

Abstract
The GRIA1 locus, encoding the GluA1 (also known as GluRA or GluR1) AMPA glutamate receptor subunit, shows genome-wide association to schizophrenia. As well as extending the evidence that glutamatergic abnormalities play a key role in the disorder, this finding draws attention to the behavioural phenotype of Gria1 knockout mice. These mice show deficits in short-term habituation. Importantly, under some conditions the attention being paid to a recently presented neutral stimulus can actually increase rather than decrease (sensitization). We propose that this mouse phenotype represents a cause of aberrant salience and, in turn, that aberrant salience (and the resulting positive symptoms) in schizophrenia may arise, at least in part, from a glutamatergic genetic predisposition and a deficit in short-term habituation. This proposal links an established risk gene with a psychological process central to psychosis, and is supported by findings of comparable deficits in short-term habituation in mice lacking the NMDAR receptor subunit Grin2a (which also shows association to schizophrenia). Since aberrant salience is primarily a dopaminergic phenomenon, the model supports the view that the dopaminergic abnormalities can be downstream of a glutamatergic aetiology. Finally, we suggest that, as illustrated here, the real value of genetically modified mice is not as ‘models of schizophrenia’, but as experimental tools which can link genomic discoveries with psychological processes, and help elucidate the underlying neural mechanisms.
Keywords: dopamine, glutamate receptor, habituation, psychosis, sensitization
There is now strong evidence that hyper-dopaminergic activity underlies the positive psychotic symptoms of schizophrenia1-3. Dopaminergic abnormalities have a similarly proximate role in aberrant salience4,5 which Kapur and others have theorized is central to the genesis and understanding of positive psychotic symptoms6-9. However, the cause of this dopamine dysregulation is unspecified in Kapur’s model and has not yet been resolved8,10. Indeed, while considerable effort has gone into investigating and describing the putative links between dopamine, aberrant salience and psychosis, comparatively little effort has been expended in identifying the possible causes of aberrant salience.
Kapur and Howes noted that although the dysregulated, hyperdopaminergic state could be the result of a primary abnormality in the mesolimbic dopamine system, it could also be a secondary consequence of some other brain disturbance (or disturbances), and thus represent a “final common pathway” in schizophrenia8. The glutamate system is a prime candidate for this upstream abnormality6, 11-15 with diverse evidence for glutamatergic dysfunction, particularly NMDAR signaling, in the pathophysiology of schizophrenia16,17, including data from post mortem18,19, neuroimaging20,21, and immunological22 studies of the disorder, as well as indirectly from pharmacological findings17 and animal models23. An aetiological role for glutamate is now also likely, based on recent genetic data. Initial evidence came from candidate gene association studies24, 25 with observations that genes involved in glutamate synapses and NMDAR-mediated signaling are over-represented among schizophrenia genes26-29. These findings were subsequently supported by pathway analyses of genome-wide association studies (GWAS), and by de novo copy number variant data30, 31 and exome sequencing32.
In the largest GWAS study of schizophrenia to date33, a genomic locus upstream of GRIA1 is now genome-wide significant (p=1.06 × 10−10 for the top SNP). A forthcoming, larger meta-analysis confirms association to the locus, and to other glutamate receptor gene loci, discussed below34. GRIA1 encodes the GluA1 (also known as GluRA or GluR1) subunit of the AMPA subtype of glutamate receptor. This association complements prior evidence that AMPARs, as well as NMDARs, are involved in the disorder18, 35-40. Of particular relevance here is evidence that there are reductions of GluA1 mRNA36, 37 and GluA135, as well as AMPAR binding sites39, in the hippocampus in schizophrenia, and which do not appear to be secondary to antipsychotic medication41-44.
In this review we describe the behavioral phenotype of Gria1−/− mice and, in particular, a deficit whereby these mice fail to reduce the amount of attention that is paid to recently presented stimuli45,46. This failure to habituate means that stimuli continue to be surprising and grab attention for longer than would be normal. In fact, under some circumstances, these mice actually display sensitization, whereby more attention is paid to a recently presented stimulus than to a non-recent stimulus47. Thus, for these mice the stimulus is treated as even more salient or intense the second time it is presented. Deficits in short-term habituation therefore represent a potential driver of aberrant salience. This provides a key, mechanistic component of our present hypothesis, that GluA1 dysfunction contributes to aberrant salience in schizophrenia; we further suggest that this is mediated via enhanced dopamine signalling. This proposal provides a plausible causal link between a robust schizophrenia risk gene locus, and a psychological process of central importance in psychosis. Moreover, it directly links glutamate and dopamine components of the disorder.
Aberrant salience, dopamine and psychosis
Psychosis has been viewed as a disorder of aberrant salience7, mediated via a hyper-dopaminergic state6,7,9,48,49. For our purposes, salience can be defined as the ability of a stimulus to grab attention and to drive behaviour5,7. Salience reflects the innate properties of the stimulus (e.g., brightness, loudness), but can also reflect its potential motivational significance. Kapur suggested that before experiencing psychosis, patients will develop an exaggerated release of dopamine, independent of, and out of synchrony with, the context.7 The cause is not specified in Kapur’s model, but the resulting hyperdopaminergic state will then lead to the persistent and inappropriate assignment of salience to stimuli.
This state of aberrant salience can lead to inappropriate associations being formed, potentially via abnormal prediction error signals in the ventral striatum and other brain regions50-54. For example, Jensen et al., (2008)50 demonstrated aberrant learning and ventral striatal activation in schizophrenic patients using an aversive, Pavlovian discriminative fear conditioning paradigm. In this task, subjects were exposed to different visual stimuli. One stimulus (the conditioned stimulus; CS+) was paired with a loud noise (the unconditioned stimulus), whereas the other visual stimulus was not paired with the aversive event (CS−). Jensen and colleagues found inappropriately strong ventral striatal activation in response to the control stimulus (CS-cue), accompanied by abnormal learning, assessed both by self-report and galvanic skin responses (see also55). Similarly, Murray et al., (2008)52 found that first episode patients with active positive symptoms responded faster to neutral stimuli than controls during a reward learning task (response latencies were not significantly different to rewarded stimuli), and that these subjects exhibited abnormal BOLD responses associated with reward prediction error in dopaminergic midbrain, striatum and limbic areas. Likewise, Roiser and colleagues have reported aberrant reward learning in symptomatic but not asymptomatic schizophrenic patients56, and in un-medicated individuals at ultra-high risk of developing the condition57. This aberrant reward learning was correlated with the severity of delusion-like symptoms, as were ventral striatal BOLD responses to irrelevant stimuli.
Thus, it has been widely argued that the psychotic symptoms associated with schizophrenia, such as delusions and hallucinations, are the result of this fundamental abnormality in learning,7,54,58,59 and that their occurrence is correlated with aberrant salience56. Kapur hypothesised that patients would begin by assigning significance or importance to an incidental, neutral stimulus and, over a period of time, build up a complex delusion as a way of explaining why this unimportant object or detail has taken on such great meaning. Delusions are therefore a “cognitive effort by the patient to make sense of these aberrantly salient experiences,”7 and they reflect a maladaptive update of the patient’s world view53. Similarly, hallucinations may be experiences that result from aberrant salience being applied to internally-generated stimuli.
Kapur’s ideas build on the incentive or motivational salience hypothesis of dopamine’s actions put forward by Berridge and Robinson,60 Robbins and Everitt61 and others62-67. Kapur suggested that the mesolimbic dopamine system underlies motivational salience, and so “mediates the conversion of the neural representation of an external stimulus from a neutral, cold bit of information into an attractive or aversive entity”, (i.e. something that is of biological significance)7. In addition, dopamine can facilitate aspects of associative learning (e.g.68-71). Therefore, the ability of a stimulus to grab attention, drive action, and potentially form associations with other stimuli, are all influenced by dopamine.
Dopamine and novelty
The hypothesis that dopamine underlies the incentive or motivational salience of stimuli (and hence provides a signal of biological significance), captures a key element of what dopamine is doing, but it may not be the whole story: dopamine release is also associated with novelty. Although burst firing of dopamine cells in the ventral tegmental area (VTA) and substantia nigra pars compacta (SNc) is increased by unexpected rewards, and reduced if an expected reward is omitted, crucially dopamine neuronal activity is also triggered by novel stimuli that are not yet, and may never be, directly associated with reward or punishment, and are affectively neutral. Human fMRI studies have demonstrated that activation of the VTA/SNc can code for the absolute novelty of a stimulus, and that this haemodynamic signal exhibits repetition suppression with repeated presentation of the stimulus72. More directly, putative dopamine neurons in the VTA/SNc have been found to exhibit an increase in firing to novel, neutral stimuli73-75. Furthermore, recent evidence using dopamine voltammetry directly shows increased dopamine release in ventral striatum in response to novel, neutral stimuli69,76-78. The response to novelty may not be restricted to just the mesolimbic VTA-ventral striatal pathway, but may also include activity in the SNc-dorsal striatal circuitry72,79. Thus, dopamine release in the striatum is associated not only with incentive or motivational salience but also with novelty. As such, dopamine may signal not only stimuli of biological significance, but also stimuli of “potential” biological significance (as would be the case for any novel stimulus). Indeed, it has been argued that the coding of absolute novelty by dopamine may be treated like a signal which motivates exploration for potential reinforcers72,80.
A key point is that when stimuli are novel they grab the focus of attention and are perceived more intensely. Novel stimuli generate exploration81, and they readily enter into associations with other stimuli82. Therefore, novelty is important, both in terms of determining salience, as well as for the corresponding changes in dopamine activity5. Glutamate receptors, and in particular GluA1-containing AMPARs, play a fundamental role in the response to novel stimuli, and in the short-term habituation to such stimuli as a result of recent experience45,46.
Gria1 (GluA1) knockout mice: selective impairments in short-term habituation
The AMPAR is a hetero-oligomeric protein complex consisting of combinations of four subunits (GluA1-4, or GluRA-D), each encoded by a separate gene (GRIA1-4)83. Mice in which the gene encoding GluA1 is knocked out constitutively exhibit normal development, life expectancy, and fine structure of neuronal dendrites and synapses (Gria1−/− mice84). However, there is a reduction in the number of functional AMPA receptors, and both somatic and synaptic glutamatergic currents are reduced85-87. A number of studies have shown deficient long-term potentiation in hippocampal slices from Gria1−/− mice84-86, although more recent studies have indicated that GluA1 subunits may contribute primarily to short-lasting forms of synaptic plasticity87-89.
Behaviourally, Gria1−/− mice are indistinguishable from wild-type littermates in their home cage environment. However, closer inspection in experimental settings reveals a specific, but striking impairment in short-term habituation in these animals. Habituation is the decline in the tendency to respond to a stimulus that has become familiar due to prior exposure. This is likely to be an adaptive response to ensure that attentional resources are allocated to novel and potentially important stimuli. It has been argued that habituation can be fractionated into short- and long-term processes, with different underlying psychological and neural mechanisms45,90-92. Gria1−/− mice exhibit a pronounced deficit in short-term habituation. For example, on the novel object recognition (NOR) test, Gria1−/− mice are slower to habituate to a would-be familiar object (Figure 1). In this task, mice are first typically exposed to two identical copies of an object during a sample (“Exposure”) phase and allowed to explore freely. When a wild-type animal is presented with novel objects, it will begin to explore them but its exploratory activity gradually decreases or habituates as the objects become familiar. In a subsequent “Test” phase (usually conducted after a short delay), the mouse is exposed to a further copy of the original object (now familiar) and a novel object. Wild-type animals will preferentially choose to explore the novel alternative, reflecting their stimulus-specific habituation to the original object. In contrast, Gria1−/− mice display a deficit in short-term habituation:93 they fail to reduce the amount of attention that is paid to recently presented stimuli. Consequently, Gria1−/− mice show less preference than wild-type mice for a novel object compared to a familiar object that was presented recently93 (Figure 1a). Gria1−/− mice are also impaired on a purely recency-dependent version of the object recognition task93 (Figure 1b). However, Gria1−/− mice can recognise an object as familiar when it is consistently and repeatedly presented in a given, distinctive context (the object-in-context paradigm93; Figure 1c). This reflects the fact that they can use the context or place to associatively retrieve or prime the memory of that object from long-term memory, such that it feels familiar (i.e. long-term habituation is preserved). Importantly, this also shows that their deficit in short-term habituation is neither a basic perceptual problem, nor a global memory deficit.
Figure 1. Gria1−/− mice display impaired short-term habituation on the novel object recognition test.
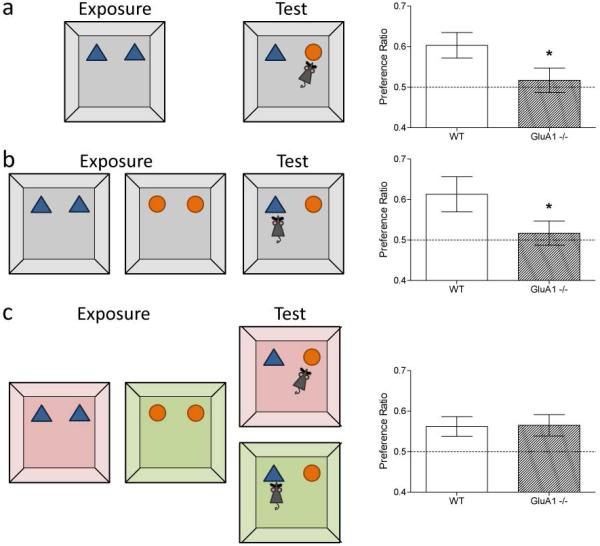
a. The top left panel shows the design of the standard novel object recognition task. In the Exposure phase (10 min duration) wild-type (WT) mice were exposed to two copies of an object and then after a 2 min interval they received a 5 min Test in which they were allowed to explore a duplicate of the familiar object and a novel object. The levels of object exploration for Gria1−/− mice for both exposure and test phases were yoked to WT mice. The times spent exploring the novel object during the test phase are shown as a ratio of the total time spent exploring both objects. The dashed line at 0.5 indicates chance performance. Gria1 deletion impaired memory on the standard object recognition task (right panel). Error bars indicate ±S.E.M. b. The middle panel shows the design of the object recency task. In the Exposure phase wild-type mice received two 10 min exposures to two different objects separated by a 2 min interval. The Test phase (5 min duration) commenced 2 min after the last exposure. Mice were allowed to explore the more recently and the less recently presented objects. The levels of object exploration for Gria1−/− mice for both exposure and test phases were yoked to WT mice. The times spent exploring the less recently experienced object are shown as a ratio of the total time spent exploring both objects. The dashed line at 0.5 indicates chance performance. Gria1 deletion impaired memory on the object recency test (right panel. Error bars indicate ±S.E.M. c. The bottom panel shows the design of the context-dependent object recognition task. In the Exposure phase two different objects were exposed in two different contexts. WT mice received four 10 min exposures to each object, one per day for four days. On the fifth day mice were simultaneously exposed to both objects in both of the contexts in two 5 min Tests. The levels of object exploration for Gria1−/− mice for both exposure and test phases were yoked to WT mice. The times spent exploring the object not previously paired with the test context (i.e. the unpaired object) are shown as a ratio of the total time spent exploring both objects. The dashed line at 0.5 indicates chance performance. Gria1 deletion did not impair context-dependent object recognition task (right panel). Error bars indicate ±S.E.M. (Data from93).
The short-term habituation deficit can also be demonstrated using a simple, spatial novelty preference task, during which animals spontaneously explore a Perspex Y-maze surrounded by extra-maze spatial cues (Figure 2; 94,95). During the sample or “Exposure” phase the animals are allowed to explore two arms of the maze; while access to the third arm is blocked off. During the subsequent choice phase all three arms of the maze are available to be explored. Wild-type mice avoid the recently visited, familiar arms and choose to explore the novel arm. In contrast, Gria1−/− mice fail to habituate to the recently visited spatial locations and therefore show no preference between the novel and familiar arms. This short-term spatial memory deficit is in marked contrast to the normal, or even enhanced, long-term associative spatial reference memory that Gria1−/− mice exhibit on tasks like the water maze or radial maze84,94,96-98, again demonstrating that their short-term habituation deficit is not due to perceptual impairments or a global memory deficit.
Figure 2. Gria1−/− mice display impaired short-term habituation on the spatial novelty preference test.
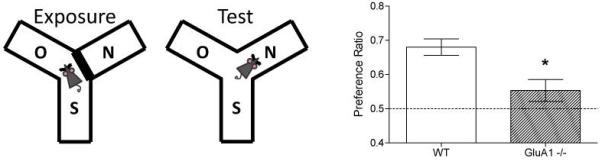
During a 5 min Exposure phase (left panel) mice were allowed to explore two arms (Start and Other) of a 3-arm, Perspex Y-maze surrounded by distal extra-maze cues. After a 1 min delay, the mice were returned to the maze for the Test phase (2 min duration), during which they were now able to explore freely all three maze arms, including the previously unvisited (novel) arm (centre panel). Gria1 deletion impaired performance on the spatial novelty preference test. Wild-type (WT) mice exhibit a preference for the previously unvisited (Novel) arm over the two familiar arms to which they have previously been exposed (Start and Other). Gria1−/− mice did not show a significant preference for the novel arm. Mean time spent in arms (±S.E.M.). (Data from95).
Thus, Gria1−/− mice are slower to habituate to both spatial and non-spatial stimuli and, as a consequence, treat stimuli as novel and salient for longer than wild-type mice. They are unable to filter out, and reduce attention to, recently experienced stimuli. This habituation deficit or attentional gating failure in Gria1−/− mice may also explain their deficit in pre-pulse inhibition (PPI), albeit the failure to habituate in the latter setting manifests over a different time scale99.
Gria1 knockout mice exhibit sensitization
Notably, under some circumstances, salience actually increases with repeated or continued exposure to a stimulus in Gria1−/− mice. This is called sensitization. For example, in a recent experiment we measured how much time mice spent looking at different light stimuli in an operant box, depending on their recent experience (Figure 3;47). Mice either received two exposures to the same light separated by 30 seconds (e.g. flashing light followed by flashing light), or received a pairing consisting of two different light stimuli (e.g. flashing light followed by a constant light), again separated by 30 seconds. In both wild-type and Gria1−/− mice, if the two light stimuli in the sequence were different then animals spent an equal amount of time looking at both lights. In contrast, if the two light stimuli were the same then wild-type mice spent less time looking at the light on its second presentation, reflecting short-term habituation to the light. However, when the same light was presented twice to Gria1−/− mice they actually spent more time attending to the light on its second exposure (relative to the first presentation of that light, and relative to the presentation of a different light that hadn’t been presented recently). Therefore, Gria1−/− mice do have a memory of the specific light stimulus that they have just experienced (their behaviour is altered by that recent prior exposure). However, they express that memory in a very different way, attending more to the recently presented light compared to a more novel light. Thus, for Gria1−/− mice a recently presented stimulus can generate exaggerated (and hence aberrant) salience, in the absence of any evidence for its motivational significance.
Figure 3. Gria1−/− mice display increased attention (sensitisation) to a recently experienced light stimulus.
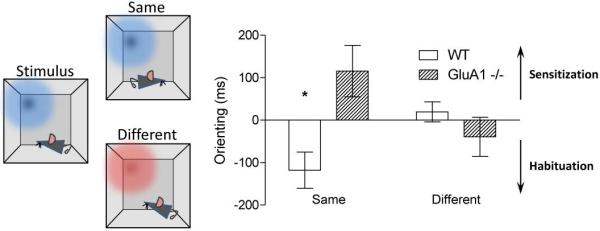
Unconditioned suppression of magazine responding to visual stimuli in an operant chamber was used as an indirect measure of the orienting response. Mice were exposed to pairs of light stimuli. Each stimulus in the pair was presented 30 sec apart (e.g., flashing vs. constant light, depicted graphically as red vs. blue; left panel). For half of trials the first light in the pair was the same as the second (Same condition). For the remaining trials the first light was different from the second (Different condition). Orienting to the first light in the pair was subtracted from orienting to the second light to give a difference score (Orienting; ms). In the Same condition Wild-type (WT) mice exhibited a reduced orienting response to the second stimulus in the pair. In contrast, Gria1−/− mice exhibited greater responding to the second stimulus. Both groups showed similar levels of orienting to both stimuli in the pair in the Different condition. This demonstrates the reduction in orienting in wild-type mice and the increase in orienting in Gria1−/− mice in the Same conditions are stimulus-specific (Data from47).
Importantly, this attentional deficit in Gria1−/− mice is set against an intact ability to form associations between stimuli. Associative learning is not impaired in these animals in a variety of experimental settings, including both maze and operant tasks, and in both spatial and non-spatial paradigms84,96,97,100. Indeed, in some situations Gria1−/− mice may actually form associations more readily than their wild-type controls97,101,102. This potentially reflects the fact that Gria1−/− mice, by finding a given stimulus more salient, and by paying more attention to that stimulus, are more likely to associate other events or consequences (e.g. such as reward) with its presence, thus facilitating long-term memory formation.
Notably, we have also shown that Gria1−/− mice can exhibit long-term memory under conditions where there is no evidence of long-term memory in wild-type controls94. Thus, in this instance Gria1−/− mice could be said to demonstrate “inappropriate” learning (where “inappropriate” learning is defined as learning that isn’t exhibited by control subjects). An extension of this is the prediction that these mice will also display abnormalities in credit assignment (i.e. the forming of appropriate associations between stimuli and events in a complex, temporally dynamic environment in which there are multiple cues competing for associative strength), leading to false inferences. This would provide a further demonstration of the kind of aberrant learning that might underlie psychotic symptoms such as delusions, and will be an important further test of our hypothesis in Gria1−/− mice.
Habituation, salience and schizophrenia
How relevant is this short-term habituation deficit in Gria1−/− mice to schizophrenia? In his original descriptions, Bleuler presciently noted that patients often experienced “an absence of the feeling of familiarity.”103 Subsequently, it has been well documented that patients with schizophrenia exhibit habituation deficits over a range of timescales104, both behaviourally (e.g. in terms of habituation of the startle response105-108), and also physiologically (e.g. the reduction in evoked responses to auditory stimuli with repeated presentations109). Impairments in PPI could also be considered as a failure to reduce the attention paid to a stimulus (the startle stimulus) based on recent prior experience (the pre-pulse106,108,110). Therefore, the link between habituation deficits and schizophrenia has been made before. What is novel here is the link between deficits in short-term habituation which can lead to sensitization, and the notion that patients may experience aberrant salience, with greater attention being paid to recently presented stimuli. Indeed, it is tempting to draw parallels between the exaggerated (aberrant) salience experienced by Gria1−/− mice and the attentional abnormalities reported in schizophrenia, including during the prodrome. Kapur7 noted that patients experience a stage of heightened sensory or perceptual awareness during the prodromal phase. Although accounts are usually anecdotal and/or post-hoc, they do suggest that everything the person experiences is intense, interesting, and highly salient. For example, patients report feelings such as “I developed a greater awareness of…My senses were sharpened…I became fascinated by the little insignificant things around me…Sights and sounds possessed a keenness that he had never experienced before…It was as if part of my brain awoke which had been dormant…My senses seemed alive….Things seemed clearcut, I noticed things I had never noticed before…My capacities for aesthetic appreciation and heightened sensory receptiveness were very keen at this time. I had had the same intensity of experience at other times when I was normal, but such periods were not sustained for long…” (taken from Kapur, 20037).
In essence, people with schizophrenia appear to pay elevated levels of attention to certain stimuli in their environment, in much the same way that the Gria1−/− mice pay an increased amount of attention to the recently presented light stimulus in our operant experiment (see Figure 3;47). The fact that these feelings of heightened awareness and intensity of perceptual experience often emerge during the prodrome is consistent with the possibility that these attentional deficits may be a contributory cause of psychosis, and could provide the trigger for subsequent positive symptoms6,8,111. We suggest that sensitization in Gria1−/− mice is homologous to the heightened intensity of sensory stimulation experienced by patients during the prodromal phase of the disorder.
Psychological and neural mechanisms underlying short-term habituation and their alteration in schizophrenia
How do deficits in short-term habituation result in sensitization, and how might these attentional phenomena be represented in the brain? Short-term habituation reflects a component of short-term memory which results in less attention being paid to a recently experienced stimulus (the stimulus might be said to exist in a secondary or reduced state of attention). This is distinct from an active form of short-term memory that underlies human working memory performance (e.g. on N-back or digit span tasks) in which the stimulus representation is actively maintained at the forefront of attention (the primary state of attention). These different short-term memory states therefore map onto different attentional states, reflecting the different amounts of attention being paid to a stimulus.
Wagner proposed a theoretical and computational model of stimulus processing that can explain the relationship between attention, habituation and learning91 (Figure 4). These ideas are of fundamental importance for understanding how deficits in short-term habituation could lead to aberrant salience and the genesis of psychosis. Wagner suggested that each stimulus is represented by a set of elements. Individual elements can exist in any one of three different activity or attentional states: an inactive state (I), the primary state of attention (A1), or the secondary state of attention (A2). While proportions of elements for a given stimulus can be in different activity states, individual elements can only be in one state at any one time. When a stimulus is novel and surprising, it occupies the forefront of attention, is highly salient, and generates strong levels of responding. This corresponds to the stimulus elements being in the A1 state. Also, associations can form between elements of different stimuli that are concurrently active in the A1 state. Conversely, when the stimulus is treated as familiar, less attention is paid to the stimulus, and it is less able to enter into associations with other stimuli (i.e. associative memory formation will be weaker). This reflects the fact that the stimulus elements are in the secondary attentional (A2) state.
Figure 4. Wagner’s model of stimulus processing.

Wagner proposed that each stimulus is represented by a number of elements. When a stimulus is presented a proportion of these elements go from being inactive (I state) and enter into a primary activity or attentional state, which might be considered as the forefront of attention or active short-term memory (A1 state). Elements then rapidly decay from this A1 state into a secondary activity state (A2 state) where they remain before gradually decaying back to the inactive state (I state). Stimulus elements can also go directly from the inactive state to the A2 state (which involves an associative retrieval process based on previously formed long-term memories). This is the basis of long-term habituation and is GluA1-independent; see lower horizontal arrow between I state and A2 state). When the elements of the stimulus are in the A1 state, higher levels of attention are paid to the stimulus and it can generate strong levels of responding. Also, associations can form between elements of different stimuli that are concurrently active in the A1 state. In contrast, when elements are in the secondary, attentional or A2 state, relatively less attention is paid to the stimulus and it will generate weaker levels of responding. GluA1 deletion retards the transition of elements from the A1 state to the A2 state. This can potentially lead to their accumulation in the A1 state and hence to sensitization. For further details, see text and 93,94.
Wagner’s model posits that there are two distinct forms of habituation (short-term and long-term habituation), each supported by a separate psychological mechanism. For the purposes of this review we will concentrate on short-term habituation which reflects the recent presentation of the stimulus, and which is dependent on the GluA1 subunit. When the stimulus is first presented a proportion of elements go from being inactive (I state) and enter into the primary activity state (A1 state). Elements then rapidly decay from this A1 state into the A2 state, where they remain before gradually decaying back to the inactive I state. If elements are already in the A2 state when the stimulus is presented (e.g. during the second presentation of the same stimulus after a short interval), these elements are unable to return directly to the A1 state. As a result, there are fewer stimulus elements available for activation into the A1 state, and consequently less responding to the stimulus (i.e. there will be habituation). Thus, habitation occurs to the degree to which the stimulus elements are in the A2 state. After sufficient passage of time, the stimulus elements decay back to the inactive state, and so are once again fully available for subsequent activation into the primary A1 state. Therefore, habituation is now no longer evident (i.e. it is short-lasting).
As described above, Gria1−/− mice demonstrate that short-term habituation is GluA1-dependent. In terms of Wagner’s model, Gria1 deletion retards the normal transition of a stimulus representation from A1 to A2 (Figure 4). Hence, in Gria1−/− mice stimuli stay at the forefront of attention (i.e. in the A1 state), and remain salient, for longer than in wild-type mice. In fact, as we have seen, in Gria1−/− mice the stimulus can actually be treated as increasingly salient with its repeated or continued presentation, as the elements that comprise the stimulus gradually accumulate in the forefront of attention and are less able to exit to the secondary attentional state47. This therefore provides an account of how deficits in short-term habituation can lead to sensitization, and gives us important clues as to possible underlying neural substrates.
What are the neural mechanisms that might underlie these changes in attention? We have suggested elsewhere that Wagner’s elements could correspond to the neurons that underlie the representation of the stimulus46. When a stimulus is first presented and occupies the forefront of attention, a proportion of the neurons in the brain that represent that stimulus will fire and generate action potentials (this would correspond to the primary state of activity). Notably, only when the stimulus elements are in this A1 state can they form excitatory associations with elements of other stimuli, consistent with Hebb’s postulate that neurons that fire together wire together112-114. In contrast, the secondary state of attention, which corresponds to habituation, presumably reflects the fact that the neurons which represent the stimulus are now less excitable and less likely to fire than when the stimulus was at the forefront of attention, and they are thus also less likely to form associations with neurons representing other stimuli. This transition from the primary to secondary state of attention likely reflects a short-term plasticity process which depends on Gria1, although the precise neural circuits and synaptic mechanisms involved remain to be established (see46 for discussion).
Evidence for reduced neuronal activity with repeated presentation of the same stimulus can be found with the phenomenon of repetition suppression of the haemodynamic BOLD signal which is often observed in human fMRI experiments, and in a variety of different brain regions72,115-120. Repetition suppression occurs when a recently presented (and now familiar) stimulus is presented again115. This reduction in the BOLD response likely reflects the tuning or modulation of neuronal representations such that familiar stimuli activate fewer neurons and evoke less neuronal firing. Consistent with this possibility, single cell recordings show that repetition suppression is associated with a decrease in neuronal firing, at least in some brain regions118,120,121.
Notably, Holt and colleagues showed that repetition suppression is impaired in schizophrenia104. They showed that, in healthy individuals, medial temporal lobe activity (and in particular hippocampal activity) habituates rapidly with repeated presentations of fearful faces. In contrast, patients exhibited no suppression of BOLD activity, consistent with a failure to habituate. Crucially, there is also evidence suggestive of sensitization in patients. A PET imaging study, conducted while subjects performed a passive viewing task122 found repetition suppression of cerebral blood flow in the right hemisphere of normal individuals across presentations of the same visual image as expected, but in patients with schizophrenia the blood flow response to the visual stimulus actually increased across the session (the equivalent of repetition enhancement in fMRI115). Therefore, patients with schizophrenia fail to reduce neuronal activity with repeated presentations of the same stimulus, consistent with their inability to reduce the amount of attention that is paid to a recently presented stimulus. In some situations, neuronal activity in patients may even increase with repeated presentations of the same stimulus122, potentially consistent with sensitization to a given stimulus.
Dopamine as a mediating transmitter system
We have drawn attention to the parallels between the impaired short-term habituation seen in Gria1−/− mice and in people with schizophrenia, and suggest that these impairments may lead to aberrant salience. We now consider how these processes are linked, and the central role which dopamine plays.
Given that (i) novelty evokes activity in the striatal dopamine system, coupled with (ii) the short-term habituation deficit and sensitization seen in Gria1−/− mice, this leads to the prediction of enhanced dopamine activity in these mice. It is important to point out that this hyper-dopaminergic response would likely be both stimulus-driven and stimulus-specific, and therefore not necessarily reflected in baseline measures of the dopamine system. Indeed, tissue levels of striatal dopamine appear normal123. However, Gria1−/− mice do exhibit a marked locomotor hyperactivity when placed in a novel environment, very reminiscent of the effects of low dose amphetamine99,124. In both cases, Gria1−/− mice can exhibit levels of locomotor activity well in excess of the activity levels displayed by controls, consistent with the possibility of sensitization (e.g.99). Furthermore, this hyperactivity is blocked by the dopamine D2 receptor antagonist haloperidol99. Using high speed chronoamperometric measurements of extracellular fluid dopamine levels in anaesthetized animals, Wiedholz et al., (2008) also found that the velocity of striatal dopamine clearance was slower in Gria1−/− mice99. This would be predicted to lead to an increase in the magnitude and duration of striatal dopamine responses. Taken together, these results are consistent with a putative hyper-dopaminergic phenotype in Gria1−/− mice. To test this prediction explicitly, it will be important to assess dopamine transients in response to novel and recently presented stimuli in freely moving, behaving mice, using techniques like fast-scan cyclic voltammetry77,125, to determine what role mesolimbic and nigrostriatal dopamine pathways play in these attentional processes (e.g.126), and, more specifically, whether changes in the novelty/familiarity of stimuli are reflected differently in dopamine signals in wild-type and Gria1−/− mice.
It is worth pointing out that current antipsychotic drugs appear to dampen all salience, not just aberrant salience7,58 (i.e. their effects are not stimulus-specific), and they do not rescue deficits in habituation or its physiological correlates104-106,109,122. Thus, these drugs may effectively silence the problem without correcting the underlying impairment. The analogy might be with a broken radio that is giving out white noise. Turning down the volume will remove the immediate problem (and the distress which it causes), but will not fix the underlying malfunction. Therefore, identifying the molecular, synaptic and circuit mechanisms that support short-term habituation, may have important therapeutic implications by allowing more targeted suppression of aberrant salience.
GRIA1 and schizophrenia – the broader context
To summarize, studies in Gria1−/− mice show that the GluA1 AMPAR subunit plays a key role in short-term habituation. Gria1−/− mice can pay even more attention to a recently experienced stimulus compared to a more novel stimulus. This phenotype may be of particular interest with regard to psychosis. Firstly, because stimuli are perceived more intensely and/or remain at the forefront of attention for longer, we propose that this short-term habituation deficit can underlie aberrant salience, a process believed to be of central importance in the origin of positive psychotic symptoms. As a consequence, these stimuli are more likely to enter into inappropriate or aberrant associations, leading to the formation of delusions. Thus, we suggest that changes in stimulus processing (and the allocation of attention) caused by GluA1 deletion are an upstream cause of deficits in prediction error learning that are seen in patients. Of course, these delusions are often sustained for long periods of time, and are impervious to contradictory evidence. Corlett and colleagues have likened this tenacity of delusions to the formation of instrumental habits seen in learning experiments with over-training54, 127. In this respect, it is worth noting that Gria1−/− mice also been display an increased propensity for habitual behavior128,129. Further experiments are required to determine whether this is related to the deficits in short-term habituation and its possible consequences for rates of associative learning, or whether it reflects a role for GluA1 in other neural circuits supporting goal-directed behaviour. Secondly, since the GRIA1 locus shows genetic association to schizophrenia, these considerations take on possible aetiological significance. They also support the widely held view that dopaminergic changes in schizophrenia are downstream of an abnormality in the glutamate system8,10,12,14-16,24,26,29,130.
With regard to the plausibility of these suggestions, several issues regarding the genetics and pathogenesis of schizophrenia are relevant. Firstly, genetic evidence for GRIA1 involvement in the disorder is far from complete. The GWAS data show association to a locus which is upstream of the gene, and it remains to be proven whether risk SNP(s) within the locus do in fact impact on the biology of GRIA1 (and not, for example, on another gene in the vicinity). It is not a trivial process to move from a genetic association signal to the identification of the molecular consequences of the risk variation131-133, as illustrated by investigations of other psychosis genes134-136. And, even assuming that GRIA1 is the target, the effect of the risk variation will likely be subtle, for example by modulating transcriptional regulation, and possibly contributing to the modest reduction of hippocampal GluA1 expression seen in schizophrenia35-37. In this context, the inherent limitations of a constitutive knockout (which models a null mutation or gene deletion) in mouse models relevant to schizophrenia are apparent137-139, and indicate the value of using additional genetic models of GRIA1. Indeed, it is already clear that it may not be necessary to remove all GluA1 subunits to produce the phenotype described here, since behavioral deficits indicative of impaired short-term habituation are also seen in mice in which Gria1 is knocked out selectively in the parvalbumin-positive (PV+) population of interneurons140. Furthermore, mice in which NMDARs have been ablated selectively from PV+ cells, or mice in which PV+ cell output has been silenced, display arguably similar phenotypes141-144. Hippocampal PV+ interneurons may be particularly important for these behavioural phenotypes145, in line with a key role for this brain region in regulating attentional processes like short-term habituation72,146,147. Thus, parenthetically, this account is also potentially consistent with the central role of PV+-interneurons148-150, and the hippocampus151,152 in schizophrenia and its onset130,153.
A second important caveat when extrapolating from Gria1−/− mice to schizophrenia is that the GRIA1 locus is but one of many, each of which in isolation has a very small effect on disease risk. In this respect it is notable that the recent GWAS study and meta-analysis also implicatesother glutamatergic genes, including GRIN2A, which encodes the NMDA receptor GluN2A (NR2A) subunit34. Grin2A−/− mice have a behavioural phenotype similar to that seen in Gria1−/− mice, albeit less extensively characterised, including a deficit in short-term habituation. For example, Grin2A−/− mice are unable to discriminate between a novel arm and recently experienced, familiar arm during the spatial novelty preference Y-maze test.154 This is set against an otherwise normal ability to perceive stimuli and to form long-term associations. These Grin2A data suggest that our proposal regarding GRIA1 and its role in short-term habituation and aberrant salience may generalise to at least some other glutamatergic genes which are involved in schizophrenia, reflecting their convergent effects on pathophysiological processes. As the genomics and genetic architecture of schizophrenia become clearer, it will be of interest to ascertain the identity and nature of the interplay between risk genes, and thence whether there is a functional convergence upon short-term habituation and salience. Such convergence is plausible, given that the underlying neural processes that support short-term habituation likely utilise fundamental synaptic plasticity mechanisms and pathways involving numerous molecular targets26-29,155-158.
Conclusions
We have drawn attention to the impaired short-term habituation phenotype of Gria1−/− mice and suggest that this impairment can generate sensitization and aberrant salience, potentially via enhanced dopaminergic signalling and defective hippocampal circuits. Impaired habituation, aberrant salience, hyperdopaminergia, and the hippocampus, are all central to current models of psychosis. The recent discovery that the GRIA1 locus shows genome-wide association to schizophrenia suggests that this phenotypic overlap between Gria1−/− mice and the clinical syndrome is more than coincidence, and instead reflects a pathway of causal significance linking these phenomena in schizophrenia. Indeed, GRIA1 provides arguably the first clear link between a GWAS-positive finding in schizophrenia and a core psychological process at the heart of the disorder. Clearly, this remains a speculative notion, and requires further critical evaluation using a range of approaches. The real value of rodent models in the next decade is surely in this domain: not as models of schizophrenia per se, but as experimental tools159 which can help link genomic discoveries to psychological processes and elucidate the underlying neural mechanisms.
Acknowledgements
MEW is a Wellcome Trust Career Development Fellow. DMB is a Wellcome Trust Senior Research Fellow. PJH’s research is supported by a Wellcome Trust Strategic Award and by the Medical Research Council.
Footnotes
Declaration of Interests: DMB is a member of the Lilly Centre for Cognitive Neuroscience. The authors declare no other relevant interests.
References
- 1.Howes OD, Kambeitz J, Kim E, Stahl D, Slifstein M, Abi-Dargham A, et al. The nature of dopamine dysfunction in schizophrenia and what this means for treatment. Arch Gen Psychiatry. 2012;69(8):776–786. doi: 10.1001/archgenpsychiatry.2012.169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Laruelle M. The second revision of the dopamine theory of schizophrenia: implications for treatment and drug development. Biol Psychiatry. 2013;74(2):80–81. doi: 10.1016/j.biopsych.2013.05.016. [DOI] [PubMed] [Google Scholar]
- 3.Seeman P. Dopamine receptors and the dopamine hypothesis of schizophrenia. Synapse. 1987;1(2):133–152. doi: 10.1002/syn.890010203. [DOI] [PubMed] [Google Scholar]
- 4.Heinz A, Schlagenhauf F. Dopaminergic dysfunction in schizophrenia: salience attribution revisited. Schizophr Bull. 2010;36(3):472–485. doi: 10.1093/schbul/sbq031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Winton-Brown TT, Fusar-Poli P, Ungless MA, Howes OD. Dopaminergic basis of salience dysregulation in psychosis. Trends Neurosci. 2014;37(2):85–94. doi: 10.1016/j.tins.2013.11.003. [DOI] [PubMed] [Google Scholar]
- 6.Gray JA, Feldon J, Rawlins JNP, Hemsley DR, Smith AD. The neuropsychology of schizophrenia. Behav brain Sci. 1991;14(1):1–84. [Google Scholar]
- 7.Kapur S. Psychosis as a state of aberrant salience: a framework linking biology, phenomenology, and pharmacology in schizophrenia. Am J Psychiatry. 2003;160(1):13–23. doi: 10.1176/appi.ajp.160.1.13. [DOI] [PubMed] [Google Scholar]
- 8.Howes OD, Kapur S. The dopamine hypothesis of schizophrenia: version III--the final common pathway. Schizophr Bull. 2009;35(3):549–562. doi: 10.1093/schbul/sbp006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.van Os J. ‘Salience syndrome’ replaces ‘schizophrenia’ in DSM-V and ICD-11: psychiatry’s evidence-based entry into the 21st century? Acta Psychiatr Scand. 2009;120(5):363–372. doi: 10.1111/j.1600-0447.2009.01456.x. [DOI] [PubMed] [Google Scholar]
- 10.Tost H, Alam T, Meyer-Lindenberg A. Dopamine and psychosis: theory, pathomechanisms and intermediate phenotypes. Neurosci Biobehav Rev. 2010;34(5):689–700. doi: 10.1016/j.neubiorev.2009.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Floresco SB, Todd CL, Grace AA. Glutamatergic afferents from the hippocampus to the nucleus accumbens regulate activity of ventral tegmental area dopamine neurons. J Neurosci. 2001;21(13):4915–4922. doi: 10.1523/JNEUROSCI.21-13-04915.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lodge DJ, Grace AA. Hippocampal dysregulation of dopamine system function and the pathophysiology of schizophrenia. Trends Pharmacol Sci. 2011;32(9):507–513. doi: 10.1016/j.tips.2011.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lisman JE, Coyle JT, Green RW, Javitt DC, Benes FM, Heckers S, et al. Circuit-based framework for understanding neurotransmitter and risk gene interactions in schizophrenia. Trends Neurosci. 2008;31(5):234–242. doi: 10.1016/j.tins.2008.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Weinberger DR. Implications of normal brain development for the pathogenesis of schizophrenia. Arch Gen Psychiatry. 1987;44(7):660–669. doi: 10.1001/archpsyc.1987.01800190080012. [DOI] [PubMed] [Google Scholar]
- 15.Egerton A, Fusar-Poli P, Stone JM. Glutamate and psychosis risk. Curr Pharm Des. 2012;18(4):466–478. doi: 10.2174/138161212799316244. [DOI] [PubMed] [Google Scholar]
- 16.Coyle JT. Glutamate and schizophrenia: beyond the dopamine hypothesis. Cell Mol Neurobiol. 2006;26(4-6):365–384. doi: 10.1007/s10571-006-9062-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Moghaddam B, Javitt D. From revolution to evolution: the glutamate hypothesis of schizophrenia and its implication for treatment. Neuropsychopharmacology. 2012;37(1):4–15. doi: 10.1038/npp.2011.181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rubio MD, Drummond JB, Meador-Woodruff JH. Glutamate Receptor Abnormalities in Schizophrenia: Implications for Innovative Treatments. Biomol Ther. 2012;20(1):1–18. doi: 10.4062/biomolther.2012.20.1.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Weickert CS, Fung SJ, Catts VS, Schofield PR, Allen KM, Moore LT, et al. Molecular evidence of N-methyl-D-aspartate receptor hypofunction in schizophrenia. Mol Psychiatry. 2013;18(11):1185–1192. doi: 10.1038/mp.2012.137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Poels EM, Kegeles LS, Kantrowitz JT, Slifstein M, Javitt DC, Lieberman JA, et al. Imaging glutamate in schizophrenia: review of findings and implications for drug discovery. Mol Psychiatry. 2014;19(1):20–29. doi: 10.1038/mp.2013.136. [DOI] [PubMed] [Google Scholar]
- 21.Marsman A, van den Heuvel MP, Klomp DW, Kahn RS, Luijten PR, Hulshoff Pol HE. Glutamate in schizophrenia: a focused review and meta-analysis of (1)H-MRS studies. Schizophr Bull. 2013;39(1):120–129. doi: 10.1093/schbul/sbr069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Deakin J, Lennox BR, Zandi MS. Antibodies to the N-methyl-D-aspartate receptor and other synaptic proteins in psychosis. Biol Psychiatry. 2014;75(4):284–291. doi: 10.1016/j.biopsych.2013.07.018. [DOI] [PubMed] [Google Scholar]
- 23.Inta D, Monyer H, Sprengel R, Meyer-Lindenberg A, Gass P. Mice with genetically altered glutamate receptors as models of schizophrenia: a comprehensive review. Neurosci Biobehav Rev. 2010;34(3):285–294. doi: 10.1016/j.neubiorev.2009.07.010. [DOI] [PubMed] [Google Scholar]
- 24.Collier DA, Li T. The genetics of schizophrenia: glutamate not dopamine? Eur J Pharmacol. 2003;480(1-3):177–184. doi: 10.1016/j.ejphar.2003.08.105. [DOI] [PubMed] [Google Scholar]
- 25.Cherlyn SY, Woon PS, Liu JJ, Ong WY, Tsai GC, Sim K. Genetic association studies of glutamate, GABA and related genes in schizophrenia and bipolar disorder: a decade of advance. Neurosci Biobehav Rev. 2010;34(6):958–977. doi: 10.1016/j.neubiorev.2010.01.002. [DOI] [PubMed] [Google Scholar]
- 26.Harrison PJ, Owen MJ. Genes for schizophrenia? Recent findings and their pathophysiological implications. Lancet. 2003;361(9355):417–419. doi: 10.1016/S0140-6736(03)12379-3. [DOI] [PubMed] [Google Scholar]
- 27.Harrison PJ, West VA. Six degrees of separation: on the prior probability that schizophrenia susceptibility genes converge on synapses, glutamate and NMDA receptors. Mol Psychiatry. 2006;11(11):981–983. doi: 10.1038/sj.mp.4001886. [DOI] [PubMed] [Google Scholar]
- 28.Moghaddam B. Bringing order to the glutamate chaos in schizophrenia. Neuron. 2003;40(5):881–4. doi: 10.1016/s0896-6273(03)00757-8. [DOI] [PubMed] [Google Scholar]
- 29.Harrison PJ, Weinberger DR. Schizophrenia genes, gene expression, and neuropathology: on the matter of their convergence. Mol Psychiatry. 2005;10(1):40–68. doi: 10.1038/sj.mp.4001558. image 45. [DOI] [PubMed] [Google Scholar]
- 30.Kirov G, Pocklington AJ, Holmans P, Ivanov D, Ikeda M, Ruderfer D, et al. De novo CNV analysis implicates specific abnormalities of postsynaptic signalling complexes in the pathogenesis of schizophrenia. Mol Psychiatry. 2012;17(2):142–153. doi: 10.1038/mp.2011.154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lips ES, Cornelisse LN, Toonen RF, Min JL, Hultman CM, Holmans PA, et al. Functional gene group analysis identifies synaptic gene groups as risk factor for schizophrenia. Mol Psychiatry. 2012;17(10):996–1006. doi: 10.1038/mp.2011.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Timms AE, Dorschner MO, Wechsler J, Choi KY, Kirkwood R, Girirajan S, et al. Support for the N-methyl-D-aspartate receptor hypofunction hypothesis of schizophrenia from exome sequencing in multiplex families. JAMA Psychiatry. 2013;70(6):582–590. doi: 10.1001/jamapsychiatry.2013.1195. [DOI] [PubMed] [Google Scholar]
- 33.Ripke S, O’Dushlaine C, Chambert K, Moran JL, Kahler AK, Akterin S, et al. Genome-wide association analysis identifies 13 new risk loci for schizophrenia. Nat Genet. 2013;45(10):1150–1159. doi: 10.1038/ng.2742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Schizophrenia Working Group of the Psychiatric Genomics Consortium Common variant association meta-analysis for schizophrenia identifies 108 genomic loci and implicates postsynaptic and immune processes. Nature. in press. [Google Scholar]
- 35.Eastwood SL, Kerwin RW, Harrison PJ. Immunoautoradiographic evidence for a loss of alpha-amino-3-hydroxy-5-methyl-4-isoxazole propionate-preferring non-N-methyl-D-aspartate glutamate receptors within the medial temporal lobe in schizophrenia. Biol Psychiatry. 1997;41(6):636–643. doi: 10.1016/S0006-3223(96)00220-X. [DOI] [PubMed] [Google Scholar]
- 36.Eastwood SL, McDonald B, Burnet PW, Beckwith JP, Kerwin RW, Harrison PJ. Decreased expression of mRNAs encoding non-NMDA glutamate receptors GluR1 and GluR2 in medial temporal lobe neurons in schizophrenia. Brain Res Mol Brain Res. 1995;29(2):211–223. doi: 10.1016/0169-328x(94)00247-c. [DOI] [PubMed] [Google Scholar]
- 37.Harrison PJ, McLaughlin D, Kerwin RW. Decreased hippocampal expression of a glutamate receptor gene in schizophrenia. Lancet. 1991;337(8739):450–452. doi: 10.1016/0140-6736(91)93392-m. [DOI] [PubMed] [Google Scholar]
- 38.Sokolov BP. Expression of NMDAR1, GluR1, GluR7, and KA1 glutamate receptor mRNAs is decreased in frontal cortex of “neuroleptic-free” schizophrenics: evidence on reversible up-regulation by typical neuroleptics. J Neurochem. 1998;71(6):2454–2464. doi: 10.1046/j.1471-4159.1998.71062454.x. [DOI] [PubMed] [Google Scholar]
- 39.Kerwin R, Patel S, Meldrum B. Quantitative autoradiographic analysis of glutamate binding sites in the hippocampal formation in normal and schizophrenic brain post mortem. Neuroscience. 1990;39(1):25–32. doi: 10.1016/0306-4522(90)90219-t. [DOI] [PubMed] [Google Scholar]
- 40.Dracheva S, McGurk SR, Haroutunian V. mRNA expression of AMPA receptors and AMPA receptor binding proteins in the cerebral cortex of elderly schizophrenics. J Neurosci Res. 2005;79(6):868–878. doi: 10.1002/jnr.20423. [DOI] [PubMed] [Google Scholar]
- 41.Eastwood SL, Porter RH, Harrison PJ. The effect of chronic haloperidol treatment on glutamate receptor subunit (GluR1, GluR2, KA1, KA2, NR1) mRNAs and glutamate binding protein mRNA in rat forebrain. Neurosci Lett. 1996;212(3):163–166. doi: 10.1016/0304-3940(96)12801-9. [DOI] [PubMed] [Google Scholar]
- 42.Eastwood SL, Story P, Burnet PW, Heath P, Harrison PJ. Differential changes in glutamate receptor subunit messenger RNAs in rat brain after haloperidol treatment. J Psychopharmacol. 1994;8(4):196–203. doi: 10.1177/026988119400800402. [DOI] [PubMed] [Google Scholar]
- 43.Oretti RG, Spurlock G, Buckland PR, McGuffin P. Lack of effect of antipsychotic and antidepressant drugs on glutamate receptor mRNA levels in rat brains. Neurosci Lett. 1994;177(1-2):39–43. doi: 10.1016/0304-3940(94)90039-6. [DOI] [PubMed] [Google Scholar]
- 44.Meador-Woodruff JH, King RE, Damask SP, Bovenkerk KA. Differential regulation of hippocampal AMPA and kainate receptor subunit expression by haloperidol and clozapine. Mol Psychiatry. 1996;1(1):41–53. [PubMed] [Google Scholar]
- 45.Sanderson DJ, Bannerman DM. The role of habituation in hippocampus-dependent spatial working memory tasks: evidence from GluA1 AMPA receptor subunit knockout mice. Hippocampus. 2012;22(5):981–994. doi: 10.1002/hipo.20896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sanderson DJ, McHugh SB, Good MA, Sprengel R, Seeburg PH, Rawlins JN, et al. Spatial working memory deficits in GluA1 AMPA receptor subunit knockout mice reflect impaired short-term habituation: evidence for Wagner’s dual-process memory model. Neuropsychologia. 2010;48(8):2303–2315. doi: 10.1016/j.neuropsychologia.2010.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Sanderson DJ, Sprengel R, Seeburg PH, Bannerman DM. Deletion of the GluA1 AMPA receptor subunit alters the expression of short-term memory. Learn Mem. 2011;18(3):128–131. doi: 10.1101/lm.2014911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Howes OD, Montgomery AJ, Asselin MC, Murray RM, Valli I, Tabraham P, et al. Elevated striatal dopamine function linked to prodromal signs of schizophrenia. Arch Gen Psychiatry. 2009;66(1):13–20. doi: 10.1001/archgenpsychiatry.2008.514. [DOI] [PubMed] [Google Scholar]
- 49.Howes O, Bose S, Turkheimer F, Valli I, Egerton A, Stahl D, et al. Progressive increase in striatal dopamine synthesis capacity as patients develop psychosis: a PET study. Mol Psychiatry. 2011;16(9):885–886. doi: 10.1038/mp.2011.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Jensen J, Willeit M, Zipursky RB, Savina I, Smith AJ, Menon M, et al. The formation of abnormal associations in schizophrenia: neural and behavioral evidence. Neuropsychopharmacology. 2008;33(3):473–479. doi: 10.1038/sj.npp.1301437. [DOI] [PubMed] [Google Scholar]
- 51.Juckel G, Schlagenhauf F, Koslowski M, Wustenberg T, Villringer A, Knutson B, et al. Dysfunction of ventral striatal reward prediction in schizophrenia. Neuroimage. 2006;29(2):409–416. doi: 10.1016/j.neuroimage.2005.07.051. [DOI] [PubMed] [Google Scholar]
- 52.Murray GK, Corlett PR, Clark L, Pessiglione M, Blackwell AD, Honey G, et al. Substantia nigra/ventral tegmental reward prediction error disruption in psychosis. Mol Psychiatry. 2008;13(3):239, 267–276. doi: 10.1038/sj.mp.4002058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Corlett PR, Murray GK, Honey GD, Aitken MR, Shanks DR, Robbins TW, et al. Disrupted prediction-error signal in psychosis: evidence for an associative account of delusions. Brain. 2007;130(9):2387–2400. doi: 10.1093/brain/awm173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Corlett PR, Taylor JR, Wang XJ, Fletcher PC, Krystal JH. Toward a neurobiology of delusions. Prog Neurobiol. 2010;92(3):345–369. doi: 10.1016/j.pneurobio.2010.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Holt DJ, Lebron-Milad K, Milad MR, Rauch SL, Pitman RK, Orr SP, et al. Extinction memory is impaired in schizophrenia. Biol Psychiatry. 2009;65(6):455–463. doi: 10.1016/j.biopsych.2008.09.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Roiser JP, Stephan KE, den Ouden HE, Barnes TR, Friston KJ, Joyce EM. Do patients with schizophrenia exhibit aberrant salience? Psychol Med. 2009;39(2):199–209. doi: 10.1017/S0033291708003863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Roiser JP, Howes OD, Chaddock CA, Joyce EM, McGuire P. Neural and behavioral correlates of aberrant salience in individuals at risk for psychosis. Schizophr Bull. 2013;39(6):1328–1336. doi: 10.1093/schbul/sbs147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kapur S. How antipsychotics become anti-“psychotic”--from dopamine to salience to psychosis. Trends Pharmacol Sci. 2004;25(8):402–406. doi: 10.1016/j.tips.2004.06.005. [DOI] [PubMed] [Google Scholar]
- 59.Kapur S, Mizrahi R, Li M. From dopamine to salience to psychosis--linking biology, pharmacology and phenomenology of psychosis. Schizophr Res. 2005;79(1):59–68. doi: 10.1016/j.schres.2005.01.003. [DOI] [PubMed] [Google Scholar]
- 60.Berridge KC, Robinson TE. What is the role of dopamine in reward: hedonic impact, reward learning, or incentive salience? Brain Res Brain Res Rev. 1998;28(3):309–369. doi: 10.1016/s0165-0173(98)00019-8. [DOI] [PubMed] [Google Scholar]
- 61.Robbins TW, Everitt BJ. Neurobehavioural mechanisms of reward and motivation. Curr Opin Neurobiol. 1996;6(2):228–236. doi: 10.1016/s0959-4388(96)80077-8. [DOI] [PubMed] [Google Scholar]
- 62.Horvitz JC. Mesolimbocortical and nigrostriatal dopamine responses to salient non-reward events. Neuroscience. 2000;96(4):651–656. doi: 10.1016/s0306-4522(00)00019-1. [DOI] [PubMed] [Google Scholar]
- 63.Martin-Soelch C, Leenders KL, Chevalley AF, Missimer J, Kunig G, Magyar S, et al. Reward mechanisms in the brain and their role in dependence: evidence from neurophysiological and neuroimaging studies. Brain Res Brain Res Rev. 2001;36(2-3):139–149. doi: 10.1016/s0165-0173(01)00089-3. [DOI] [PubMed] [Google Scholar]
- 64.Di Chiara G. A motivational learning hypothesis of the role of mesolimbic dopamine in compulsive drug use. J Psychopharmacol. 1998;12(1):54–67. doi: 10.1177/026988119801200108. [DOI] [PubMed] [Google Scholar]
- 65.Bindra D. A motivational view of learning, performance, and behavior modification. Psychol Rev. 1974;81(3):199–213. doi: 10.1037/h0036330. [DOI] [PubMed] [Google Scholar]
- 66.Fibiger HC, Phillips AG. Mesocorticolimbic dopamine systems and reward. Ann N Y Acad Sci. 1988;537:206–215. doi: 10.1111/j.1749-6632.1988.tb42107.x. [DOI] [PubMed] [Google Scholar]
- 67.Bindra D. How adaptive behavior is produced: a perceptual-motivational alternative to response reinforcements. Behavioral and Brain Sciences. 1978;1(1):41–52. [Google Scholar]
- 68.Lisman JE, Grace AA. The hippocampal-VTA loop: controlling the entry of information into long-term memory. Neuron. 2005;46(5):703–713. doi: 10.1016/j.neuron.2005.05.002. [DOI] [PubMed] [Google Scholar]
- 69.Flagel SB, Clark JJ, Robinson TE, Mayo L, Czuj A, Willuhn I, et al. A selective role for dopamine in stimulus-reward learning. Nature. 2011;469(7328):53–57. doi: 10.1038/nature09588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Dalley JW, Chudasama Y, Theobald DE, Pettifer CL, Fletcher CM, Robbins TW. Nucleus accumbens dopamine and discriminated approach learning: interactive effects of 6-hydroxydopamine lesions and systemic apomorphine administration. Psychopharmacology. 2002;161(4):425–433. doi: 10.1007/s00213-002-1078-2. [DOI] [PubMed] [Google Scholar]
- 71.Steinberg EE, Keiflin R, Boivin JR, Witten IB, Deisseroth K, Janak PH. A causal link between prediction errors, dopamine neurons and learning. Nat Neurosci. 2013;16(7):966–973. doi: 10.1038/nn.3413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Bunzeck N, Duzel E. Absolute coding of stimulus novelty in the human substantia nigra/VTA. Neuron. 2006;51(3):369–379. doi: 10.1016/j.neuron.2006.06.021. [DOI] [PubMed] [Google Scholar]
- 73.Horvitz JC, Stewart T, Jacobs BL. Burst activity of ventral tegmental dopamine neurons is elicited by sensory stimuli in the awake cat. Brain Res. 1997;759(2):251–258. doi: 10.1016/s0006-8993(97)00265-5. [DOI] [PubMed] [Google Scholar]
- 74.Steinfels GF, Heym J, Strecker RE, Jacobs BL. Response of dopaminergic neurons in cat to auditory stimuli presented across the sleep-waking cycle. Brain Res. 1983;277(1):150–154. doi: 10.1016/0006-8993(83)90917-4. [DOI] [PubMed] [Google Scholar]
- 75.Ljungberg T, Apicella P, Schultz W. Responses of monkey dopamine neurons during learning of behavioral reactions. J Neurophysiol. 1992;67(1):145–163. doi: 10.1152/jn.1992.67.1.145. [DOI] [PubMed] [Google Scholar]
- 76.Rebec GV, Christensen JR, Guerra C, Bardo MT. Regional and temporal differences in real-time dopamine efflux in the nucleus accumbens during free-choice novelty. Brain Res. 1997;776(1-2):61–67. doi: 10.1016/s0006-8993(97)01004-4. [DOI] [PubMed] [Google Scholar]
- 77.Clark JJ, Sandberg SG, Wanat MJ, Gan JO, Horne EA, Hart AS, et al. Chronic microsensors for longitudinal, subsecond dopamine detection in behaving animals. Nat Methods. 2010;7(2):126–129. doi: 10.1038/nmeth.1412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Robinson DL, Wightman RM. Nomifensine amplifies subsecond dopamine signals in the ventral striatum of freely-moving rats. J Neurochem. 2004;90(4):894–903. doi: 10.1111/j.1471-4159.2004.02559.x. [DOI] [PubMed] [Google Scholar]
- 79.Schiemann J, Schlaudraff F, Klose V, Bingmer M, Seino S, Magill PJ, et al. K-ATP channels in dopamine substantia nigra neurons control bursting and novelty-induced exploration. Nat Neurosci. 2012;15(9):1272–1280. doi: 10.1038/nn.3185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Kakade S, Dayan P. Dopamine: generalization and bonuses. Neural Netw. 2002;15(4-6):549–559. doi: 10.1016/s0893-6080(02)00048-5. [DOI] [PubMed] [Google Scholar]
- 81.Berlyne DE. Novelty and curiosity as determinants of exploratory behaviour. British Journal of Psychology. 1950;41:68–80. [Google Scholar]
- 82.Lubow RE, Moore AU. Latent inhibition: the effect of nonreinforced pre-exposure to the conditional stimulus. J Comp Physiol Psychol. 1959;52:415–419. doi: 10.1037/h0046700. [DOI] [PubMed] [Google Scholar]
- 83.Keinanen K, Wisden W, Sommer B, Werner P, Herb A, Verdoorn TA, et al. A family of AMPA-selective glutamate receptors. Science. 1990;249(4968):556–560. doi: 10.1126/science.2166337. [DOI] [PubMed] [Google Scholar]
- 84.Zamanillo D, Sprengel R, Hvalby O, Jensen V, Burnashev N, Rozov A, et al. Importance of AMPA receptors for hippocampal synaptic plasticity but not for spatial learning. Science. 1999;284(5421):1805–1811. doi: 10.1126/science.284.5421.1805. [DOI] [PubMed] [Google Scholar]
- 85.Andrasfalvy BK, Smith MA, Borchardt T, Sprengel R, Magee JC. Impaired regulation of synaptic strength in hippocampal neurons from GluR1-deficient mice. J Physiol. 2003;552(Pt 1):35–45. doi: 10.1113/jphysiol.2003.045575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Jensen V, Kaiser KM, Borchardt T, Adelmann G, Rozov A, Burnashev N, et al. A juvenile form of postsynaptic hippocampal long-term potentiation in mice deficient for the AMPA receptor subunit GluR-A. J Physiol. 2003;553(Pt 3):843–856. doi: 10.1113/jphysiol.2003.053637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Romberg C, Raffel J, Martin L, Sprengel R, Seeburg PH, Rawlins JN, et al. Induction and expression of GluA1 (GluR-A)-independent LTP in the hippocampus. Eur J Neurosci. 2009;29(6):1141–1152. doi: 10.1111/j.1460-9568.2009.06677.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Erickson MA, Maramara LA, Lisman J. A single brief burst induces GluR1-dependent associative short-term potentiation: a potential mechanism for short-term memory. J Cogn Neurosci. 2010;22(11):2530–2540. doi: 10.1162/jocn.2009.21375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Hoffman DA, Sprengel R, Sakmann B. Molecular dissection of hippocampal theta-burst pairing potentiation. Proc Natl Acad Sci U S A. 2002;99(11):7740–7745. doi: 10.1073/pnas.092157999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Sanderson DJ, Bannerman DM. Competitive short-term and long-term memory processes in spatial habituation. J Exp Psychol Anim Behav Process. 2011;37(2):189–199. doi: 10.1037/a0021461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Wagner AR. SOP: A model of automatic memory processing in animal behavior. In: Spear NE, Miller RR, editors. Information processing in animals: Memory mechanisms. Lawrence Erlbaum Associates, Inc.; Hillsdale, NJ: 1981. pp. 5–47. [Google Scholar]
- 92.Kandel ER, Schwartz JH, Jessell TM, Siegelbaum SA, Hudspeth AJ. Princliples of Neural Science. Fifth edn. McGraw-Hill Medical; New York: 2012. [Google Scholar]
- 93.Sanderson DJ, Hindley E, Smeaton E, Denny N, Taylor A, Barkus C, et al. Deletion of the GluA1 AMPA receptor subunit impairs recency-dependent object recognition memory. Learn Mem. 2011;18(3):181–190. doi: 10.1101/lm.2083411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Sanderson DJ, Good MA, Skelton K, Sprengel R, Seeburg PH, Rawlins JN, et al. Enhanced long-term and impaired short-term spatial memory in GluA1 AMPA receptor subunit knockout mice: evidence for a dual-process memory model. Learn Mem. 2009;16(6):379–386. doi: 10.1101/lm.1339109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Sanderson DJ, Gray A, Simon A, Taylor AM, Deacon RM, Seeburg PH, et al. Deletion of glutamate receptor-A (GluR-A) AMPA receptor subunits impairs one-trial spatial memory. Behav Neurosci. 2007;121(3):559–569. doi: 10.1037/0735-7044.121.3.559. [DOI] [PubMed] [Google Scholar]
- 96.Reisel D, Bannerman DM, Schmitt WB, Deacon RM, Flint J, Borchardt T, et al. Spatial memory dissociations in mice lacking GluR1. Nat Neurosci. 2002;5(9):868–873. doi: 10.1038/nn910. [DOI] [PubMed] [Google Scholar]
- 97.Schmitt WB, Deacon RM, Seeburg PH, Rawlins JN, Bannerman DM. A within-subjects, within-task demonstration of intact spatial reference memory and impaired spatial working memory in glutamate receptor-A-deficient mice. J Neurosci. 2003;23(9):3953–3959. doi: 10.1523/JNEUROSCI.23-09-03953.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Schmitt WB, Sprengel R, Mack V, Draft RW, Seeburg PH, Deacon RM, et al. Restoration of spatial working memory by genetic rescue of GluR-A-deficient mice. Nat Neurosci. 2005;8(3):270–272. doi: 10.1038/nn1412. [DOI] [PubMed] [Google Scholar]
- 99.Wiedholz LM, Owens WA, Horton RE, Feyder M, Karlsson RM, Hefner K, et al. Mice lacking the AMPA GluR1 receptor exhibit striatal hyperdopaminergia and ‘schizophrenia-related’ behaviors. Mol Psychiatry. 2008;13(6):631–640. doi: 10.1038/sj.mp.4002056. [DOI] [PubMed] [Google Scholar]
- 100.Mead AN, Stephens DN. Selective disruption of stimulus-reward learning in glutamate receptor gria1 knock-out mice. J Neurosci. 2003;23(3):1041–1048. doi: 10.1523/JNEUROSCI.23-03-01041.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Barkus C, Feyder M, Graybeal C, Wright T, Wiedholz L, Izquierdo A, et al. Do GluA1 knockout mice exhibit behavioral abnormalities relevant to the negative or cognitive symptoms of schizophrenia and schizoaffective disorder? Neuropharmacology. 2012;62(3):1263–1272. doi: 10.1016/j.neuropharm.2011.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Taylor AM, Niewoehner B, Seeburg PH, Sprengel R, Rawlins JN, Bannerman DM, et al. Dissociations within short-term memory in GluA1 AMPA receptor subunit knockout mice. Behav Brain Res. 2011;224(1):8–14. doi: 10.1016/j.bbr.2011.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Bleuler E. Dementia Praecox or the Group of Schizophrenias. International Universities Press; New York: 1950. [Google Scholar]
- 104.Holt DJ, Weiss AP, Rauch SL, Wright CI, Zalesak M, Goff DC, et al. Sustained activation of the hippocampus in response to fearful faces in schizophrenia. Biol Psychiatry. 2005;57(9):1011–1019. doi: 10.1016/j.biopsych.2005.01.033. [DOI] [PubMed] [Google Scholar]
- 105.Akdag SJ, Nestor PG, O’Donnell BF, Niznikiewicz MA, Shenton ME, McCarley RW. The startle reflex in schizophrenia: habituation and personality correlates. Schizophr Res. 2003;64(2-3):165–173. doi: 10.1016/s0920-9964(03)00059-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Braff DL, Grillon C, Geyer MA. Gating and habituation of the startle reflex in schizophrenic patients. Arch Gen Psychiatry. 1992;49(3):206–215. doi: 10.1001/archpsyc.1992.01820030038005. [DOI] [PubMed] [Google Scholar]
- 107.Geyer MA, Braff DL. Habituation of the Blink reflex in normals and schizophrenic patients. Psychophysiology. 1982;19(1):1–6. doi: 10.1111/j.1469-8986.1982.tb02589.x. [DOI] [PubMed] [Google Scholar]
- 108.Ludewig K, Geyer MA, Vollenweider FX. Deficits in prepulse inhibition and habituation in never-medicated, first-episode schizophrenia. Biol Psychiatry. 2003;54(2):121–128. doi: 10.1016/s0006-3223(02)01925-x. [DOI] [PubMed] [Google Scholar]
- 109.Freedman R, Adler LE, Myles-Worsley M, Nagamoto HT, Miller C, Kisley M, et al. Inhibitory gating of an evoked response to repeated auditory stimuli in schizophrenic and normal subjects. Human recordings, computer simulation, and an animal model. Arch Gen Psychiatry. 1996;53(12):1114–1121. doi: 10.1001/archpsyc.1996.01830120052009. [DOI] [PubMed] [Google Scholar]
- 110.Braff D, Stone C, Callaway E, Geyer M, Glick I, Bali L. Prestimulus effects on human startle reflex in normals and schizophrenics. Psychophysiology. 1978;15(4):339–343. doi: 10.1111/j.1469-8986.1978.tb01390.x. [DOI] [PubMed] [Google Scholar]
- 111.McGhie A, Chapman J. Disorders of attention and perception in early schizophrenia. Br J Med Psychol. 1961;34:103–116. doi: 10.1111/j.2044-8341.1961.tb00936.x. [DOI] [PubMed] [Google Scholar]
- 112.Bliss TV, Lomo T. Long-lasting potentiation of synaptic transmission in the dentate area of the anaesthetized rabbit following stimulation of the perforant path. J Physiol. 1973;232(2):331–356. doi: 10.1113/jphysiol.1973.sp010273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Hebb DO. The Organization of Behavior. Wiley & Sons; New York: 1949. [Google Scholar]
- 114.Konorski J. Conditioned Reflexes and Neuron Organization. Cambridge University Press; Cambridge: 1948. [Google Scholar]
- 115.Turk-Browne NB, Scholl BJ, Chun MM. Babies and brains: habituation in infant cognition and functional neuroimaging. Front Hum Neurosci. 2008;2:16. doi: 10.3389/neuro.09.016.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Henson RN, Rugg MD. Neural response suppression, haemodynamic repetition effects, and behavioural priming. Neuropsychologia. 2003;41(3):263–270. doi: 10.1016/s0028-3932(02)00159-8. [DOI] [PubMed] [Google Scholar]
- 117.Ranganath C, Rainer G. Neural mechanisms for detecting and remembering novel events. Nat Rev Neurosci. 2003;4(3):193–202. doi: 10.1038/nrn1052. [DOI] [PubMed] [Google Scholar]
- 118.Grill-Spector K, Henson R, Martin A. Repetition and the brain: neural models of stimulus-specific effects. Trends Cogn Sci. 2006;10(1):14–23. doi: 10.1016/j.tics.2005.11.006. [DOI] [PubMed] [Google Scholar]
- 119.Kumaran D, Maguire EA. An unexpected sequence of events: mismatch detection in the human hippocampus. PLoS Biol. 2006;4(12):e424. doi: 10.1371/journal.pbio.0040424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Brown MW, Aggleton JP. Recognition memory: what are the roles of the perirhinal cortex and hippocampus? Nat Rev Neurosci. 2001;2(1):51–61. doi: 10.1038/35049064. [DOI] [PubMed] [Google Scholar]
- 121.Brown MW, Wilson FA, Riches IP. Neuronal evidence that inferomedial temporal cortex is more important than hippocampus in certain processes underlying recognition memory. Brain Res. 1987;409(1):158–162. doi: 10.1016/0006-8993(87)90753-0. [DOI] [PubMed] [Google Scholar]
- 122.Heckers S, Goff D, Weiss AP. Reversed hemispheric asymmetry during simple visual perception in schizophrenia. Psychiatry Res. 2002;116(1-2):25–32. doi: 10.1016/s0925-4927(02)00067-7. [DOI] [PubMed] [Google Scholar]
- 123.Fitzgerald PJ, Barkus C, Feyder M, Wiedholz LM, Chen YC, Karlsson RM, et al. Does gene deletion of AMPA GluA1 phenocopy features of schizoaffective disorder? Neurobiol Dis. 2010;40(3):608–621. doi: 10.1016/j.nbd.2010.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Bannerman DM, Deacon RM, Brady S, Bruce A, Sprengel R, Seeburg PH, et al. A comparison of GluR-A-deficient and wild-type mice on a test battery assessing sensorimotor, affective, and cognitive behaviors. Behav Neurosci. 2004;118(3):643–647. doi: 10.1037/0735-7044.118.3.643. [DOI] [PubMed] [Google Scholar]
- 125.Robinson DL, Venton BJ, Heien ML, Wightman RM. Detecting subsecond dopamine release with fast-scan cyclic voltammetry in vivo. Clin Chem. 2003;49(10):1763–1773. doi: 10.1373/49.10.1763. [DOI] [PubMed] [Google Scholar]
- 126.Totah NK, Kim Y, Moghaddam B. Distinct prestimulus and poststimulus activation of VTA neurons correlates with stimulus detection. J Neurophysiol. 2013;110(1):75–85. doi: 10.1152/jn.00784.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Corlett PR, Krystal JH, Taylor JR, Fletcher PC. Why do delusions persist? Front Hum Neurosci. 2009;3:12. doi: 10.3389/neuro.09.012.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Johnson AW, Bannerman DM, Rawlins NP, Sprengel R, Good MA. Impaired outcome-specific devaluation of instrumental responding in mice with a targeted deletion of the AMPA receptor glutamate receptor 1 subunit. J Neurosci. 2005;25(9):2359–2365. doi: 10.1523/JNEUROSCI.4146-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Johnson AW, Bannerman D, Rawlins N, Sprengel R, Good MA. Targeted deletion of the GluR-1 AMPA receptor in mice dissociates general and outcome-specific influences of appetitive rewards on learning. Behav Neurosci. 2007;121(6):1192–1202. doi: 10.1037/0735-7044.121.6.1192. [DOI] [PubMed] [Google Scholar]
- 130.Schobel SA, Chaudhury NH, Khan UA, Paniagua B, Styner MA, Asllani I, et al. Imaging patients with psychosis and a mouse model establishes a spreading pattern of hippocampal dysfunction and implicates glutamate as a driver. Neuron. 2013;78(1):81–93. doi: 10.1016/j.neuron.2013.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Edwards SL, Beesley J, French JD, Dunning AM. Beyond GWASs: illuminating the dark road from association to function. Am J Hum Genet. 2013;93(5):779–797. doi: 10.1016/j.ajhg.2013.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Maurano MT, Humbert R, Rynes E, Thurman RE, Haugen E, Wang H, et al. Systematic localization of common disease-associated variation in regulatory DNA. Science. 2012;337(6099):1190–1195. doi: 10.1126/science.1222794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Mowry BJ, Gratten J. The emerging spectrum of allelic variation in schizophrenia: current evidence and strategies for the identification and functional characterization of common and rare variants. Mol Psychiatry. 2013;18(1):38–52. doi: 10.1038/mp.2012.34. [DOI] [PubMed] [Google Scholar]
- 134.Law AJ, Lipska BK, Weickert CS, Hyde TM, Straub RE, Hashimoto R, et al. Neuregulin 1 transcripts are differentially expressed in schizophrenia and regulated by 5′ SNPs associated with the disease. Proc Natl Acad Sci U S A. 2006;103(17):6747–6752. doi: 10.1073/pnas.0602002103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Rueckert EH, Barker D, Ruderfer D, Bergen SE, O’Dushlaine C, Luce CJ, et al. Cis-acting regulation of brain-specific ANK3 gene expression by a genetic variant associated with bipolar disorder. Mol Psychiatry. 2013;18(8):922–929. doi: 10.1038/mp.2012.104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Tao R, Cousijn H, Jaffe AE, Burnet PWJ, Edwards F, Eastwood SL, et al. ZNF804A expression in human brain: a novel transcript fetally regulated by the psychosis risk SNP rs1344706, and alterations in schizophrenia, bipolar disorder and major depression. JAMA Psychiatry. doi: 10.1001/jamapsychiatry.2014.1079. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Harrison PJ, Pritchett D, Stumpenhorst K, Betts JF, Nissen W, Schweimer J, et al. Genetic mouse models relevant to schizophrenia: taking stock and looking forward. Neuropharmacology. 2012;62(3):1164–1167. doi: 10.1016/j.neuropharm.2011.08.009. [DOI] [PubMed] [Google Scholar]
- 138.Lin CY, Sawa A, Jaaro-Peled H. Better understanding of mechanisms of schizophrenia and bipolar disorder: from human gene expression profiles to mouse models. Neurobiol Dis. 2012;45(1):48–56. doi: 10.1016/j.nbd.2011.08.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Kvajo M, McKellar H, Gogos JA. Avoiding mouse traps in schizophrenia genetics: lessons and promises from current and emerging mouse models. Neuroscience. 2012;211:136–164. doi: 10.1016/j.neuroscience.2011.07.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Fuchs EC, Zivkovic AR, Cunningham MO, Middleton S, Lebeau FE, Bannerman DM, et al. Recruitment of parvalbumin-positive interneurons determines hippocampal function and associated behavior. Neuron. 2007;53(4):591–604. doi: 10.1016/j.neuron.2007.01.031. [DOI] [PubMed] [Google Scholar]
- 141.Belforte JE, Zsiros V, Sklar ER, Jiang Z, Yu G, Li Y, et al. Postnatal NMDA receptor ablation in corticolimbic interneurons confers schizophrenia-like phenotypes. Nat Neurosci. 2010;13(1):76–83. doi: 10.1038/nn.2447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Carlen M, Meletis K, Siegle JH, Cardin JA, Futai K, Vierling-Claassen D, et al. A critical role for NMDA receptors in parvalbumin interneurons for gamma rhythm induction and behavior. Mol Psychiatry. 2012;17(5):537–548. doi: 10.1038/mp.2011.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Korotkova T, Fuchs EC, Ponomarenko A, von Engelhardt J, Monyer H. NMDA receptor ablation on parvalbumin-positive interneurons impairs hippocampal synchrony, spatial representations, and working memory. Neuron. 2010;68(3):557–569. doi: 10.1016/j.neuron.2010.09.017. [DOI] [PubMed] [Google Scholar]
- 144.Murray AJ, Sauer JF, Riedel G, McClure C, Ansel L, Cheyne L, et al. Parvalbumin-positive CA1 interneurons are required for spatial working but not for reference memory. Nat Neurosci. 2011;14(3):297–299. doi: 10.1038/nn.2751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Caputi A, Fuchs EC, Allen K, Le Magueresse C, Monyer H. Selective reduction of AMPA currents onto hippocampal interneurons impairs network oscillatory activity. PLoS One. 2012;7(6):e37318. doi: 10.1371/journal.pone.0037318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Honey RC, Good M. Associative components of recognition memory. Curr Opin Neurobiol. 2000;10(2):200–204. doi: 10.1016/s0959-4388(00)00069-6. [DOI] [PubMed] [Google Scholar]
- 147.Marshall VJ, McGregor A, Good M, Honey RC. Hippocampal lesions modulate both associative and nonassociative priming. Behav Neurosci. 2004;118(2):377–382. doi: 10.1037/0735-7044.118.2.377. [DOI] [PubMed] [Google Scholar]
- 148.Konradi C, Yang CK, Zimmerman EI, Lohmann KM, Gresch P, Pantazopoulos H, et al. Hippocampal interneurons are abnormal in schizophrenia. Schizophr Res. 2011;131(1-3):165–173. doi: 10.1016/j.schres.2011.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Lewis DA, Curley AA, Glausier JR, Volk DW. Cortical parvalbumin interneurons and cognitive dysfunction in schizophrenia. Trends Neurosci. 2012;35(1):57–67. doi: 10.1016/j.tins.2011.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Marin O. Interneuron dysfunction in psychiatric disorders. Nat Rev Neurosci. 2012;13(2):107–120. doi: 10.1038/nrn3155. [DOI] [PubMed] [Google Scholar]
- 151.Harrison PJ. The hippocampus in schizophrenia: a review of the neuropathological evidence and its pathophysiological implications. Psychopharmacology. 2004;174(1):151–162. doi: 10.1007/s00213-003-1761-y. [DOI] [PubMed] [Google Scholar]
- 152.Tamminga CA, Stan AD, Wagner AD. The hippocampal formation in schizophrenia. Am J Psychiatry. 2010;167(10):1178–1193. doi: 10.1176/appi.ajp.2010.09081187. [DOI] [PubMed] [Google Scholar]
- 153.Stone JM, Howes OD, Egerton A, Kambeitz J, Allen P, Lythgoe DJ, et al. Altered relationship between hippocampal glutamate levels and striatal dopamine function in subjects at ultra high risk of psychosis. Biol Psychiatry. 2010;68(7):599–602. doi: 10.1016/j.biopsych.2010.05.034. [DOI] [PubMed] [Google Scholar]
- 154.Bannerman DM, Niewoehner B, Lyon L, Romberg C, Schmitt WB, Taylor A, et al. NMDA receptor subunit NR2A is required for rapidly acquired spatial working memory but not incremental spatial reference memory. J Neurosci. 2008;28(14):3623–3630. doi: 10.1523/JNEUROSCI.3639-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Stephan KE, Friston KJ, Frith CD. Dysconnection in schizophrenia: from abnormal synaptic plasticity to failures of self-monitoring. Schizophr Bull. 2009;35(3):509–527. doi: 10.1093/schbul/sbn176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Frost DO, Tamminga CA, Medoff DR, Caviness V, Innocenti G, Carpenter WT. Neuroplasticity and schizophrenia. Biol Psychiatry. 2004;56(8):540–543. doi: 10.1016/j.biopsych.2004.01.020. [DOI] [PubMed] [Google Scholar]
- 157.Daskalakis ZJ, Christensen BK, Fitzgerald PB, Chen R. Dysfunctional neural plasticity in patients with schizophrenia. Arch Gen Psychiatry. 2008;65(4):378–385. doi: 10.1001/archpsyc.65.4.378. [DOI] [PubMed] [Google Scholar]
- 158.Lewis DA, Gonzalez-Burgos G. Neuroplasticity of neocortical circuits in schizophrenia. Neuropsychopharmacology. 2008;33(1):141–165. doi: 10.1038/sj.npp.1301563. [DOI] [PubMed] [Google Scholar]
- 159.Nestler EJ, Hyman SE. Animal models of neuropsychiatric disorders. Nat Neurosci. 2010;13(10):1161–1169. doi: 10.1038/nn.2647. [DOI] [PMC free article] [PubMed] [Google Scholar]