

Abstract
Hereditary spastic paraplegias are a large, diverse group of neurological disorders (SPG1-71) with the unifying feature of prominent lower extremity spasticity, owing to a length-dependent axonopathy of corticospinal motor neurons. The most common early-onset form of pure, autosomal dominant hereditary spastic paraplegia is caused by mutation in the ATL1 gene encoding the atlastin-1 GTPase, which mediates homotypic fusion of ER tubules to form the polygonal ER network. We have identified a p.Pro342Ser mutation in a young girl with pure SPG3A. This residue is in a critical hinge region of atlastin-1 between its GTPase and assembly domains, and it is conserved in all known eukaryotic atlastin orthologs. We produced induced pluripotent stem cells from skin fibroblasts and differentiated these into forebrain neurons to generate a human neuronal model for SPG3A. Axons of these SPG3A neurons showed impaired growth, recapitulating axonal defects in atlastin-1-depleted rat cortical neurons and impaired root hair growth in loss-of-function mutants of the ATL1 ortholog rhd3 in the plant Arabidopsis. Both the microtubule cytoskeleton and tubular ER are important for mitochondrial distribution and function within cells, and SPG3A neurons showed alterations in mitochondrial motility. Even so, it is not clear whether this change is involved in disease pathogenesis. The SPG3A axon growth defects could be rescued with microtubule-binding agents, emphasizing the importance of tubular ER interactions with the microtubule cytoskeleton in hereditary spastic paraplegia pathogenesis. The prominent alterations in axon growth in SPG3A neurons may represent a particularly attractive target for suppression in screens for novel pharmacologic agents.
INTRODUCTION
Hereditary spastic paraplegias (HSPs) are a diverse group of inherited neurological disorders unified by the defining feature of a length-dependent axonopathy of corticospinal motor neurons, resulting in prominent lower extremity spasticity and gait difficulties (1,2). These disorders have been historically classified as pure or complex based upon the presence (complex) or absence (pure) of associated clinical features such as cognitive impairment, distal amyotrophy, retinopathy, neuropathy and thin corpus callosum. More recently, a genetic classification scheme has predominated, and HSPs are commonly identified by their affected genes and spastic gait (SPG) loci, SPG1-71, assigned in order of locus identification (1–4). Despite the remarkable genetic heterogeneity of HSPs, a relatively small number of common cellular themes have emerged, including perturbations in mitochondrial function, endoplasmic reticulum shaping/distribution, myelination, lipid/cholesterol metabolism, protein/membrane trafficking, bone morphogenetic protein (BMP) signaling and endolysosomal function (1,4,5).
Disruption in the formation of the tubular ER network in cells has emerged as a key pathogenic theme underlying the HSPs, because over half of patients with autosomal dominant, pure HSP have SPG3A, SPG4, SPG12 or SPG31, each of which is caused by mutations in genes encoding proteins that bind one another and function in the organization of the tubular ER network (1,5,6). Thus, the development of animal and cellular models for these disorders of ER network formation is increasingly important.
The SPG3A protein atlastin-1 along with its human paralogs atlastin-2 and atlastin-3, yeast ortholog Sey1p and plant ortholog RHD3 are large, oligomeric GTPases that harbor two very closely spaced transmembrane domains required for proper ER localization; these GTPases mediate homotypic fusion of ER tubules to form the polygonal ER network (7–13). The SPG31 and SPG12 proteins REEP1 and reticulon 2, respectively, are members of the reticulon/REEP/Yop1p superfamily of proteins that harbor partially membrane spanning, hydrophobic hairpin domains and shape the high-curvature ER tubules (14,15). Last, the SPG4 protein spastin is an ATPase associated with a variety of activities (AAA), and the larger M1 spastin isoform contains a hydrophobic hairpin and localizes to ER tubules. These hydrophobic domains are important for self-interactions among these proteins as well as for interactions with one another (16).
To date no mouse model has been reported for SPG3A, the second most common cause of HSP and the most common early-onset form; there have been a few cellular as well as fly and zebrafish models described. For instance, depletion of atlastin-1 from rat cortical neurons in primary culture using shRNA causes prominent axon growth and branching defects (17). Disruption of the single atlastin ortholog in Drosophila causes synapse and muscle defects that can be rescued with the microtubule-destabilizing drug vinblastine (18). In zebrafish, knockdown of the atlastin gene atl1 causes a decrease in larval mobility that is preceded by abnormal architecture of spinal motor axons (19).
Here, we have identified a de novo SPG3A mutation (c.1024C>T; p.Pro342Ser) in a 2-year-old girl with early-onset, pure HSP. This Pro residue is at a critical position for atlastin-1 within a small linker region between the GTPase domain and three-helical bundle (3HB) assembly domain. In fact, this Pro residue is conserved in all eukaryotic atlastin orthologs (20) and is mutated in ATL1 in SPG3A [Ref. 21 (p.Pro342Gln) and the present study] as well as in ATL3 in hereditary sensory neuropathy type 1 [Ref. 22, p.Pro338Arg]. We used skin fibroblasts from our SPG3A patient to generate induced pluripotent stem cells (iPSCs), which were then differentiated into forebrain neurons to generate the first human neuronal model of SPG3A. These neurons have prominent axonal growth defects that can be partially rescued with microtubule-binding agents, emphasizing the importance of interactions of tubular ER with the microtubule cytoskeleton in HSP pathogenesis (16).
RESULTS
Cellular effects of SPG3A ATL1 mutation
A 2-year-old girl with a clinical presentation of early-onset, pure HSP had a de novo, heterozygous mutation in ATL1, c.1024C>T (p.Pro342Ser), identified by commercial DNA sequencing (Athena Diagnostics). Although this particular mutation has not been previously reported, it occurs at a residue mutated in other patients with SPG3A (21) and is conserved among all eukaryotes in the linker region of atlastin GTPases (Fig. 1A); this linker plays a key role in a conformational switch of atlastin important for the ER tubule fusion process (Fig. 1B; Refs. 20,23). This mutation also results in a modest reduction in GTPase activity (Fig. 1C and D).
Figure 1.

Atlastin-1 P342S SPG3A mutation affects a critical residue for conformational shifts. (A) Schematic diagram of atlastin-1 domain organization (top) and sequence comparisons among different species in the atlastin-1 linker region (bottom). Asterisks denote conserved residues, and the conserved Pro342 residue is shown in red. 3HB, three-helical bundle; TM, transmembrane domains. (B) Left, superposition of the cytosolic domain of atlastin-1 in two different crystal forms, denoted 1 and 2, with the respective GTPase domains as the reference. Right, major differences in conformation between these forms affect a GTPase domain-internal helix and the position of the 3HB domain, accompanied by restructuring of the linker region; the different positions of Pro342 are shown. This image with labels modified slightly is reproduced with permission from Byrnes and Sondermann (20). (C and D) Myc-tagged wild-type atlastin-1 or the P342 missense mutant were immunopurified from COS7 cells, and GTP hydrolysis was performed in vitro and plotted as a function of time (C). Representative thin-layer chromatography plates show conversion of GTP to GDP (D). Means ± SD are graphed.
Skin fibroblasts were cultured to assess the effects of this mutation on atlastin-1 protein levels. Immunoblots of SPG3A and control fibroblasts indicate that the levels of the atlastin-1 protein are not diminished in the SPG3A cells, suggesting that the mutant protein is present in cells. The levels of a number of other ER proteins, including the atlastin-1 paralogs atlastin-2 and atlastin-3, which are abundant in these cells (8), are also not significantly affected (Fig. 2A). The atlastin-1 P342S protein is also present in puncta throughout the peripheral ER, which is mostly tubular ER in contrast to the more perinuclear ER sheets labeled with CLIMP-63 (Fig. 2B). The distributions of endogenous atlastin-1-positive puncta appeared similar in control and atlastin-1 P342S fibroblasts (Fig. 2C). As atlastin-2 and -3 are likely the predominant atlastin GTPases in skin fibroblasts and may thus provide adequate protein for ER fusion even in the presence of atlastin-1 dysfunction, we examined effects of atlastin-1 P342S overexpression in these cells. Heterologous expression of Myc-tagged, wild-type atlastin in COS7 cells did not result in changes in ER morphology, but the expression of the P342S mutant markedly altered ER morphology in all cells examined (Fig. 2D).
Figure 2.

Atlastin P342S localizes to the tubular ER and disrupts ER morphology. (A) Aliquots of total membranes (10 μg of protein per lane) from control or SPG3A P342S human fibroblasts were immunblotted for the indicated ER proteins. Sizes of molecular weight standards (in kDa) are at the left. (B) Fibroblasts were co-immunostained for endogenous atlastin-1 (green) and CLIMP-63 (red). (C) Control or SPG3A P342S human fibroblasts were co-immunostained for atlastin-1 (green) and calreticulin (red). (D) COS7 cells were transfected with Myc-tagged wild-type or P342S mutant atlastin-1 and co-immunostained for Myc-epitope (green) and β-tubulin (red). Scale bars: 10 μm in (B) and 20 μm in (C and D).
Characterization and neural differentiation of control and SPG3A iPSCs
SPG3A iPSCs were generated from skin fibroblasts cultured from the patient with this P342S mutation in atlastin-1. The fibroblasts were reprogrammed using an integration-free episomal method (24), and multiple iPSC clones were generated from both SPG3A fibroblasts and wild-type controls. These clones had a typical ESC colony morphology, with no obvious differences between SPG3A and control lines, and expressed the characteristic human pluripotent stem cell markers Nanog, SSEA4 and Tra1-60 (Fig. 3A). To confirm that the SPG3A iPSCs did not contain chromosomal abnormalities after passaging, karyotype analysis was performed; this did not reveal any defects (Fig. 3B). To confirm the presence of the heterozygous p.Pro342Ser mutation in the ATL1 gene, this region was sequenced in both the iPSCs and neural cells after differentiation (Fig. 3C).
Figure 3.

Generation of SPG3A iPSC lines. (A) Control and SPG3A iPSC lines expressed the human ESC markers Tra-1-60 (red), Nanog (red) and SSEA-4 (green), as visualized by immunofluorescence microscopy. Hoechst nuclear staining is blue. Scale bars: 100 µm. (B) SPG3A iPSC lines maintained a normal 46,XX female karyotype after 10 passages as shown by G-banded analysis. (C) DNA sequencing electropherograms confirm the presence of a heterozygous c.1024C>T, p.Pro342Ser mutation before and after neural differentiation.
We differentiated the iPSCs to forebrain glutamatergic neurons using a well-established differentiation protocol (25–27). Representative images are shown for cells at different stages during differentiation (Fig. 4A). No apparent alterations were observed in the abilities of control and SPG3A cells to generate neurons. Although atlastins are expressed in all eukaryotic cells, atlastin-1 is by far the most abundant member in the central nervous system (7,8,17). To examine how atlastin-1 levels in human neurons change during the course of differentiation in culture, we performed immunoblot analysis (Fig. 4B). Very low levels of atlastin-1 protein were present in iPSCs, but by the neurosphere stage at Day 28 of differentiation, there was a dramatic increase. Atlastin-1 levels remained high in early generated neurons (Week 6) and in more mature neurons (Week 15). Conversely, the levels of atlastin-2 and atlastin-3 were high at the iPSC stage and decreased dramatically following neural differentiation (Fig. 4C). The levels of atlastin-1 were comparable between control and SPG3A lines in week 8 neurons (Fig. 4D). Thus, similar to what was observed in patient fibroblasts (Fig. 2A), the p.Pro342Ser mutation did not affect total atlastin-1 protein levels. In addition, the levels of the microtubule-severing ATPase spastin, a direct binding partner of atlastin-1 (28,29) mutated in the SPG4 subtype of HSP (30), were not dramatically different between control and SPG3A neurons (Fig. 4E). To confirm that forebrain glutamatergic neurons were generated, immunostaining was performed for Tbr1 and the neuron-specific βIII-tubulin isoform (Fig. 4F). Quantification of Tbr1+ cells did not reveal significant differences between control and SPG3A iPSC lines (Fig. 4G).
Figure 4.

Neural differentiation of SPG3A iPSCs. (A) Representative differential interference contrast images of control and SPG3A cells during neural differentiation at various stages. EB, embryoid body; NE, neuroepithelial. Scale bars: 100 µm. (B) Cell aliquots were immunoblotted for atlastin-1 during different stages (Weeks 0, 4, 6 and 15) of neural differentiation. Actin levels were monitored as a control for protein loading. (C) Atlastin-2 and -3 levels decrease following neural differentiation. Cell aliquots were immunoblotted during different stages (Weeks 0, 4, 6 and 15) of neural differentiation as shown. Actin levels were monitored as a control for protein loading. (D) Immunoblot analysis of atlastin-1 protein levels in extracts from week 15 neurons derived from wild-type (iWT-1 & iWT-3) and SPG3A iPSCs (iSPG3A-6 & iSPG3A-8). Actin levels were monitored as a control for protein loading. (E) Spastin levels are not dramatically altered in SPG3A neurons. Protein samples from week 15 neurons were immunoblotted for spastin. Actin levels were monitored as a control for protein loading. (F) Both control and SPG3A neurons efficiently generated Tbr1+ (red) glutamatergic neurons. Nuclei are identified with Hoechst staining (blue), and βIII-tubulin and Tbr1 were visualized by immunofluorescence microscopy. Scale bars: 50 µm. (G) Quantification of Tbr1+ neurons did not reveal any significant differences between the indicated iPSC lines. Data are presented as means ± SD, N = 3. In (B–E), migrations of molecular mass standards in kDa are indicated to the left.
Axonal outgrowth of neurons derived from control and SPG3A iPSCs
In a zebrafish model of SPG3A, knocking down atl1 resulted in abnormal outgrowth of spinal motor axons (19). To investigate axonal morphology in SPG3A iPSC-derived forebrain neurons, neurospheres were plated onto coverslips and axons were allowed to extend for 48 h, then fixed and stained for the axon marker tau. The tau staining extended to the end of the axons, as shown by co-localization with F-actin, which is enriched in growth cones (Supplementary Material, Fig. S1). In addition, the long neurites that extend from the neurospheres were established to be axons by co-localization of tau with the presynaptic marker bassoon (Supplementary Material, Fig. S2). This revealed an apparent reduction in the length of axons in the SPG3A axons compared with control neurons (Fig. 5A), and quantification of neurite outgrowth revealed a significant reduction in the average length of axons (Fig. 5B). Analysis of dissociated neurons revealed a similar reduction in axon outgrowth in SPG3A neurons derived from two different iPSC clones (Fig. 6). Together these data suggest that SPG3A neurons have impaired axon outgrowth with reduced axon length.
Figure 5.
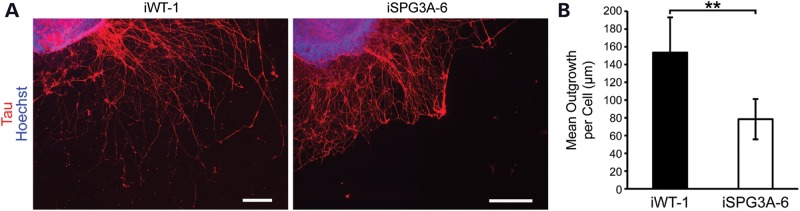
SPG3A neurons display reduced axon outgrowth. (A) Fluorescence images of tau+ axons from iPSC-derived neurons 2 days after plating. Scale bars: 200 µm. (B) Quantification of tau+ axon length shows a significant reduction in SPG3A neurons. Data are presented as means ± SD, N = 3–6 coverslips, with at least 1100 cells analyzed per group. **P < 0.01 versus control (iWT-1).
Figure 6.
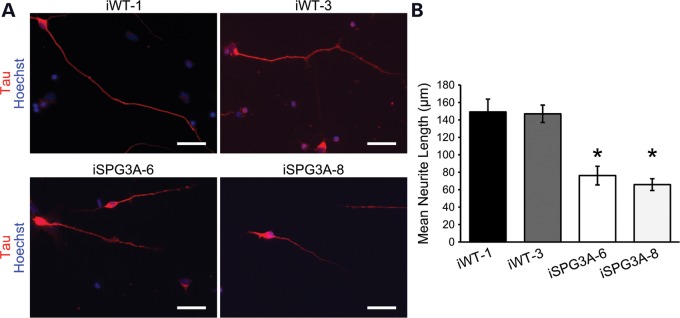
Reduced axon outgrowth in dissociated SPG3A neurons. (A) Control and SPG3A iPSC-derived neurons were plated and cultured for 48 h prior to staining for tau. Nuclei are identified with Hoechst staining (blue). (B) Quantification of mean tau+ process length for each group. N = 3 coverslips, with at least 50 cells analyzed per group. Data are presented as means ± SD. *P < 0.05 versus controls.
Fast axonal transport defects in SPG3A neurons
There is abundant evidence that links other forms of dominant, pure HSP to defects in fast axonal transport (31–35). To investigate whether transport is disrupted in SPG3A neurons, we analyzed mitochondrial fast axonal transport in week 12 iPSC-derived neurons with live-cell imaging, using the MitoTracker CMXRos dye. After the cells were stained, fluorescent images of proximal axons were obtained every 5 s for 5 min, and individual trajectories for each mitochondrion within the field were acquired, allowing detailed analysis of multiple transport parameters. Representative position versus time kymographs are shown in Figure 7A, revealing the nature of fast axonal transport in the two neuron groups. Comparison of mitochondrial transport velocities in both anterograde (Fig. 7B) and retrograde (Fig. 7C) directions revealed no significant differences between control and SPG3A neurons. The frequency of motile events was calculated by counting the number of times each mitochondrion moved with a velocity of >300 nm/s. This velocity threshold was used to exclude transport events mediated by actin, which fall well below this threshold (36). Calculation of the percentage of motile mitochondria for each cell revealed a significant reduction in SPG3A neurons compared with controls (Fig. 7D), and the frequency of motile events was lower in the SPG3A neurons as compared with controls (Fig. 7E). These events were separated based on the direction of transport, and while there was a non-significant trend toward the reduction of events in the retrograde direction (Fig. 7F), a significant reduction in anterograde motile events was observed in SPG3A cells (Fig. 7G).
Figure 7.

Decreased mitochondrial transport in SPG3A neurons. (A) Representative distance versus time kymographs show decreased motile mitochondria. Scale bar: 5 µm. (B and C) Velocities of movement events in anterograde (B) and retrograde (C) directions were not significantly affected in SPG3A neurons. (D) The percentage of motile mitochondria was significantly decreased in SPG3A neurons. (E) The number of motile movement events (velocity >300 nm/s) was also decreased in SPG3A neurons. (F and G) There was a non-significant trend toward a decreased frequency of motile movement events (per mitochondrion over 5 min) in the retrograde direction (F), whereas the frequency of events in the anterograde direction (G) was significantly reduced in SPG3A neurons. Data are presented as means ± SEM, N = 10–20 cells. *P < 0.05 versus control group (iWT-1); ns, not significant.
Treatment with microtubule-targeting drugs rescues the axon outgrowth phenotype
In a Drosophila model of SPG3A, where the single atlastin ortholog was disrupted, neuromuscular junction defects were linked to abnormal accumulation of stable microtubules (18). It was suggested that this effect is mediated through atlastin's interactions with the SPG4 protein spastin, a microtubule-severing AAA ATPase. Importantly, treatment with the microtubule-destabilizing drug vinblastine partially rescued neuromuscular junction defects and improved the survival of mutant flies (18). To test whether treatment with microtubule-targeting drugs is similarly effective on human SPG3A neurons and whether this iPSC-based model may have the potential for drug screening, we treated week 8 SPG3A neurons with several compounds. Briefly, both control and SPG3A neural progenitors were plated onto coverslips and allowed to attach for 24 h. The next day, neurons were treated with 10 nm vinblastine for 48 h, followed by analysis of tau+ axon growth. While vinblastine treatment did not have a significant effect on axon outgrowth in control neurons, it significantly increased outgrowth in SPG3A neurons (Fig. 8), suggesting that alterations in microtubule dynamics may be involved in this phenotype.
Figure 8.
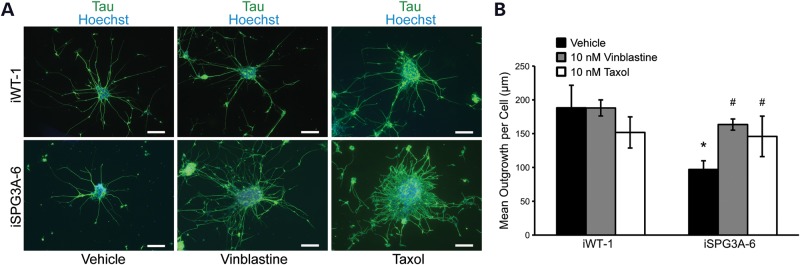
Vinblastine or taxol treatment partially rescues the axon outgrowth phenotype in SPG3A-derived neurons. (A) Immunofluorescence images of control (iWT-1) and SPG3A (iSPG3A-6)-plated neural clusters treated with vehicle, 10 nm vinblastine or 10 nm taxol for 48 h. Scale bars: 100 µm. (B) Quantification of axon length showed that vinblastine and taxol treatment significantly increased the length of axons in SPG3A-derived neurons. Data are presented as means ± SD, with a minimum of 1100 cells analyzed per group. *P < 0.05 versus controls (iWT-1); #P < 0.05 versus vehicle-treated SPG3A group.
A previous report investigating a mouse model of SPG4 found that treatment with nanomolar concentrations of both vinblastine and taxol, which are thought to alter only microtubule dynamic instability at these concentrations, significantly reversed the presence of axonal swellings in cortical neurons (37). Treatment of wild-type iPSC-derived neurons with taxol or vinblastine did not alter axon outgrowth significantly. However, treatment with either taxol or vinblastine significantly increased axon outgrowth in the SPG3A cells (Fig. 8). Together, these results reveal that microtubule-targeting drugs are able to rescue axon outgrowth defects in SPG3A patient-iPSC-derived neurons.
DISCUSSION
We have used skin fibroblasts from an SPG3A patient with a novel p.Pro342Ser mutation in the ATL1 gene to generate iPSC lines and model this common, early-onset, pure form of HSP in human neurons. The SPG3A iPSCs were able to differentiate into forebrain glutamatergic neurons with an efficiency similar to that of control neurons, and the levels of atlastin-1 protein were not decreased by the p.Pro342Ser mutation. As overexpression of this mutant alters ER morphology in a manner similar to atlastin depletion, a dominant-negative, loss-of-function disease mechanism appears most likely, with mutant proteins binding and impairing the function of the wild-type protein in atlastin oligomers. Previously, lymphoblasts from a patient with a p.N436del in-frame deletion in ATL1 were shown to have markedly decreased atlastin-1 levels, which were postulated to result from increased susceptibility of wild-type/p.del436N heteromers to degrade in a dominant-negative manner (38). Very recently, multiple affected members of a consanguineous Pakistani family were reported with a homozygous p.Arg118Gln mutation in ATL1; heterozygotes had minimal or no clinical signs, consistent with recessive inheritance, though the functional effects of this mutation are unknown (39). In these cases, a loss-of-function pathogenesis seems most plausible, while also illustrating that loss of atlastin function can arise in a number of ways.
Consistent with a dominant-negative, loss-of-function disease mechanism, SPG3A p.Pro342Ser neurons displayed markedly reduced axon outgrowth, similar to results from shRNA knockdown of rat cortical neurons (17). This phenotype is also highly similar to that seen in loss-of-function mutations in the atlastin-1 ortholog RHD3 of the flowering plant Arabidopsis, where there are prominent defects in root hair growth (40). These root hairs are long protrusions from root cells that are reminiscent of axons (1,41). Thus, these defects in polarized cell expansion are a broadly conserved function of atlastin proteins across phylogeny and consequently may represent an important phenotype for rescue in pharmacologic screens. In fact, these axon growth defects in human SPG3A neurons could be rescued by treatment with both vinblastine and taxol at nanomolar concentrations, indicating an important role for ER interactions with dynamic microtubules.
In addition to its interactions with microtubules, the ER has extensive contacts with other organelles, which could be also impacted by changes in ER shape (42). We investigated fast axonal transport of mitochondria because such defects have already been observed in autosomal dominant SPG4 (31,34,37), which is caused by mutations in the spastin AAA ATPase that binds atlastin-1. There were no significant differences in the transport velocities of mitochondria in either anterograde or retrograde directions, suggesting that activities of the molecular motors were unaffected. However, there were decreases in the frequency of mitochondrial motile events. As the axoplasm accounts for >99% of total cellular volume in the longest neurons, they are particularly susceptible to defects in intracellular transport (5). In fact, many neurodegenerative disorders have been linked to deficits in axonal transport, including amyotrophic lateral sclerosis (ALS), Parkinson disease, Alzheimer disease and Huntington disease (43). The proper distribution of mitochondria and likely other organelles throughout the axon thus appears critical for maintaining neuronal health.
Defects in axonal transport of mitochondria, other organelles or proteins may underlie the axon outgrowth defect that is observed in these SPG3A cells. A majority of proteins in the axon and at synapses are generated in the cell body and must be transported down axons via anterograde transport (44). Mitochondrial localization, which is dependent on fast axonal transport, is also important for regulating the variability of presynaptic strength as well as for axon formation and outgrowth (45,46). In fact, mitochondria accumulate in the growth cones of axons during outgrowth (47). In the SGP3A neurons, reduction in the number of anterograde motile events could conceivably reduce the net flux of mitochondria toward the distal portions of the axons, negatively affecting axon outgrowth, presynaptic regulation or both. However, selectively increasing mitochondrial axonal mobility does not slow ALS-like impairment in the SOD1(G93A) mutant mouse model for ALS (48). Alternatively, other consequences of atlastin dysfunction such as aberrant ER distribution (which could secondarily affect mitochondrial motility) or BMP signaling (5) may play key roles in pathogenesis.
Studies in Drosophila have previously shown that there are increased stabilized microtubules in atl mutant flies and that vinblastine treatment can rescue several neuromuscular defects (18). Loss of atlastin function was hypothesized to result in altered microtubules by disrupting the interaction between atlastin and the microtubule-severing, SPG4 ATPase spastin. We and others have previously shown that treatment with microtubule-targeting drugs can rescue defects in cultured SPG4 neurons, suggesting that altered microtubules may be a common finding among different forms of HSP (35,37,49). One role of atlastin-1 may be to distribute spastin's microtubule-severing activity within neurons, because spastin is a direct binding partner (16,28,29). Treatment with sub-stoichiometric concentrations of either vinblastine or taxol increased axon outgrowth to levels comparable with wild-type neurons. It will be interesting to examine further whether axon growth and transport deficits can be rescued by these drugs in longer-term cultures.
On initial consideration, it seems hard to reconcile a primary defect in axon growth as a mediator of the pathology in HSPs, because most HSPs are progressive disorders characterized by distal, dying-back axonal degeneration (1–5,50). However, SPG3A in particular is notable for not only being the most common early-onset, pure form of HSP but also for having a large number of affected individuals presenting with apparently non-progressive forms (51). Thus, abnormal axon development may play a key role in SPG3A pathogenesis. Our findings suggest that iPSC-based models of SPG3A will be useful for drug screening trials. In future studies, it will be particularly interesting to assess how rescue of axon growth defects owing to SPG3A mutations correlates with rescue of known intracellular functions of atlastins, such as BMP signaling, ER morphology, organelle distribution and lipid droplet formation (19,52,53).
MATERIALS AND METHODS
Clinical studies
All study procedures were performed under an Institutional Review Board-approved clinical research protocol (NINDS protocol 00-N-0043) at the National Institutes of Health Clinical Center. The parents of the SPG3A patient provided informed consent.
Cell culture, transfection and GTPase activity assays
Heterologous expression studies in COS7 cells and in vitro GTPase assays of Myc-atlastin-1 proteins were performed as described previously (7,54).
Reprogramming fibroblasts into iPSC lines
Human fibroblast cell lines were established from skin punch biopsies and maintained using standard procedures. To generate iPSC lines using episomal transduction, ∼200,000 cells were dissociated and transfected with episomal plasmids (Addgene) containing pluripotency factors (Oct3/4, Sox2, L-Myc, Klf4 and Lin 28), as reported previously (24). At around 1 week after electroporation transduction, cells were plated onto a 35-mm dish in DMEM supplemented with 10% fetal bovine serum. After culturing for 7 days, cells were dissociated and seeded onto a mouse embryonic fibroblast (MEF) feeder at ∼105 cells/100-mm dish. Two weeks later, colonies with morphologies similar to human embryonic stem cells (ESCs) were observed. These colonies were split onto MEF feeder cells to derive iPSC lines. Following several passages, homogenous colonies with ESC-like morphology were generated. The iPSC lines used in this study were iWT-1, iWT-3, iSPG3A-6 and iSPG3A-8.
Human iPSC neural differentiation
To generate telencephalic neurons from iPSCs, stem cells were cultured on a feeder layer of irradiated MEFs in 6-well tissue culture-treated plates for ∼6 days, with the human ESC media [+10 ng/ml of fibroblast growth factor (FGF)-2] changed daily. When nearly confluent, cells were detached from the feeder layer to initiate neural differentiation, as previously described (25–27). Briefly, iPSC aggregates were cultured in suspension for 4 days in human ESC media and were then transferred to neural induction media (NIM). After 3 additional days in suspension, iPSC aggregates were plated onto 6-well tissue culture-treated plates in NIM with 10% fetal bovine serum. After 12 h, the media was replaced with fresh NIM. The media was then changed every other day until Day 17, when the generated neuroepithelial (NE) cells were isolated. Mechanically isolated NE cells were cultured in suspension with NIM [+B27, +cyclic AMP, +insulin-like growth factor 1 (IGF-1)] to generate neurospheres for at least 10 additional days. On about Day 28, neurospheres were dissociated and plated onto polyornithine- and laminin-coated coverslips in neural differentiation media containing N2, B27, ascorbic acid, cyclic AMP, laminin, IGF-1, brain-derived neurotrophic factor and glial-derived neurotrophic factor. Half of the media was changed every other day for 6–12 weeks, depending on the analysis to be performed. For the treatment of cells with microtubule-targeting drugs, the media was replaced with standard neural differentiation media with either 10 nm vinblastine (Sigma–Aldrich) or 10 nm taxol dissolved in water.
Immunoblotting and immunocytochemistry
Immunofluorescence microscopy and immunoblotting were performed as described previously (7,25,55). Quantification of immunoblots was performed using ImageJ (56), normalized against actin as a loading control. The percentage of Tbr1+ cells among total cells (Hoechst) was determined by taking three randomly selected fields per coverslip and blindly counting cells using MetaMorph software (25). Three coverslips were analyzed for each group.
Antibodies used in this study included mouse monoclonal IgM anti-Tra-1-60 (Santa Cruz, 1:50), goat polyclonal IgG anti-Nanog (R&D Systems, 1:500), mouse monoclonal IgG3 anti-SSEA-4 (Developmental Studies Hybridoma Bank, 1:100), rabbit IgG anti-tau (Sigma–Aldrich, 1:100), rabbit polyclonal anti-Tbr1 (Proteintech, 1:1000), mouse monoclonal IgG βIII-tubulin (TuJ-1; Developmental Studies Hybridoma Bank, 1:100), rabbit polyclonal anti-REEP5 (Proteintech, 1:2000), rabbit polyclonal anti-atlastin-1, -2 and -3 (8), rabbit polyclonal anti-calreticulin (Abcam, 1:1000), mouse monoclonal Myc-epitope (Santa Cruz Biotechnology, 1:1000), mouse monoclonal anti-β-tubulin (Sigma–Aldrich, 1:2000) and rabbit polyclonal anti-spastin (Sigma–Aldrich, 1:1,000). Rhodamine-phalloidin (100 nm) was from Cytoskeleton.
Axon length measurements
Axon outgrowth properties of neurons plated in small clusters were quantified with MetaMorph software using the Neurite Outgrowth application as described by the manufacturer; this program identifies cell bodies and their neurites and determines the length of each neurite. Three to six coverslips for each group, comprising at least 1100 cells per group, were analyzed for neurite outgrowth. Axonal outgrowth of dissociated neurons was quantified using the NeuronJ plugin for ImageJ, and the length of the longest process that also had the greatest tau intensity was measured. At least 50 cells were quantified per cell line, for a total of 123 control and 175 SPG3A cells from at least 3 coverslips.
Live-cell imaging
Neurospheres were plated onto polyornithine- and laminin-coated 35-mm dishes (MatTek). At 8 weeks of total differentiation, the cells were stained with 50 nm MitoTracker Red CMXRos (Invitrogen) for 3 min to allow visualization of mitochondria, after which the media was replaced with fresh neural differentiation media. Live-cell imaging was performed using a Zeiss Axiovert 200M microscope equipped with an incubation chamber. Cells were kept at 37°C with 5% CO2 while imaging. Axons identified according to morphological criteria (constant thin diameter, long neurites, no branching and direct emergence from the cell body) were imaged every 5 s for 5 min, yielding 60 frames. Quantifications were performed using a protocol described previously (36). In short, the location of each mitochondrion was manually selected using the Track Points function in MetaMorph, and parameters such as distance from cell body and velocity were recorded. A velocity threshold of 300 nm/s was used to select microtubule-based transport events (57). To determine the percentage of motile mitochondria, the total number of mitochondria that were present along the imaged neurite was counted, and those that changed position (velocity >300 nm/s) in at least three consecutive frames were considered motile.
Statistical analysis
Statistical significance of mean values among multiple sample groups was analyzed with Tukey's studentized range test after ANOVA. Two-sided t-tests were used to examine the statistical significance between two sample groups. The significance level was defined as P < 0.05, and significance tests were conducted using SAS 9.1 (SAS Institute).
SUPPLEMENTARY MATERIAL
FUNDING
This work was supported by the Intramural Research Program of the NINDS, National Institutes of Health, the Spastic Paraplegia Foundation, and a Connecticut Stem Cell Research Grant (11SCB24).
Supplementary Material
ACKNOWLEDGEMENTS
We thank Dr Benoit Renvoisé for assistance with the skin fibroblasts and for critical review of the manuscript.
Conflict of Interest Statement. None declared.
REFERENCES
- 1.Blackstone C. Cellular pathways of hereditary spastic paraplegia. Annu. Rev. Neurosci. 2012;35:25–47. doi: 10.1146/annurev-neuro-062111-150400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Finsterer J., Löscher W., Quasthoff S., Wanschitz J., Auer-Grumbach M., Stevanin G. Hereditary spastic paraplegias with autosomal dominant, recessive, X-linked, or maternal trait of inheritance. J. Neurol. Sci. 2012;318:1–18. doi: 10.1016/j.jns.2012.03.025. [DOI] [PubMed] [Google Scholar]
- 3.Schüle R., Schöls L. Genetics of hereditary spastic paraplegias. Semin. Neurol. 2011;31:484–493. doi: 10.1055/s-0031-1299787. [DOI] [PubMed] [Google Scholar]
- 4.Novarino G., Fenstermaker A.G., Zaki M.S., Hofree M., Silhavy J.L., Heiberg A.D., Abdellateef M., Rosti B., Scott E., Mansour L., et al. Exome sequencing links corticospinal motor neuron disease to common neurodegenerative disorders. Science. 2014;343:506–511. doi: 10.1126/science.1247363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Blackstone C., O'Kane C.J., Reid E. Hereditary spastic paraplegias: membrane traffic and the motor pathway. Nat. Rev. Neurosci. 2011;12:31–42. doi: 10.1038/nrn2946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Park S.H., Blackstone C. Further assembly required: construction and dynamics of the endoplasmic reticulum network. EMBO Rep. 2010;11:515–521. doi: 10.1038/embor.2010.92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhu P.-P., Patterson A., Lavoie B., Stadler J., Shoeb M., Patel R., Blackstone C. Cellular localization, oligomerization, and membrane association of the hereditary spastic paraplegia 3A (SPG3A) protein atlastin. J. Biol. Chem. 2003;278:49063–49071. doi: 10.1074/jbc.M306702200. [DOI] [PubMed] [Google Scholar]
- 8.Rismanchi N., Soderblom C., Stadler J., Zhu P.-P., Blackstone C. Atlastin GTPases are required for Golgi apparatus and ER morphogenesis. Hum. Mol. Genet. 2008;17:1591–1604. doi: 10.1093/hmg/ddn046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hu J., Shibata Y., Zhu P.-P., Voss C., Rismanchi N., Prinz W.A., Rapoport T.A., Blackstone C. A class of dynamin-like GTPases involved in the generation of the tubular ER network. Cell. 2009;138:549–561. doi: 10.1016/j.cell.2009.05.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Orso G., Pendin D., Liu S., Tosetto J., Moss T.J., Faust J.E., Micaroni M., Egorova A., Martinuzzi A., McNew J.A., Daga A. Homotypic fusion of ER membranes requires the dynamin-like GTPase atlastin. Nature. 2009;460:978–983. doi: 10.1038/nature08280. [DOI] [PubMed] [Google Scholar]
- 11.Anwar K., Klemm R.W., Condon A., Severin K.N., Zhang M., Ghirlando R., Hu J., Rapoport T.A., Prinz W.A. The dynamin-like GTPase Sey1p mediates homotypic ER fusion in S. cerevisiae. J. Cell Biol. 2012;197:209–217. doi: 10.1083/jcb.201111115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chen J., Stefano G., Brandizzi F., Zheng H. Arabidopsis RHD3 mediates the generation of the tubular ER network and is required for Golgi distribution and motility in plant cells. J Cell Sci. 2011;124:2241–2252. doi: 10.1242/jcs.084624. [DOI] [PubMed] [Google Scholar]
- 13.Zhang M., Wu F., Shi J., Zhu Y., Zhu Z., Gong Q., Hu J. ROOT HAIR DEFECTIVE3 family of dynamin-like GTPases mediates homotypic endoplasmic reticulum fusion and is essential for Arabidopsis development. Plant Physiol. 2013;163:713–720. doi: 10.1104/pp.113.224501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Voeltz G.K., Prinz W.A., Shibata Y., Rist J.M., Rapoport T.A. A class of membrane proteins shaping the endoplasmic reticulum. Cell. 2006;124:573–586. doi: 10.1016/j.cell.2005.11.047. [DOI] [PubMed] [Google Scholar]
- 15.Hu J., Shibata Y., Voss C., Shemesh T., Li Z., Coughlin M., Kozlov M.M., Rapoport T.A., Prinz W.A. Membrane proteins of the endoplasmic reticulum induce high-curvature tubules. Science. 2008;319:1247–1250. doi: 10.1126/science.1153634. [DOI] [PubMed] [Google Scholar]
- 16.Park S.H., Zhu P.-P., Parker R.L., Blackstone C. Hereditary spastic paraplegia proteins REEP1, spastin, and atlastin-1 coordinate microtubule interactions with the tubular ER network. J. Clin. Invest. 2010;120:1097–1110. doi: 10.1172/JCI40979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhu P.-P., Soderblom C., Tao-Cheng J.-H., Stadler J., Blackstone C. SPG3A protein atlastin-1 is enriched in growth cones and promotes axon elongation during neuronal development. Hum. Mol. Genet. 2006;15:1343–1353. doi: 10.1093/hmg/ddl054. [DOI] [PubMed] [Google Scholar]
- 18.Lee M., Paik S.K., Lee M.-J., Kim Y.-J., Kim S., Nahm M., Oh S.-J., Kim H.-M., Yim J., Lee C.J., et al. Drosophila Atlastin regulates the stability of muscle microtubules and is required for synapse development. Dev. Biol. 2009;330:250–262. doi: 10.1016/j.ydbio.2009.03.019. [DOI] [PubMed] [Google Scholar]
- 19.Fassier C., Hutt J.A., Scholpp S., Lumsden A., Giros B., Nothias F., Schneider-Maunoury S., Houart C., Hazan J. Zebrafish atlastin controls motility and spinal motor axon architecture via inhibition of the BMP pathway. Nat. Neurosci. 2010;13:1380–1387. doi: 10.1038/nn.2662. [DOI] [PubMed] [Google Scholar]
- 20.Byrnes L.J., Sondermann H. Structural basis for the nucleotide-dependent dimerization of the large G protein atlastin-1/SPG3A. Proc. Natl. Acad. Sci. USA. 2011;108:2216–2221. doi: 10.1073/pnas.1012792108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.de Bot S.T., Veldink J.H., Vermeer S., Mensenkamp A.R., Brugman F., Scheffer H., van den Berg L.H., Kremer H.P.H., Kamsteeg E.J., van de Warrenburg B.P. ATL1 and REEP1 mutations in hereditary and sporadic upper motor neuron syndromes. J. Neurol. 2013;260:869–875. doi: 10.1007/s00415-012-6723-z. [DOI] [PubMed] [Google Scholar]
- 22.Fischer D., Schabhüttl M., Wieland T., Windhager R., Strom T.M., Auer-Grumbach M. A novel missense mutation confirms ATL3 as a gene for hereditary sensory neuropathy type 1. Brain. 2014 doi: 10.1093/brain/awu091. in press. doi:10.1093/brain/awu091. [DOI] [PubMed] [Google Scholar]
- 23.Byrnes L.J., Singh A., Szeto K., Benvin N.M., O'Donnell J.P., Zipfel W.R., Sondermann H. Structural basis for conformational switching and GTP loading of the large G protein atlastin. EMBO J. 2013;32:369–384. doi: 10.1038/emboj.2012.353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Okita K., Matsumura Y., Sato Y., Okada A., Morizane A., Okamoto S., Hong H., Nakagawa M., Tanabe K., Tezuka K., et al. A more efficient method to generate integration-free human iPS cells. Nat. Methods. 2011;8:409–412. doi: 10.1038/nmeth.1591. [DOI] [PubMed] [Google Scholar]
- 25.Li X.-J., Zhang X., Johnson M.A., Wang Z.-B., LaVaute T., Zhang S.-C. Coordination of sonic hedgehog and Wnt signaling determines ventral and dorsal telencephalic neuron types from human embryonic stem cells. Development. 2009;136:4055–4063. doi: 10.1242/dev.036624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zeng H., Guo M., Martins-Taylor K., Wang X., Zhang Z., Park J.W., Zhan S., Kronenberg M.S., Lichtler A., Liu H.-X., et al. Specification of region-specific neurons including forebrain glutamatergic neurons from human induced pluripotent stem cells. PLoS One. 2010;5:e11853. doi: 10.1371/journal.pone.0011853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Boisvert E.M., Denton K., Lei L., Li X.-J. The specification of telencephalic glutamatergic neurons from human pluripotent stem cells. J. Vis. Exp. 2013;74:e50321. doi: 10.3791/50321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Evans K., Keller C., Pavur K., Glasgow K., Conn B., Lauring B. Interaction of two hereditary spastic paraplegia gene products, spastin and atlastin, suggests a common pathway for axonal maintenance. Proc. Natl. Acad. Sci. USA. 2006;103:10666–10671. doi: 10.1073/pnas.0510863103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sanderson C.M., Connell J.W., Edwards T.L., Bright N.A., Duley S., Thompson A., Luzio J.P., Reid E. Spastin and atlastin, two proteins mutated in autosomal-dominant hereditary spastic paraplegia, are binding partners. Hum. Mol. Genet. 2006;15:307–318. doi: 10.1093/hmg/ddi447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hazan J., Fonknechten N., Mavel D., Paternotte C., Samson D., Artiguenave F., Davoine C.-S., Cruaud C., Dürr A., Wincker P., et al. Spastin, a new AAA protein, is altered in the most frequent form of autosomal dominant spastic paraplegia. Nat. Genet. 1999;23:296–303. doi: 10.1038/15472. [DOI] [PubMed] [Google Scholar]
- 31.Tarrade A., Fassier C., Courageot S., Charvin D., Vitte J., Peris L., Thorel A., Mouisel E., Fonknechten N., Roblot N., et al. A mutation of spastin is responsible for swellings and impairment of transport in a region of axon characterized by changes in microtubule composition. Hum. Mol. Genet. 2006;15:3544–3558. doi: 10.1093/hmg/ddl431. [DOI] [PubMed] [Google Scholar]
- 32.Ebbing B., Mann K., Starosta A., Jaud J., Schöls L., Schüle R., Woehlke G. Effect of spastic paraplegia mutations in KIF5A kinesin on transport activity. Hum. Mol. Genet. 2008;17:1245–1252. doi: 10.1093/hmg/ddn014. [DOI] [PubMed] [Google Scholar]
- 33.Solowska J.M., Morfini G., Falnikar A., Himes B.T., Brady S.T., Huang D., Baas P.W. Quantitative and functional analyses of spastin in the nervous system: implications for hereditary spastic paraplegia. J. Neurosci. 2008;28:2147–2157. doi: 10.1523/JNEUROSCI.3159-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kasher P.R., De Vos K.J., Wharton S.B., Manser C., Bennett E.J., Bingley M., Wood J.D., Milner R., McDermott C.J., Miller C.C., et al. Direct evidence for axonal transport defects in a novel mouse model of mutant spastin-induced hereditary spastic paraplegia (HSP) and human HSP patients. J. Neurochem. 2009;110:34–44. doi: 10.1111/j.1471-4159.2009.06104.x. [DOI] [PubMed] [Google Scholar]
- 35.Denton K.R., Lei L., Grenier J., Rodionov V., Blackstone C., Li X.-J. Loss of spastin function results in disease-specific axonal defects in human pluripotent stem cell-based models of hereditary spastic paraplegia. Stem Cells. 2014;32:414–423. doi: 10.1002/stem.1569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.De Vos K.J., Chapman A.L., Tennant M.E., Manser C., Tudor E.L., Lau K.-F., Brownlees J., Ackerley S., Shaw P.J., McLoughlin D.M., et al. Familial amyotrophic lateral sclerosis-linked SOD1 mutants perturb fast axonal transport to reduce axonal mitochondria content. Hum. Mol. Genet. 2007;16:2720–2728. doi: 10.1093/hmg/ddm226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fassier C., Tarrade A., Peris L., Courageot S., Mailly P., Dalard C., Delga S., Roblot N., Lefèvre J., Job D., et al. Microtubule-targeting drugs rescue axonal swellings in cortical neurons from spastin knockout mice. Dis. Model Mech. 2013;6:72–83. doi: 10.1242/dmm.008946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Meijer I.A., Dion P., Laurent S., Dupré N., Brais B., Levert A., Puymirat J., Rioux M.F., Sylvain M., Zhu P.-P., et al. Characterization of a novel SPG3A deletion in a French-Canadian family. Ann. Neurol. 2007;61:599–603. doi: 10.1002/ana.21114. [DOI] [PubMed] [Google Scholar]
- 39.Khan T.N., Klar J., Tariq M., Baig S.A., Malik N.A., Yousaf R., Baig S.M., Dahl N. Evidence for autosomal recessive inheritance in SPG3A caused by homozygosity for a novel ATL1 missense mutation. Eur. J. Hum. Genet. 2014 doi: 10.1038/ejhg.2014.5. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wang H., Lockwood S.K., Hoeltzel M.F., Schiefelbein J.W. The ROOT HAIR DEFECTIVE3 gene encodes an evolutionarily conserved protein with GTP-binding motifs and is required for regulated cell enlargement in Arabidopsis. Genes Dev. 1997;11:799–811. doi: 10.1101/gad.11.6.799. [DOI] [PubMed] [Google Scholar]
- 41.Zheng H., Chen J. Emerging aspects of ER organization in root hair tip growth: lessons from RHD3 and Atlastin. Plant Signal. Behav. 2011;6:1710–1713. doi: 10.4161/psb.6.11.17477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.English A.R., Voeltz G.K. Endoplasmic reticulum structure and interconnections with other organelles. Cold Spring Harb. Perspect. Biol. 2013;5:a013227. doi: 10.1101/cshperspect.a013227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Morfini G.A., Burns M., Binder L.I., Kanaan N.M., LaPointe N., Bosco D.A., Brown R.H., Jr, Brown H., Tiwari A., Hayward L., et al. Axonal transport defects in neurodegenerative diseases. J. Neurosci. 2009;29:12776–12786. doi: 10.1523/JNEUROSCI.3463-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hirokawa N., Niwa S., Tanaka Y. Molecular motors in neurons: transport mechanisms and roles in brain function, development, and disease. Neuron. 2010;68:610–638. doi: 10.1016/j.neuron.2010.09.039. [DOI] [PubMed] [Google Scholar]
- 45.Mattson M.P., Partin J. Evidence for mitochondrial control of neuronal polarity. J. Neurosci. Res. 1999;56:8–20. doi: 10.1002/(SICI)1097-4547(19990401)56:1<8::AID-JNR2>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]
- 46.Sun T., Qiao H., Pan P.-Y., Chen Y., Sheng Z.-H. Mobile axonal mitochondria contribute to the variability of presynaptic strength. Cell Rep. 2013;4:413–419. doi: 10.1016/j.celrep.2013.06.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ruthel G., Hollenbeck P.J. Response of mitochondrial traffic to axon determination and differential branch growth. J. Neurosci. 2003;23:8618–8624. doi: 10.1523/JNEUROSCI.23-24-08618.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhu Y.-B., Sheng Z.-H. Increased axonal mobility does not slow ALS-like disease in mutant SOD1 mice. J. Biol. Chem. 2011;286:23432–23440. doi: 10.1074/jbc.M111.237818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Abrahamsen G., Fan Y., Matigian N., Wali G., Bellette B., Sutharsan R., Raju J., Wood S.A., Veivers D., Sue C.M., Mackay-Sim A. A patient-derived stem cell model of hereditary spastic paraplegia with SPAST mutations. Dis. Model. Mech. 2013;6:489–502. doi: 10.1242/dmm.010884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Soderblom C., Blackstone C. Traffic accidents: molecular genetic insights into the pathogenesis of the hereditary spastic paraplegias. Pharmacol. Ther. 2006;109:42–56. doi: 10.1016/j.pharmthera.2005.06.001. [DOI] [PubMed] [Google Scholar]
- 51.Fink J.K. Advances in the hereditary spastic paraplegias. Exp. Neurol. 2003;184(Suppl. 1):S106–S110. doi: 10.1016/j.expneurol.2003.08.005. [DOI] [PubMed] [Google Scholar]
- 52.Klemm R.W., Norton J.P., Cole R.A., Li C.S., Park S.H., Crane M.M., Li L., Jin D., Boye-Doe A., Liu T.Y., et al. A conserved role for atlastin GTPases in regulating lipid droplet size. Cell Rep. 2013;3:1465–1475. doi: 10.1016/j.celrep.2013.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zhao J., Hedera P. Hereditary spastic paraplegia-causing mutations in atlastin-1 interfere with BMPRII trafficking. Mol. Cell. Neurosci. 2013;52:87–96. doi: 10.1016/j.mcn.2012.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Guelly C., Zhu P.-P., Leonardis L., Papić L., Zidar J., Schabhüttl M., Strohmaier H., Weis J., Strom T.M., Baets J., et al. Targeted high-throughput sequencing identifies mutations in atlastin-1 as a cause of hereditary sensory neuropathy type I. Am. J. Hum. Genet. 2011;88:99–105. doi: 10.1016/j.ajhg.2010.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wang Z.-B., Zhang X., Li X.-J. Recapitulation of spinal motor neuron-specific disease phenotypes in a human cell model of spinal muscular atrophy. Cell Res. 2013;23:378–393. doi: 10.1038/cr.2012.166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Schneider C.A., Rasband W.S., Eliceiri K.W. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods. 2012;9:671–675. doi: 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.De Vos K.J., Sheetz M.P. Visualization and quantification of mitochondrial dynamics in living animal cells. Methods Cell Biol. 2007;80:627–682. doi: 10.1016/S0091-679X(06)80030-0. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.