cells. J Control Release. 1998;53(1–3):39–48. doi: 10.1016/s0168-3659(97)00236-8. [DOI] [PubMed] [Google Scholar]
- 10.Henne WA, Doorneweerd DD, Hilgenbrink AR, Kularatne SA, Low PS. Synthesis and activity of a folate peptide camptothecin prodrug. Bioorg Med Chem Lett. 2006;16(20):5350–5355. doi: 10.1016/j.bmcl.2006.07.076. [DOI] [PubMed] [Google Scholar]
- 11.Bae Y, Nishiyama N, Kataoka K. In vivo antitumor activity of the folate-conjugated pH-sensitive polymeric micelle selectively releasing adriamycin in the intracellular acidic compartments. Bioconjug Chem. 2007;18(4):1131–1139. doi: 10.1021/bc060401p. [DOI] [PubMed] [Google Scholar]
- 12.Sanhai WR, Sakamoto JH, Canady R, Ferrari M. Seven challenges for nanomedicine. Nat Nanotechnol. 2008;3(5):242–244. doi: 10.1038/nnano.2008.114. [DOI] [PubMed] [Google Scholar]
- 13.Rahman A, Husain SR, Siddiqui J, et al. Liposome-mediated modulation of multidrug resistance in human HL-60 leukemia cells. J Natl Cancer Inst. 1992;84(24):1909–1915. doi: 10.1093/jnci/84.24.1909. [DOI] [PubMed] [Google Scholar]
- 14.Gabizon A, Shmeeda H, Horowitz AT, Zalipsky S. Tumor cell targeting of liposome-entrapped drugs with phospholipid-anchored folic acid-PEG conjugates. Adv Drug Deliv Rev. 2004;56(8):1177–1192. doi: 10.1016/j.addr.2004.01.011. [DOI] [PubMed] [Google Scholar]
- 15.Perez AT, Domenech GH, Frankel C, Vogel CL. Pegylated liposomal doxorubicin (Doxil) for metastatic breast cancer: the Cancer Research Network, Inc., experience. Cancer Invest. 2002;20(Suppl 2):22–29. doi: 10.1081/cnv-120014883. [DOI] [PubMed] [Google Scholar]
- 16.Theodoulou M, Hudis C. Cardiac profiles of liposomal anthracyclines: greater cardiac safety versus conventional doxorubicin? Cancer. 2004;100(10):2052–2063. doi: 10.1002/cncr.20207. [DOI] [PubMed] [Google Scholar]
- 17.Safra T. Cardiac safety of liposomal anthracyclines. Oncologist. 2003;8(Suppl 2):17–24. doi: 10.1634/theoncologist.8-suppl_2-17. [DOI] [PubMed] [Google Scholar]
- 18.Batist G. Cardiac safety of liposomal anthracyclines. Cardiovasc Toxicol. 2007;7(2):72–74. doi: 10.1007/s12012-007-0014-4. [DOI] [PubMed] [Google Scholar]
- 19.Leamon CP, Cooper SR, Hardee GE. Hardee, Folate-liposome-mediated antisense oligodeoxynucleotide targeting to cancer cells: evaluation in vitro and in vivo. Bioconjug Chem. 2003;14(4):738–747. doi: 10.1021/bc020089t. [DOI] [PubMed] [Google Scholar]
- 20.Gabizon A, Horowitz AT, Goren D, Tzemach D, Shmeeda H, Zalipsky S. In vivo fate of folate-targeted polyethylene-glycol liposomes in tumor-bearing mice. Clin Cancer Res. 2003;9(17):6551–6559. [PubMed] [Google Scholar]
- 21.Lee RJ, Low PS. Folate-mediated tumor cell targeting of liposome-entrapped doxorubicin in vitro. Biochim Biophys Acta. 1995;1233(2):134–144. doi: 10.1016/0005-2736(94)00235-h. [DOI] [PubMed] [Google Scholar]
- 22.Gabizon A, Horowitz AT, Goren D, et al. Targeting folate receptor with folate linked to extremities of poly(ethylene glycol)-grafted liposomes: in vitro studies. Bioconjug Chem. 1999;10(2):289–298. doi: 10.1021/bc9801124. [DOI] [PubMed] [Google Scholar]
- 23.Xiang G, Wu J, Lu Y, Liu Z, Lee RJ. Synthesis and evaluation of a novel ligand for folate-mediated targeting liposomes. Int J Pharm. 2008;356:29–36. doi: 10.1016/j.ijpharm.2007.12.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Liu Y, Xu S, Teng L, et al. Synthesis and evaluation of a novel lipophilic folate receptor targeting ligand. Anticancer Res. 2011;31(5):1521–1525. [PubMed] [Google Scholar]
- 25.Wu J, Liu Q, Lee RJ. A folate receptor-targeted liposomal formulation for paclitaxel. Int J Pharm. 2006;316(1–2):148–153. doi: 10.1016/j.ijpharm.2006.02.027. [DOI] [PubMed] [Google Scholar]
- 26.Masuda T, Akita H, Nishio T, et al. Development of lipid particles targeted via sugar-lipid conjugates as novel nuclear gene delivery system. Biomaterials. 2008;29(6):709–723. doi: 10.1016/j.biomaterials.2007.09.039. [DOI] [PubMed] [Google Scholar]
- 27.Haran G, Cohen R, Bar LK, Barenholz Y. Transmembrane ammonium sulfate gradients in liposomes produce efficient and stable entrapment of amphipathic weak bases. Biochim Biophys Acta. 1993;1151(2):201–215. doi: 10.1016/0005-2736(93)90105-9. [DOI] [PubMed] [Google Scholar]
- 28.Stevens PJ, Sekido M, Lee RJ. A folate receptor-targeted lipid nanoparticle formulation for a lipophilic paclitaxel prodrug. Pharm Res. 2004;21(12):2153–2157. doi: 10.1007/s11095-004-7667-5. [DOI] [PubMed] [Google Scholar]
- 29.Hattori Y, Maitani Y. Folate-linked lipid-based nanoparticle for targeted gene delivery. Curr Drug Deliv. 2005;2(3):243–252. doi: 10.2174/1567201054368002. [DOI] [PubMed] [Google Scholar]
- 30.Chen C, Han D, Cai C, Tang X. An overview of liposome lyophilization and its future potential. J Control Release. 2010;142(3):299–311. doi: 10.1016/j.jconrel.2009.10.024. [DOI] [PubMed] [Google Scholar]
- 31.Zhao XB, Muthusamy N, Byrd JC, Lee RJ. Cholesterol as a bilayer anchor for PEGylation and targeting ligand in folate-receptor-targeted liposomes. J Pharm Sci. 2007;96(9):2424–2435. doi: 10.1002/jps.20885. [DOI] [PubMed] [Google Scholar]
- 32.Weitman SD, Lark RH, Coney LR. Distribution of the folate receptor GP38 in normal and malignant cell lines and tissues. Cancer Res. 1992;52(12):3396–3401. [PubMed] [Google Scholar]
- 33.Muller C, Forrer F, Schibli R, Krenning EP, de Jong M. SPECT study of folate receptor-positive malignant and normal tissues in mice using a novel 99mTc-radiofolate. J Nucl Med. 2008;49(2):310–317. doi: 10.2967/jnumed.107.045856. [DOI] [PubMed] [Google Scholar]
- 34.Chang HI, Yeh MK. Clinical development of liposome-based drugs: formulation, characterization, and therapeutic efficacy. Int J Nanomedicine. 2012;7:49–60. doi: 10.2147/IJN.S26766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chao TC, Wang WS, Yen CC, Chiou TJ, Liu JH, Chen PM. A dose-escalating pilot study of sterically stabilized, pegylated liposomal doxorubicin (Lipo-Dox) in patients with metastatic breast cancer. Cancer Invest. 2003;21(6):837–847. doi: 10.1081/cnv-120025086. [DOI] [PubMed] [Google Scholar]
- 36.Lyass O, Uziely B, Ben-Yosef R, et al. Correlation of toxicity with pharmacokinetics of pegylated liposomal doxorubicin (Doxil) in metastatic breast carcinoma. Cancer. 2000;89(5):1037–1047. doi: 10.1002/1097-0142(20000901)89:5<1037::aid-cncr13>3.0.co;2-z. [DOI] [PubMed] [Google Scholar]
- 37.Dromi S, Frenkel V, Luk A, et al. Pulsed-high intensity focused ultrasound and low temperature-sensitive liposomes for enhanced targeted drug delivery and antitumor effect. Clin Cancer Res. 2007;13(9):2722–2727. doi: 10.1158/1078-0432.CCR-06-2443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lu Y, Wu J, Gonit M, et al. Role of formulation composition in folate receptor-targeted liposomal doxorubicin delivery to acute myelogenous leukemia cells. Mol Pharm. 2007;4(5):707–712. doi: 10.1021/mp070058l. [DOI] [PubMed] [Google Scholar]
- 39.Zhai G, Wu J, Xiang G, et al. Preparation, characterization and pharmacokinetics of folate receptor-targeted liposomes for docetaxel delivery. J Nanosci Nanotechnol. 2009;9(3):2155–2161. doi: 10.1166/jnn.2009.450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhang Q, Xiang G, Zhang Y, et al. Increase of doxorubicin sensitivity for folate receptor positive cells when given as the prodrug N-(phenylacetyl) doxorubicin in combination with folate-conjugated PGA. J Pharm Sci. 2006;95(10):2266–2275. doi: 10.1002/jps.20714. [DOI] [PubMed] [Google Scholar]
- 41.Qu X, Yang C, Zhang K, et al. In vitro evaluation of a Folate-bovine serum albumin-doxorubicin conjugate. J Drug Target. 2010;18(5):351–361. doi: 10.3109/10611860903450049. [DOI] [PubMed] [Google Scholar]
- 42.White SC, Lorigan P, Margison GP, et al. Phase II study of SPI-77 (sterically stabilised liposomal cisplatin) in advanced non-small-cell lung cancer. Br J Cancer. 2006;95(7):822–828. doi: 10.1038/sj.bjc.6603345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Booser DJ, Esteva FJ, Rivera E, et al. Phase II study of liposomal annamycin in the treatment of doxorubicin-resistant breast cancer. Cancer Chemother Pharmacol. 2002;50(1):6–8. doi: 10.1007/s00280-002-0464-0. [DOI] [PubMed] [Google Scholar]
- 44.Gabizon A, Shmeeda H, Barenholz Y. Pharmacokinetics of pegylated liposomal Doxorubicin: review of animal and human studies. Clin Pharmacokinet. 2003;42(5):419–436. doi: 10.2165/00003088-200342050-00002. [DOI] [PubMed] [Google Scholar]
- 45.Gabizon A, Catane R, Uziely B, et al. Prolonged circulation time and enhanced accumulation in malignant exudates of doxorubicin encapsulated in polyethylene-glycol coated liposomes. Cancer Res. 1994;54(4):987–992. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Mass spectrum of F-CONH-PEG-NH-Chol (folate-CONH-PEG-NH-Cholesterol).
Notes: The calculated molecular weight of F-CONH-PEG-NH-Chol is 4,158 Da and the mass spectrum was determined by matrix-assisted laser desorption/ionization/time-of-flight mass spectroscopy. The fragment of F-CONH-PEG-NH-Chol was distributed in a bell shape centered at 4,140 Da, consistent with calculated molecular weight.