

Abstract
Cardiac hypertrophy, an adaptive process that responds to increased wall stress, is characterized by the enlargement of cardiomyocytes and structural remodeling. It is stimulated by various growth signals, of which the mTORC1 pathway is a well-recognized source. Here, we show that loss of Flcn, a novel AMPK–mTOR interacting molecule, causes severe cardiac hypertrophy with deregulated energy homeostasis leading to dilated cardiomyopathy in mice. We found that mTORC1 activity was upregulated in Flcn-deficient hearts, and that rapamycin treatment significantly reduced heart mass and ameliorated cardiac dysfunction. Phospho-AMP-activated protein kinase (AMPK)-alpha (T172) was reduced in Flcn-deficient hearts and nonresponsive to various stimulations including metformin and AICAR (5-amino-1-β-D-ribofuranosyl-imidazole-4-carboxamide). ATP levels were elevated and mitochondrial function was increased in Flcn-deficient hearts, suggesting that excess energy resulting from up-regulated mitochondrial metabolism under Flcn deficiency might attenuate AMPK activation. Expression of Ppargc1a, a central molecule for mitochondrial metabolism, was increased in Flcn-deficient hearts and indeed, inactivation of Ppargc1a in Flcn-deficient hearts significantly reduced heart mass and prolonged survival. Ppargc1a inactivation restored phospho-AMPK-alpha levels and suppressed mTORC1 activity in Flcn-deficient hearts, suggesting that up-regulated Ppargc1a confers increased mitochondrial metabolism and excess energy, leading to inactivation of AMPK and activation of mTORC1. Rapamycin treatment did not affect the heart size of Flcn/Ppargc1a doubly inactivated hearts, further supporting the idea that Ppargc1a is the critical element leading to deregulation of the AMPK–mTOR-axis and resulting in cardiac hypertrophy under Flcn deficiency. These data support an important role for Flcn in cardiac homeostasis in the murine model.
INTRODUCTION
Cardiovascular disease is the leading cause of mortality in industrialized countries (1,2). Cardiac hypertrophy, characterized by cardiomyocyte enlargement, induction of a fetal gene expression pattern and cytoskeletal remodeling, develops in response to increased wall stress caused by physiological stimuli such as exercise, or pathological stimuli from pressure or volume overload induced by hypertension, myocardial infarction or valvular heart disease (3–5). Cardiac hypertrophy is an important adaptation to compensate for increased wall stress; however, with prolonged mechanical stress, the hypertrophic response can no longer compensate, leading to dilated cardiomyopathy and heart failure (3,4,6,7). Therefore, it is of clinical importance to decipher the underlying mechanisms that lead to the development of cardiac hypertrophy.
In hypertrophied cardiomyocytes, protein synthesis that supports the growth of individual cardiomyocytes is increased (8–10). Protein synthesis is regulated by the PI3K-AKT and AMPK–mTOR signaling pathways. Indeed, manipulations of genes within these pathways in mice, including amplification (gain of function) of AKT (11–14) and inactivation of LKB1 (15), PTEN (16) or TSC1 (10), have been reported to result in a cardiac hypertrophy phenotype. Importantly, treatment of some of these mouse models with rapamycin was effective in abrogating cardiac hypertrophy (10,11,14,15), suggesting that deregulation of the AMPK–mTOR pathway and subsequent increased protein synthesis may play an important role in the development of cardiac hypertrophy.
Birt–Hogg–Dubé (BHD) syndrome is an inherited kidney cancer syndrome, which predisposes patients to renal tumors, fibrofolliculomas and lung cysts (17–21). The causative gene, FLCN, is a tumor suppressor, which encodes folliculin (FLCN), a 64 kDa protein (22). Although little was known about FLCN function until recently, current studies have shown that FLCN may orchestrate a number of intracellular signaling pathways (23,24). In particular, the association between the FLCN pathway and metabolism has been highlighted, and most studies have suggested that FLCN is an important molecule for nutrient and energy sensing. Importantly, two FLCN interacting proteins, FNIP1 and FNIP2, bind to AMPK, a critical energy-sensing molecule that broadly regulates metabolic pathways (22,25,26). Furthermore, inactivation of FLCN causes deregulation of the mTOR pathway in mouse and human kidney (19,27), while upregulation of PPARGC1A drives increased mitochondrial biogenesis in Flcn knockout mouse skeletal muscle and kidney (28). Additionally, FLCN-null mouse embryonic fibroblasts (MEFs) and embryonic stem (ES) cells, and a FLCN-deficient renal cancer cell line all display aberrant TFE3 localization (29,30), and the FLCN-null renal cancer cell line also shows activation of the HIF–VEGF pathway (31). Based on the recently reported FLCN crystal structure, the C-terminus of FLCN is distantly related to differentially expressed in normal cells and neoplasia (DENN) domain proteins and possesses guanine exchange factor (GEF) activity towards Rab35, suggesting that FLCN might be related to the Rab GEF family of proteins and be involved in membrane trafficking (32). Biochemical studies from two research groups support functions for the FLCN/FNIP1/FNIP2 complex in sensing amino acid sufficiency, regulating GDP/GTP status of Rag proteins through physical interaction and mediating mTORC1 translocation to the lysosome surface where mTORC1 becomes activated, suggesting that FLCN might be necessary for the activation of mTORC1 (33,34). However, considering that FLCN is a tumor suppressor, there should be a critical mechanism for activation of mTORC1 under FLCN deficiency (34). Although a precise mechanism by which FLCN deficiency alters cell metabolism remains elusive, the previous report of high FLCN expression in human heart (22), recent reports of a role for FLCN in regulating metabolic pathways, and earlier reports of murine cardiac hypertrophy models that developed as a consequence of inactivation of genes in the AMPK–mTOR pathway, raise the possibility that FLCN has a potential role in cardiac homeostasis.
Here, we uncover an important role for FLCN in cardiac homeostasis in which inactivation of Flcn in murine hearts causes cardiac hypertrophy resulting in a fatal dilated cardiomyopathy. Our in vivo and in vitro data demonstrate that cardiac hypertrophy resulting from mTORC1 activation is dependent on Ppargc1a, an important molecule for energy homeostasis. Decreased phospho-AMPKα (T172) and subsequent increased activation of mTORC1, which were observed in Flcn-deficient hearts, were reversed in Flcn/Ppargc1a doubly inactivated hearts. Taken together these data support an important role for FLCN in cardiac homeostasis involving modulation of cellular energy levels and the AMPK–mTOR pathway.
RESULTS
FLCN inactivation in murine hearts causes hypertrophy leading to heart failure
To determine the role of FLCN in heart function, we crossbred mice carrying loxP-flanked Flcn alleles (floxed, f) with alpha-myosin heavy chain (αMHC)-Cre transgenic mice in which Cre recombinase expression is driven by the αMHC promoter (35), thereby deleting Flcn gene sequences specifically in cardiomyocytes. Unfortunately, cardiomyocyte-targeted Flcn knockout mice generated with the αMHC-Cre mice died within 3 weeks of birth from heart failure (Supplementary Material, Fig. S1), and this deleterious phenotype made it difficult to conduct further cardiac functional studies. Muscle creatine kinase (CKM)-Cre transgenic mice also express Cre transgenes in heart and skeletal muscle at a later stage of embryogenesis relative to αMHC-Cre transgenes (36–38), and this mouse model (Flcn f/f, CKM-Cre; Flcn KO) developed an enlarged heart but survived on average to 3 months of age (Fig. 1A and B). We evaluated cardiac function by echocardiography, and Flcn KO mice showed severe cardiac dysfunction, including decreased cardiac output (CO), decreased ejection fraction (EF) and decreased fractional shortening (FS) (Fig. 1C). mRNA expression of atrial natriuretic peptide (ANP), an indicator of pathological hypertrophy, was increased in Flcn KO mouse hearts (Fig. 1D). Kaplan–Meier survival analysis revealed a median survival of 85.5 days for the Flcn KO mice, whereas Flcn f/f control littermates (CT) survived until termination of the 150 day observation period (Fig. 1E, n = 20 for each group). Since we did not observe any arrhythmias such as ventricular tachycardia or atrial fibrillation by electrocardiography (data not shown), and the Flcn KO mice displayed severe lung and liver congestion at necropsy, we concluded that FLCN knockout mice died due to dilated cardiomyopathy.
Figure 1.

Flcn inactivation in the heart causes dilated cardiomyopathy. (A) Representative western blot image of Flcn f/f littermate control (CT) heart and Flcn f/f, CKM-Cre (Flcn KO) heart at 6 weeks of age shows the inactivation of Flcn in the mouse heart. (B) Representative gross morphology (left panel) and H&E staining (middle panel) of hearts and heart weight-to-body weight (%HW/BW) ratio (right panel) from Flcn f/f littermate control (CT) mice and Flcn f/f, CKM-Cre (Flcn KO) mice at 6 weeks of age. Scale bars = 10 mm (left panel) and 1 mm (middle panel). Heart weight-to-body weight (%HW/BW) ratio was greater in Flcn KO mice (mean = 1.67%) than in CT mice (mean = 0.45%). Bar, mean ± SD, P<0.0001, unpaired t-test with Welch's correction. (C) Representative echocardiographs (left panel) and parameters obtained in echocardiographs (right panel) for CT and Flcn KO mice at 6 weeks of age. Flcn KO mice showed severely decreased contractile function including decreased CO, EF and FS (t-test: CO, P = 0.043; EF, P = 0.0044; FS, P = 0.0044). Three animals per group were analyzed. (D) mRNA expression for ANP relative to 36B4 in CT and Flcn KO mouse hearts (t-test:P = 0.0002; n = 8 per group). (E) Kaplan–Meier survival analysis demonstrated shortened life span for Flcn KO mice. Median survival was 85.5 days for Flcn KO mice and undetermined for CT mice (n = 20 for each group, P < 0.0001).
FLCN inactivation in hearts causes cardiac hypertrophy with increased mTORC1 activity
To elucidate the underlying mechanism by which Flcn knockout mice develop dilated cardiomyopathy, we examined the histology of the Flcn KO hearts. The cross-sectional diameter of cardiomyocytes in Flcn KO mice was increased suggesting that Flcn loss caused hypertrophy of cardiomyocytes (Fig. 2A). Deregulation of the mTORC1 pathway is known to contribute to development of cardiac hypertrophy (3), and indeed, we observed activation of mTORC1 and its downstream pathway targets, including p-4EBP1 and p-S6R, in hearts from Flcn KO mice relative to CT hearts by immunoblotting (Fig. 2B) as well as increased protein synthesis in Flcn-null MEFs (Fig. 2C), suggesting that the up-regulated mTORC1 pathway might cause increased protein synthesis in Flcn-null cells and contribute to cardiac hypertrophy of Flcn KO hearts. mTORC1 is an important regulator of autophagy (39) and indeed we observed suppressed autophagic induction (Fig. 2D and E), further supporting activation of mTORC1 in Flcn KO hearts.
Figure 2.

Cardiac hypertrophy phenotype in the Flcn KO heart and up-regulation of mTORC1 signaling under FLCN deficiency. (A) H&E staining of cross-sectional cardiac muscle (left panel, scale bar = 50 mm) and muscle fiber diameter (right panel) of Flcn f/f (CT) and Flcn f/f, CKM-Cre (Flcn KO) hearts. Muscle fiber diameter of Flcn KO heart was significantly greater than CT heart (P < 0.0001, unpaired t-test with Welch's correction). (B) Western blotting showed activation of downstream components of the mTOR signaling pathway in the Flcn KO heart relative to the CT heart. Protein levels of three independent experiments were quantified by Odyssey imager (Li-Cor) and shown as mean values (±SD) in right panels. (C) Protein synthesis of Flcn-null MEFs (Dox-) and doxycycline-induced FLCN-expressing MEFs (Dox+; cultured with doxycycline for 24 h) was determined by BCA method and shown as mean values + SD. Flcn-null MEFs (Dox−) showed increase protein synthesis compared with FLCN-expressing MEFs (Dox+). n = 3, ****Significance at P < 0.0001; unpaired t-test. (D) CT and Flcn KO mice were starved for 24 h and autophagic activity in the hearts was investigated. Induction of LC3 II, an indicator of autophagic activity was suppressed in Flcn KO mice (NS, non-starved; S, starved). (E) Aberrant accumulation of SQSTM1, which monitors autophagic degradation, was observed in Flcn-deficient heart.
Rapamycin treatment significantly reduces heart size and improves cardiac function of FLCN-deficient hearts
To assess the role of mTOR activation in the cardiac hypertrophy phenotype, we treated Flcn KO mice with rapamycin. Rapamycin treatment significantly decreased heart weight-to-body weight ratio (%HW/BW) and the diameter of cardiomyocytes in Flcn KO mice (Fig. 3A and B). A marked improvement of the contractile function was seen by echocardiogram in rapamycin-treated Flcn KO mice (Fig. 3C) suggesting that increased mTORC1 activity promotes cardiomyocyte cell growth resulting in dilated cardiomyopathy.
Figure 3.

Rapamycin treatment significantly reduces heart size and improves cardiac function of the Flcn KO heart. (A) Representative gross morphology (left panel) and H&E staining (middle panel) of hearts, and heart weight-to-body weight (%HW/BW) ratio (right panel) from rapamycin-treated Flcn f/f (CT + rapamycin) mice, rapamycin-treated Flcn f/f, CKM-Cre (Flcn KO + rapamycin) mice and vehicle-treated Flcn f/f, CKM-Cre (Flcn KO + vehicle) mice. Scale bars = 10 mm (left panel) and 1 mm (middle panel). Rapamycin treatment significantly decreased HW/BW ratio in Flcn KO hearts (P < 0.0001), but had no effect on CT hearts (NS, non-significance; one-way ANOVA). (B) The average cardiac muscle diameter was reduced in rapamycin-treated Flcn KO mice. (C) Representative echocardiographs (left panel) and parameters obtained in echocardiographs (right panel) for rapamycin-treated Flcn CT and KO mice, and vehicle-treated Flcn KO mice at 6 weeks of age. Rapamycin treatment significantly improved cardiac function of Flcn KO mice. EF and FS are shown here. *Significance at P < 0.05; **Significance at P < 0.01; ****Significance at P < 0.0001; one-way ANOVA. At least three animals per group were analyzed.
Phosphorylation of AMPK at T172 is impaired under FLCN deficiency
To clarify how mTORC1 is up-regulated in Flcn KO hearts, we investigated the PI3K-AKT pathway. However, we observed neither an elevation of growth factors (i.e. IGF-1 and insulin) in the serum of Flcn KO mice nor an increase of signaling molecules in the PI3K-AKT pathway (i.e. AKT and PDK1) in Flcn KO hearts (Fig. 4A and B). AMPK is an important energy-sensing molecule that responds to an elevated AMP/ATP ratio by negatively regulating mTORC1 through phosphorylation of Raptor (40) and TSC2-mediated inhibition of Rheb (41). Immunoblotting of Flcn KO heart lysates showed reduced phospho-AMPKα (T172) (indicative of decreased AMPK activity) and decreased phospho-ULK1 (S555) [a direct target of AMPK (42)] in Flcn-deficient hearts compared with CT hearts (Fig. 4C). Therefore, we hypothesized that cardiac hypertrophy resulting in severe cardiac dysfunction in Flcn-deficient hearts was caused by attenuated AMPK activation and that restoration of AMPK activity might inhibit development of cardiac hypertrophy by suppressing mTORC1 activity. However, treatment with AMPK activators including AICAR (5-amino-1-β-D-ribofuranosyl-imidazole-4-carboxamide) and metformin did not improve cardiac phenotypes of Flcn-deficient hearts (Fig. 4D and Supplementary Material, Fig. S2). Notably, although phosphorylation was increased in CT hearts and in quadriceps muscle, we observed no effect of AICAR or metformin on phospho-AMPKα (T172) in either tissue from Flcn KO mice (Fig. 4E). Although both Flcn-null and doxycycline-induced Flcn-expressing MEFs responded to amino acid starvation by activation of AMPK, relative phospho-AMPKα levels were reduced in Flcn-null MEFs compared with Flcn-expressing MEFs as assessed by immunoblotting (Fig. 4F). Furthermore, phosphorylation of Raptor at S792 (a direct target of AMPK) (40) mirrored activation of AMPK in Flcn-null MEFs relative to Flcn-expressing MEFs (Fig. 4F). These data suggest that the cardiac hypertrophy associated with increased mTORC1 activity during FLCN deficiency might be due to impaired AMPK activation.
Figure 4.

Impaired phosphorylation of AMPKα at T172 under FLCN deficiency. (A) IGF-1 and insulin in mouse serum were measured using ELISA and radioimmunoassay (RIA), respectively. There was no significant difference between Flcn f/f, CKM-Cre (Flcn KO) mice and Flcn f/f littermate control (CT) mice (NS, non-significance; one-way ANOVA, n = 6). (B) Signaling molecules in PI3K-AKT pathway in Flcn KO hearts were investigated by immunoblotting. AKT was not up-regulated whereas PDK1 was down-regulated, indicating a potential negative feedback loop from increased mTORC1 activity. (C) Western blotting showed decreased phospho-AMPKα (T172) in Flcn KO heart relative to CT heart. Immunoblotting revealed decreased p-ULK1 (S555), a direct target of AMPK, in Flcn KO heart relative to CT heart as shown in the right panel. (D) AICAR treatment had no effect on heart size in CT or Flcn KO mice. ****Significance at P < 0.0001; NS, non-significance; one-way ANOVA, n = 12. (E) Immunoblotting of quadriceps muscle showed AICAR treatment increased phospho-AMPKα (T172) in CT mice, but not in Flcn KO mice (upper panel). Immunoblotting of heart lysates showed metformin treatment increased phospho-AMPKα (T172) in CT mice, but not in Flcn KO mice (lower panel). (F) Maximum stimulation of phospho-AMPKα (T172) and its downstream phospho-Raptor (S792) activation by amino acid starvation was reduced in Flcn-null MEFs (Dox−) compared with FLCN-expressing MEFs (Dox+).
FLCN deficiency results in increased PPARGC1A expression and mitochondrial biogenesis leading to overproduction of ATP
Because phosphorylation of AMPKα at T172 is regulated by the AMP/ATP ratio (43), we examined ATP levels in Flcn-deficient hearts. We observed markedly elevated ATP levels in Flcn KO hearts, compared with CT hearts (Fig. 5A). Reconstitution of wild-type FLCN decreased ATP levels in Flcn-null MEFs (Fig. 5A). Electron microscopic images of cardiac muscle showed an increased mitochondrial mass in the Flcn KO heart. The ratio of mitochondrial area to muscle fiber area was significantly higher in the Flcn-deficient heart relative to the control heart (Fig. 5B). Furthermore, we observed increased respiratory capacity in mitochondria isolated from Flcn KO hearts (Fig. 5C). Interestingly, immunoblotting experiments demonstrated that protein expression of Ppargc1a, an important transcriptional co-activator for energy homeostasis, which controls mitochondrial biogenesis, angiogenesis and generation of reactive oxygen species (44–47), was increased in Flcn KO hearts (Fig. 5D). Furthermore, we observed that doxycycline-induced FLCN expression in FLCN-null UOK257 cells decreased PPARGC1A at the protein level (Fig. 5E). These observations are consistent with our previous report showing that loss of Flcn in skeletal muscle causes increased mitochondrial biogenesis and subsequent elevated levels of ATP through increasing PPARGC1A expression (28), and led us to hypothesize that excess ATP resulting from increased Ppargc1a expression in Flcn-deficient hearts might continually suppress AMPK activity and drive mTORC1 hyperactivation resulting in cardiac hypertrophy.
Figure 5.

FLCN deficiency leads to increased PPARGC1A expression and mitochondrial biogenesis leading to overproduction of ATP. (A) ATP content was measured in Flcn f/f (CT) and Flcn f/f, CKM-Cre (Flcn KO) hearts (left panel, n = 7, *at P < 0.05, unpaired t-test with Welch's correction), and in MEFs (right panel, D2, Flcn f/d MEF (Flcn+); DA2-2, Flcn d/d MEF (Flcn−); T, DA2-2 only with tet regulator (Flcn−); T + Dox, T treated with doxycycline (Flcn-); F, T with wild-type FLCN cassette under tet promoter (Flcn-); F + Dox, F treated with doxycycline (FLCN+).) (NS, non-significance; ***at P < 0.001; unpaired t-test). (B) Representative electron micrograph images of cardiac muscle from CT and Flcn KO mice at 6 weeks of age (left panel). Flcn KO cardiac muscle has more mitochondrial mass (scale bar, 2 mm). Insert panels show well-preserved mitochondrial structure in both CT and Flcn KO hearts (scale bar, 500 nm). Bar graph shows the ratio of mitochondrial area to cardiac fiber area in CT and Flcn KO hearts (right panel). Three pairs of images from CT and Flcn KO hearts were analyzed using ImageJ software (P = 0.0002, unpaired t-test). (C) Maximum respiration capacity was measured with 1 mg of mitochondria isolated from Flcn f/f (CT) and Flcn f/f, CKM-Cre (Flcn KO) hearts using Seahorse XF96 analyzer. Bars represent mean + SD of four animals of each genotype (*at P < 0.05, **at P < 0.01, unpaired t-test). (D) Ppargc1a protein expression was increased in the Flcn KO heart relative to the CT littermate heart. Non-specific band was shown as a loading control (*at P < 0.05, unpaired t-test). (E) Restoration of wild-type FLCN in FLCN-null UOK257 cells (Dox+) decreased PPARGC1A protein expression compared with uninduced cells (Dox−). Lower panel represents densitometry of western blot bands from three independent experiments, indicated as mean values (±SD) (***at P < 0.001, unpaired t-test).
Cardiac hypertrophy of FLCN-deficient hearts associated with increased mTORC1 is PPARGC1A dependent
To test this hypothesis, we crossbred Flcn f/f, CKM-Cre mice with Ppargc1a conditional knockout mice to delete both genes in the double knockout heart (DKO). In support of this hypothesis, inactivation of Ppargc1a significantly decreased the heart size of Flcn KO mice (Fig. 6A). Kaplan–Meier survival analysis confirmed a statistically significant increase in survival of Flcn knockout mice with Pparc1a inactivation (Fig. 6B). Furthermore, in Flcn /Ppargc1a doubly inactivated hearts, we observed increased phospho-AMPKα (T172) levels and suppression of mTORC1 pathway components (Fig. 7A) relative to Flcn-deficient hearts. To further confirm the role of PPARGC1A in deregulation of the AMPK–mTOR axis under FLCN deficiency, we performed PPARGC1A knockdown experiments using Flcn-null MEFs and UOK257 cells. siRNA knockdown of PPARGC1A resulted in up-regulation of phospho-AMPKα (T172) and phospho-Raptor(S792) in FLCN-null UOK257 cells, and suppression of mTOR downstream effectors in Flcn-null MEFs (Fig. 7B). Immunoblotting experiments demonstrated that overexpression of a doxycycline-inducible constitutively active form of AMPKα (CA), which is a truncated form of AMPKα at residue 312 and therefore retains significant kinase activity (48) in FLCN-null UOK257 cells, suppressed mTORC1 (Fig. 7C), indicating that up-regulated mTORC1 under FLCN deficiency is due to impaired activation of AMPK. Taken together, we concluded that increased energy production driven by upregulated PPARGC1A continually suppresses AMPK activity and the impaired activation of AMPK can no longer attenuate mTORC1 under FLCN deficiency. Finally, we subjected mice with both Flcn and Ppargc1a inactivation in hearts to rapamycin treatment to determine whether or not cardiac hypertrophy resulting from increased mTORC1 activity was PPARGC1A dependent. Interestingly, rapamycin treatment did not produce a statistically significant change in the size of Flcn/Ppargc1a doubly inactivated hearts compared with untreated DKO mice (Fig. 8A). Histologic analysis of cardiomyocyte diameter revealed no additional effect of rapamycin treatment on cardiac hypertrophy in DKO mice (Fig. 8B), suggesting that cardiac hypertrophy resulting from increased mTORC1 activation during FLCN deficiency was totally PPARGC1A dependent.
Figure 6.
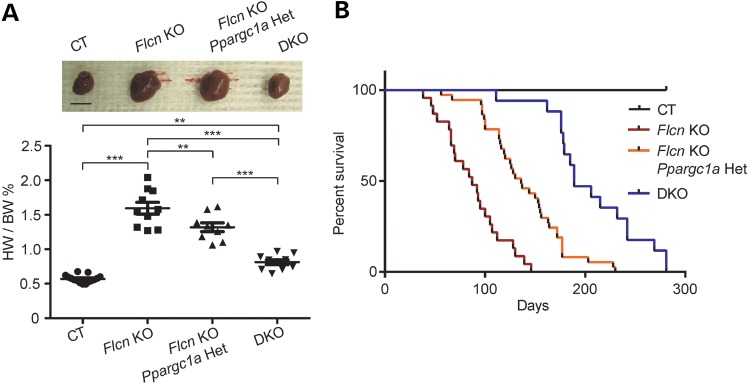
Inactivation of PPARGC1A decreased the heart size and prolonged the survival of Flcn KO mice. (A) Muscle-targeted Flcn/Ppargc1a DKO mice show dramatic reduction in heart size. The HW/BW ratio of each genotype at 8 weeks of age. Mean ± SD are shown. Haploinsufficient effect was observed in Flcn f/f, Ppargc1a f/+, CKM-Cre mice (Flcn KO, Ppargc1a Het). Upper panel shows representative gross morphology of hearts for each genotype (scale bar = 1 cm). ***Significance at P < 0.001, **significance at P < 0.01, NS, non-significance; one-way ANOVA. (B) Kaplan–Meier survival analysis shows prolonged survival for DKO mice compared with Flcn f/f, CKM-Cre (Flcn KO) mice. Median survival for each of the genotypes is as follows: Flcn f/f (CT) = undefined; Flcn f/f, CKM-Cre (Flcn KO) = 87 days; Flcn f/f, Ppargc1a f/+, CKM-Cre (Flcn KO, Ppargc1a Het) = 136 days and Flcn f/f, Ppargc1a f/f, CKM-Cre (DKO) = 189 days, respectively (P < 0.0001, n > = 17).
Figure 7.

Inactivation of PPARGC1A reactivates AMPK and suppresses mTORC1 in the Flcn KO heart and FLCN-null cells. (A) Western blotting of the heart lysates shows AMPK was reactivated (left panel) and mTOR signaling was suppressed (right panel) in muscle-targeted Flcn/Ppargc1a DKO mice. (B) FLCN-null UOK257 cells were transfected with siPPARGC1A or scrambled control (con) and harvested after 48 h, and showed up-regulation of phospho-AMPKα (T172) and phospho-Raptor (S792) (upper panel). Suppression of Ppargc1a for 72 h resulted in suppression of mTOR downstream components in Flcn-null MEFs (lower panel). Protein levels of three independent experiments were quantified by Odyssey imager (Li-Cor) and shown as mean values (±SD) in right panels. (C) Overexpression of doxycycline-induced constitutively active (CA) AMPKα, which is a truncated form of AMPKα at residue 312 and therefore retains significant kinase activity, in FLCN-null UOK257 cells suppressed mTOR signaling by phosphorylating Raptor. Both AMPKα and AMPKα (CA) were blotted with AMPKα antibody in the same membrane. Protein levels of three independent experiments were quantified by Odyssey imager (Li-Cor) and shown as mean values (±SD) in lower panels.
Figure 8.

Cardiac hypertrophy of Flcn KO mice associated with increased mTORC1 is PPARGC1A dependent. (A) Inhibition of mTOR by rapamycin had no additional effect on heart size of muscle-targeted Flcn/Ppargc1a knockout (DKO) mice. No significant difference was seen in HW/BW ratio between DKO and rapamycin-treated DKO mice (NS, non-significance; ****P < 0.0001, ***P < 0.001, one-way ANOVA). (B) Histologic analysis of DKO hearts with and without rapamycin treatment revealed no change in muscle fiber thickness. Scale bar = 50 mm. (NS, non-significance; **** P < 0.0001, one-way ANOVA). (C) Hypothetical scheme showing how FLCN inactivation leads to cardiac hypertrophy.
DISCUSSION
In this study, we demonstrate that Flcn inactivation in murine hearts leads to cardiac hypertrophy, resulting in a fatal dilated cardiomyopathy. mTORC1 was activated in Flcn-deficient hearts, and rapamycin treatment significantly reduced cardiac hypertrophy and ameliorated cardiac dysfunction. Phospho-AMPKα (T172) was decreased in Flcn-deficient hearts and was not restored by AICAR/metformin treatment. We observed increased ATP in Flcn-deficient hearts and an attenuated response of AMPKα T172 phosphorylation to amino acid starvation in Flcn-null MEFs. Inactivation of Ppargc1a, an essential molecule for ATP production, significantly reduced cardiac hypertrophy and prolonged survival of Flcn knockout mice. In Flcn/Ppargc1a doubly inactivated hearts, we observed the restoration of phospho-AMPKα (T172) levels, which were decreased in Flcn-deficient hearts, and the suppression of mTORC1 activity that was increased in Flcn-deficient hearts. Rapamycin treatment did not further affect the size of the Flcn/Ppargc1a doubly inactivated murine hearts, indicating that FLCN deficiency causes energy overproduction that might lead to cardiac hypertrophy by deregulation of the AMPK–mTOR pathway.
The AMPK–mTOR pathway plays an important role in cardiac homeostasis (49–51). Germline mutation in AMPKγ2 predisposes patients to cardiac hypertrophy with increased glycogen storage (52,53). Inactivation of AMPKα2, a predominant isoform in heart, exacerbated cardiac hypertrophy and increased mTORC1 activity induced by aortic constriction (54). Murine hearts with kinase dead mTOR displayed significantly decreased heart function (55) and a heart-targeted mTOR knockout mouse model displayed dilated cardiomyopathy with an impaired hypertrophic response when subjected to pressure overload (56). In this report, Flcn-deficient hearts displayed decreased phospho-AMPKα (T172) and increased mTORC1 activation, and rapamycin treatment significantly reduced cardiac hypertrophy and improved heart function. Because the exogenous expression of a constitutively active form of AMPK in FLCN-null cell lines suppressed mTORC1 signaling, the increased mTORC1 activity in Flcn-deficient hearts might be attributed to decreased activity of AMPK. Our data indicate a critical role for Flcn in maintaining the integrity of the AMPK–mTOR signaling pathway in heart muscle, the failure of which leads to development of cardiac hypertrophy. The Flcn KO mice died at 6 weeks of age due to dilated cardiomyopathy, even though ATP levels in Flcn-deficient hearts were increased. Recent reports have highlighted the significance of autophagy in pathologic cardiac hypertrophy (57,58). mTORC1 is a negative regulator of autophagy through phosphorylation of the ULK1/ATG13/FIP200 complex (59). Lack of autophagy to maintain the quality of proteins and organelles might exacerbate the severe cardiomyopathy in our model, underscoring the detrimental effect of upregulated mTORC1 in pathologic cardiac hypertrophy.
A number of in vivo animal studies have reported that inactivation of causative genes responsible for hamartoma syndromes leads to increased susceptibility for developing cardiac hypertrophy. Cardiac-specific deletion of Lkb1 leads to cardiac hypertrophy with cardiac dysfunction (15). Smooth muscle protein-22-mediated deletion of Tsc1 results in cardiac hypertrophy that is mTORC1-mediated and reversed by rapamycin (10). Cardiomyocyte-specific deletion of Pten causes cardiac hypertrophy (16). In this study, we report that cardiac inactivation of Flcn, the gene responsible for the hamartoma syndrome BHD, caused cardiac hypertrophy in the mouse model through modulation of the AMPK–mTOR axis. However, studies with genetically modified AMPK-null mice have shown that reduced AMPK activity does not cause basal cardiac hypertrophy (60–63). This suggests that an as yet uncharacterized mechanism, which is not through AMPK might regulate mTORC1 and contribute to the development of cardiac hypertrophy under deficiency of hamartoma syndrome causative genes in heart muscle. We observed suppressed mTORC1 in Flcn/Ppargc1a doubly inactivated hearts, which was activated in Flcn-deficient hearts. Taken together with the fact that AMPK-null mice do not develop cardiac hypertrophy, this implies a potential mTORC1 regulation by Ppargc1a that is not necessarily through AMPK in Flcn-deficient hearts, further highlighting the importance of Ppargc1a under Flcn deficiency.
The Bateman domain in the AMPKγ subunit detects the AMP/ATP ratio and triggers a conformational change resulting in phosphorylation of the AMPKα subunit at T172 (64). In Flcn-deficient hearts, we observed decreased phospho-AMPKα (T172) levels, which were most likely due to increased levels of ATP. Consequently, AMPK failed to suppress the mTORC1 signaling pathway. In order to stimulate phospho-AMPKα (T172), treatment with metformin was introduced, which was expected to have a cardioprotective effect that might be beneficial for patients at risk for myocardial ischemia (65,66). Indeed, we observed significantly increased cardiac function in metformin-treated control animals accompanied by elevated AMPKα phosphorylation on T172, but metformin treatment had no effect on Flcn KO heart function or AMPKα activation. Inactivation of Ppargc1a decreases energy production (44,67), and we saw that phosphorylation of AMPKα (T172) was restored in Flcn/Ppargc1a double KO mice. Taken together, excess energy (ATP) might compete with metformin, which has been suggested to activate AMPK by increasing cytosolic AMP (68). Our data support the concept that the cardioprotective effect of metformin might be attenuated under conditions of energy overproduction.
To date, an association between BHD syndrome and cardiac manifestations has not been documented. Furthermore, we have never observed dilated cardiomyopathy in whole-body heterozygous Flcn knockout mice, indicating that haploinsufficiency of Flcn alleles does not lead to a heart phenotype. However, although genetic studies of familial dilated cardiomyopathy have revealed that some of the genes linked to dilated cardiomyopathy encode proteins of the sarcomere, costamere, Z band and nuclear membrane, others have functions distinct from these broad cell biological classifications, including succinate dehydrogenase A (SDHA), lysosomal-associated membrane protein 2 (LAMP2) and AMPKα2 (69), which are involved in mitochondrial metabolism, autophagy and energy-sensing pathways. Since FLCN is essential for energy homeostasis through PPARGC1A-dependent regulation of mitochondrial metabolism, its interaction with Rag proteins that facilitate mTOR activation at the lysosomal surface, and modulation of energy-sensing pathways through FLCN/FNIP interaction with AMPK, it would be of interest to investigate the status of the FLCN pathway in human dilated cardiomyopathy.
PPARGC1A regulates energy homeostasis by modulating mitochondrial biogenesis, angiogenesis and reactive oxygen species (45–47,70). Interestingly, PPARGC1A overexpression in murine heart under the αMHC promoter caused dilated cardiomyopathy with increased mitochondrial biogenesis (71). Postnatal induction of Ppargc1a overexpression using a doxycycline-inducible murine system caused cardiac hypertrophy, which resulted in terminal dilated cardiomyopathy. Cessation of Ppargc1a overexpression by the removal of doxycycline reversed most of the myofibrillar structural abnormalities and cardiac dysfunction induced by doxycycline administration (72). However, the molecular mechanism by which cardiac hypertrophy developed was not fully elucidated in these two studies. In our study, we observed decreased phospho-AMPKα (T172) with increased ATP production and elevated mTORC1 activity in Flcn-deficient hearts, which was reversed by inactivation of Ppargc1a. Our results suggest a possible molecular mechanism for these Ppargc1a overexpression models, in which excessive ATP production might suppress AMPK activity and trigger protein synthesis initiated by mTORC1 activation, resulting in cardiac hypertrophy. Therefore, it would be of interest to investigate the activities of AMPK and mTORC1 in the previously reported models of heart-directed Ppargc1a overexpression.
In summary, we have identified FLCN as a novel molecule with a critical role in cardiac homeostasis. Our suggested model is summarized in Figure 8C. These data highlight the importance of the FLCN pathway in regulating cardiomyocyte cell growth through modulation of the AMPK–mTOR axis, which might be triggered by improper energy homeostasis regulated by PPARGC1A. These important findings underscore a role for Flcn in cardiac homeostasis in the murine model. Further study will be required to determine if the FLCN pathway may also play a role in human cardiac disease.
MATERIALS AND METHODS
Mice and in vivo drug treatment
The mice carrying Flcn alleles flanked by loxP sites (floxed, f) were generated as previously described (27) and backcrossed at least four times with B6 129SVJ mice. Control littermate mice were always used as controls. Cardiac-specific deletion of Flcn was achieved by crossing with αMHC-Cre mice kindly provided by Dr Michael D. Schneider (35). Muscle creatine kinase (CKM)-Cre transgenic mice [FVB-Tg(Ckmm-cre) 5Khn/J, stock number: 006405] were obtained from Jackson Laboratories (Bar Harbor, ME). Mice carrying Ppargc1a alleles flanked by loxP sites (floxed, f) were kindly provided by Dr Bruce Spiegelman at Dana-Farber Cancer Institute, Harvard Medical School (70). We confirmed that the CKM-Cre and αMHC-Cre transgenes had no detectable effect on mouse phenotypes. We are aware that the CKM-Cre transgene could be affecting the heart indirectly via respiratory muscle dysfunction (73). However, because of the difficulty to analyze heart-targeted Flcn knockout mice carrying the αMHC-Cre transgene that survived only 3 weeks, we decided to conduct further cardiac analyses on muscle-targeted Flcn knockout mice carrying the CKM-Cre transgene. We also confirmed the absence of cardiac phenotype in other skeletal muscle-targeted Flcn knockout mouse models generated with myogenin (MYOG)-Cre or myocyte enhancer factor 2C (MEF2C)-Cre transgenic mice (data not shown). For in vivo drug treatment, 2 mg/kg rapamycin (LC Laboratories, Woburn, MA), 200 mg/kg metformin (Sigma-Aldrich, St Louis MO) or 0.5 mg/g AICAR (LC Laboratories, Woburn, MA) were administered daily by intraperitoneal injection from postnatal Day 7 for 2 months. All mice used in these experiments were housed in the National Cancer Institute animal facilities according to the National Cancer Institute Animal Care and Use Committee guidelines. The aged mice were killed by carbon dioxide asphyxiation, and the heart was removed, cut into small pieces, snap frozen in liquid nitrogen, and stored at −80°C for further analysis.
Cell culture
FLCN-null UOK257, a cancer cell line originally derived from a renal tumor of a BHD syndrome patient surgically treated at the Urologic Oncology Branch, National Institutes of Health, Bethesda, MD, with written patient permission under National Institutes of Health Institutional Review Board-approved protocol 97-C-0147, was reconstituted with HA tagged wild-type FLCN gene expression by two sequential lentiviral transductions using the full-length Tet-on 3G gene (Clontech, Mountain View, CA) and the HA tagged wild-type FLCN gene under the TRE3G promoter. Doxycycline was added at a concentration of 1.0 ng/ml, unless otherwise indicated. Flcn-null MEFs derived from conditional Flcn targeted mice (29) were also reconstituted with HA tagged wild-type FLCN expression using Tet-on 3G doxycycline-inducible system. These cells were analyzed after addition of doxycycline. MEFs were established from Flcn conditional knockout mice as previously described (29). Briefly, MEFs were established from Flcn f/d mice (Flcn f/d, with one allele of wild-type Flcn gene; cell line name, D2) and treated with adenoviral Cre recombinase (Flcn d/d, without Flcn gene; cell line name, DA2-2). Then, DA2-2 was reconstituted with HA tagged wild-type FLCN gene expression by two sequential lentiviral transductions using the full-length Tet-on 3G gene (only with Tet regulator and without FLCN expression; cell line name, T) and the HA tagged wild-type FLCN gene under the TRE3G promoter (with HA-tagged wild-type FLCN expression; cell line name, F). For protein synthesis analysis, same numbers of cells cultured with or without doxycycline for 24 h were lysed with radio immunoprecipitation assay (RIPA) buffer and protein concentration was determined by bicinchoninic acid (BCA) method. For amino acid starvation, cells were cultured with or without doxycycline for 24 h prior to starvation, and then replaced with medium containing the following: phosphate-buffered saline, 10% dialyzed fetal bovine serum, sodium bicarbonate 3.7 g/l, minimum essential media (MEM) vitamin solution(100×), d-glucose 4.5 g/l and incubated for the indicated period. MEM essential amino acid solution (50×) and l-glutamine 0.584 g/l were supplemented for control medium (all from Invitrogen, Carlsbad, CA).
siRNA transfection
The siRNA oligos for PPARGC1A and scramble control were purchased from Invitrogen. siRNA was transfected with Xfect siRNA transfection reagent (Clontech) following the manufacturer's instructions. Cells were harvested 48 h (UOK257) or 72 h (MEFs) post-transfection and analyzed by western blot to confirm the knockdown of PPARGC1A. siRNA sequences for PPARGC1A are as follows: sense-5′-CUCUGGAUUUUGAUAGUUUtt-3′, anti-sense-5′-AAACUAUCAAAAUCCAGAGag-3′.
Transthoracic echocardiography
Mice at the age of 2–8 weeks old were anesthetized with 1–2% isoflurane in 100% oxygen and restrained on a temperature-controlled mouse board (Indus Instruments, Webster, TX), and echocardiography was performed on Flcn KO mice or CT mice using a Vevo 770 ultrasound system (Visual Sonics, Toronto, ON, Canada) equipped with a 30 MHz transducer. An echocardiographer blind to animal genotype captured two-dimensional parasternal long axis views of the left ventricle (B mode) and short axis views of the left ventricle (M mode). The following measurements were obtained for systole and diastole using the average of three cardiac cycles: heart rate (HR), stroke volume (SV), CO, LV end-diastolic volume (EDV), LV end-systolic volume (ESV), EF, LV end-diastolic diameter, LV end-systolic diameter and fractional shortening (FS). CO was calculated as SV × HR; EF was calculated as (EDV − ESV)/EDV × 100; FS was calculated as (LV end-diastolic dimension − LV end-systolic dimension)/LV end-diastolic dimension × 100.
RNA isolation and quantification
Total RNA was isolated from flash frozen mouse hearts and from cell cultures using TRIzol reagent (Invitrogen Carlsbad, CA), and total RNA was reverse transcribed to cDNA using a Superscript III reverse transcriptase kit (Invitrogen). Quantitative real-time polymerase chain reaction (PCR) was performed with the 7300 real-time PCR System (Applied Biosystems, Foster City, CA) using SyBr Green PCR master mix (Fermentas, Glen Burnie, CA). Primer sequences for ANP are as follows: mouse ANP-forward 5′- ATCTGCCCTCTTGAAAAGCA-3′, mouse ANP-reverse 5′- AAGCTGTTGCAGCCTAGTCC-3′. Signal intensity obtained from real-time PCR System was described in relative units; each value was normalized to 36B4 with the following sequences: mouse 36B4-forward 5′- GCAGACAACGTGGGCTCCAAGCAGAT-3′, mouse 36B4-reverse 5′- GGTCCTCCTTGGTGAACACGAAGCCC-3′.
Protein isolation and western blotting
Heart tissues were flash frozen in liquid nitrogen immediately after dissection. Frozen tissues were homogenized with a polytron homogenizer on ice in RIPA buffer [20 mm tris–HCl, pH 7.5, 150 mm NaCl, 1 mm ethylenediamine tetraacetic acid, 1.0% Triton X-100, 0.5% deoxycholate, 0.1% sodium dodecylsulfate] supplemented with PhosSTOP phosphatase inhibitor cocktail and Complete protease inhibitor cocktail (Roche, Indianapolis, IN), followed by centrifugation at 13,200 g for 30 min. Protein concentrations of cleared supernatants were measured with BCA protein assay kit (Pierce Biotechnology, Rockford, IL) and adjusted to 1.33 mg/ml. Four times sodium dodecyl sulphate-polyacrylamide gel electrophoresis (SDS) sample buffer was then added and samples were boiled for 5 min to produce 1 μg/μl sample lysates. A total of 20 μg of protein was loaded onto 4–20% tris–glycine SDS-PAGE gels. Immunoblotting was performed as previously described (27) with minor modifications optimized for the Odyssey infrared imaging system (LI-COR, Lincoln, NE). In brief, separated proteins were transferred to Immobilon-FL polyvinylidene difluoride (PVDF) membrane (Millipore, Billerica, MA), blocked with Odyssey blocking buffer (LI-COR) or 5% milk at room temperature for 1 h. Blocked membranes were incubated overnight at 4°C with primary antibodies, diluted with 0.1% bovine serum albumin in Tris-buffered saline with Tween 20 (TBST; 20 mm Tris–HCl pH8.0, 150 mm NaCl, 0.05% Tween 20) as follows: p-mTOR (S2448) 1:1000, total mTOR 1:500, p-Raptor(S792) 1:1000, p-AMPKα (T172) 1:1000, AMPKα 1:1000, p-S6 ribosomal protein (S240/244) 1:1000, total S6R 1:500, p-P70-S6 Kinase (T421/S424) 1:1000, p-4EBP1 (T37/46) 1:1000, total 4EBP1 1:500, glyceraldehyde-3-phosphate dehydrogenase 1:1000, p-AKT (T308) 1:1000, p-PDK1 (S241), p-ULK1 (S555) 1:1000, SQSTM1 1:1000, and LC3 (D11XP) 1;1000 (Cell Signaling, Danvers, MA). FLCN mouse monoclonal antibody was raised against recombinant FLCN protein (22). α-tubulin antibody was purchased from Sigma-Aldrich (St Louis MO). After three times washing with TBST, membranes were incubated at room temperature for 1 h with infrared dye-conjugated secondary antibodies (IRDye 800CW goat anti-rabbit or mouse IgG) (LI-COR), diluted 1:15000 with 0.2% Tween 20/Odyssey blocking buffer. After three additional washes with TBST and brief soaking in phosphate-buffered saline, images were acquired with an Odyssey infrared imaging system (LI-COR). All the experiments were repeated at least three times and a representative picture is shown.
Histological analysis
Mouse hearts were obtained immediately after euthanasia, fixed with 10% neutral buffered formalin overnight, embedded in paraffin block, sectioned at 5 μm thickness, and stained with hematoxylin and eosin by standard methods. Each section was scanned using the Aperio Image Scope system (Aperio, Vista, CA) and analyzed.
Respiratory capacity of isolated mitochondria from mice hearts
Maximum oxygen consumption of mitochondria isolated from mouse heart tissue was measured as previously described (28). Mitochondria were isolated from mouse hearts using standard Nagarse method (74) and 1 μg of mitochondria was attached to the bottom of XF96 V3 PET plate (Seahorse Bioscience) at 936 g for 10 min which was precoated overnight with 1:15000 polyethyleneimine solution/assay buffer (137 mm potassium chloride, 2 mm monopotassium phosphate, 2.5 mm magnesium chloride, 20 mm HEPES (4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid), 0.5 mm EGTA, 0.2% fatty acid-free BSA). The plate was warmed at 37°C for 10 min and transferred to the Seahorse XF96 analyzer. State 3 respiration (maximum adenosine diphosphate-stimulated oxygen consumption ratio; oxygen consumption ratio under sufficient substrate for mitochondrial complex) of complex I was measured immediately after addition of 5 mm glutamate, 5 mm malate, and 0.5 mm adenosine diphosphate, and that of complex II was measured immediately after addition of 5 mm succinate, 0.28 μm rotenone and 0.5 mm adenosine diphosphate.
ATP measurement
ATP concentration was measured using ATP Colorimetric/Fluorometric Assay Kit (Biovision, Milpitas, CA). Tissues or cultured cells were lysed in Nucleotide releasing buffer, adjusted to 1 mg/ml of protein concentration, treated with ATP converting enzyme, and read with a VICTOR™ X3 Multilabel Plate Reader (PerkinElmer, Waltham MA). Fifty microliters of lysate were used for one assay. Three independent assays were performed for each sample.
IGF-1 and insulin measurement
IGF-1 in mouse serum was measured by enzyme-linked immunosorbent assay (ELISA) using IGF-1 Mouse/Rat ELISA kit (BioVendor, Asheville, NC) and insulin in mouse serum was measured by radioimmunoassay using Rat Insulin RIA (Millipore, Billerica, MA) which has 100% cross reactivity with mouse insulin.
Electron microscopy
Mouse hearts were removed and immersed in 4% formaldehyde/2% glutaraldehyde (Electron Microscopy Sciences, Hartfield, PA)/PBS immediately after euthanasia. Small blocks were cut, osmicated, and dehydrated before embedding. The blocks were sectioned and observed in a Jeol 1200 transmission electron microscope equipped with an XR-100 CCD camera (Advanced Microscopy Techniques Corporation, Danvers, MA). Percentage of mitochondrial area was analyzed with Image J software (National Institutes of Health, Bethesda, MD). Three pairs of independent sections were analyzed, and the mean value with ±SD was shown.
Statistical analysis
Experimental data were summarized as the mean value with ±SD. Statistical analyses were performed using unpaired t-test with or without Welch's correction for two group comparison, or one-way ANOVA followed by Tukey post tests for multiple comparison. Kaplan–Meier survival analysis was performed using Graphpad Prism version 6.01 (GraphPad Software, Inc. La Jolla, CA), and differences were considered to be statistically significant at a value of P <0.05.
SUPPLEMENTARY MATERIAL
FUNDING
This word was supported by the Intramural Research Program of NIH, Frederick National Lab, Center for Cancer Research. This project has been funded in whole or in part with federal funds from the Frederick National Laboratory for Cancer Research, NIH, under contract HHSN261200800001E. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products or organizations imply endorsement by the US Government.
Supplementary Material
ACKNOWLEDGEMENTS
The authors thank Dr Bruce Spiegelman for distributing PPARGC1A knockout mice; Dr Michael D Schneider for distributing αMHC transgenic mice; Drs Mary Anne Conti and Douglas Rosing, at National Heart, Lung and Blood Institute, NIH for helpful discussions; Protein Expression Laboratory, Leidos Biomedical Research, Inc., for generating lentiviral constructs; Antoine Smith for his technical assistance on mouse electrocardiogram, Lisa Riffle for her excellent technical support for mouse echocardiograms and Louise Cromwell for excellent technical support with the mouse studies. Frederick National Laboratory for Cancer Research is accredited by AAALAC International and follows the Public Health Service Policy for the Care and Use of Laboratory Animals. Animal care was provided in accordance with the procedures outlined in the ‘Guide for Care and Use of Laboratory Animals’ (National Research Council; 1996; National Academy Press; Washington, DC).
Conflict of Interest statement. None declared.
REFERENCES
- 1.Braunwald E. Cardiovascular science: opportunities for translating research into improved care. J Clin Invest. 2013;123:6–10. doi: 10.1172/JCI67541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sciarretta S., Zhai P., Shao D., Maejima Y., Robbins J., Volpe M., Condorelli G., Sadoshima J. Rheb is a critical regulator of autophagy during myocardial ischemia: pathophysiological implications in obesity and metabolic syndrome. Circulation. 2012;125:1134–1146. doi: 10.1161/CIRCULATIONAHA.111.078212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Maillet M., van Berlo J.H., Molkentin J.D. Molecular basis of physiological heart growth: fundamental concepts and new players. Nat. Rev. Mol. Cell Biol. 2013;14:38–48. doi: 10.1038/nrm3495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sadoshima J., Izumo S. The cellular and molecular response of cardiac myocytes to mechanical stress. Annu. Rev. Physiol. 1997;59:551–571. doi: 10.1146/annurev.physiol.59.1.551. [DOI] [PubMed] [Google Scholar]
- 5.Shende P., Plaisance I., Morandi C., Pellieux C., Berthonneche C., Zorzato F., Krishnan J., Lerch R., Hall M.N., Ruegg M.A., et al. Cardiac raptor ablation impairs adaptive hypertrophy, alters metabolic gene expression, and causes heart failure in mice. Circulation. 2011;123:1073–1082. doi: 10.1161/CIRCULATIONAHA.110.977066. [DOI] [PubMed] [Google Scholar]
- 6.Bueno O.F., De Windt L.J., Tymitz K.M., Witt S.A., Kimball T.R., Klevitsky R., Hewett T.E., Jones S.P., Lefer D.J., Peng C.F., et al. The MEK1-ERK1/2 signaling pathway promotes compensated cardiac hypertrophy in transgenic mice. EMBO J. 2000;19:6341–6350. doi: 10.1093/emboj/19.23.6341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Purcell N.H., Wilkins B.J., York A., Saba-El-Leil M.K., Meloche S., Robbins J., Molkentin J.D. Genetic inhibition of cardiac ERK1/2 promotes stress-induced apoptosis and heart failure but has no effect on hypertrophy in vivo. Proc. Natl. Acad. Sci. U. S. A. 2007;104:14074–14079. doi: 10.1073/pnas.0610906104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wang Y., Huang B.P., Luciani D.S., Wang X., Johnson J.D., Proud C.G. Rheb activates protein synthesis and growth in adult rat ventricular cardiomyocytes. J. Mol. Cell Cardiol. 2008;45:812–820. doi: 10.1016/j.yjmcc.2008.07.016. [DOI] [PubMed] [Google Scholar]
- 9.McMullen J.R., Shioi T., Zhang L., Tarnavski O., Sherwood M.C., Dorfman A.L., Longnus S., Pende M., Martin K.A., Blenis J., et al. Deletion of ribosomal S6 kinases does not attenuate pathological, physiological, or insulin-like growth factor 1 receptor-phosphoinositide 3-kinase-induced cardiac hypertrophy. Mol. Cell Biol. 2004;24:6231–6240. doi: 10.1128/MCB.24.14.6231-6240.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Malhowski A.J., Hira H., Bashiruddin S., Warburton R., Goto J., Robert B., Kwiatkowski D.J., Finlay G.A. Smooth muscle protein-22-mediated deletion of Tsc1 results in cardiac hypertrophy that is mTORC1-mediated and reversed by rapamycin. Hum. Mol. Genet. 2011;20:1290–1305. doi: 10.1093/hmg/ddq570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Shiojima I., Sato K., Izumiya Y., Schiekofer S., Ito M., Liao R., Colucci W.S., Walsh K. Disruption of coordinated cardiac hypertrophy and angiogenesis contributes to the transition to heart failure. J. Clin. Invest. 2005;115:2108–2118. doi: 10.1172/JCI24682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tian R., Balschi J.A. Interaction of insulin and AMPK in the ischemic heart: another chapter in the book of metabolic therapy? Circ. Res. 2006;99:3–5. doi: 10.1161/01.RES.0000233142.26369.f6. [DOI] [PubMed] [Google Scholar]
- 13.Condorelli G., Drusco A., Stassi G., Bellacosa A., Roncarati R., Iaccarino G., Russo M.A., Gu Y., Dalton N., Chung C., et al. Akt induces enhanced myocardial contractility and cell size in vivo in transgenic mice. Proc Natl Acad Sci U. S. A. 2002;99:12333–12338. doi: 10.1073/pnas.172376399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shioi T., McMullen J.R., Kang P.M., Douglas P.S., Obata T., Franke T.F., Cantley L.C., Izumo S. Akt/protein kinase B promotes organ growth in transgenic mice. Mol. Cell Biol. 2002;22:2799–2809. doi: 10.1128/MCB.22.8.2799-2809.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ikeda Y., Sato K., Pimentel D.R., Sam F., Shaw R.J., Dyck J.R., Walsh K. Cardiac-specific deletion of LKB1 leads to hypertrophy and dysfunction. J. Biol. Chem. 2009;284:35839–35849. doi: 10.1074/jbc.M109.057273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Crackower M.A., Oudit G.Y., Kozieradzki I., Sarao R., Sun H., Sasaki T., Hirsch E., Suzuki A., Shioi T., Irie-Sasaki J., et al. Regulation of myocardial contractility and cell size by distinct PI3K-PTEN signaling pathways. Cell. 2002;110:737–749. doi: 10.1016/s0092-8674(02)00969-8. [DOI] [PubMed] [Google Scholar]
- 17.Linehan W.M., Ricketts C.J. The metabolic basis of kidney cancer. Semin. Cancer Biol. 2013;23:46–55. doi: 10.1016/j.semcancer.2012.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nickerson M.L., Warren M.B., Toro J.R., Matrosova V., Glenn G., Turner M.L., Duray P., Merino M., Choyke P., Pavlovich C.P., et al. Mutations in a novel gene lead to kidney tumors, lung wall defects, and benign tumors of the hair follicle in patients with the Birt–Hogg–Dube syndrome. Cancer Cell. 2002;2:157–164. doi: 10.1016/s1535-6108(02)00104-6. [DOI] [PubMed] [Google Scholar]
- 19.Hasumi Y., Baba M., Ajima R., Hasumi H., Valera V.A., Klein M.E., Haines D.C., Merino M.J., Hong S.B., Yamaguchi T.P., et al. Homozygous loss of BHD causes early embryonic lethality and kidney tumor development with activation of mTORC1 and mTORC2. Proc. Natl. Acad. Sci. U. S. A. 2009;106:18722–18727. doi: 10.1073/pnas.0908853106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Singh S.R., Zhen W., Zheng Z., Wang H., Oh S.W., Liu W., Zbar B., Schmidt L.S., Hou S.X. The Drosophila homolog of the human tumor suppressor gene BHD interacts with the JAK-STAT and Dpp signaling pathways in regulating male germline stem cell maintenance. Oncogene. 2006;25:5933–5941. doi: 10.1038/sj.onc.1209593. [DOI] [PubMed] [Google Scholar]
- 21.Furuya M., Nakatani Y. Birt–Hogg–Dube syndrome: clinicopathological features of the lung. J Clin Pathol. 2013;66:178–186. doi: 10.1136/jclinpath-2012-201200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Baba M., Hong S.B., Sharma N., Warren M.B., Nickerson M.L., Iwamatsu A., Esposito D., Gillette W.K., Hopkins R.F., Hartley J.L., et al. Folliculin encoded by the BHD gene interacts with a binding protein, FNIP1, and AMPK, and is involved in AMPK and mTOR signaling. Proc. Natl. Acad. Sci. U. S. A. 2006;103:15552–15557. doi: 10.1073/pnas.0603781103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schmidt L.S. Birt–Hogg–Dube syndrome: from gene discovery to molecularly targeted therapies. Fam. Cancer. 2013;12:357–364. doi: 10.1007/s10689-012-9574-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tee A.R., Pause A. Birt–Hogg–Dube: tumour suppressor function and signalling dynamics central to folliculin. Fam. Cancer. 2013;12:367–372. doi: 10.1007/s10689-012-9576-9. [DOI] [PubMed] [Google Scholar]
- 25.Hasumi H., Baba M., Hong S.B., Hasumi Y., Huang Y., Yao M., Valera V.A., Linehan W.M., Schmidt L.S. Identification and characterization of a novel folliculin-interacting protein FNIP2. Gene. 2008;415:60–67. doi: 10.1016/j.gene.2008.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Takagi Y., Kobayashi T., Shiono M., Wang L., Piao X., Sun G., Zhang D., Abe M., Hagiwara Y., Takahashi K., Hino O. Interaction of folliculin (Birt–Hogg–Dube gene product) with a novel Fnip1-like (FnipL/Fnip2) protein. Oncogene. 2008;27:5339–5347. doi: 10.1038/onc.2008.261. [DOI] [PubMed] [Google Scholar]
- 27.Baba M., Furihata M., Hong S.B., Tessarollo L., Haines D.C., Southon E., Patel V., Igarashi P., Alvord W.G., Leighty R., et al. Kidney-targeted Birt–Hogg–Dube gene inactivation in a mouse model: Erk1/2 and Akt-mTOR activation, cell hyperproliferation, and polycystic kidneys. J. Natl. Cancer Inst. 2008;100:140–154. doi: 10.1093/jnci/djm288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hasumi H., Baba M., Hasumi Y., Huang Y., Oh H., Hughes R.M., Klein M.E., Takikita S., Nagashima K., Schmidt L.S., Linehan W.M. Regulation of mitochondrial oxidative metabolism by tumor suppressor FLCN. J. Natl. Cancer. Inst. 2012;104:1750–1764. doi: 10.1093/jnci/djs418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hong S.B., Oh H., Valera V.A., Baba M., Schmidt L.S., Linehan W.M. Inactivation of the FLCN tumor suppressor gene induces TFE3 transcriptional activity by increasing its nuclear localization. PLoS One. 2010;5:e15793. doi: 10.1371/journal.pone.0015793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Betschinger J., Nichols J., Dietmann S., Corrin P.D., Paddison P.J., Smith A. Exit from pluripotency is gated by intracellular redistribution of the bHLH transcription Factor Tfe3. Cell. 2013;153:335–347. doi: 10.1016/j.cell.2013.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Preston R.S., Philp A., Claessens T., Gijezen L., Dydensborg A.B., Dunlop E.A., Harper K.T., Brinkhuizen T., Menko F.H., Davies D.M., et al. Absence of the Birt–Hogg–Dube gene product is associated with increased hypoxia-inducible factor transcriptional activity and a loss of metabolic flexibility. Oncogene. 2011;30:1159–1173. doi: 10.1038/onc.2010.497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nookala R.K., Langemeyer L., Pacitto A., Ochoa-Montano B., Donaldson J.C., Blaszczyk B.K., Chirgadze D.Y., Barr F.A., Bazan J.F., Blundell T.L. Crystal structure of folliculin reveals a hidDENN function in genetically inherited renal cancer. Open Biol. 2012;2:120071. doi: 10.1098/rsob.120071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Petit C.S., Roczniak-Ferguson A., Ferguson S.M. Recruitment of folliculin to lysosomes supports the amino acid-dependent activation of Rag GTPases. J. Cell Biol. 2013;202:1107–1122. doi: 10.1083/jcb.201307084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tsun Z.Y., Bar-Peled L., Chantranupong L., Zoncu R., Wang T., Kim C., Spooner E., Sabatini D.M. The folliculin tumor suppressor is a GAP for the RagC/D GTPases that signal amino acid levels to mTORC1. Mol. Cell. 2013;52:495–505. doi: 10.1016/j.molcel.2013.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Agah R., Frenkel P.A., French B.A., Michael L.H., Overbeek P.A., Schneider M.D. Gene recombination in postmitotic cells. Targeted expression of Cre recombinase provokes cardiac-restricted, site-specific rearrangement in adult ventricular muscle in vivo. J. Clin. Invest. 1997;100:169–179. doi: 10.1172/JCI119509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wang J., Wilhelmsson H., Graff C., Li H., Oldfors A., Rustin P., Bruning J.C., Kahn C.R., Clayton D.A., Barsh G.S., et al. Dilated cardiomyopathy and atrioventricular conduction blocks induced by heart-specific inactivation of mitochondrial DNA gene expression. Nat Genet. 1999;21:133–137. doi: 10.1038/5089. [DOI] [PubMed] [Google Scholar]
- 37.Li H., Wang J., Wilhelmsson H., Hansson A., Thoren P., Duffy J., Rustin P., Larsson N.G. Genetic modification of survival in tissue-specific knockout mice with mitochondrial cardiomyopathy. Proc. Natl. Acad. Sci. U. S. A. 2000;97:3467–3472. doi: 10.1073/pnas.97.7.3467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Beedle A.M., Turner A.J., Saito Y., Lueck J.D., Foltz S.J., Fortunato M.J., Nienaber P.M., Campbell K.P. Mouse fukutin deletion impairs dystroglycan processing and recapitulates muscular dystrophy. J. Clin. Invest. 2012;122:3330–3342. doi: 10.1172/JCI63004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Diaz-Troya S., Perez-Perez M.E., Florencio F.J., Crespo J.L. The role of TOR in autophagy regulation from yeast to plants and mammals. Autophagy. 2008;4:851–865. doi: 10.4161/auto.6555. [DOI] [PubMed] [Google Scholar]
- 40.Gwinn D.M., Shackelford D.B., Egan D.F., Mihaylova M.M., Mery A., Vasquez D.S., Turk B.E., Shaw R.J. AMPK phosphorylation of raptor mediates a metabolic checkpoint. Mol. Cell. 2008;30:214–226. doi: 10.1016/j.molcel.2008.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Inoki K., Corradetti M.N., Guan K.L. Dysregulation of the TSC-mTOR pathway in human disease. Nat. Genet. 2005;37:19–24. doi: 10.1038/ng1494. [DOI] [PubMed] [Google Scholar]
- 42.Egan D.F., Shackelford D.B., Mihaylova M.M., Gelino S., Kohnz R.A., Mair W., Vasquez D.S., Joshi A., Gwinn D.M., Taylor R., et al. Phosphorylation of ULK1 (hATG1) by AMP-activated protein kinase connects energy sensing to mitophagy. Science. 2011;331:456–461. doi: 10.1126/science.1196371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mihaylova M.M., Shaw R.J. The AMPK signalling pathway coordinates cell growth, autophagy and metabolism. Nat. Cell Biol. 2011;13:1016–1023. doi: 10.1038/ncb2329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Arany Z., He H., Lin J., Hoyer K., Handschin C., Toka O., Ahmad F., Matsui T., Chin S., Wu P.H., et al. Transcriptional coactivator PGC-1 alpha controls the energy state and contractile function of cardiac muscle. Cell Metab. 2005;1:259–271. doi: 10.1016/j.cmet.2005.03.002. [DOI] [PubMed] [Google Scholar]
- 45.Arany Z., Foo S.Y., Ma Y., Ruas J.L., Bommi-Reddy A., Girnun G., Cooper M., Laznik D., Chinsomboon J., Rangwala S.M., et al. HIF-independent regulation of VEGF and angiogenesis by the transcriptional coactivator PGC-1alpha. Nature. 2008;451:1008–1012. doi: 10.1038/nature06613. [DOI] [PubMed] [Google Scholar]
- 46.Lin J., Wu H., Tarr P.T., Zhang C.Y., Wu Z., Boss O., Michael L.F., Puigserver P., Isotani E., Olson E.N., et al. Transcriptional co-activator PGC-1 alpha drives the formation of slow-twitch muscle fibres. Nature. 2002;418:797–801. doi: 10.1038/nature00904. [DOI] [PubMed] [Google Scholar]
- 47.St-Pierre J., Drori S., Uldry M., Silvaggi J.M., Rhee J., Jager S., Handschin C., Zheng K., Lin J., Yang W., et al. Suppression of reactive oxygen species and neurodegeneration by the PGC-1 transcriptional coactivators. Cell. 2006;127:397–408. doi: 10.1016/j.cell.2006.09.024. [DOI] [PubMed] [Google Scholar]
- 48.Woods A., Azzout-Marniche D., Foretz M., Stein S.C., Lemarchand P., Ferre P., Foufelle F., Carling D. Characterization of the role of AMP-activated protein kinase in the regulation of glucose-activated gene expression using constitutively active and dominant negative forms of the kinase. Mol. Cell Biol. 2000;20:6704–6711. doi: 10.1128/mcb.20.18.6704-6711.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Viollet B., Athea Y., Mounier R., Guigas B., Zarrinpashneh E., Horman S., Lantier L., Hebrard S., Devin-Leclerc J., Beauloye C., et al. AMPK: Lessons from transgenic and knockout animals. Front. Biosci. 2009;14:19–44. doi: 10.2741/3229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Steinberg G.R., Kemp B.E. AMPK in health and disease. Physiol. Rev. 2009;89:1025–1078. doi: 10.1152/physrev.00011.2008. [DOI] [PubMed] [Google Scholar]
- 51.Yang Z., Ming X.F. mTOR signalling: the molecular interface connecting metabolic stress, aging and cardiovascular diseases. Obes. Rev. 2012;13(Suppl 2):58–68. doi: 10.1111/j.1467-789X.2012.01038.x. [DOI] [PubMed] [Google Scholar]
- 52.Blair E., Redwood C., Ashrafian H., Oliveira M., Broxholme J., Kerr B., Salmon A., Ostman-Smith I., Watkins H. Mutations in the gamma(2) subunit of AMP-activated protein kinase cause familial hypertrophic cardiomyopathy: evidence for the central role of energy compromise in disease pathogenesis. Hum. Mol. Genet. 2001;10:1215–1220. doi: 10.1093/hmg/10.11.1215. [DOI] [PubMed] [Google Scholar]
- 53.Zaha V.G., Young L.H. AMP-activated protein kinase regulation and biological actions in the heart. Circ. Res. 2012;111:800–814. doi: 10.1161/CIRCRESAHA.111.255505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zhang P., Hu X., Xu X., Fassett J., Zhu G., Viollet B., Xu W., Wiczer B., Bernlohr D.A., Bache R.J., Chen Y. AMP activated protein kinase-alpha2 deficiency exacerbates pressure-overload-induced left ventricular hypertrophy and dysfunction in mice. Hypertension. 2008;52:918–924. doi: 10.1161/HYPERTENSIONAHA.108.114702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Shen W.H., Chen Z., Shi S., Chen H., Zhu W., Penner A., Bu G., Li W., Boyle D.W., Rubart M., et al. Cardiac restricted overexpression of kinase-dead mammalian target of rapamycin (mTOR) mutant impairs the mTOR-mediated signaling and cardiac function. J. Biol. Chem. 2008;283:13842–13849. doi: 10.1074/jbc.M801510200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zhang D., Contu R., Latronico M.V., Zhang J., Rizzi R., Catalucci D., Miyamoto S., Huang K., Ceci M., Gu Y., et al. MTORC1 regulates cardiac function and myocyte survival through 4E-BP1 inhibition in mice. J Clin Invest. 2010;120:2805–2816. doi: 10.1172/JCI43008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hariharan N., Ikeda Y., Hong C., Alcendor R.R., Usui S., Gao S., Maejima Y., Sadoshima J. Autophagy plays an essential role in mediating regression of hypertrophy during unloading of the heart. PLoS One. 2013;8:e51632. doi: 10.1371/journal.pone.0051632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zhu H., Tannous P., Johnstone J.L., Kong Y., Shelton J.M., Richardson J.A., Le V., Levine B., Rothermel B.A., Hill J.A. Cardiac autophagy is a maladaptive response to hemodynamic stress. J. Clin. Invest. 2007;117:1782–1793. doi: 10.1172/JCI27523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kim J., Kundu M., Viollet B., Guan K.L. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat. Cell Biol. 2011;13:132–141. doi: 10.1038/ncb2152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Russell R.R., III, Li J., Coven D.L., Pypaert M., Zechner C., Palmeri M., Giordano F.J., Mu J., Birnbaum M.J., Young L.H. AMP-activated protein kinase mediates ischemic glucose uptake and prevents postischemic cardiac dysfunction, apoptosis, and injury. J. Clin. Invest. 2004;114:495–503. doi: 10.1172/JCI19297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Xing Y., Musi N., Fujii N., Zou L., Luptak I., Hirshman M.F., Goodyear L.J., Tian R. Glucose metabolism and energy homeostasis in mouse hearts overexpressing dominant negative alpha2 subunit of AMP-activated protein kinase. J. Biol. Chem. 2003;278:28372–28377. doi: 10.1074/jbc.M303521200. [DOI] [PubMed] [Google Scholar]
- 62.Zarrinpashneh E., Carjaval K., Beauloye C., Ginion A., Mateo P., Pouleur A.C., Horman S., Vaulont S., Hoerter J., Viollet B., et al. Role of the alpha2-isoform of AMP-activated protein kinase in the metabolic response of the heart to no-flow ischemia. Am. J. Physiol. Heart Circ. Physiol. 2006;291:H2875–H2883. doi: 10.1152/ajpheart.01032.2005. [DOI] [PubMed] [Google Scholar]
- 63.Demeulder B., Zarrinpashneh E., Ginion A., Viollet B., Hue L., Rider M.H., Vanoverschelde J.L., Beauloye C., Horman S., Bertrand L. Differential regulation of eEF2 and p70S6 K by AMPKalpha2 in heart. Biochim. Biophys. Acta. 2013;1832:780–790. doi: 10.1016/j.bbadis.2013.02.015. [DOI] [PubMed] [Google Scholar]
- 64.Hardie D.G., Ross F.A., Hawley S.A. AMPK: a nutrient and energy sensor that maintains energy homeostasis. Nat. Rev. Mol. Cell Biol. 2012;13:251–262. doi: 10.1038/nrm3311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.El M.S., Rongen G.A., de Boer R.A., Riksen N.P. The cardioprotective effects of metformin. Curr. Opin. Lipidol. 2011;22:445–453. doi: 10.1097/MOL.0b013e32834ae1a7. [DOI] [PubMed] [Google Scholar]
- 66.Eurich D.T., McAlister F.A., Blackburn D.F., Majumdar S.R., Tsuyuki R.T., Varney J., Johnson J.A. Benefits and harms of antidiabetic agents in patients with diabetes and heart failure: systematic review. BMJ. 2007;335:497. doi: 10.1136/bmj.39314.620174.80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Spiegelman B.M. Transcriptional control of mitochondrial energy metabolism through the PGC1 coactivators. Novartis Found Symp. 2007;287:60–69. [PubMed] [Google Scholar]
- 68.Zhang L., He H., Balschi J.A. Metformin and phenformin activate AMP-activated protein kinase in the heart by increasing cytosolic AMP concentration. Am. J. Physiol Heart Circ. Physiol. 2007;293:H457–H466. doi: 10.1152/ajpheart.00002.2007. [DOI] [PubMed] [Google Scholar]
- 69.McNally E.M., Golbus J.R., Puckelwartz M.J. Genetic mutations and mechanisms in dilated cardiomyopathy. J. Clin. Invest. 2013;123:19–26. doi: 10.1172/JCI62862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Lin J., Wu P.H., Tarr P.T., Lindenberg K.S., St-Pierre J., Zhang C.Y., Mootha V.K., Jager S., Vianna C.R., Reznick R.M., et al. Defects in adaptive energy metabolism with CNS-linked hyperactivity in PGC-1alpha null mice. Cell. 2004;119:121–135. doi: 10.1016/j.cell.2004.09.013. [DOI] [PubMed] [Google Scholar]
- 71.Lehman J.J., Barger P.M., Kovacs A., Saffitz J.E., Medeiros D.M., Kelly D.P. Peroxisome proliferator-activated receptor gamma coactivator-1 promotes cardiac mitochondrial biogenesis. J. Clin. Invest. 2000;106:847–856. doi: 10.1172/JCI10268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Russell L.K., Mansfield C.M., Lehman J.J., Kovacs A., Courtois M., Saffitz J.E., Medeiros D.M., Valencik M.L., McDonald J.A., Kelly D.P. Cardiac-specific induction of the transcriptional coactivator peroxisome proliferator-activated receptor gamma coactivator-1alpha promotes mitochondrial biogenesis and reversible cardiomyopathy in a developmental stage-dependent manner. Circ. Res. 2004;94:525–533. doi: 10.1161/01.RES.0000117088.36577.EB. [DOI] [PubMed] [Google Scholar]
- 73.McNally E.M., Goldstein J.A. Interplay between heart and skeletal muscle disease in heart failure: the 2011 George E. Brown Memorial Lecture. Circ. Res. 2012;110:749–754. doi: 10.1161/CIRCRESAHA.111.256776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Bhattacharya S.K., Thakar J.H., Johnson P.L., Shanklin D.R. Isolation of skeletal muscle mitochondria from hamsters using an ionic medium containing ethylenediaminetetraacetic acid and nagarse. Anal. Biochem. 1991;192:344–349. doi: 10.1016/0003-2697(91)90546-6. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.