

Abstract
Autophagy is a highly regulated catabolic process that involves lysosomal degradation of proteins and organelles, mostly mitochondria, for the maintenance of cellular homeostasis and reduction of metabolic stress. Problems in the execution of this process are linked to different pathological conditions, such as neurodegeneration, aging, and cancer. Many of the proteins that regulate autophagy are either oncogenes or tumor suppressor proteins. Specifically, tumor suppressor genes that negatively regulate mTOR, such as PTEN, AMPK, LKB1, and TSC1/2 stimulate autophagy while, conversely, oncogenes that activate mTOR, such as class I PI3K, Ras, Rheb, and AKT, inhibit autophagy, suggesting that autophagy is a tumor suppressor mechanism. Consistent with this hypothesis, the inhibition of autophagy promotes oxidative stress, genomic instability, and tumorigenesis. Nevertheless, autophagy also functions as a cytoprotective mechanism under stress conditions, including hypoxia and nutrient starvation, that promotes tumor growth and resistance to chemotherapy in established tumors. Here, in this brief review, we will focus the discussion on this ambiguous role of autophagy in the development and progression of cancer.
1. Introduction
According to the World Health Organization (WHO), noncommunicable diseases (NCDs) or chronic diseases (CDs), such as cardiovascular diseases, cancer, diabetes, and chronic respiratory diseases, are the leading causes of global mortality. Moreover, because average life-expectation is increasing, their incidence is on the rise and approaching epidemic proportions. The resulting public health burden is spiraling out of control and doing so at an accelerated rate particularly among lower income countries [1]. Despite this rather bleak outlook, the good news is that the impact of these sdiseases could be significantly reduced and considerable suffering avoided by changes in lifestyle to reduce associated risk factors and by the implementation of easy measures for early detection and timely treatment. Specifically, NCDs could be avoided to a considerable extent by reducing four main behavioral risk factors: tobacco use, physical inactivity, harmful use of alcohol, and unhealthy diet. Of interest, particularly in the context of this review series, the latter three risk factors result in a chronic systemic imbalance between caloric intake and consumption, thereby positioning metabolic alterations at the core of chronic disease development. Importantly, although perhaps not as immediately obvious as for diabetes and obesity, cancer is no exception in this respect.
Cancer, a group of diseases generally characterized by abnormal and uncontrolled growth of a population of cells (tumor cells), which eventually invade tissues and form metastases, is one of the leading causes of death worldwide. The latest cancer statistics according to GLOBOCAN 2012 (http://globocan.iarc.fr/Default.aspx) reveal that the global burden of cancer increased in 2012 to 14.1 million new cases and 8.2 million deaths, up from 12.7 million and 7.6 million, respectively, in 2008. Furthermore, these figures are expected to continue increasing to a worrisome 26.4 million new cases and 17 million cancer-related deaths by 2030. In the more developed world (MDW) cancers of the lung, breast, prostate, and colon are the most prevalent types encountered. In contrast, in less developed world (LDW), stomach, liver, oral cavity, and cervical cancers are a more significant concern. These notable differences can be attributed to variations in lifestyles and habits. However, patterns are gradually changing in the LDW and beginning to resemble those of the MDW due to the aging of the population, as well as the acquisition of similar lifestyles and associated risk factors [1, 2].
Thus, despite the many scientific and technological advances that have been developed since the “War on Cancer” was declared by Richard Nixon in 1971, cancer not only remains one of the leading causes of morbidity and mortality worldwide, but it is in fact predicted to become the leading cause of human demise in the coming 20–30 years. In large part, the complexity associated with successful treatment is directly linked to the incredible variety of molecular changes implicated in disease development. The cancer hallmarks defined by Hanahan and Weinberg [3, 4] helped enormously in identifying the general nature of the changes that are required to convert normal cells into tumor cells (transformation). Amongst these, metabolic changes, including the famous Warburg effect, are now recognized as crucial to the development of the transformed phenotype. Bearing this in mind, it should come as no surprise that processes that facilitate cell survival under conditions of metabolic stress are likely to be important in the development of tumors. In this context, we will focus our discussion here on how an evolutionarily ancient response to cellular stress, coined autophagy, may contribute to the pathogenesis of a wide range of cancers. A better understanding of the role of autophagy in tumorigenesis may open up opportunities for more successful treatment of the disease.
2. Autophagy: General Aspects and Regulation
Autophagy is a crucial biological process for the survival of unicellular and multicellular eukaryotic organisms under conditions of nutrient deprivation that participates in the maintenance of cellular homeostasis by controlling the quality of proteins and cytoplasmic organelles. The term autophagy (“self-eating”) was introduced by Christian De Duve in the decade of the sixties, based on the observation, by transmission electron microscopy, of double membrane vacuoles containing cytoplasmic material [5]. Nowadays, autophagy is defined as a cellular pathway by which cytoplasmic macromolecules and organelles are delivered to the lysosomes for degradation [6].
At least three different forms of autophagy have been identified to date [6], macroautophagy, microautophagy, and chaperone-mediated autophagy (CMA). These differ with respect to their function and the mode of delivery of the cargo to the lysosomes. In this review, we will focus the discussion on macroautophagy (hereafter referred to as autophagy) and its role in cancer. During macroautophagy, the cargo is sequestered within a de novo formed double membrane vesicle, the autophagosome, which fuses with the lysosome to generate autolysosomes, in which lysosomal enzymes degrade the vesicle content. Not surprisingly, autophagy represents an important catabolic mechanism that cancer cells activate in response to cellular stress and/or increased metabolic demands imposed by rapid cell proliferation. In this scenario, autophagy should favor tumor cell survival. Interestingly, however, autophagy also acts as a tumor suppressor mechanism by preventing the accumulation of damaged organelles and proteins. Here, we will discuss our current understanding of the apparently contradictory role that autophagy plays in cancer development and progression.
The autophagosome is the double membrane vesicle that represents the morphological hallmark of autophagy. Autophagosomes originate from the phagophore, an isolation membrane that most likely derives from the endoplasmic reticulum (ER) [7, 8]. However, the source of the membrane still remains a matter of debate and recent findings indicate that both the ER and mitochondria may provide the membranes required [9, 10]. The phagophore then expands and surrounds the material destined for degradation and finally forms the characteristic double membrane vesicle, known as autophagosome. The mature autophagosome then fuses with the lysosome generating the autolysosomes, where the internal membrane and material enclosed in the autolysosome are degraded by the activity of the lysosomal hydrolases and acidification of the luminal microenvironment. The degradation products generated by autophagy are then transferred back to the cytosol by permeases in the autolysosomal membrane and recycled into different metabolic pathways.
The molecular execution of the autophagic pathway—generation, maturation and degradation of the autophagosomes—requires the participation of specific autophagy-related (ATG) proteins [11] that were first described in yeast before orthologs in higher eukaryotes were identified. The ATG proteins organize into multiprotein complexes that function in a nonredundant manner in the different steps of the process. Thus, although many ATGs exist, inhibition of just one ATG suffices to block execution of the autophagic cascade.
In mammalian cells, nucleation of the phagophore is regulated by a protein serine/threonine kinase complex that responds to the mammalian target of rapamycin (mTOR), a key regulator of the autophagic pathway, which shuts down autophagy in the presence of nutrients and growth factors [12]. Phagophore nucleation [7, 13] is regulated by the balance between class I and class III phosphatidylinositol 3-kinase (PI3K) enzymatic activities [14]. The active enzyme VPS34, a class III PI3K, together with the counterparts of yeast Vps15 and Vps30/Atg6, identified in mammals as p150 and Beclin-1, and ATG14 form a PI3K complex that catalyzes the production of phosphatidylinositol-3-phosphate, thereby generating a signal to initiate the recruitment of effectors proteins, such as double FYVE-containing protein 1 (DFCP1) and WD-repeat domain phosphoinositide-interacting (WIPI) family proteins [15–18]. The elongation of the isolation membrane and subsequent closure of the autophagosome require the formation of two ubiquitin-like conjugates. First, ATG12 is conjugated to ATG5 by the sequential activity of ATG7 and ATG10. The resulting ATG5-ATG12 complex interacts with ATG16L, which then oligomerizes to form the ATG16L complex [19]. Second, LC3 (the mammalian homologue of yeast Atg8) is cleaved by the protease ATG4 and then conjugated to the lipid phosphatidylethanolamine via the activity of ATG7 and ATG3 [19, 20]. While the unprocessed form of LC3 (LC3I) is diffusely distributed throughout the cytoplasm, the lipidated form of LC3 (LC3II) specifically accumulates on nascent autophagosomes and thus represents a marker to monitor autophagy [21]. The autophagosome eventually seals off and fuses with lysosomes through mechanisms that remain poorly characterized in mammalian cells [11]. Some regulators of the autophagosome-lysosome fusion process include LC3, the lysosomal proteins LAMP-1 and LAMP-2, the small GTP-binding protein RAB7, and the AAA-type ATPase SKD1 [22–24]. Autophagosome-lysosome fusion then results in the activation of the hydrolases which completely degrade the autophagosomal cargo.
Different signaling mechanisms are known to modulate autophagy in mammalian cells [25]. The best characterized pathways are those that modulate autophagy in response to nutritional changes and, as previously mentioned, mTOR is critical for sensing the nutritional status of the cell and regulating the initiation of autophagy [12]. In higher eukaryotes, mTOR can be found in at least two distinct multiprotein complexes, referred to as mTOR complex 1 (mTORC1) and mTOR complex 2 (mTORC2) [26, 27]. The former is considered the principal regulator of autophagy [28]. When nutrients and growth factors are available, mTORC1 inhibits autophagy by phosphorylating and maintaining in an inactive state ULK1, which is required for the formation of the phagophore [29, 30]. As indicated (see Figure 1), mTOR activity is controlled by different signaling pathways triggered via cues from the extracellular and intracellular microenvironment.
Figure 1.
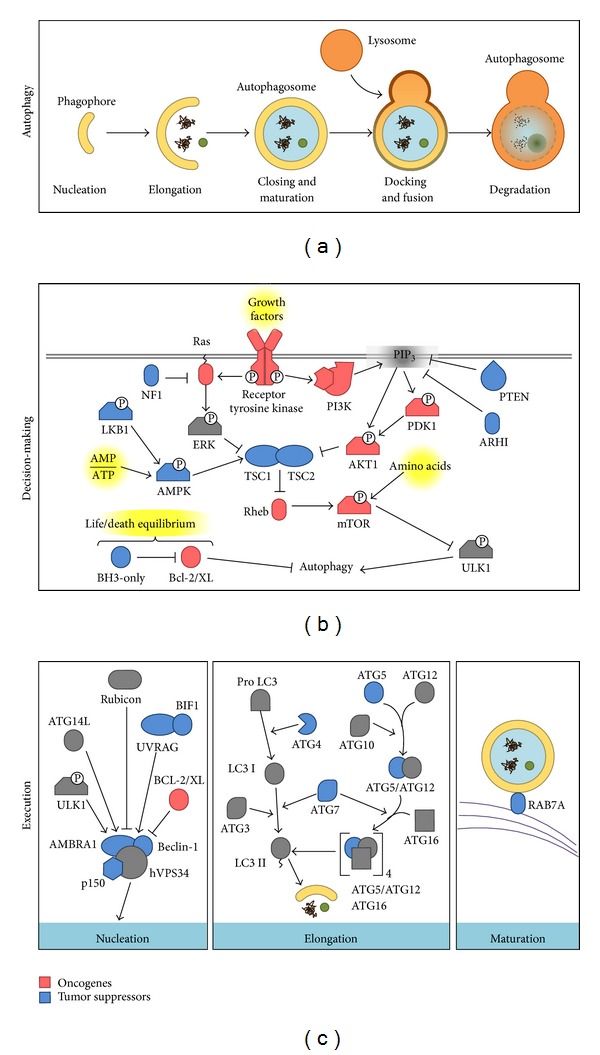
Phases of autophagy and its regulation by oncogenes and tumor suppressors. In (a), the five stages of autophagy are summarized. In (b), inhibition of autophagy by oncogenes (in red) and activation by tumor suppressors (in blue) is shown. Finally, (c) summarizes details of the complex regulation and interplay between different proteins in each stage of autophagy (see text for more details).
AMP-activated protein kinase (AMPK), another sensor of the cellular energy status, responds to decreases in ATP/AMP ratios [31]. In conditions of nutrient deprivation, AMPK directly phosphorylates and inhibits mTOR (not shown in Figure 1). The ensuing reduction in mTOR activity decreases ULK1 phosphorylation and promotes autophagosome formation [31, 32]. Moreover, AMPK also directly activates autophagy by phosphorylation of TSC2 [33, 34]. In summary, autophagy is a highly regulated process that involves a large number of modulators and the complexity of these events will increase as novel components continue to be identified in both mTOR dependent and independent pathways [35].
3. Control of Autophagy by Oncogenes and Tumor Suppressors
Most of the proteins that participate in the regulation of autophagy are either tumor suppressor proteins or oncogenes. Perhaps not surprisingly, mechanisms involved in the regulation of autophagy largely overlap with signaling pathways implicated in the control of cancer. Thus, tumor suppressor genes that negatively regulate mTOR, such as PTEN, AMPK, LKB1, and TSC1/2 stimulate autophagy while, conversely, oncogenes that activate mTOR, such as class I PI3K, Ras, RHEB, and AKT, inhibit autophagy [51] (see Table 1). In the following paragraphs, the role of Beclin-1, DAPK, Bcl2/Bcl-XL, and mTOR will be discussed briefly (see Figure 1).
Table 1.
Summary of oncogenes and tumor suppressors involved in autophagy regulation.
| Oncogenes | Role in autophagy | Evidences of oncogenesis | Reference |
|---|---|---|---|
| AKT1 | Upstream inhibitor of autophagy via mTOR activation | Gain-of-function mutations in several cancer types | [36] |
| BCL-2, BCL-XL | Sequester Beclin-1 into inactive complexes | Overexpressed in several cancer types | [37] |
| PI3K | Upstream inhibitors of autophagy via AKT1 activation | Gain-of-function mutations in many cancer types | [36, 38] |
| Ras | Upstream inhibitors of autophagy via mTOR activation | Hyperactivated in several cancer types | [37] |
|
| |||
| Tumor suppressors | Role in autophagy | Evidences of tumor suppression | |
|
| |||
| ATG4 | Converts LC3 into LC3 I during stress conditions | Mutations in ATG4C increase susceptibility to carcinogens | [39] |
| ARHI/DIRAS3, PTEN | Relieve autophagy inhibition mediated by PI3K-AKT1 | Downregulated in ovarian cancer | [36, 40] |
| Beclin-1, p150 | Required in the nucleation complex for autophagy initiation | Deleted in breast, ovarian, and prostate cancer | [41] |
| BH3-only proteins | Relieve autophagy inhibition mediated by BCL-2/BCL-XL | Mutated or silenced in many cancer types | [42–44] |
| UVRAG, BIF1 | Positive regulator of the nucleation complex | Deleted or downregulated in colorectal cancer | [45] |
| DAPK1 | Relieve autophagy inhibition mediated by BCL-2/BCL-XL | Silenced in many tumor types | [46] |
| LKB1/STK11 | Promotes autophagy via AMPK activation | Mutated in Peutz-Jeghers syndrome and non-small cell lung carcinomas | [47, 48] |
| NF1 | Relieve autophagy inhibition mediated by Ras | Mutated in neurofibromatosis, juvenile myelomonocytic leukemia | [37] |
| RAB7A | Modulates endosomal trafficking involved in autophagosome maturation | Rearranged in leukemia, deleted in solid tumors | [49] |
| TSC1, TSC2 | Stimulate Rheb GTPase, thus inhibiting the PI3K-AKT1-mTOR pathway | Mutated in TSC | [50] |
Consistent with this view, Beclin-1, which is part of the class III PI3K complex that promotes autophagy, functions as a tumor suppressor in mammalian cells. Interestingly, monoallelic mutations in the beclin-1 gene are frequently observed in prostate, ovarian, and breast cancers in humans. In addition, studies in mice have demonstrated that the animals are more sensitive to spontaneous tumor development when beclin-1 is monoallelically disrupted. These observations provide direct evidence for a role of beclin-1 as a haploinsufficient tumour suppressor gene implicated in the pathogenesis of several human cancers [41, 52–54]. Additionally, the death-associated protein kinase, DAPK, a protein that phosphorylates Beclin-1 thereby disrupting Beclin-1/BCL-2 complex and favoring autophagy, is another inducer of autophagy that is commonly silenced in different types of human cancers by methylation [55].
BCL-2 and BCL-XL are antiapoptotic members of the BCL-2 family that modulate cell death in an autophagy-independent manner and are overexpressed in several hematological malignancies [56]. There, BCL-2 and BCL-XL suppress cell death and promote survival and growth of cancer cells by suppression of BAK/BAX-dependent pore formation during mitochondrial outer membrane permeabilization (MOMP) [57]. In addition to the role of BCL-2 and BCL-XL in the inhibition of apoptosis, they have also been implicated in oncogenesis as negative regulators of autophagy. Although these proteins do not directly participate in mTOR signaling, they can interact with the Beclin-1 BH3 domain and sequester Beclin-1 into an inactive complex in the ER [58, 59].
The protein kinase mTOR is the major negative regulator of autophagy [60]. This kinase participates in multiple signaling pathways that regulate cell growth, especially downstream of growth factor receptors with tyrosine kinase activity. Interestingly, both the constitutive activation of these receptors, as well as activating mutations of downstream elements in these pathways (Ras, PI3-K, AKT, and PDK-1) or mutations that inactivate negative regulators (TSC1/2, LKB1, and PTEN) are common in the development of cancer [36, 38, 47, 48, 50], suggesting that inhibition of autophagy likely contributes to the onset to tumor development.
4. Autophagy as a Tumor Suppressor Mechanism
The first data pointing towards the possible tumor suppressor role of autophagy were obtained in studies of Beclin-1. Monoallelic loss of the beclin-1 gene on chromosome 17q21 has been reported in 40% to 75% of human breast, ovary, and prostate tumors, suggesting that autophagy represents a tumor suppressor mechanism [41]. Also, a reduction in Beclin-1 protein levels has been observed in various brain cancers [61]. Accordingly, Beclin-1+/− mice have a high incidence of spontaneous tumors, especially lymphoma and hepatocellular carcinoma. Furthermore, the evidence provided suggests that Beclin-1 functions as a haploinsufficient tumor suppressor gene, given that the tumors continued to express Beclin-1 [53, 54]. Moreover, immortalized breast epithelial cells with a monoallelic deletion of Beclin-1 form tumors more rapidly after inoculation into nude mice [62]. More recently, phosphorylation of Beclin-1 on multiple tyrosine residues in an EGFR-dependent manner was found to decrease the activity of the Beclin-1/PI3KC3 complex and therefore decreased autophagy in non-small-cell lung carcinoma cells (NSCLC) and that this effect was reduced in the presence of an inhibitor of EGFR kinase activity. Alternatively, the expression of a tyrosine phosphomimetic mutant of Beclin-1 reduces autophagy and increases tumor growth [63]. Similarly, several proteins that interact with Beclin-1 and positively regulate autophagy, such as AMBRA 1 [64], BIF-1 [45], and UVRAG [65], have been shown to display antiproliferative or tumor suppressor effects. However, a complication here is that all these proteins have other functions that are independent of their role in autophagy, for example in regulating the endocytic pathway [66]. Moreover, the Beclin-1/PI3KC3 complex also controls the ubiquitination and degradation of p53 by regulating the stability and activity of the deubiquitinating enzymes USP13 and USP10 [67]. Given these additional functions, the contribution of such autophagy-independent mechanisms to the observed tumor suppressor phenotype cannot be excluded.
In agreement with the tumor suppressor hypothesis, the generation of knockout mice for specific genes involved in autophagy (ATGs) has shown that defects in specific regulators of this process are associated with the development of a tumorigenic phenotype. Because systemic deletion of Atg3, Atg5, Atg7, Atg9, or Atg16L1 causes neonatal death [68–72], long-term effects of the inhibition of autophagy could not be assessed until mice with systemic mosaic Atg5 deletion were generated. In this background, systemic mosaic Atg5 deletion or liver-specific deletion of Atg7 results in mice that spontaneously develop benign liver adenomas [73]. While these data suggest that defects in autophagy promote the development of benign tumors in this tissue, they also indicate that, in the absence of autophagy, progression to a malignant phenotype is prevented. Similarly, Strohecker et al. showed that the deletion of Atg7 in mice expressing an activating mutation of B-Raf (Braf V600E/+) promotes early tumor development in the lung but also inhibits the progression to a more malignant phenotype and increases mouse survival [74]. Additional autophagy-promoting factors that have tumor suppressor functions are Atg4C and RAB7A. For animals deficient in Atg4C, increased susceptibility to the development of fibrosarcomas induced by chemical carcinogens was detected [39]. RAB7A has been shown to prevent growth factor-independent survival by inhibiting cell-autonomous nutrient transporter expression and the RAB7A gene is frequently rearranged in different types of leukemia [49, 75].
Despite this evidence that favors a role for autophagy in tumor suppression, some more recent findings concerning Beclin-1 contrast with the previous interpretation of data. The beclin-1 gene lies close to BRCA1 on chromosome 17q21 raising the specter that the relevance of the loss of Beclin-1 in ovarian, breast, and prostate cancer may have been overinterpreted. Indeed, deletions encompassing both genes (BRCA1 and beclin-1) and deletions of only BRCA1 but not beclin-1 were found in breast and ovarian cancers, which is consistent with BRCA1 loss representing the primary driver mutation in these cancers. Furthermore, no evidence for beclin-1 mutations or loss have been detected in any other cancer, which questions whether beclin-1 is indeed a tumor suppressor in various human cancers [76]. Taken together, the evidence presented supports the hypothesis that autophagy may play an important role in tumor suppression at early stages. However, the findings discussed also reveal the potentially dual nature of this process in tumor development and progression.
4.1. Mechanisms Involved in Tumor Suppression by Autophagy
4.1.1. Oxidative Stress and Genomic Instability
One of the most important connections between autophagy and tumor suppression is via the regulation of reactive oxygen species (ROS). Increased ROS production accelerates mutagenesis, increasing the activation of oncogenes, thus stimulating carcinogenesis [77, 78]. Mitochondria are considered the main source of intracellular ROS and their production increases as these organelles age or become damaged [79]. In this context, autophagy helps to avoid damage through selective degradation of defective mitochondria, a process known as mitophagy. Consequently, inhibition of autophagy facilitates genomic instability by promoting the activation of oncogenes [62, 80] and genotoxic effects observed in autophagy-defective cells seem to be dependent on ROS generation [81]. Thus, the selective removal of potentially damaged mitochondria (mitophagy) reduces excessive ROS production and thereby limits tumor-promoting effects dependent on the production of such species [82]. Accordingly, inhibition of autophagy in different models leads to accumulation of defective mitochondria [69, 73, 74, 83–85].
Autophagy also permits the degradation of protein aggregates. Defects in the autophagic process have been associated with the accumulation of protein aggregates and the autophagy substrate p62/SQSTM1. Such events are associated with increased production of ROS, ER stress, and activation of the DNA damage response [81]. The p62 protein is a selective autophagy substrate that accumulates when autophagy is reduced. This scaffolding protein contains a PB1 domain that permits protein oligomerization, an UBA domain required for binding to polyubiquitinated proteins and an LIR domain (LC3-interacting region) necessary for association with LC3. For these reasons, p62 favors selective degradation of both polyubiquitinated proteins and organelles (i.e., mitochondria) [86, 87]. Interestingly, p62 levels are commonly elevated in human tumors. In addition, tumorigenic development observed in autophagy-deficient cells is reversed by genetic inactivation of p62 in various models, suggesting that the accumulation of p62 promotes tumor formation in this context [73, 81, 83, 88]. Moreover, p62 accumulation stabilizes and activates the transcription via NRF-2, by binding to Keap-1, the main negative regulator of NRF-2. In doing so, antioxidant defense is upregulated and may contribute to tumor development [88–90]. Specifically, overexpression of p62 and activation of NRF-2 are critical for anchorage-independent growth observed in hepatocellular carcinoma cells [88].
4.1.2. Inflammation and Necrosis
The tumor microenvironment is defined by complex interactions between various cell types that coexist within tumors (tumor and stromal cells) and crosstalk between these cells regulates both tumor growth and progression. In this context, it is important to note that both inflammatory cells and cytokines are extremely relevant because a proinflammatory environment promotes proliferation and survival of malignant cells, stimulates angiogenesis, metastasis, and modifies the response to drugs [91]. In different models, autophagy inhibition in apoptosis-deficient tumor cells has been shown to promote necrotic cell death, local inflammation, and tumor growth [92]. These results suggest that autophagy may contribute to tumor suppression by restricting tumor necrosis and local inflammation [60]. The anti-inflammatory effect of autophagy has been suggested to be linked to the removal of cell corpses [93] because of findings in Atg5 −/− embryonic stem cells, where defects in the clearance of apoptotic bodies during embryonic development are observed [94]. Moreover, a complex connection between autophagy and different aspects of the immune response has been noted, which could contribute to the tumor suppressor role of autophagy, as has been reviewed elsewhere [95].
4.1.3. Autophagic Cell Death and Senescence
Although autophagy is primarily considered a mechanism that permits survival under stress conditions, some reports indicate that, under specific conditions, an increase in autophagic flux may cause cell death due to autophagy and explain in part the tumor suppressor effects [96]. The findings of Pattingre et al. revealed that the expression of a mutant Beclin-1, unable to interact with BCL-2, induced autophagy to a greater extent than wild-type Beclin-1, and unlike the latter, it promoted cell death [58]. More recently, studies in an ovarian cancer cell line showed that ectopic expression of Ras induces autophagic cell death through the upregulation of Beclin-1 and Noxa, a BH3-only protein, which ultimately limits the oncogenic potential of Ras [97]. Similarly, Zhao and colleagues demonstrated that the transcription factor FoxO1 promotes autophagy in a manner independent of its transcriptional activity and induces autophagic cell death in tumor cells, suppressing tumor growth of xenografts in nude mice. These results suggest that autophagy promoted by cytosolic FoxO1 is a tumor suppressor mechanism [98, 99].
Another controversial mechanism that may potentially explain the tumor suppressor activity of autophagy is its role in senescence. Young et al. [100] showed that autophagy is activated during senescence induced by the oncogene Ras in fibroblasts and that inhibition of autophagy in this context delays but does not abrogate the development of the oncogene-mediated senescence phenotype. This finding is important because senescence represents a major intrinsic barrier to malignant transformation [101] although this barrier function may only be transient. In studies of senescence induced by chemotherapy in breast and colon cancer cell lines, autophagy and senescence occur in parallel but not in an interdependent manner. In fact, senescence was only transiently subdued and subsequently recovered despite prolonged inhibition of autophagy [102]. Similar results have been obtained in different experimental settings and are discussed in [103].
5. Autophagy as a Promoter of Tumor Growth
5.1. Autophagy Promotes Cell Survival under Conditions of Stress
The notion that autophagy represents a mechanism that promotes tumor growth is based on the necessity of tumoral cells to adapt to ischemia in an environment that is hypoxic, as well as growth factor and nutrient deprived. Consistent with this conundrum, autophagy is activated in hypoxic regions of tumors and inhibition of autophagy by monoallelic deletion of beclin-1 (Bcn1 +/−) promotes cell death specifically in those regions. These observations suggest a role for autophagy in promoting survival of tumor cells under conditions of metabolic stress [92].
Furthermore, tumor cells generally have high proliferation rates, which translate into higher bioenergetic and biosynthetic needs than is the case for non-transformed cells. These requirements can be satisfied by increasing autophagy as a mechanism that permits obtaining both ATP and metabolic intermediates [66]. Importantly, for tumor cells in which the oncogene Ras is activated, high levels of basal autophagy and dependence on this mechanism for survival are observed [83, 85]. For these reasons, autophagy is thought to promote tumor cell survival by increasing stress tolerance and providing a pathway that permits obtaining the nutrients necessary to meet the enhanced energetic requirements of these cells [66].
5.2. Ras-Dependent Tumor Progression and Autophagy Addiction
Small GTPases of the Ras family are involved in signaling pathways important for proliferation, cell survival, and metabolism. RAS-activating mutations are present in 33% of all human cancers, whereby mutations in KRas are most prevalent and linked to the development of some of the most lethal cancers, including those of the lung, colon, and pancreas [104, 105]. In human pancreatic cancer, enhanced levels of active autophagy and LC3 correlate with poor patient prognosis [106]. In vitro studies shown in several cell lines with Ras-activating mutations revealed that these cells exhibit high levels of basal autophagy and marked autophagy-dependent survival under conditions of nutrient deprivation. Moreover, silencing of proteins involved in autophagy promotes the accumulation of dysfunctional mitochondria, low oxygen consumption, and decreased cell growth [83, 85].
In vivo studies confirm the aforementioned results. In a KRas-driven pancreatic cancer model, inhibition of autophagy by Atg5 deletion, decreased the capacity of preneoplastic lesions to progress to invasive cancer, in a manner that was independent of the p53 status [107]. Rosenfeldt et al. reported similar results using a mouse model of humanized pancreatic cancer, but they demonstrated that p53 deletion precludes tumor progression promoted by autophagy. Therefore, the role of autophagy in pancreatic cancer progression may depend on the presence of p53 [108]. In a model of KRas-dependent NSCLC, the inhibition of autophagy through inducible Atg7 deletion leads to abnormal accumulation of mitochondria and decreases in proliferation and necrotic cell death, which in combination translated into reduced tumor burden. Moreover, in this same study, the absence of Atg7 resulted in progression of Ras-induced adenomas and adenocarcinomas to oncocytomas, benign tumors characterized by accumulation of dysfunctional mitochondria [84]. In studies following up on the role of the Ras pathway in tumor promotion and its dependence on autophagy, Strohecker et al. investigated whether pulmonar carcinogenesis driven by an activating B-Raf mutation was dependent on autophagy. Atg7 deletion increased oxidative stress and enhanced tumor growth at early stages, but promoted abnormal mitochondria accumulation, proliferation defects, a decrease in tumor burden, and increased survival of animals in more advanced stages of tumorigenesis. Furthermore, when cell lines derived from these tumors were supplemented with glutamine, nutrient deprivation-induced cell death was prevented, suggesting that metabolic stress due to mitochondrial dysfunction may be related to the sensitivity of autophagy-deficient cells to nutrient deprivation [74]. Finally, an in vitro study demonstrated that adhesion-independent growth promoted by Ras was dependent on autophagy. Specifically, upon inhibition of autophagy in different cell lines with Ras-activating mutations, the ability of cells to grow in an anchorage-independent manner was lost. These observations underscore the importance of autophagy in maintaining glycolytic activity, which facilitates Ras-mediated anchorage-independent cell growth [109].
5.3. Mechanisms Involved in Autophagy Addiction in Ras-Driven Tumors
The dependence on autophagy in Ras-/Raf-driven tumoral cells is explained by the decrease in the acetyl-CoA pool necessary to fuel the tricarboxylic acid (TCA) cycle. Ras activation modulates mitochondrial metabolism by inducing hypoxia-inducible factor (HIF)-1α-dependent expression of lactate dehydrogenase (LDH) expression [110], which converts pyruvate to lactate resulting in reduced acetyl-CoA synthesis and, hence, acetyl-CoA depletion. In addition, the Raf/Erk pathway promotes inactivation of LKB1 and subsequently AMPK, thereby preventing β-oxidation [111] and decreasing the available mitochondrial acetyl-CoA pool. Due to acetyl-CoA depletion, Ras/Raf-driven tumors require autophagy in order to obtain TCA cycle intermediates. These in turn promote mitochondrial activity, which provides reductive equivalents necessary for oxidative phosphorylation and mitochondrial respiration. Moreover, in Ras-driven tumors, autophagy inhibition promotes the accumulation of dysfunctional mitochondria. This kind of cancer requires autophagy to maintain a pool of functional mitochondria necessary for enhanced energetic requirements of tumoral cells [83, 85].
Beyond the requirement of autophagy for survival of Ras-driven cancer cells, Ras activation also promotes cell signaling events involved in the induction of autophagy by the upregulation of Noxa and Beclin-1 expression [97]. Furthermore, Ras can directly stimulate BNIP3 expression through activation of the Ras/Raf/Erk pathway or indirectly through HIF-1α induction [112–114].
5.4. Autophagy in Ras-Independent Tumor Progression
The role of autophagy also has been studied in different contexts that are independent of Ras. For instance, in a model of breast cancer driven by the PyMT oncogene, the inhibition of autophagy by FIP200 deletion suppresses mammary tumor initiation and progression. Here, FIP200 ablation increased the number of mitochondria with abnormal morphology in tumor cells and reduced significantly proliferation, but it did not affect apoptosis of mammary tumor cells [115]. Although PyMT requires Ras activation to initiate cell transformation, PI3-kinase and Src activation are also involved [116]. Thus, it would be interesting to determine whether these kinases contribute to dependence on autophagy for cell proliferation. Another study employed a Palb2 knockout model specific to epithelial breast cells to determine the role of autophagy in breast cancer progression. PALB2 is a protein that cooperates with BRCA1 and BRCA2 in DNA repair via homolog recombination and helps maintain genomic stability. Palb2 knockout mice develop breast adenocarcinoma when p53 is mutated. Partial inhibition of autophagy by monoallelic loss of Beclin-1 (Bcn1 +/−) increased apoptosis and delayed tumor growth in a manner dependent on p53. The authors proposed that autophagy promotes tumor growth by p53 suppression when DNA is damaged [117]. These studies indicate that autophagy can promote tumor progression in a manner independent of Ras activation and that autophagy could be a more general mechanism involved in cancer cell survival and tumor progression.
In summary, current evidence points towards autophagy as a mechanism that ensures adequate mitochondrial metabolism in Ras-driven cancers by supplying mitochondrial intermediates via the degradation of macromolecules under basal and starvation conditions [118]. Particularly, Ras-driven tumorigenesis appears to be “addicted to autophagy” for metabolic support and maintenance of rapid tumor growth. All these data explain why autophagy is required in Ras-driven cancers to promote tumor cell survival and tumor progression. Interestingly, however, some more recent evidence indicates that autophagy is also important for tumoral cell survival of other cancers, independent of the Ras activation status.
6. Caveolin-1, a Connection to Autophagy?
Caveolin-1 (CAV1) is a scaffolding protein that is essential for caveolae formation, is expressed in a wide variety of tissues, and is involved in many biological processes, including cholesterol homeostasis, vesicular transport, and signal transduction. Moreover, similar to autophagy, CAV1 plays a dual role in cancer, functioning both as a tumor suppressor and promoter of tumor metastasis [119–121]. Although, E-cadherin has been identified as important in determining CAV1 function in this context [122–125], the molecular mechanisms explaining such ambiguous behavior remain largely undefined.
Given the parallels between the roles of CAV1 and autophagy in cancer, it is intriguing to speculate that there might be a connection between the two. Indeed, Martinez-Outschoorn et al. demonstrated, using a coculture system, that CAV1 is degraded via lysosomes in stromal fibroblasts subjected to hypoxia and that this correlated with increased levels of autophagic markers such as LC3, ATG16L, BNIP3, BNIP3L, HIF-1α, and NF-kB. Moreover, knockdown of CAV1 in stromal fibroblasts was sufficient to induce the upregulation of lysosomal and autophagic markers, suggesting that the loss of CAV1 in the stromal compartment induces autophagy [126]. Also, loss of CAV1 leads to metabolic reprogramming of stromal cells to support the growth of adjacent tumor cells by delivering energy-rich metabolites and essential building blocks [127]. Consistent with the notion that CAV1 is a negative regulator of autophagy, CAV1 depletion in HCT116 colorectal cancer cells was shown to reduce glucose uptake and ATP production, which then triggered autophagy via activation of AMPK-p53 signaling [128]. Moreover, both in vitro cell growth and in vivo xenograft tumor growth were attenuated to a greater extent by CAV1 depletion in p53+/+ than in p53−/− cells [128].
An inverse relationship between autophagy and CAV1 has also been observed in models of nontransformed cells. For instance, metabolomic profiling of endothelial cell lysates following transfection with si-CAV1 or si-control resulted in marked increases in dipeptide levels for the CAV1 knockdown cells, which was attributed to an increase in autophagy [129]. To corroborate these results, the authors evaluated the processing of LC3 I to LC3 II by western blotting and showed that siRNA-mediated CAV1 knockdown led to an increase in the presence of the autophagy marker LC3-II. Also, treatment with the lysosomal inhibitor bafilomycin A1 markedly increased LC3-II levels, indicating that reduced CAV1 expression leads to an increase in autophagy flux [129]. Recently, CAV1 was also shown to regulate autophagy in cigarette smoking-induced injury of lung epithelium [130]. Specifically, CAV1 depletion increased basal and starvation-induced levels of ATG12-ATG5 and autophagy. Biochemical analysis revealed that CAV1 interacted with ATG5, ATG12, and the active ATG12-ATG5 complex to suppress autophagy in lung epithelial cells, thereby providing new insights as to how CAV1 modulates autophagy in this model [130]. However, details of the molecular mechanisms by which CAV1 regulates autophagy in cancer cells remain to be determined. A rather speculative idea is that the dual role of CAV1 in cancer may be linked to its participation in the control of autophagy. However, further experimentation is required to corroborate this intriguing hypothesis.
In summary, CAV1, a membrane protein typically implicated in the formation of cell surface structures like caveolae and regulation of signalling, also plays a dual role in cancer, functioning as a tumor suppressor at early stages and a tumor promoter later on. The future will reveal how the seemingly opposing roles of autophagy in tumor development and progression are controlled, and to what extent the ambiguous role of CAV1 in cancer may be linked to the control of autophagy.
7. Conclusions
Autophagy is an evolutionarily conserved mechanism that developed in eukaryotes to ensure protein and organelle homeostasis. A hallmark of cancer cells is their increased proliferation and as a consequence their demand for energy equivalents and specific metabolites, which can be provided by autophagy. In this context, autophagy favors tumor cell development, adaptation, and progression, and particularly some oncogene-driven tumors are “addicted” to autophagy in this respect. However, autophagy also appears to have a tumor suppressor function early in cancer development by eliminating damaged mitochondria and reducing ROS-mediated genotoxic damage (see Figure 2). Accordingly, pharmacological modulation of autophagy in established tumors may represent an important anticancer therapy, as is supported by the use of autophagy inhibitors (chloroquine or hydroxychloroquine) in a large number of clinical trials and currently as a treatment for various kinds of cancers that are generally very aggressive or resistant to therapy (see Table 2).
Figure 2.
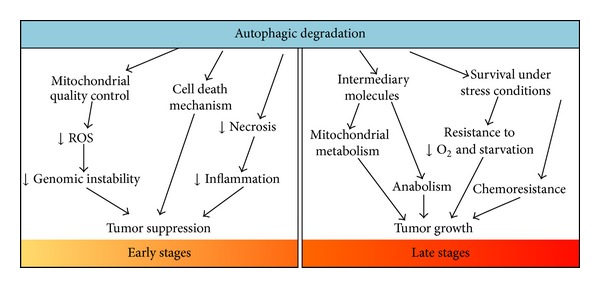
The two facets of autophagy in cancer. At early stages, autophagy acts as a tumor suppressor mechanism by enhancing the degradation of damaged proteins and organelles, mostly mitochondria. In doing so, autophagy acts as a quality control system that decreases ROS production and genomic instability. Moreover, autophagy prevents necrotic cell death in apoptosis-defective cells, thereby reducing local inflammation and tumor growth. Also, autophagy may serve (in some cases) as a mechanism that leads to cell death. On the other hand, at later stages of tumor development, activation of autophagy supplies tumor cells under metabolic stress conditions with nutrients and also maintains mitochondrial metabolism by providing metabolic intermediates, which promote cell survival and tumor growth. Finally, autophagy acts as a mechanism that promotes resistance to cancer therapy.
Table 2.
Summary of clinical trials involving autophagy inhibitors (chloroquine or hydroxychloroquine) for cancer treatment (data obtained from http://www.cancer.gov/clinicaltrials).
| Cancer type | Therapy | Phase | Status | Protocol ID |
|---|---|---|---|---|
| Relapsed and refractory multiple myeloma | Cyclophosphamide and pulse dexamethasone with hydroxychloroquine or rapamycin | 0 | Completed | NCT01396200 |
|
| ||||
| Glioblastoma multiforme | Hydroxychloroquine, radiation, and temozolimide | I, II | Closed | NCT00486603 |
|
| ||||
| Pancreas adenocarcinoma | Hydroxychloroquine, gemcitabine | I, II | Closed | NCT01128296 |
|
| ||||
| Prostate cancer | Hydroxychloroquine after prostate cancer treatment | II | Closed | NCT00726596 |
|
| ||||
| Non-small cell lung cancer | Erlotinib with or without hydroxychloroquine | II | Closed | NCT00977470 |
|
| ||||
| Metastatic pancreatic cancer | Hydroxychloroquine after prostate cancer treatment | II | Closed | NCT01273805 |
|
| ||||
| Relapsed and refractory multiple myeloma | Chloroquine, bortezomib, and cyclophosphamide | II | Closed | NCT01438177 |
|
| ||||
| Advanced solid tumors irresponsive to chemotherapy | Hydroxychloroquine, sunitinib | I | Closed | NCT00813423 |
|
| ||||
| B-cell chronic lymphocytic leukemia | Hydroxychloroquine | II | Temporarily Closed | NCT00771056 |
|
| ||||
| Surgery removable Stage III or Stage IV melanoma | Hydroxychloroquine | 0 | Temporarily Closed | NCT00962845 |
|
| ||||
| Relapsed and refractory multiple myeloma | Hydroxychloroquine, bortezomib | I, II | Active | NCT00568880 |
|
| ||||
| Lung cancer | Hydroxychloroquine, gefitinib | I, II | Active | NCT00809237 |
|
| ||||
| Ductal carcinoma in situ | Chloroquine | I, II | Active | NCT01023477 |
|
| ||||
| Colorectal cancer | Hydroxychloroquine, folinic acid, 5-fluorouracil, oxaliplatin, and bevacizumab | I, II | Active | NCT01206530 |
|
| ||||
| Pancreatic cancer | Hydroxychloroquine, protein-bound paclitaxel, and gemcitabine | I, II | Active | NCT01506973 |
|
| ||||
| Previously treated renal cell carcinoma | Hydroxychloroquine, everolimus | I, II | Active | NCT01510119 |
|
| ||||
| Renal cell carcinoma | Hydroxychloroquine, aldesleukin | I, II | Active | NCT01550367 |
|
| ||||
| Unresectable hepatocellular carcinoma | Hydroxychloroquine, transarterial chemoembolization (TACE) | I, II | Active | NCT02013778 |
|
| ||||
| Metastatic colorectal cancer | Hydroxychloroquine, capecitabine, oxaliplatin, and bevacizumab | II | Active | NCT01006369 |
|
| ||||
| Chronic myeloid leukemia | Imatinib mesylate with or without hydroxychloroquine | II | Active | NCT01227135 |
|
| ||||
| Breast cancer | Hydroxychloroquine | II | Active | NCT01292408 |
|
| ||||
| Advanced or metastatic breast cancer | Chloroquine, taxane | II | Active | NCT01446016 |
|
| ||||
| Resectable pancreatic cancer | Hydroxychloroquine, capecitabine, and radiation | II | Active | NCT01494155 |
|
| ||||
| High grade gliomas | Hydroxychloroquine, radiation | II | Active | NCT01602588 |
|
| ||||
| Advanced/recurrent non-small cell lung cancer | Hydroxychloroquine, paclitaxel, carboplatin, and bevacizumab | II | Active | NCT01649947 |
|
| ||||
| Progressive metastatic castrate refractory prostate cancer | Navitoclax, abiraterone acetate with or without hydroxychloroquine | II | Active | NCT01828476 |
|
| ||||
| Soft tissue sarcoma | Hydroxychloroquine, rapamycin | II | Active | NCT01842594 |
|
| ||||
| Potentially resectable pancreatic cancer | Protein-bound paclitaxel, gemcitabine with or without hydroxychloroquine | II | Active | NCT01978184 |
|
| ||||
| Metastatic or unresectable solid tumors | Hydroxychloroquine, temozolomide | I | Active | NCT00714181 |
|
| ||||
| Irresponsive metastatic solid tumors | Hydroxychloroquine, temsirolimus | I | Active | NCT00909831 |
|
| ||||
| Stage IV small cell lung cancer | Chloroquine | I | Active | NCT00969306 |
|
| ||||
| Advanced solid tumors | Hydroxychloroquine, vorinostat | I | Active | NCT01023737 |
|
| ||||
| Primary renal cell carcinoma | Hydroxychloroquine before surgery | I | Active | NCT01144169 |
|
| ||||
| Advanced cancer | Hydroxychloroquine sirolimus, or vorinostat | I | Active | NCT01266057 |
|
| ||||
| Solid tumors | Hydroxychloroquine, radiation | I | Active | NCT01417403 |
|
| ||||
| Melanoma | Chloroquine, radiation, DT01 | I | Active | NCT01469455 |
|
| ||||
| Advanced solid tumors, melanoma, prostate, or kidney cancer | Hydroxychloroquine, Akt inhibitor MK2206 | I | Active | NCT01480154 |
|
| ||||
| Stages I–III small cell lung cancer | Chloroquine, radiation | I | Active | NCT01575782 |
|
| ||||
| Refractory or relapsed solid tumors | Hydroxychloroquine, sorafenib | I | Active | NCT01634893 |
|
| ||||
| Lymphangioleiomyomatosis in women | Hydroxychloroquine sirolimus | I | Active | NCT01687179 |
|
| ||||
| Relapsed or refractory multiple myeloma | Hydroxychloroquine, cyclophosphamide, dexamethasone, and sirolimus | I | Active | NCT01689987 |
|
| ||||
| Nonresectable pancreatic adenocarcinoma | Chloroquine, gemcitabine | I | Active | NCT01777477 |
|
| ||||
| BRAF mutant metastatic melanoma | Hydroxychloroquine, vemurafenib | I | Active | NCT01897116 |
|
| ||||
| Advanced solid tumors | Chloroquine, carboplatin, and gemcitabine | I | Active | NCT02071537 |
|
| ||||
| Brain metastasis | Chloroquine, radiation | 0 | Active | NCT01727531 |
Alternatively, considering the potential tumor suppressor role of autophagy in early stages of cancer development, one may speculate that stimulation of this process could be useful as a preventive mechanism against the development of cancer. Consistent with this notion, caloric restriction has been shown to prolong life span and reduce cancer incidence in several animal models [131]. Also, treatments with metformin, an activator of the AMPK pathway that stimulates autophagy, are associated with lower risk of different kinds of cancers [132].
Clearly, the role of autophagy in cancer depends on many factors like tissue type, tumor stage, and the type of oncogenic mutation involved. Because of these dramatic differences, more research is required to understand the role of autophagy in cancer biology and how we may harness such knowledge to improve cancer therapies and patient survival.
Acknowledgments
This work was supported by CONICYT-FONDAP 15130011 (Andrew F. G. Quest and Sergio Lavandero), Anillo Project ACT1111 (Andrew F. G. Quest and Sergio Lavandero), FONDECYT 1130250 (Andrew F. G. Quest), FONDECYT 1140908 (Alfredo Criollo), and CONICYT PhD scholarships (Yenniffer Ávalos, Jimena Canales, and Roberto Bravo-Sagua).
Abbreviations
- AMBRA1:
Autophagy/Beclin-1 regulator 1
- AMPK:
AMP-activated protein kinase
- ATG:
Autophagy-related
- BCL-2:
B-cell lymphoma 2
- BCL-XL:
B-cell lymphoma-extra large
- BIF-1:
BAX-interacting factor 1
- BNIP3:
BCL2/adenovirus E1B 19 kDa interacting protein 3
- BNIP3L:
BCL2/adenovirus E1B 19 kDa interacting protein 3-like
- BRCA1:
Breast cancer 1, early onset
- BRCA2:
Breast cancer 2, early onset
- ER:
Endoplasmic reticulum
- FIP200:
FAK family-interacting protein of 200 kDa
- FoxO:
Forkhead box O
- LAMP:
Lysosomal-associated membrane protein
- LC3:
Microtubule-associated protein 1 light chain 3 (homolog of yeast Atg8)
- LKB1:
Liver kinase B1
- mTOR:
Mammalian target of rapamycin
- NF-κB:
Nuclear factor kappa-light-chain-enhancer of activated B cells
- NRF-2:
Nuclear factor erythroid 2-related factor 2
- PALB2:
Partner and localizer of BRCA2
- PI3KC3/VSP34:
Phosphatidylinositol 3-kinase, catalytic subunit type 3
- PI3K:
Phosphatidylinositol 3-kinase
- PTEN:
Phosphatase and tensin homolog
- PyMT:
Polyoma middle T-antigen
- ULK:
Unc-51 like autophagy activating kinase
- UVRAG:
Ultraviolet radiation resistance-associated gene.
Conflict of Interests
The authors declare that there is no conflict of interests regarding the publication of this paper.
References
- 1.Vineis P, Wild CP. Global cancer patterns: causes and prevention. The Lancet. 2014;383(9916):549–557. doi: 10.1016/S0140-6736(13)62224-2. [DOI] [PubMed] [Google Scholar]
- 2.Are C, Rajaram S, Are M, et al. A review of global cancer burden: trends, challenges, strategies, and a role for surgeons. Journal of Surgical Oncology. 2013;107(2):221–226. doi: 10.1002/jso.23248. [DOI] [PubMed] [Google Scholar]
- 3.Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100(1):57–70. doi: 10.1016/s0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]
- 4.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144(5):646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 5.Klionsky DJ. Autophagy revisited: a conversation with Christian de Duve. Autophagy. 2008;4(6):740–743. doi: 10.4161/auto.6398. [DOI] [PubMed] [Google Scholar]
- 6.Jardon MA, Rothe K, Bortnik S, et al. Autophagy: from structure to metabolism to therapeutic regulation. Autophagy. 2013;9(12):2180–2182. doi: 10.4161/auto.26378. [DOI] [PubMed] [Google Scholar]
- 7.Hayashi-Nishino M, Fujita N, Noda T, Yamaguchi A, Yoshimori T, Yamamoto A. A subdomain of the endoplasmic reticulum forms a cradle for autophagosome formation. Nature Cell Biology. 2009;11(12):1433–1437. doi: 10.1038/ncb1991. [DOI] [PubMed] [Google Scholar]
- 8.Hayashi-Nishino M, Fujita N, Noda T, Yamaguchi A, Yoshimori T, Yamamoto A. Electron tomography reveals the endoplasmic reticulum as a membrane source for autophagosome formation. Autophagy. 2010;6(2):301–303. doi: 10.4161/auto.6.2.11134. [DOI] [PubMed] [Google Scholar]
- 9.Hamasaki M, Furuta N, Matsuda A, et al. Autophagosomes form at ER-mitochondria contact sites. Nature. 2013;495(7441):389–393. doi: 10.1038/nature11910. [DOI] [PubMed] [Google Scholar]
- 10.Tooze SA, Yoshimori T. The origin of the autophagosomal membrane. Nature Cell Biology. 2010;12(9):831–835. doi: 10.1038/ncb0910-831. [DOI] [PubMed] [Google Scholar]
- 11.Parzych KR, Klionsky DJ. An overview of autophagy: morphology, mechanism, and regulation. Antioxidants and Redox Signaling. 2014;20(3):460–473. doi: 10.1089/ars.2013.5371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Efeyan A, Zoncu R, Sabatini DM. Amino acids and mTORC1: from lysosomes to disease. Trends in Molecular Medicine. 2012;18(9):524–533. doi: 10.1016/j.molmed.2012.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kroemer G, Mariño G, Levine B. Autophagy and the integrated stress response. Molecular Cell. 2010;40(2):280–293. doi: 10.1016/j.molcel.2010.09.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.O'Farrell F, Rusten TE, Stenmark H. Phosphoinositide 3-kinases as accelerators and brakes of autophagy. FEBS Journal. 2013;280(24):6322–6337. doi: 10.1111/febs.12486. [DOI] [PubMed] [Google Scholar]
- 15.Funderburk SF, Wang QJ, Yue Z. The Beclin 1-VPS34 complex—at the crossroads of autophagy and beyond. Trends in Cell Biology. 2010;20(6):355–362. doi: 10.1016/j.tcb.2010.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhong Y, Wang QJ, Li X, et al. Distinct regulation of autophagic activity by Atg14L and Rubicon associated with Beclin 1-phosphatidylinositol-3-kinase complex. Nature Cell Biology. 2009;11(4):468–476. doi: 10.1038/ncb1854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Polson HEJ, De Lartigue J, Rigden DJ, et al. Mammalian Atg18 (WIPI2) localizes to omegasome-anchored phagophores and positively regulates LC3 lipidation. Autophagy. 2010;6(4):506–522. doi: 10.4161/auto.6.4.11863. [DOI] [PubMed] [Google Scholar]
- 18.Proikas-Cezanne T, Robenek H. Freeze-fracture replica immunolabelling reveals human WIPI-1 and WIPI-2 as membrane proteins of autophagosomes. Journal of Cellular and Molecular Medicine. 2011;15(9):2007–2010. doi: 10.1111/j.1582-4934.2011.01339.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Klionsky DJ, Schulman BA. Dynamic regulation of macroautophagy by distinctive ubiquitin-like proteins. Nature Structural and Molecular Biology. 2014;21(4):336–345. doi: 10.1038/nsmb.2787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nakatogawa H. Two ubiquitin-like conjugation systems that mediate membrane formation during autophagy. Essays in Biochemistry. 2013;55(1):39–50. doi: 10.1042/bse0550039. [DOI] [PubMed] [Google Scholar]
- 21.Tasdemir E, Galluzzi L, Maiuri MC, et al. Methods for assessing autophagy and autophagic cell death. Methods in Molecular Biology. 2008;445:29–76. doi: 10.1007/978-1-59745-157-4_3. [DOI] [PubMed] [Google Scholar]
- 22.Eskelinen E-L, Illert AL, Tanaka Y, et al. Role of LAMP-2 in lysosome biogenesis and autophagy. Molecular Biology of the Cell. 2002;13(9):3355–3368. doi: 10.1091/mbc.E02-02-0114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ao X, Zou L, Wu Y. Regulation of autophagy by the Rab GTPase network. Cell Death and Differentiation. 2014;21(3):348–358. doi: 10.1038/cdd.2013.187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nara A, Mizushima N, Yamamoto A, Kabeya Y, Ohsumi Y, Yoshimori T. SKD1 AAA ATPase-dependent endosomal transport is involved in autolysosome formation. Cell Structure and Function. 2002;27(1):29–37. doi: 10.1247/csf.27.29. [DOI] [PubMed] [Google Scholar]
- 25.Mehrpour M, Esclatine A, Beau I, Codogno P. Overview of macroautophagy regulation in mammalian cells. Cell Research. 2010;20(7):748–762. doi: 10.1038/cr.2010.82. [DOI] [PubMed] [Google Scholar]
- 26.Zoncu R, Efeyan A, Sabatini DM. MTOR: from growth signal integration to cancer, diabetes and ageing. Nature Reviews Molecular Cell Biology. 2011;12(1):21–35. doi: 10.1038/nrm3025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hsu PP, Kang SA, Rameseder J, et al. The mTOR-regulated phosphoproteome reveals a mechanism of mTORC1-mediated inhibition of growth factor signaling. Science. 2011;332(6035):1317–1322. doi: 10.1126/science.1199498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Efeyan A, Zoncu R, Chang S, et al. Regulation of mTORC1 by the Rag GTPases is necessary for neonatal autophagy and survival. Nature. 2013;493(7434):679–683. doi: 10.1038/nature11745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chan EY. MTORC1 phosphorylates the ULK1-mAtg13-FIP200 autophagy regulatory complex. Science Signaling. 2009;2(84, article pe51) doi: 10.1126/scisignal.284pe51. [DOI] [PubMed] [Google Scholar]
- 30.Hosokawa N, Hara T, Kaizuka T, et al. Nutrient-dependent mTORCl association with the ULK1-Atg13-FIP200 complex required for autophagy. Molecular Biology of the Cell. 2009;20(7):1981–1991. doi: 10.1091/mbc.E08-12-1248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shang L, Chen S, Du F, Li S, Zhao L, Wang X. Nutrient starvation elicits an acute autophagic response mediated by Ulk1 dephosphorylation and its subsequent dissociation from AMPK. Proceedings of the National Academy of Sciences of the United States of America. 2011;108(12):4788–4793. doi: 10.1073/pnas.1100844108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kim J, Kundu M, Viollet B, Guan K-L. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nature Cell Biology. 2011;13(2):132–141. doi: 10.1038/ncb2152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lee JW, Park S, Takahashi Y, Wang H-G. The association of AMPK with ULK1 regulates autophagy. PLoS ONE. 2010;5(11) doi: 10.1371/journal.pone.0015394.e15394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tripathi DN, Chowdhury R, Trudel LJ, et al. Reactive nitrogen species regulate autophagy through ATM-AMPK-TSC2-mediated suppression of mTORC1. Proceedings of the National Academy of Sciences of the United States of America. 2013;110(32):E2950–E2957. doi: 10.1073/pnas.1307736110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sarkar S. Regulation of autophagy by mTOR-dependent and mTOR-independent pathways: autophagy dysfunction in neurodegenerative diseases and therapeutic application of autophagy enhancers. Biochemical Society Transactions. 2013;41(5):1103–1130. doi: 10.1042/BST20130134. [DOI] [PubMed] [Google Scholar]
- 36.Cully M, You H, Levine AJ, Mak TW. Beyond PTEN mutations: the PI3K pathway as an integrator of multiple inputs during tumorigenesis. Nature Reviews Cancer. 2006;6(3):184–192. doi: 10.1038/nrc1819. [DOI] [PubMed] [Google Scholar]
- 37.Levine B, Sinha S, Kroemer G. Bcl-2 family members: dual regulators of apoptosis and autophagy. Autophagy. 2008;4(5):600–606. doi: 10.4161/auto.6260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Shaw RJ, Cantley LC. Ras, PI(3)K and mTOR signalling controls tumour cell growth. Nature. 2006;441(7092):424–430. doi: 10.1038/nature04869. [DOI] [PubMed] [Google Scholar]
- 39.Mariño G, Salvador-Montoliu N, Fueyo A, Knecht E, Mizushima N, López-Otín C. Tissue-specific autophagy alterations and increased tumorigenesis in mice deficient in Atg4C/autophagin-3. Journal of Biological Chemistry. 2007;282(25):18573–18583. doi: 10.1074/jbc.M701194200. [DOI] [PubMed] [Google Scholar]
- 40.Lu Z, Luo RZ, Lu Y, et al. The tumor suppressor gene ARHI regulates autophagy and tumor dormancy in human ovarian cancer cells. The Journal of Clinical Investigation. 2008;118(12):3917–3929. doi: 10.1172/JCI35512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Liang XH, Jackson S, Seaman M, et al. Induction of autophagy and inhibition of tumorigenesis by beclin 1. Nature. 1999;402(6762):672–676. doi: 10.1038/45257. [DOI] [PubMed] [Google Scholar]
- 42.Labi V, Erlacher M, Kiessling S, Villunger A. BH3-only proteins in cell death initiation, malignant disease and anticancer therapy. Cell Death and Differentiation. 2006;13(8):1325–1338. doi: 10.1038/sj.cdd.4401940. [DOI] [PubMed] [Google Scholar]
- 43.Lomonosova E, Chinnadurai G. BH3-only proteins in apoptosis and beyond: an overview. Oncogene. 2008;27(1):S2–S19. doi: 10.1038/onc.2009.39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Maiuri MC, Criollo A, Tasdemir E, et al. BH3-only proteins and BH3 mimetics induce autophagy by competitively disrupting the interaction between Beclin 1 and Bcl-2/Bcl-XL. Autophagy. 2007;3(4):374–376. doi: 10.4161/auto.4237. [DOI] [PubMed] [Google Scholar]
- 45.Takahashi Y, Coppola D, Matsushita N, et al. Bif-1 interacts with Beclin 1 through UVRAG and regulates autophagy and tumorigenesis. Nature Cell Biology. 2007;9(10):1142–1151. doi: 10.1038/ncb1634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Harrison B, Kraus M, Burch L, et al. DAPK-1 binding to a linear peptide motif in MAP1B stimulates autophagy and membrane blebbing. The Journal of Biological Chemistry. 2008;283(15):9999–10014. doi: 10.1074/jbc.M706040200. [DOI] [PubMed] [Google Scholar]
- 47.Liang J, Shao SH, Xu Z-X, et al. The energy sensing LKB1-AMPK pathway regulates p27kip1 phosphorylation mediating the decision to enter autophagy or apoptosis. Nature Cell Biology. 2007;9(2):218–224. doi: 10.1038/ncb1537. [DOI] [PubMed] [Google Scholar]
- 48.Ji H, Ramsey MR, Hayes DN, et al. LKB1 modulates lung cancer differentiation and metastasis. Nature. 2007;448(7155):807–810. doi: 10.1038/nature06030. [DOI] [PubMed] [Google Scholar]
- 49.Edinger AL, Cinalli RM, Thompson CB. Rab7 prevents growth factor-independent survival by inhibiting cell-autonomous nutrient transporter expression. Developmental Cell. 2003;5(4):571–582. doi: 10.1016/s1534-5807(03)00291-0. [DOI] [PubMed] [Google Scholar]
- 50.Schwartz RA, Fernández G, Kotulska K, Jóźwiak S. Tuberous sclerosis complex: advances in diagnosis, genetics, and management. Journal of the American Academy of Dermatology. 2007;57(2):189–202. doi: 10.1016/j.jaad.2007.05.004. [DOI] [PubMed] [Google Scholar]
- 51.Choi AMK, Ryter SW, Levine B. Mechanisms of disease: autophagy in human health and disease. The New England Journal of Medicine. 2013;368(7):651–662. doi: 10.1056/NEJMra1205406. [DOI] [PubMed] [Google Scholar]
- 52.Aita VM, Liang XH, Murty VVVS, et al. Cloning and genomic organization of beclin 1, a candidate tumor suppressor gene on chromosome 17q21. Genomics. 1999;59(1):59–65. doi: 10.1006/geno.1999.5851. [DOI] [PubMed] [Google Scholar]
- 53.Qu X, Yu J, Bhagat G, et al. Promotion of tumorigenesis by heterozygous disruption of the beclin 1 autophagy gene. Journal of Clinical Investigation. 2003;112(12):1809–1820. doi: 10.1172/JCI20039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Yue Z, Jin S, Yang C, Levine AJ, Heintz N. Beclin 1, an autophagy gene essential for early embryonic development, is a haploinsufficient tumor suppressor. Proceedings of the National Academy of Sciences of the United States of America. 2003;100(25):15077–15082. doi: 10.1073/pnas.2436255100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Huang Y, Chen L, Guo L, Hupp TR, Lin Y. Evaluating DAPK as a therapeutic target. Apoptosis. 2014;19(2):371–386. doi: 10.1007/s10495-013-0919-2. [DOI] [PubMed] [Google Scholar]
- 56.Khan KH, Blanco-Codesido M, Molife LR. Cancer therapeutics: targeting the apoptotic pathway. Critical Reviews in Oncology/Hematology. 2014;90(3):200–219. doi: 10.1016/j.critrevonc.2013.12.012. [DOI] [PubMed] [Google Scholar]
- 57.Galluzzi L, Kepp O, Kroemer G. Mitochondria: master regulators of danger signalling. Nature Reviews Molecular Cell Biology. 2012;13(12):780–788. doi: 10.1038/nrm3479. [DOI] [PubMed] [Google Scholar]
- 58.Pattingre S, Tassa A, Qu X, et al. Bcl-2 antiapoptotic proteins inhibit Beclin 1-dependent autophagy. Cell. 2005;122(6):927–939. doi: 10.1016/j.cell.2005.07.002. [DOI] [PubMed] [Google Scholar]
- 59.Maiuri MC, le Toumelin G, Criollo A, et al. Functional and physical interaction between Bcl-XL and a BH3-like domain in Beclin-1. EMBO Journal. 2007;26(10):2527–2539. doi: 10.1038/sj.emboj.7601689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Morselli E, Galluzzi L, Kepp O, et al. Anti- and pro-tumor functions of autophagy. Biochimica et Biophysica Acta: Molecular Cell Research. 2009;1793(9):1524–1532. doi: 10.1016/j.bbamcr.2009.01.006. [DOI] [PubMed] [Google Scholar]
- 61.Miracco C, Cosci E, Oliveri G, et al. Protein and mRNA expression of autophagy gene Beclin 1 in human brain tumours. International Journal of Oncology. 2007;30(2):429–436. [PubMed] [Google Scholar]
- 62.Karantza-Wadsworth V, Patel S, Kravchuk O, et al. Autophagy mitigates metabolic stress and genome damage in mammary tumorigenesis. Genes & Development. 2007;21(13):1621–1635. doi: 10.1101/gad.1565707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Wei Y, Zou Z, Becker N, et al. EGFR-mediated beclin 1 phosphorylation in autophagy suppression, tumor progression, and tumor chemoresistance. Cell. 2013;154(6):1269–1284. doi: 10.1016/j.cell.2013.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Maria Fimia G, Stoykova A, Romagnoli A, et al. Ambra1 regulates autophagy and development of the nervous system. Nature. 2007;447(7148):1121–1125. doi: 10.1038/nature05925. [DOI] [PubMed] [Google Scholar]
- 65.Liang C, Feng P, Ku B, et al. Autophagic and tumour suppressor activity of a novel Beclin1-binding protein UVRAG. Nature Cell Biology. 2006;8(7):688–698. doi: 10.1038/ncb1426. [DOI] [PubMed] [Google Scholar]
- 66.White E. Deconvoluting the context-dependent role for autophagy in cancer. Nature Reviews Cancer. 2012;12(6):401–410. doi: 10.1038/nrc3262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Liu J, Xia H, Kim M, et al. Beclin1 controls the levels of p53 by regulating the deubiquitination activity of USP10 and USP13. Cell. 2011;147(1):223–234. doi: 10.1016/j.cell.2011.08.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kuma A, Hatano M, Matsui M, et al. The role of autophagy during the early neonatal starvation period. Nature. 2004;432(7020):1032–1036. doi: 10.1038/nature03029. [DOI] [PubMed] [Google Scholar]
- 69.Komatsu M, Waguri S, Ueno T, et al. Impairment of starvation-induced and constitutive autophagy in Atg7-deficient mice. Journal of Cell Biology. 2005;169(3):425–434. doi: 10.1083/jcb.200412022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Saitoh T, Fujita N, Jang MH, et al. Loss of the autophagy protein Atg16L1 enhances endotoxin-induced IL-1β production. Nature. 2008;456(7219):264–268. doi: 10.1038/nature07383. [DOI] [PubMed] [Google Scholar]
- 71.Saitoh T, Naonobu F, Yoshimori T, Akira S. Regulation of inflammation by autophagy. Tanpakushitsu kakusan koso. 2009;54(supplement 8):1119–1124. [PubMed] [Google Scholar]
- 72.Sou Y-S, Waguri S, Iwata J-I, et al. The Atg8 conjugation system is indispensable for proper development of autophagic isolation membranes in mice. Molecular Biology of the Cell. 2008;19(11):4762–4775. doi: 10.1091/mbc.E08-03-0309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Takamura A, Komatsu M, Hara T, et al. Autophagy-deficient mice develop multiple liver tumors. Genes and Development. 2011;25(8):795–800. doi: 10.1101/gad.2016211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Strohecker AM, Guo JY, Karsli-Uzunbas G, et al. Autophagy sustains mitochondrial glutamine metabolism and growth of BrafV600E -driven lung tumors. Cancer Discovery. 2013;3(11):1272–1285. doi: 10.1158/2159-8290.CD-13-0397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Kashuba VI, Gizatullin RZ, Protopopov AI, et al. NotI linking/jumping clones of human chromosome 3: mapping of the TFRC, RAB7 and HAUSP genes to regions rearranged in leukemia and deleted in solid tumors. FEBS Letters. 1997;419(2-3):181–185. doi: 10.1016/s0014-5793(97)01449-x. [DOI] [PubMed] [Google Scholar]
- 76.Laddha SV, Ganesan S, Chan CS, White E. Mutational landscape of the essential autophagy gene BECN1 in human cancers. Molecular Cancer Research. 2014;12(4):485–490. doi: 10.1158/1541-7786.MCR-13-0614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Marnett LJ. Oxyradicals and DNA damage. Carcinogenesis. 2000;21(3):361–370. doi: 10.1093/carcin/21.3.361. [DOI] [PubMed] [Google Scholar]
- 78.Goetz ME, Luch A. Reactive species: a cell damaging rout assisting to chemical carcinogens. Cancer Letters. 2008;266(1):73–83. doi: 10.1016/j.canlet.2008.02.035. [DOI] [PubMed] [Google Scholar]
- 79.Galluzzi L, Morselli E, Kepp O, et al. Mitochondrial gateways to cancer. Molecular Aspects of Medicine. 2010;31(1):1–20. doi: 10.1016/j.mam.2009.08.002. [DOI] [PubMed] [Google Scholar]
- 80.Mathew R, Kongara S, Beaudoin B, et al. Autophagy suppresses tumor progression by limiting chromosomal instability. Genes and Development. 2007;21(11):1367–1381. doi: 10.1101/gad.1545107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Mathew R, Karp CM, Beaudoin B, et al. Autophagy suppresses tumorigenesis through elimination of p62. Cell. 2009;137(6):1062–1075. doi: 10.1016/j.cell.2009.03.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Morselli E, Galluzzi L, Kepp O, et al. Oncosuppressive functions of autophagy. Antioxidants and Redox Signaling. 2011;14(11):2251–2269. doi: 10.1089/ars.2010.3478. [DOI] [PubMed] [Google Scholar]
- 83.Guo JY, Chen H-Y, Mathew R, et al. Activated Ras requires autophagy to maintain oxidative metabolism and tumorigenesis. Genes and Development. 2011;25(5):460–470. doi: 10.1101/gad.2016311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Guo JY, Karsli-Uzunbas G, Mathew R, et al. Autophagy suppresses progression of K-ras-induced lung tumors to oncocytomas and maintains lipid homeostasis. Genes and Development. 2013;27(13):1447–1461. doi: 10.1101/gad.219642.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Yang SH, Wang X, Contino G, et al. Pancreatic cancers require autophagy for tumor growth. Genes and Development. 2011;25(7):717–729. doi: 10.1101/gad.2016111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Geisler S, Holmström KM, Skujat D, et al. PINK1/Parkin-mediated mitophagy is dependent on VDAC1 and p62/SQSTM1. Nature Cell Biology. 2010;12(2):119–131. doi: 10.1038/ncb2012. [DOI] [PubMed] [Google Scholar]
- 87.Moscat J, Diaz-Meco MT. p62 at the crossroads of autophagy, apoptosis, and cancer. Cell. 2009;137(6):1001–1004. doi: 10.1016/j.cell.2009.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Inami Y, Waguri S, Sakamoto A, et al. Persistent activation of Nrf2 through p62 in hepatocellular carcinoma cells. Journal of Cell Biology. 2011;193(2):275–284. doi: 10.1083/jcb.201102031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Komatsu M, Kurokawa H, Waguri S, et al. The selective autophagy substrate p62 activates the stress responsive transcription factor Nrf2 through inactivation of Keap1. Nature Cell Biology. 2010;12(3):213–223. doi: 10.1038/ncb2021. [DOI] [PubMed] [Google Scholar]
- 90.Lau A, Wang X-J, Zhao F, et al. A noncanonical mechanism of Nrf2 activation by autophagy deficiency: direct interaction between keap1 and p62. Molecular and Cellular Biology. 2010;30(13):3275–3285. doi: 10.1128/MCB.00248-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Candido J, Hagemann T. Cancer-related inflammation. Journal of Clinical Immunology. 2013;33, supplement 1:S79–S84. doi: 10.1007/s10875-012-9847-0. [DOI] [PubMed] [Google Scholar]
- 92.Degenhardt K, Mathew R, Beaudoin B, et al. Autophagy promotes tumor cell survival and restricts necrosis, inflammation, and tumorigenesis. Cancer Cell. 2006;10(1):51–64. doi: 10.1016/j.ccr.2006.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.White E, DiPaola RS. The double-edged sword of autophagy modulation in cancer. Clinical Cancer Research. 2009;15(17):5308–5316. doi: 10.1158/1078-0432.CCR-07-5023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Qu X, Zou Z, Sun Q, et al. Autophagy gene-dependent clearance of apoptotic cells during embryonic development. Cell. 2007;128(5):931–946. doi: 10.1016/j.cell.2006.12.044. [DOI] [PubMed] [Google Scholar]
- 95.Levine B, Mizushima N, Virgin HW. Autophagy in immunity and inflammation. Nature. 2011;469(7330):323–335. doi: 10.1038/nature09782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Kroemer G, Levine B. Autophagic cell death: the story of a misnomer. Nature Reviews Molecular Cell Biology. 2008;9(12):1004–1010. doi: 10.1038/nrm2527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Elgendy M, Sheridan C, Brumatti G, Martin SJ. Oncogenic Ras-induced expression of Noxa and Beclin-1 promotes autophagic cell death and limits clonogenic survival. Molecular Cell. 2011;42(1):23–35. doi: 10.1016/j.molcel.2011.02.009. [DOI] [PubMed] [Google Scholar]
- 98.Zhao Y, Yang J, Liao W, et al. Cytosolic FoxO1 is essential for the induction of autophagy and tumour suppressor activity. Nature Cell Biology. 2010;12(7):665–675. doi: 10.1038/ncb2069. [DOI] [PubMed] [Google Scholar]
- 99.Zhao Y, Wang L, Yang J, et al. Anti-neoplastic activity of the cytosolic FoxO1 results from autophagic cell death. Autophagy. 2010;6(7):988–990. doi: 10.4161/auto.6.7.13289. [DOI] [PubMed] [Google Scholar]
- 100.Young ARJ, Narita M, Ferreira M, et al. Autophagy mediates the mitotic senescence transition. Genes and Development. 2009;23(7):798–803. doi: 10.1101/gad.519709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Kimmelman AC. The dynamic nature of autophagy in cancer. Genes and Development. 2011;25(19):1999–2010. doi: 10.1101/gad.17558811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Goehe RW, Di X, Sharma K, et al. The autophagy-senescence connection in chemotherapy: must tumor cells (self) eat before they sleep. Journal of Pharmacology and Experimental Therapeutics. 2012;343(3):763–778. doi: 10.1124/jpet.112.197590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Gewirtz DA. Autophagy and senescence in cancer therapy. Journal of cellular physiology. 2014;229(1):6–9. doi: 10.1002/jcp.24420. [DOI] [PubMed] [Google Scholar]
- 104.Karnoub AE, Weinberg RA. Ras oncogenes: split personalities. Nature Reviews Molecular Cell Biology. 2008;9(7):517–531. doi: 10.1038/nrm2438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Jemal A, Siegel R, Xu J, Ward E. Cancer statistics, 2010. CA Cancer Journal for Clinicians. 2010;60(5):277–300. doi: 10.3322/caac.20073. [DOI] [PubMed] [Google Scholar]
- 106.Fujii S, Mitsunaga S, Yamazaki M, et al. Autophagy is activated in pancreatic cancer cells and correlates with poor patient outcome. Cancer Science. 2008;99(9):1813–1819. doi: 10.1111/j.1349-7006.2008.00893.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Yang A, Rajeshkumar NV, Wang X, et al. Autophagy is critical for pancreatic tumor growth and progression in tumors with p53 alterations. Cancer Discovery. 2014;4(8):905–913. doi: 10.1158/2159-8290.CD-14-0362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Rosenfeldt MT, O’Prey J, Morton JP, et al. P53 status determines the role of autophagy in pancreatic tumour development. Nature. 2013;504(7479):296–300. doi: 10.1038/nature12865. [DOI] [PubMed] [Google Scholar]
- 109.Lock R, Roy S, Kenific CM, et al. Autophagy facilitates glycolysis during Ras-mediated oncogenic transformation. Molecular Biology of the Cell. 2011;22(2):165–178. doi: 10.1091/mbc.E10-06-0500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Chun SY, Johnson C, Washburn JG, Cruz-Correa MR, Dang DT, Dang LH. Oncogenic KRAS modulates mitochondrial metabolism in human colon cancer cells by inducing HIF-1α and HIF-2α target genes. Molecular Cancer. 2010;9, article 293 doi: 10.1186/1476-4598-9-293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Zheng B, Jeong JH, Asara JM, et al. Oncogenic B-RAF negatively regulates the tumor suppressor LKB1 to promote melanoma cell proliferation. Molecular Cell. 2009;33(2):237–247. doi: 10.1016/j.molcel.2008.12.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Kalas W, Swiderek E, Rapak A, Kopij M, Rak J, Strzadala L. H-ras Up-regulates expression of BNIP3. Anticancer Research. 2011;31(9):2869–2875. [PubMed] [Google Scholar]
- 113.Wu S-Y, Lan S-H, Cheng D-E, et al. Ras-related tumorigenesis is suppressed by BNIP3-mediated autophagy through inhibition of cell proliferation. Neoplasia. 2011;13(12):1171–1182. doi: 10.1593/neo.11888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.An H-J, Maeng O, Kang K-H, et al. Activation of Ras up-regulates pro-apoptotic BNIP3 in nitric oxide-induced cell death. Journal of Biological Chemistry. 2006;281(45):33939–33948. doi: 10.1074/jbc.M605819200. [DOI] [PubMed] [Google Scholar]
- 115.Wei H, Wei S, Gan B, Peng X, Zou W, Guan J-L. Suppression of autophagy by FIP200 deletion inhibits mammary tumorigenesis. Genes and Development. 2011;25(14):1510–1527. doi: 10.1101/gad.2051011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Smith BA, Shelton DN, Kieffer C, et al. Targeting the PyMT oncogene to diverse mammary cell populations enhances tumor heterogeneity and generates rare breast cancer subtypes. Genes and Cancer. 2012;3(9-10):550–563. doi: 10.1177/1947601913475359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Huo Y, Cai H, Teplova I, et al. Autophagy opposes p53-mediated tumor barrier to facilitate tumorigenesis in a model of PALB2 -associated hereditary breast cancer. Cancer Discovery. 2013;3(8):894–907. doi: 10.1158/2159-8290.CD-13-0011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Mathew R, White E. Autophagy, stress, and cancer metabolism: what doesn't kill you makes you stronger. Cold Spring Harbor Symposia on Quantitative Biology. 2011;76:389–396. doi: 10.1101/sqb.2012.76.011015. [DOI] [PubMed] [Google Scholar]
- 119.Quest AFG, Gutierrez-Pajares JL, Torres VA. Caveolin-1: an ambiguous partner in cell signalling and cancer. Journal of Cellular and Molecular Medicine. 2008;12(4):1130–1150. doi: 10.1111/j.1582-4934.2008.00331.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Quest AFG, Leyton L, Párraga M. Caveolins, caveolae, and lipid rafts in cellular transport, signaling, and disease. Biochemistry and Cell Biology. 2004;82(1):129–144. doi: 10.1139/o03-071. [DOI] [PubMed] [Google Scholar]
- 121.Quest AFG, Lobos-González L, Nuñez S, et al. The Caveolin-1 connection to cell death and survival. Current Molecular Medicine. 2013;13(2):266–281. doi: 10.2174/156652413804810745. [DOI] [PubMed] [Google Scholar]
- 122.Lobos-González L, Aguilar L, Diaz J, et al. E-cadherin determines Caveolin-1 tumor suppression or metastasis enhancing function in melanoma cells. Pigment Cell and Melanoma Research. 2013;26(4):555–570. doi: 10.1111/pcmr.12085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Rodriguez DA, Tapia JC, Fernandez JG, et al. Caveolin-1-mediated suppression of cyclooxygenase-2 via a β-catenin-Tcf/Lef-dependent transcriptional mechanism reduced prostaglandin E2 production and survivin expression. Molecular Biology of the Cell. 2009;20(8):2297–2310. doi: 10.1091/mbc.E08-09-0939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Torres VA, Tapia JC, Rodríguez DA, et al. Caveolin-1 controls cell proliferation and cell death by suppressing expression of the inhibitor of apoptosis protein survivin. Journal of Cell Science. 2006;119(part 9):1812–1823. doi: 10.1242/jcs.02894. [DOI] [PubMed] [Google Scholar]
- 125.Torres VA, Tapia JC, Rodriguez DA, et al. E-cadherin is required for caveolin-1-mediated down-regulation of the inhibitor of apoptosis protein survivin via reduced β-catenin-Tcf/Lef- dependent transcription. Molecular and Cellular Biology. 2007;27(21):7703–7717. doi: 10.1128/MCB.01991-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Martinez-Outschoorn UE, Trimmer C, Lin Z, et al. Autophagy in cancer associated fibroblasts promotes tumor cell survival: role of hypoxia, HIF1 induction and NFκB activation in the tumor stromal microenvironment. Cell Cycle. 2010;9(17):3515–3533. doi: 10.4161/cc.9.17.12928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Sotgia F, Martinez-Outschoorn UE, Howell A, Pestell RG, Pavlides S, Lisanti MP. Caveolin-1 and cancer metabolism in the tumor microenvironment: markers, models, and mechanisms. Annual Review of Pathology: Mechanisms of Disease. 2012;7:423–467. doi: 10.1146/annurev-pathol-011811-120856. [DOI] [PubMed] [Google Scholar]
- 128.Ha T-K, Her N-G, Lee M-G, et al. Caveolin-1 increases aerobic glycolysis in colorectal cancers by stimulating HMGA1-mediated GLUT3 transcription. Cancer Research. 2012;72(16):4097–4109. doi: 10.1158/0008-5472.CAN-12-0448. [DOI] [PubMed] [Google Scholar]
- 129.Shiroto T, Romero N, Sugiyama T, et al. Caveolin-1 is a critical determinant of autophagy, metabolic switching, and oxidative stress in vascular endothelium. PLoS ONE. 2014;9(2) doi: 10.1371/journal.pone.0087871.e87871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Chen Z-H, Cao J-F, Zhou J-S, et al. Interaction of caveolin-1 with ATG12-ATG5 system suppresses autophagy in lung epithelial cells. The American Journal of Physiology: Lung Cellular and Molecular Physiology. 2014;306(11):L1016–L1025. doi: 10.1152/ajplung.00268.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Meynet O, Ricci J-E. Caloric restriction and cancer: molecular mechanisms and clinical implications. Trends in Molecular Medicine. 2014;20(8):419–427. doi: 10.1016/j.molmed.2014.05.001. [DOI] [PubMed] [Google Scholar]
- 132.Malek M, Aghili R, Emami Z, Khamseh ME. Risk of cancer in diabetes: the effect of metformin. ISRN Endocrinology. 2013;2013:9 pages. doi: 10.1155/2013/636927.636927 [DOI] [PMC free article] [PubMed] [Google Scholar]