

Abstract
Retinitis pigmentosa (RP), a genetically heterogeneous group of retinopathies that occur in both non-syndromic and syndromic forms, is caused by mutations in ∼100 genes. Although recent advances in next-generation sequencing have aided in the discovery of novel RP genes, a number of the underlying contributing genes and loci remain to be identified. We investigated three siblings, born to asymptomatic parents of Italian–American descent, who each presented with atypical RP with systemic features, including facial dysmorphologies, psychomotor developmental delays recognized since early childhood, learning disabilities and short stature. RP-associated ophthalmological findings included salt-and-pepper retinopathy, attenuation of the arterioles and generalized rod–cone dysfunction as determined by almost extinguished electroretinogram in 2 of 3 siblings. Atypical for RP features included mottled macula at an early age and peripapillary sparing of the retinal pigment epithelium. Whole-exome sequencing data, queried under a recessive model of inheritance, identified compound heterozygous stop mutations, c.C199T:p.R67* and c.C322T:p.R108*, in the retinol dehydrogenase 11 (RDH11) gene, resulting in a non-functional protein, in all affected children. In summary, deleterious mutations in RDH11, an important enzyme for vision-related and systemic retinoic acid metabolism, cause a new syndrome with RP.
INTRODUCTION
Retinitis pigmentosa (RP) is a genetically and clinically heterogeneous group of inherited retinopathies (1). It includes both non-syndromic and syndromic forms of all modes of inheritance, autosomal recessive, autosomal dominant and X-linked, and is caused by mutations in close to 100 genes. By current estimates known genes explain between 60 and 80% of RP cases suggesting many RP loci remain to be found (1).
Syndromic forms of RP present with many heterogeneous phenotypes, the two best characterized of which are Usher syndrome, caused by 12 genes (2) and Bardet–Biedl syndrome (BBS), which is caused by mutations in at least 17 genes (https://sph.uth.edu/retnet/) (3,4). Most prominent systemic phenotypes of Usher syndrome include congenital or early-onset hearing loss (5), although BBS presents often with obesity, developmental delay and polydactyly (6,7). Other forms of syndromic RP include those associated with mitochondrial diseases (Kearns–Sayre, Wolfram syndromes, etc.) (8,9) and some forms of renal or neurodegenerative phenotypes (Joubert, Jeune syndromes, etc.; https://sph.uth.edu/retnet/) (10–12). The presence of specific systemic phenotypic features in patients vary and often mutations in the same gene can cause different phenotypes (allelic affinity), sometimes classified as separate diseases (13) or the same syndrome can be caused by mutations in different genes (locus heterogeneity) (14).
Recent advances in the next-generation sequencing (NGS), the high-throughput, ‘deep’ sequencing technology, have enabled several novel RP genes to be identified (15–17) or found new mutations in known genes; nevertheless, a substantial fraction of unsolved cases still remain. Here, we investigated the clinical phenotype and the genetic cause of RP with syndromic features in a family in which all three children presented with atypical RP and a unique combination of systemic features potentially describing a novel syndrome.
RESULTS
Physical evaluation
A family with three affected children of Italian–American descent (Fig. 1) presented to the clinic for evaluation of retinal degeneration. The oldest sibling is a 19-year-old woman (Case 1), the middle sibling a 17-year-old boy (Case 2) and youngest an 8-year-old boy (Case 3). Ophthalmic examinations in both parents were unremarkable, and they did not exhibit any physical or systemic abnormalities. The siblings were born at term after uneventful pregnancies but had psychomotor developmental delays since early childhood. At presentation, the main complaints were lack of fine motor skills and coordination (writing, drawing) for which they are receiving physical and occupational therapy. Each sibling is enrolled in special educational programs due to learning difficulties. The three affected siblings share a striking resemblance with distinctive facial features (Fig. 2). Each appear to be abnormally short in stature relative to their age groups, the oldest and middle sibling were measured to be 57″ and 61″ tall, respectively, and the youngest sibling 47″ tall. The two older siblings fall below the 5th percentile for stature within their respective age and gender groups (Fig. 2C and D). The youngest sibling is just above the 5th percentile (Fig. 2D). Older siblings consistently exhibited short stature throughout development until early puberty (9–10 years) and then again around the early teenage years. The youngest sibling appears to be following a similar trend (Fig. 2C and D). Skeletal X-rays of the hands from an earlier age in Cases 1 and 2 were reviewed with no suggestive evidence for skeletal dysplasias; e.g. brachydactyly.
Figure 1.
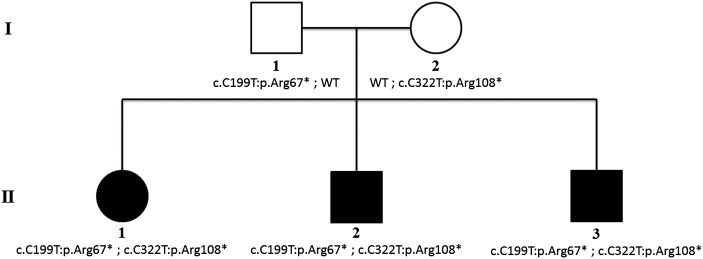
Pedigree of the family and segregation of the RDH11 mutations with the disease phenotype. Open circles and squares represent the unaffected female and male family members, respectively; closed circles and squares represent the affected female and male patients.
Figure 2.

Clinical presentation of the syndromic features in affected family members. Affected siblings shared distinct craniofacial and physical dysmorphologies. (A) Case 2, middle sibling, presented with prominent alae nasae and malar hypoplasia; the oldest and youngest sibling also exhibited these features in a less pronounced manner. (B) The body frame of each sibling suggests irregular physical development according to their respective ages. Growth curves (C, Case 1 and D, Cases 2 and 3) documenting stature from age 2 to the present illustrate a consistent pattern of abnormal height, falling below the 5th percentile of age- and gender-matched reference population.
Excessive dental spacing and malocclusion were observed in Case 3, which occurred similarly in Cases 1 and 2 but was corrected with dental braces according to their reported health history. All exhibited apparent facial dysmorphologies which included distinct formation of the nose with hypoplasia of the alae nasae (Fig. 2A). Cases 2 and 3 exhibited malar hypoplasia, whereas Cases 1 and 2 had attached ear lobes. The palpebral fissures in each sibling appeared slightly upslanted. An extensive review of pediatric health records from birth for each sibling did not reveal any additional significant systemic abnormalities.
Ophthalmic evaluation
The progressive visual acuity decrease and reported difficulties with night vision developed at the age of 10 years in Case 1 and at the age of 8 years in Cases 2 and 3, and an onset of juvenile cataracts was diagnosed and operated within the same year. After cataract surgery, night vision became progressively worse and RP was diagnosed at the age of 16 years (Case 1), at the age of 13 years (Case 2) and at the age of 8 years (Case 3) according to the fundus appearance and electroretinogram (ERG) findings (Fig. 3). The oldest sibling presented at a later disease stage. The best-corrected visual acuity (BCVA) was 20/20RE and 20/20LE. Fundus examination revealed attenuation and narrowing of the retinal arterioles in both eyes (Fig. 3A). The retina exhibited a mottled, pigmented appearance consistent with localized retinal pigment epithelium (RPE) atrophy, but relatively spared fovea. Particular areas of the mid-periphery had an abnormal, grayish sheen. A confluent pattern of bone-spicule pigmentation was observed in the periphery (Fig. 3B). Autofluorescence imaging revealed the presence of an amorphous hyperfluorescent ring beyond the peripheral vascular arcades with sparing of RPE in the peripapillary region of the optic nerve (Fig. 3C). Spectral domain-optical coherence tomography (SD-OCT) cross sections revealed intact retinal lamination in the central macula proceeding to an abrupt absence or disruption of outer layers in the parafoveal and peripheral retina (Fig. 3D). The parafoveal outer retinal layers were progressively atrophic or absent resulting in a loss of laminar architecture of the retina with increasing spatial eccentricity. Such affected areas also exhibited decreased retinal thickening in the mid- and far-peripheral retina. Cases 2 and 3 presented at a relatively milder stage on fundus examination. BCVA in Case 2 was 20/30RE and 20/25 LE, and in Case 3 was 20/25 in both eyes. Optic discs had a pinkish waxy appearance with distinct borders. The retina appeared mottled and was marked with diffuse pigment clumping without bone-spicule pigmentation in the periphery. A similar but more uniformly symmetric hyperfluorescent ring just beyond the superior and inferior vascular arcades and around the optic disc was seen in autofluorescence imaging. SD-OCT scans through the macula revealed relatively spared foveae but abnormal outer retinal lamination in parafoveal area. Areas of pigment clumping seen in fundus examination spatially correspond to hyperreflective deposits which bulged up anteriorly to an otherwise undisrupted photoreceptor layer. The external limiting membrane (ELM) underlying these lesions appeared to be thinner.
Figure 3.

Ophthalmic examination of affected family members consistent with an early-onset retinal dystrophy. (A) Funduscopy of Case 1 revealed an abnormal, mottled appearance of the retina in addition to narrowing and attenuation of the retinal vessels (white arrows). (B) A more peripheral examination of the same eye revealed a confluent pattern of bone-spicule pigment deposition (white arrows). (C) Autofluorescence imaging of the same eye uncovered a large amorphous ring (white arrows) situated just beyond the vascular arcades surrounding an area of granular RPE atrophy with sparing of the fovea and regions surrounding the optic nerve. (D) SD-OCT scans revealed relatively intact retina layers in areas immediately adjacent to the fovea, which proceed to an abrupt loss (white arrow) of the ELM, inner segment ellipsoid layer (ISe) and thinning of the RPE with increasing eccentricity from the fovea (inset). (E) Full-field ERG results in each sibling were compared with an age-matched control (dotted gray line). Case 1 (solid red line) and exhibited generalized retinal dysfunction with severe waveform reductions and delays in both the scotopic and photopic systems. Case 2 (solid green line) and Case 3 (solid blue line) appeared to be less affected with mild delays in the 30 Hz flicker response and amplitudinal reductions in scotopic function.
Full-field ERG conducted in each affected sibling showed changes mostly consistent with their retinal pathology (Fig. 3E). Severe amplitude reductions and delays of the a- and b-waves in both the rod (scotopic) and cone (photopic) system were apparent, especially in the oldest sibling. The scotopic system was clearly more affected than photopic, consistent with the RP phenotype (Fig. 3E).
The 30-2 Humphrey visual field for Case 1 was constricted (Supplementary Material, Fig. S1), consistent with the RP phenotype. There was a marked decline in visual sensitivity with increasing eccentricity from the fovea. Foveal sensitivity was slightly decreased compared with values for age-similar healthy observers. The two-color dark-adapted threshold results also showed decreased sensitivity with increasing eccentricity to both chromatic stimuli compared with the mean value for healthy observers, and at 15° nasal field 650 nm stimuli at the maximum intensity were not detected (Supplementary Material, Fig. S1). The differences in sensitivity of ≥2 log units between the 500 and 650 nm test lights along the horizontal meridian indicate rod mediation.
Genetic analyses
Considering the apparent autosomal recessive inheritance of the disease (Fig. 1), the variants detected by NGS in affected children and parents were selected for further analysis when they met all of the following criteria: (1) variants were present in <0.5% in ESP6500 (http://evs.gs.washington.edu/EVS/; last accessed 17 April 2014), 1000 Genomes (18) and Atherosclerosis risk in communities study (ARIC), an internal control database of 3996 exomes at the Baylor College of Medicine Human Genome Sequencing Center, (2) variants were in protein coding regions and/or in canonical splice sites, (3) variants were either missense, nonsense, frameshift or splice site variants and (4) variants were compound heterozygous or homozygous in the same gene in all affected children. The filtering strategy and numbers of identified variants and genes at each step are provided in Supplementary Material, Table S1. After assessing the phase of remaining variants using the sequence data of the parents and the segregation analyses in the entire family, only two genes, dual specificity (all-trans- and 11-cis-) retinol dehydrogenase (RDH11) (19) and cadherin 23 (CDH23) (20), remained as possible candidates.
The CDH23 gene encodes a Ca2+-dependent cell–cell adhesion glycoprotein that is involved in stereocilia organization and hair bundle formation (21). Mutations in CDH23 are causal for two allelic recessive disorders: Usher syndrome, Type 1D (USH1D)—presenting with congenital deafness with a variable degree of retinal degeneration and non-syndromic autosomal recessive deafness 12 (DFNB12)—high-frequency progressive sensorineural hearing loss with normal retinal and vestibular function. Specifically, missense mutations in CDH23 mostly cause DFNB12, whereas nonsense, frameshift, splice site and some more severe missense mutations of CDH23 cause USH1D (22,23). All affected children in the family were compound heterozygous for two rare missense variants, c.G574C:p.E192Q and c.T9185C:p.M3062T. Predictive programs suggested that the c.G574C:p.E192Q variant may be possibly disease associated and the c.T9185C:p.M3062T is likely benign. This information coupled with the normal hearing in all siblings suggested that the CDH23 gene is not associated with the RP and syndromic phenotype in the family.
This left RDH11 as the only plausible candidate gene for the disease phenotype segregating with the two variants. The two RDH11 variants shared by the affected siblings, c.C199T:p.R67* and c.C322T:p.R108*, are predicted to either result in a truncated protein and, consequently, an inactive enzyme due to severed di-nucleotide-binding domain which is necessary for catalysis (19), or in a null allele due to nonsense-mediated decay. Neither variant is present in 1000 Genome database, in the Exome Sequencing Project (ESP) database, or in the ARIC database of 3996 control exomes at the Baylor Genome Center, where both specific nucleotide positions were well covered in all control exomes. In fact, truncating mutations (nonsense or frameshift alleles) in RDH11 are extremely rare, since only one deleterious allele has been described in the ESP database of 6500 exomes and none was present in 3996 ARIC and 2940 BH-CMG exomes in the Baylor Human Genome Sequencing Center database. Moreover, we also did not find any bi-allelic events in both datasets at Baylor Genome Center which further confirms that disease-associated variants in RDH11 are exceptionally rare.
RDH11 encodes a dual specificity retinol dehydrogenase which is ubiquitously expressed and its expression is hormonally regulated (e.g. by androgens) (19,24). In the eye, RDH11 has an oxidoreductive function in the visual cycle (19). In the mouse eyes, the protein is expressed in the RPE and Muller cells (25). In the RPE, RDH11 is proposed to aid RDH5 in the regeneration of chromophore by oxidizing 11-cis-retinol to 11-cis-retinal (26,27). Rdh11−/− mice have a mild phenotype, a normal retinal morphology and ERGs under baseline conditions and normal retinoid profiles. The only RP-related defect in Rdh11−/− mice is a delayed dark adaptation (28).
DISCUSSION
A new form of syndromic RP was investigated in a family presenting with a previously undescribed constellation of phenotypic features. Several common features characteristic for RP, such as salt-and-pepper retinopathy, attenuation of the arterioles and generalized rod–cone dysfunction based on ERG, were mixed with relatively atypical for RP features, e.g. mottled macula at early age and peripapillary sparing of RPE. The array of systemic features included developmental delay, which can often be seen in cases with RP, but also short stature and craniofacial features; the latter phenotypes not usually associated with syndromic RP. Overall, the combination of retinal and systemic features was quite unique. There has been one family described where two affected siblings presented with a remarkably similar phenotype (29); however, no mutations in RDH11 were detected in that family suggesting that the phenocopy could be due to mutations in genes from the same, retinoic acid metabolism, pathway.
The assessment of RDH11 (dual all-trans- and 11-cis-retinol-specific dehydrogenase) as the likely causal gene for the syndromic RP phenotype in this family was aided by two main facts: first, the protein has a well-characterized, albeit auxiliary role in the visual cycle and second, the compound heterozygous nonsense mutations predict null alleles and these render the protein non-functional. Although the RP phenotype is clearly due to deleterious mutations in RDH11, the systemic features were somewhat unexpected, because the role of RDH11 in all-trans-retinoic acid metabolism and thus development in other organs remains obscure. However, there were no other variants in any genes which segregated with the phenotype and could plausibly explain the phenotype and mutations in several genes from the retinoic acid metabolism pathway have been associated with severe developmental disorders. The prominent examples include retinol transporter STRA6 (30) and another retinol dehydrogenase, RDH10 (31) which, in addition to developmental phenotypes have also specific, albeit different from RP, eye phenotype.
RDH11 is one of the 11-cis-retinol dehydrogenases which catalyzes the last oxidation step, the conversion of 11-cis-retinol to 11-cis-retinal, in the retinoid cycle in the RPE (28). In mice, RDH11 plays an auxiliary role in this enzymatic reaction, because most of it is carried out by RDH5 (32). Mutations in RDH5 cause autosomal recessive fundus albipunctatus, a relatively stationary night blindness which can, in some patients, develop into progressive cone dystrophy (33,34). Interestingly, knockout mouse models of either Rdh5 or Rdh11 do not replicate the human phenotypes, except for the night blindness/delayed dark adaptation (27,32). In a double knockout, the Rdh5−/−Rdh11−/− mice, the phenotype was enhanced, the animals exhibited enhanced delay of dark adaptation and increased accumulation of cis-retinols and retinyl esters, suggesting epistasis and a cooperative role between RDH5 and RDH11 (32). Slowed recovery of rod responses and abnormal retinoid profiling suggests that RDH11 plays a complementary role to RDH5 in the flow of retinoids during dark adaptation (26). It is also possible that not a lack of 11-cis-retinol visual cycle activity of RDH11 is detrimental, but all-trans-retinol activity related to retinoic acid metabolism abnormalities in the eye causes the RP phenotype.
In conclusion, we describe a new syndromic phenotype with RP caused by deleterious mutations in the RDH11 gene. Given that the Rdh11−/− mouse phenotype is mild, the phenotype in all three affected siblings was surprisingly and uniformly severe.
MATERIALS AND METHODS
Patients and clinical evaluation
Three affected siblings, together with an unaffected mother and father (Fig. 1), were enrolled in the study under the protocol IRB-AAAB6560 after obtaining informed consent. The protocol was approved by the Institutional Review Board at Columbia University and adheres to tenets of the Declaration of Helsinki. Each patient underwent a complete ophthalmic examination by a retina specialist (S.H.T.), which included color fundus photography with an FF 450plus Fundus Camera (Carl Zeiss Meditec AG, Jena, Germany). Fundus autofluorescence (FAF) images were obtained using a confocal scanning laser ophthalmoscope (Heidelberg Retina Angiograph 2, Heidelberg Engineering, Dossenheim, Germany) by illuminating the fundus with argon laser light (488 nm) and viewing the resultant fluorescence through a band-pass filter with a short wavelength cutoff at 495 nm. Simultaneous FAF and SD-OCT images were acquired using a Spectralis HRA+OCT (Heidelberg Engineering, Heidelberg, Germany). Electroretinography was carried out using the Diagnosys Espion Electrophysiology System (Diagnosys LLC, Littleton, MA, USA). For all recordings, the pupils were maximally dilated before full-field ERG testing using guttate tropicamide (1%) and phenylephrine hydrochloride (2.5%); and the corneas were anesthetized with guttate proparacaine 0.5%. Silver impregnated fiber electrodes (DTL; Diagnosys LLC) were used with a ground electrode on the forehead. Full-field ERGs to test generalized retinal function were performed using extended testing protocols incorporating the International Society for Clinical Electrophysiology of Vision standard (35). Standard automated perimetry was attempted on all three siblings using the 30-2 Swedish Interactive Threshold Algorithm field program in the Humphrey Visual Field Analyzer (Carl Zeiss Meditec, Inc., Dublin, CA, USA). Reliable visual fields (global indices <33%) were obtained only for the oldest sibling. In addition, to assess the loss in rod- and cone-mediated sensitivity, a two-color dark-adapted threshold technique on a modified Octopus perimeter (Haag-Streit AG, Köniz, Switzerland) was used. Following pupil dilation and 45 min of dark adaptation, sensitivities to 500 and 650 nm size V, 200 ms targets were obtained along the horizontal meridian (2° intervals) through the fovea to 15° eccentricity. The sensitivity difference to the two chromatic test stimuli determined whether rods, cone or both photoreceptor systems mediated threshold at a given location (36). A comprehensive review of pediatric health records in each sibling was conducted to assess systemic health.
Genetic analyses
The proband was initially screened for variants in the ABCA4 gene and on an array containing most known arRP genes (Asper Biotech, Inc., Tartu, Estonia; www.asperbio.com; last accessed 12 February 2013) revealing no disease-associated variants. Subsequently, whole-exome sequencing was performed on family members at the Baylor College of Medicine Human Genome Sequencing Center. Genomic DNA samples were constructed into Illumina paired-end pre-capture libraries according to the manufacturer's protocol (Illumina Multiplexing_SamplePrep_Guide_1005361_D) with modifications as described in the BCM-HGSC Illumina Barcoded Paired-End Capture Library Preparation protocol. Libraries were prepared using Beckman robotic workstations (Biomek NXp and FXp models). The complete protocol and oligonucleotide sequences are accessible from the HGSC website (https://hgsc.bcm.edu/sites/default/files/documents/Illumina_Barcoded_Paired-End_Capture_Library_Preparation.pdf; last accessed 20 October 2013). Pre-captured libraries were pooled together and hybridized in solution to the HGSC CORE design (52 Mb, NimbleGen), and exome capture was performed according to the manufacturer's protocol NimbleGen SeqCap EZ Exome Library SR User's Guide (Version 2.2) with minor revisions. Library templates were prepared for sequencing using Illumina's cBot cluster generation system with TruSeq PE Cluster Generation Kits (Part no. PE-401-3001). Real-time analysis software was used to process the image analysis and base calling. Sequencing runs generated ∼300–400 million successful reads on each lane of a flow cell, yielding 9–10 Gb per sample. With these sequencing yields, samples achieved an average of 90% of the targeted exome bases covered to a depth of 20× or greater.
Illumina sequence analysis was performed using the HGSC Mercury analysis pipeline (https://github.com/dsexton2/Mercury-Pipeline; last accessed 12 May 2013) that addresses all aspects of data processing and analyses, moving data step by step through various analysis tools from the initial sequence generation on the instrument to annotated variant calls [single nucleotide polymorphisms (SNPs) and intra-read in/dels]. Pathogenicity of novel variants was assessed with predictive programs for splice sites and coding sequences, accessed via Alamut software (Alamut 2.3; http://www.interactive-biosoftware.com/alamut-visual/; last accessed 20 April 2014). All variants of interest were confirmed by Sanger sequencing, and segregation analyses were performed on all members of the family.
SUPPLEMENTARY MATERIAL
FUNDING
This work was supported in part by National Institutes of Health grants EY021163, EY019861, HG006542 and EY019007 (Core Support for Vision Research), by research grants FIS PI13/00226, FIS PS09/00459, RD09-0076-00101 (Retics Biobank), CIBERER Intra/07/704.1 and Intra/09/702.1, ONCE, Fundaluce and Fundacion Conchita Rabago de Jimenez Diaz; and unrestricted funds from Research to Prevent Blindness (New York, NY) to the Department of Ophthalmology, Columbia University.
Supplementary Material
ACKNOWLEDGEMENTS
The authors thank the family members for participating in this study and acknowledge the insightful discussions, advice and the critical reading of the manuscript by Drs Krzysztof Palczewski and Uta Francke.
Conflict of Interest Statement. None declared.
REFERENCES
- 1.Daiger S.P., Sullivan L.S., Bowne S.J. Genes and mutations causing retinitis pigmentosa. Clin. Genet. 2013;84:132–141. doi: 10.1111/cge.12203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bonnet C., El-Amraoui A. Usher syndrome (sensorineural deafness and retinitis pigmentosa): pathogenesis, molecular diagnosis and therapeutic approaches. Curr. Opin. Neurol. 2012;25:42–49. doi: 10.1097/WCO.0b013e32834ef8b2. [DOI] [PubMed] [Google Scholar]
- 3.Leitch C.C., Zaghloul N.A., Davis E.E., Stoetzel C., Diaz-Font A., Rix S., Alfadhel M., Lewis R.A., Eyaid W., Banin E., et al. Hypomorphic mutations in syndromic encephalocele genes are associated with Bardet-Biedl syndrome. Nat. Genet. 2008;40:443–448. doi: 10.1038/ng.97. [DOI] [PubMed] [Google Scholar]
- 4.Zaghloul N.A., Katsanis N. Mechanistic insights into Bardet-Biedl syndrome, a model ciliopathy. J. Clin. Invest. 2009;119:428–437. doi: 10.1172/JCI37041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Friedman T.B., Schultz J.M., Ahmed Z.M., Tsilou E.T., Brewer C.C. Usher syndrome: hearing loss with vision loss. Adv. Otorhinolaryngol. 2011;70:56–65. doi: 10.1159/000322473. [DOI] [PubMed] [Google Scholar]
- 6.Guo D.F., Rahmouni K. Molecular basis of the obesity associated with Bardet-Biedl syndrome. Trends Endocrinol. Metab. 2011;22:286–293. doi: 10.1016/j.tem.2011.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Putoux A., Attie-Bitach T., Martinovic J., Gubler M.C. Phenotypic variability of Bardet–Biedl syndrome: focusing on the kidney. Pediatr. Nephrol. 2012;27:7–15. doi: 10.1007/s00467-010-1751-3. [DOI] [PubMed] [Google Scholar]
- 8.Puddu P., Barboni P., Mantovani V., Montagna P., Cerullo A., Bragliani M., Molinotti C., Caramazza R. Retinitis pigmentosa, ataxia, and mental retardation associated with mitochondrial DNA mutation in an Italian family. Br. J. Ophthalmol. 1993;77:84–88. doi: 10.1136/bjo.77.2.84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Inoue H., Tanizawa Y., Wasson J., Behn P., Kalidas K., Bernal-Mizrachi E., Mueckler M., Marshall H., Donis-Keller H., Crock P., et al. A gene encoding a transmembrane protein is mutated in patients with diabetes mellitus and optic atrophy (Wolfram syndrome) Nat. Genet. 1998;20:143–148. doi: 10.1038/2441. [DOI] [PubMed] [Google Scholar]
- 10.Keeler L.C., Marsh S.E., Leeflang E.P., Woods C.G., Sztriha L., Al-Gazali L., Gururaj A., Gleeson J.G. Linkage analysis in families with Joubert syndrome plus oculo-renal involvement identifies the CORS2 locus on chromosome 11p12-q13.3. Am. J. Hum. Genet. 2003;73:656–662. doi: 10.1086/378206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dixon-Salazar T., Silhavy J.L., Marsh S.E., Louie C.M., Scott L.C., Gururaj A., Al-Gazali L., Al-Tawari A.A., Kayserili H., Sztriha L., et al. Mutations in the AHI1 gene, encoding jouberin, cause Joubert syndrome with cortical polymicrogyria. Am. J. Hum. Genet. 2004;75:979–987. doi: 10.1086/425985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bredrup C., Saunier S., Oud M.M., Fiskerstrand T., Hoischen A., Brackman D., Leh S.M., Midtbo M., Filhol E., Bole-Feysot C., et al. Ciliopathies with skeletal anomalies and renal insufficiency due to mutations in the IFT-A gene WDR19. Am. J. Hum. Genet. 2011;89:634–643. doi: 10.1016/j.ajhg.2011.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Coppieters F., Lefever S., Leroy B.P., De Baere E. CEP290, a gene with many faces: mutation overview and presentation of CEP290base. Hum. Mutat. 2010;31:1097–1108. doi: 10.1002/humu.21337. [DOI] [PubMed] [Google Scholar]
- 14.Katsanis N. The oligogenic properties of Bardet-Biedl syndrome. Hum. Mol. Genet. 2004;13(Spec No. 1):R65–R71. doi: 10.1093/hmg/ddh092. [DOI] [PubMed] [Google Scholar]
- 15.Siemiatkowska A.M., van den Born L.I., van Hagen P.M., Stoffels M., Neveling K., Henkes A., Kipping-Geertsema M., Hoefsloot L.H., Hoyng C.B., Simon A., et al. Mutations in the mevalonate kinase (MVK) gene cause nonsyndromic retinitis pigmentosa. Ophthalmology. 2013;120:2697–2705. doi: 10.1016/j.ophtha.2013.07.052. [DOI] [PubMed] [Google Scholar]
- 16.Davidson A.E., Schwarz N., Zelinger L., Stern-Schneider G., Shoemark A., Spitzbarth B., Gross M., Laxer U., Sosna J., Sergouniotis P.I., et al. Mutations in ARL2BP, encoding ADP-ribosylation-factor-like 2 binding protein, cause autosomal-recessive retinitis pigmentosa. Am. J. Hum. Genet. 2013;93:321–329. doi: 10.1016/j.ajhg.2013.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zuchner S., Dallman J., Wen R., Beecham G., Naj A., Farooq A., Kohli M.A., Whitehead P.L., Hulme W., Konidari I., et al. Whole-exome sequencing links a variant in DHDDS to retinitis pigmentosa. Am. J. Hum. Genet. 2011;88:201–206. doi: 10.1016/j.ajhg.2011.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Abecasis G.R., Altshuler D., Auton A., Brooks L.D., Durbin R.M., Gibbs R.A., Hurles M.E., McVean G.A. 1000 Genomes Project Consortium. A map of human genome variation from population-scale sequencing. Nature. 2010;467:1061–1073. doi: 10.1038/nature09534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Haeseleer F., Jang G.F., Imanishi Y., Driessen C.A., Matsumura M., Nelson P.S., Palczewski K. Dual-substrate specificity short chain retinol dehydrogenases from the vertebrate retina. J. Biol. Chem. 2002;277:45537–45546. doi: 10.1074/jbc.M208882200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bolz H., Reiners J., Wolfrum U., Gal A. Role of cadherins in Ca2+-mediated cell adhesion and inherited photoreceptor degeneration. Adv. Exp. Med. Biol. 2002;514:399–410. doi: 10.1007/978-1-4615-0121-3_24. [DOI] [PubMed] [Google Scholar]
- 21.Shin J.B., Gillespie P.G. Unraveling cadherin 23’s role in development and mechanotransduction. Proc. Natl. Acad. Sci. U. S. A. 2009;106:4959–4960. doi: 10.1073/pnas.0902008106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bork J.M., Peters L.M., Riazuddin S., Bernstein S.L., Ahmed Z.M., Ness S.L., Polomeno R., Ramesh A., Schloss M., Srisailpathy C.R., et al. Usher syndrome 1D and nonsyndromic autosomal recessive deafness DFNB12 are caused by allelic mutations of the novel cadherin-like gene CDH23. Am. J. Hum. Genet. 2001;68:26–37. doi: 10.1086/316954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schultz J.M., Bhatti R., Madeo A.C., Turriff A., Muskett J.A., Zalewski C.K., King K.A., Ahmed Z.M., Riazuddin S., Ahmad N., et al. Allelic hierarchy of CDH23 mutations causing non-syndromic deafness DFNB12 or Usher syndrome USH1D in compound heterozygotes. J. Med. Genet. 2011;48:767–775. doi: 10.1136/jmedgenet-2011-100262. [DOI] [PubMed] [Google Scholar]
- 24.Lin B., White J.T., Ferguson C., Wang S., Vessella R., Bumgarner R., True L.D., Hood L., Nelson P.S. Prostate short-chain dehydrogenase reductase 1 (PSDR1): a new member of the short-chain steroid dehydrogenase/reductase family highly expressed in normal and neoplastic prostate epithelium. Cancer Res. 2001;61:1611–1618. [PubMed] [Google Scholar]
- 25.Kasus-Jacobi A., Ou J., Bashmakov Y.K., Shelton J.M., Richardson J.A., Goldstein J.L., Brown M.S. Characterization of mouse short-chain aldehyde reductase (SCALD), an enzyme regulated by sterol regulatory element-binding proteins. J. Biol. Chem. 2003;278:32380–32389. doi: 10.1074/jbc.M304969200. [DOI] [PubMed] [Google Scholar]
- 26.Parker R.O., Crouch R.K. Retinol dehydrogenases (RDHs) in the visual cycle. Exp. Eye Res. 2010;91:788–792. doi: 10.1016/j.exer.2010.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kiser P.D., Golczak M., Maeda A., Palczewski K. Key enzymes of the retinoid (visual) cycle in vertebrate retina. Biochim. Biophys. Acta. 2012;1821:137–151. doi: 10.1016/j.bbalip.2011.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kasus-Jacobi A., Ou J., Birch D.G., Locke K.G., Shelton J.M., Richardson J.A., Murphy A.J., Valenzuela D.M., Yancopoulos G.D., Edwards A.O. Functional characterization of mouse RDH11 as a retinol dehydrogenase involved in dark adaptation in vivo. J. Biol. Chem. 2005;280:20413–20420. doi: 10.1074/jbc.M413789200. [DOI] [PubMed] [Google Scholar]
- 29.Lorda-Sanchez I., Trujillo M.J., Gimenez A., Garcia-Sandoval B., Franco A., Sanz R., Rodriguez de Alba M., Ramos C., Ayuso C. Retinitis pigmentosa, mental retardation, marked short stature, and brachydactyly in two sibs. Ophthalmic Genet. 1999;20:127–131. doi: 10.1076/opge.20.2.127.2289. [DOI] [PubMed] [Google Scholar]
- 30.Berry D.C., Noy N. Signaling by vitamin A and retinol-binding protein in regulation of insulin responses and lipid homeostasis. Biochim. Biophys. Acta. 2012;1821:168–176. doi: 10.1016/j.bbalip.2011.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ashique A.M., May S.R., Kane M.A., Folias A.E., Phamluong K., Choe Y., Napoli J.L., Peterson A.S. Morphological defects in a novel Rdh10 mutant that has reduced retinoic acid biosynthesis and signaling. Genesis. 2012;50:415–423. doi: 10.1002/dvg.22002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kim T.S., Maeda A., Maeda T., Heinlein C., Kedishvili N., Palczewski K., Nelson P.S. Delayed dark adaptation in 11-cis-retinol dehydrogenase-deficient mice: a role of RDH11 in visual processes in vivo. J. Biol. Chem. 2005;280:8694–8704. doi: 10.1074/jbc.M413172200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nakamura M., Hotta Y., Tanikawa A., Terasaki H., Miyake Y. A high association with cone dystrophy in Fundus albipunctatus caused by mutations of the RDH5 gene. Invest. Ophthalmol. Vis. Sci. 2000;41:3925–3932. [PubMed] [Google Scholar]
- 34.Cideciyan A.V., Haeseleer F., Fariss R.N., Aleman T.S., Jang G.F., Verlinde C.L., Marmor M.F., Jacobson S.G., Palczewski K. Rod and cone visual cycle consequences of a null mutation in the 11-cis-retinol dehydrogenase gene in man. Vis. Neurosci. 2000;17:667–678. doi: 10.1017/s0952523800175029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Marmor M.F., Fulton A.B., Holder G.E., Miyake Y., Brigell M., Bach M. International Society for Clinical Electrophysiology of Vision. ISCEV Standard for full-field clinical electroretinography (2008 update) Doc. Ophthalmol. 2009;118:69–77. doi: 10.1007/s10633-008-9155-4. [DOI] [PubMed] [Google Scholar]
- 36.Jacobson S.G., Voigt W.J., Parel J.M., Apathy P.P., Nghiem-Phu L., Myers S.W., Patella V.M. Automated light- and dark-adapted perimetry for evaluating retinitis pigmentosa. Ophthalmology. 1986;93:1604–1611. doi: 10.1016/s0161-6420(86)33522-x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.