

Summary
Background
The epilepsies are a clinically heterogeneous group of neurological disorders. Despite strong evidence for heritability, genome-wide association studies have had little success in identification of risk loci associated with epilepsy, probably because of relatively small sample sizes and insufficient power. We aimed to identify risk loci through meta-analyses of genome-wide association studies for all epilepsy and the two largest clinical subtypes (genetic generalised epilepsy and focal epilepsy).
Methods
We combined genome-wide association data from 12 cohorts of individuals with epilepsy and controls from population-based datasets. Controls were ethnically matched with cases. We phenotyped individuals with epilepsy into categories of genetic generalised epilepsy, focal epilepsy, or unclassified epilepsy. After standardised filtering for quality control and imputation to account for different genotyping platforms across sites, investigators at each site conducted a linear mixed-model association analysis for each dataset. Combining summary statistics, we conducted fixed-effects meta-analyses of all epilepsy, focal epilepsy, and genetic generalised epilepsy. We set the genome-wide significance threshold at p<1·66 × 10−8.
Findings
We included 8696 cases and 26 157 controls in our analysis. Meta-analysis of the all-epilepsy cohort identified loci at 2q24.3 (p=8·71 × 10−10), implicating SCN1A, and at 4p15.1 (p=5·44 × 10−9), harbouring PCDH7, which encodes a protocadherin molecule not previously implicated in epilepsy. For the cohort of genetic generalised epilepsy, we noted a single signal at 2p16.1 (p=9·99 × 10−9), implicating VRK2 or FANCL. No single nucleotide polymorphism achieved genome-wide significance for focal epilepsy.
Interpretation
This meta-analysis describes a new locus not previously implicated in epilepsy and provides further evidence about the genetic architecture of these disorders, with the ultimate aim of assisting in disease classification and prognosis. The data suggest that specific loci can act pleiotropically raising risk for epilepsy broadly, or can have effects limited to a specific epilepsy subtype. Future genetic analyses might benefit from both lumping (ie, grouping of epilepsy types together) or splitting (ie, analysis of specific clinical subtypes).
Funding
International League Against Epilepsy and multiple governmental and philanthropic agencies.
Introduction
Epilepsy is a common disorder, affecting up to 4% of people at some time in life.1 The disorder includes a group of heterogeneous syndromes defined by clinical, electroencephalographic (EEG), and brain imaging criteria.2 Broadly, the epilepsies are divided clinically into generalised and focal forms. Genetic factors contribute to both, as shown by findings from familial aggregation and twin studies.3 Causative mutations in many genes, including some genes coding for ion channel subunits and others involved in synaptic function or brain development, have been reported.3, 4 Most of these findings were reported in patients with fairly rare familial epilepsies segregating in a Mendelian way or epilepsies arising from de-novo mutations (particularly in patients with severe infantile epilepsies).5, 6, 7
The genetic determinants underlying the common epilepsies, for which clinical genetic data suggest complex inheritance, remain largely unknown. Some evidence suggests a role for rare sequence and copy number variants,8, 9, 10 whereas the contribution of common polymorphisms is still unclear,11, 12 partly as a result of the relatively small sample sizes analysed to date.
Findings from the largest genome-wide association study (GWAS) in epilepsy so far, including 3445 patients with focal epilepsy,13 showed no variants of genome-wide significance. More recently, findings from a study of 1018 patients with mesial temporal lobe epilepsy with hippocampal sclerosis (a subtype of focal epilepsy) implicated the 2q24.3 region around the gene encoding the sodium channel SCN1A,14 and findings from an independent study of Han Chinese patients with known or suspected lesional focal epilepsy showed evidence for a risk allele at 1q32 on the basis of a discovery sample of 504 cases.15
For generalised epilepsy, a GWAS included 1527 European patients with genetic generalised epilepsies in the discovery analysis and 1493 patients in the replication cohort; investigators reported evidence for common risk alleles at 2p16.1 and 17q21.32, and suggestive evidence at the SCN1A locus.16 Additionally, associations were reported for the juvenile myoclonic subtype of genetic generalised epilepsy at 1q43 and for absence epilepsy at 2q22.3.16
In a large multicentre collaboration, we undertook a meta-analysis to detect variants that could increase risk for common epilepsies. In view of clinical evidence that some genetic factors might increase risk for epilepsy broadly and in a syndrome-specific manner,17, 18, 19 we prespecified three analyses as part of the study. Variants were sought that affected risk for all epilepsies, genetic generalised epilepsy (previously known as idiopathic generalised epilepsy),2, 20 or focal epilepsy.
Methods
Study design and participants
We did a meta-analysis of data from 12 previously published or unpublished genetic cohort studies from EPICURE,16 EPIGEN,13 Philadelphia (PA, USA), the Imperial-Liverpool-Melbourne Collaboration,21 GenEpa,13 and Hong Kong (China)15 (appendix). We identified these studies from the scientific literature (through searches of PubMed in December, 2011, with the terms “epilepsy”, “seizures”, and “association studies”), through publicity via Chapters of the International League Against Epilepsy, and during international conferences. All participants in these 12 case cohorts (and their associated controls) were of European, Asian, or African ancestry (table 1, appendix).
Table 1.
Cases and controls, by index GWAS
| Ethnic origin* | All epilepsy (n=8696) | Genetic generalised epilepsy (n=2606) | Focal epilepsy (n=5310) | Population controls‡(n=26 157) | |
|---|---|---|---|---|---|
| EPIGEN-Dublin | Irish | 638 | .. | 520 | 2232 |
| EPIGEN-Brussels | Belgian | 505 | 48 | 406 | 1675 |
| EPIGEN-Duke† | African-American and European-American | 760 | 102 | 551 | 504 |
| EPIGEN-London | British and other | 1007 | 93 | 773 | 2494 |
| ILM Collaboration | European descent | 1703 | 212 | 1263 | 2699 |
| GenEpa | Finnish | 422 | .. | 422 | 1963 |
| EPICURE | Northwest European | 1440 | 1440 | .. | 2454 |
| Philadelphia_550_AA† | African-American | 324 | 81 | 222 | 2746 |
| Philadelphia_550_CAU | European-American | 819 | 440 | 378 | 5736 |
| Philadelphia_Omni_AA§ | African-American | 106 | .. | .. | 97 |
| Philadelphia_Omni_CAU | European-American | 485 | 190 | 288 | 682 |
| Hong Kong | Asian-Han | 487 | .. | 487 | 2875 |
Numbers of cases and controls are after quality control filtering. GWAS=genome-wide association study. ILM=Imperial-Liverpool-Melbourne.
Broad ethnic origin of the cohort. Other indicates people of mixed ethnic origin, as would be expected in a cosmopolitan population. European descent refers to white European.
EPIGEN-Duke individuals of African-American ancestry were merged with participants in the Philadelphia_550_AA cohort.
See appendix for further details about control cohorts.
Small sample size prohibited epilepsy subtype analysis in this cohort.
The genetic cohort studies used a combination of population-based datasets as controls. These control cohorts were either screened or unscreened by questionnaire for neurological disorders (table 1, appendix).
All study participants provided written informed consent for DNA analysis. Local institutional review boards reviewed and approved study protocols at each site.
Procedures
We classified seizures and epilepsy syndromes according to the International League Against Epilepsy terminology.2, 20 For all cases, epilepsy specialists assessed phenotype at the source centre. Patients with epilepsy were assigned to one of three phenotypic categories: genetic generalised epilepsy, focal epilepsy, or unclassified epilepsy.
Criteria for genetic generalised epilepsy were tonic-clonic, absence, or myoclonic seizures with generalised spike–wave discharges on EEG and no evidence of an acquired cause. In rare instances the criterion for a diagnostic EEG was waived when clear clinical evidence suggested myoclonic or absence seizures with tonic-clonic seizures, and no evidence for an acquired cause. The International League Against Epilepsy has adopted the term genetic generalised epilepsy for syndromes previously known as idiopathic or primary generalised epilepsies, in view of strong evidence for a genetic basis from genetic epidemiological and twin studies and an absence of identified acquired factors.2, 20
In the phenotypic category of focal epilepsy, we included patients with a confirmed diagnosis of focal epilepsy, including cases with focal structural brain lesions. These cases were predominantly adults, and as such, cases of benign epilepsy of childhood with centro-temporal spikes were not specifically included.
Unclassified epilepsy consisted of patients in whom there was neither electroclinical evidence for generalised epilepsy nor evidence for a focal seizure onset. Additionally, cases with evidence for both generalised and focal epilepsy were included here.
The phenotyping committee curated patient phenotypes into a single database. Details relating to individual case cohorts are provided in the appendix. Analyses were done for three phenotypic groups: genetic generalised epilepsy, focal epilepsy, and all epilepsy (consisting of all patients with a confirmed diagnosis of epilepsy, including genetic generalised epilepsy, focal epilepsy, and unclassified epilepsy).
Statistical analysis
We used prespecified criteria for quality control to filter cases and controls from the 12 cohorts (appendix). Because contributing sites had used different genotyping platforms, we did imputation to infer genotypes for common genetic variants that were not directly genotyped, allowing us to combine results across sites. Each of the five sites imputed their study datasets according to a standardised protocol. This protocol used IMPUTE2 to infer and impute haplotypes, with the 1000 Genomes Phase I (interim) June, 2011, reference panel (appendix).
Investigators at each site did a linear mixed-model association analysis for each of their datasets with FaSTLMM (version 1.09).22 This analysis uses linear regression, including a polygenic term designed to account for the contributions of population stratification and causal variants aside from the one being tested. Although we were assessing a binary trait, we used linear regression (rather than logistic regression) because we expected effect sizes to be small. We did this analysis separately for each of the preselected phenotypic categories of epilepsy (all epilepsy, genetic generalised epilepsy, and focal epilepsy). Sex was included as a covariate.
We did a fixed-effects meta-analysis with METAL (version generic-metal-2011-03-25).23 Because almost all epilepsy cases were of European descent (table 1), we chose a fixed-effects model to optimise power. Single nucleotide polymorphisms showing significant amounts of heterogeneity (p<0·05) were removed before application of the fixed-effects analysis. We applied genomic correction to the association analysis results for each dataset before combining for meta-analysis. These steps were done separately for each of the three phenotypic tests.
We set our genome-wide threshold for statistical significance at 1·66 × 10−8, representing an empirical Bonferroni correction of the 5 × 10−8 genome-wide significance threshold for three tests. We regarded signals with p values between 1·66 × 10−8 and 5 × 10−7 as suggestive evidence of association.
We calculated the proportion of phenotypic variance a variant must explain (heritability) for the detection power to be at least 80%. We used variance explained on the liability scale,24 for which we assumed a point prevalence of 0·5% for all epilepsy, 0·2% for genetic generalised epilepsy, and 0·3% for focal epilepsy.25 The required heritability was 0·07% or greater for all epilepsy, 0·17% or greater for genetic generalised epilepsy, and 0·10% or greater for focal epilepsy (appendix).
In addition to the main association analysis, we did logistic regression for variants in a 1 megabase window centred on each variant that showed suggestive evidence of association (p<5 × 10−7) from any of the three meta-analyses (all epilepsy, genetic generalised epilepsy, or focal epilepsy). The purpose of this analysis was technical validation and to estimate odds ratios (ORs). We analysed the dosage data, including sex and the first 20 principal components, with PLINK (version 1.07),26 and then combined the results from each site again with a fixed-effect meta-analysis.
Conditional analysis was done with FaSTLMM (version 2.0) on variants in the same regions as those defined for the logistic regression. The purpose of the conditional analysis was to establish whether any other genetic variants in the region were associated with the disease phenotype, independent of the strongest signal from that region. We conditioned on the most significant variants within each of the three regions. Sex was included as a covariate in the conditional analysis. We applied Bonferroni correction to control for multiple testing in the conditional analysis and set the threshold for significance at 5 × 10−6 (each 1 megabase region contained approximately 10 000 single nucleotide polymorphisms).
To assess the accuracy of the imputation across regions showing signals satisfying genome-wide significance, we did genotyping in a subset of patients included in the meta-analysis and compared hard genotypes with imputation dosage files. We selected a subset of individuals to represent each of the three broad ethnic origins included in our analysis (ie, European ancestry, African-American, and Asian). Genotyping was done with TaqMan (Life Technologies, Carlsbad, CA, USA) for rs28498976, Sanger sequencing for rs6732655, and Kasper KASP (LGC Genomics, Hoddesdon, Hertfordshire, UK) for rs2947349 (appendix), because differences in sequence context required specific genotyping platforms for each single nucleotide polymorphism.
We did enrichment analysis with the interval-based enrichment analysis tool as integrated in the package INRICH (version 1.0).27 Briefly, INRICH takes a set of independent, nominally associated genomic intervals and tests for enrichment of predefined gene sets with permutation. We analysed variants with p values less than 1 × 10−5 and defined the interval around index single nucleotide polymorphisms with an r2 threshold of 0·2. Gene sets as defined by gene ontology pathways were tested for enrichment.
Role of the funding source
The funders had no role in the study design, data collection, data analysis, data interpretation, or writing of the report. The members of the strategy and analysis committees of the International League Against Epilepsy Consortium on Complex Epilepsies had full access to all data in the study. The strategy committee (appendix) of the Consortium takes final responsibility for the decision to submit for publication.
Results
40 789 participants, comprising 10 064 people with epilepsy from 12 cohorts and 30 725 controls, were studied. After application of our quality control criteria (appendix), we included a total of 34 853 individuals (8696 with epilepsy and 26 157 controls) in the meta-analysis for all epilepsies (table 1).
Principal component analysis suggested that the cohorts clustered in three broad ethnic origins (European, Asian, and admixed African-American), as expected (appendix). We noted an inflation factor of 1·031, suggesting adequate control for possible cryptic stratification (appendix).
In the all-epilepsy analysis, we identified two loci with genome-wide significance (p<1·66 × 10−8; figure 1). The first signal was located at 2q24.3 (figure 2). This signal was centred on the voltage-gated sodium channel gene SCN1A, which is a known gene associated with some monogenic epilepsies.7, 28, 29 The most strongly associated variant in this interval was rs6732655 (p=8·71 × 10−10, OR 0·89, 95% CI 0·86–0·93; table 2, appendix), located in intron 16 of SCN1A. Seventy other variants in this region satisfied the threshold for genome-wide significance. Logistic regression validated the association with 2q24.3 (appendix). The direction of effect was consistent across most cohorts, and there was no evidence of substantial heterogeneity.
Figure 1.
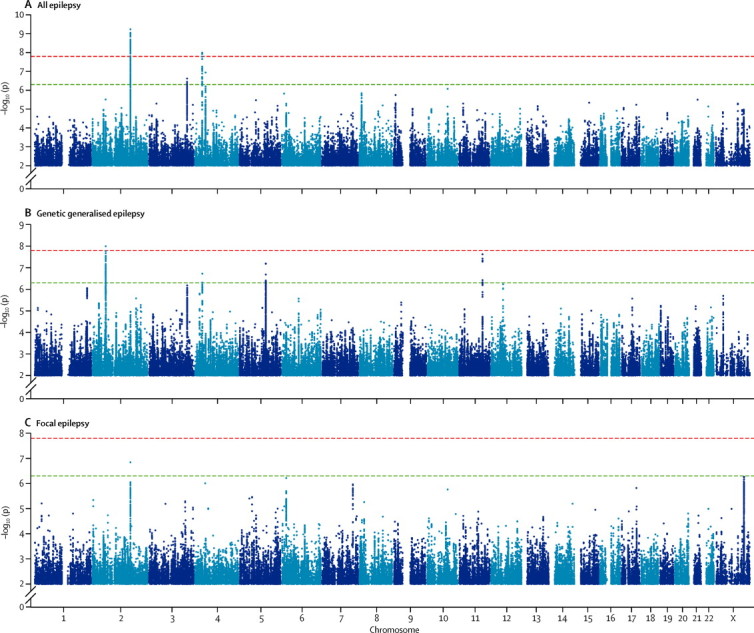
Manhattan plots for meta-analyses of all epilepsy (A), genetic generalised epilepsy (B), and focal epilepsy (C)
The red line shows our threshold of significance set at p=1·66 × 10−8, and the green line shows the suggestive threshold of p=5 × 10−7. Y axis is broken in all graphs.
Figure 2.
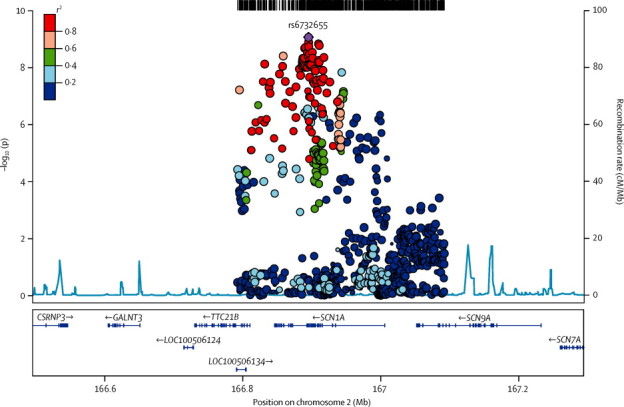
Genomic context of 2q24.3 signal from all-epilepsy analysis
Plot created with LocusZoom (version 1.1). Linkage disequilibrium data taken from the 1000 Genomes Project, HG19, March, 2012.
Table 2.
Genome-wide associated loci at p<5·0 × 10−7
| Cytogenetic band | Base pair position | Allele 1, allele 2 | Minor allele frequency | Candidate gene | Annotation | Phenotype | OR (95% CI) | pLMM | pcond | |
|---|---|---|---|---|---|---|---|---|---|---|
| rs6732655 | 2q24.3 | 166895066 | T*, A | 0·22 (A) | SCN1A | Intronic | All epilepsy | 0·89 (0·86–0·93) | 8·71 × 10−10 | 4·95 × 10−7 |
| rs28498976 | 4p15.1 | 31151357 | A, G* | 0·46 (A) | PCDH7 | Intergenic | All epilepsy | 0·90 (0·87–0·94) | 5·44 × 10−9 | 2·29 × 10−4 |
| rs111577701 | 3q26.2 | 167861408 | T, C* | 0·09 (T) | GOLIM4 | Intergenic | All epilepsy | 1·16 (1·09–1·24) | 4·42 × 10−7 | .. |
| rs535066 | 4p12 | 46240287 | T, G* | 0·40 (G) | GABRA2 | Intergenic | All epilepsy | 1·10 (1·05–1·16) | 1·71 × 10−7 | .. |
| rs2947349 | 2p16.1 | 58059803 | A*, C | 0·26 (C) | VRK2/FANCL | Intergenic | GGE | 1·23 (1·16–1·31) | 9·99 × 10−9 | 1 × 10−4 |
| rs1939012 | 11q22.2 | 102595135 | C, T* | 0·40 (T) | MMP8 | Intronic | GGE | 1·12 (1·07–1·17) | 2·37 × 10−8 | .. |
| rs1044352 | 4p15.1 | 31147874 | T*, G | 0·50 (T) | PCDH7 | Synonymous | GGE | 0·88 (0·82–0·93) | 1·87 × 10−7 | .. |
| rs55670112 | 5q22.3 | 114268470 | A, C* | 0·47 (C) | None | Intergenic | GGE | 1·18 (1·1–1·26) | 6·34 × 10−8 | .. |
| rs12987787 | 2q24.3 | 166858391 | C, T* | 0·21 (C) | SCN1A | Intronic | Focal epilepsy | 1·12 (1·01–1·14) | 1·45 × 10−7 | .. |
Base pair position refers to build 37 (hg19). Minor allele frequency is from all poulations from the 1000 Genomes Project. Candidate gene refers to the most plausible candidate gene attributable to the signal. OR corresponds to allele 2, computed from logistic regression. Annotation refers to type of SNP. pLMM refers to p value from linear mixed-model meta-analysis. pcond refers to p value when conditioning on this specific SNP to determine independent signals from same locus. OR=odds ratio. GGE=genetic generalised epilepsy. SNP=single nucleotide polymorphism.
Ancestral or chimpanzee allele.
In view of the extent of linkage disequilibrium between the variants associated with all epilepsy in the 2q24.3 region (figure 2), we did logistic regression conditioned on the most significant variant identified from the univariate analysis (rs6732655). Our results suggested a tentative independent signal, coming from rs13406236, in an intronic variant in SCN9A (p=1·39 × 10−4 on conditioning; appendix). We did not identify any further significant signals.
A second signal for the all-epilepsy phenotype was located at 4p15.1 and included the 3′ end of the protocadherin gene, PCDH7 (figure 3). The most strongly associated variant in this region was rs28498976 (p=5·44 × 10−9, OR 0·90, 95% CI 0·87–0·94; table 2), located 2·5 kilobases from the 3′ end of PCDH7. Logistic regression across PCDH7 supported the association with this locus (appendix). We noted no additional significant signals from 4p15.1 on conditioning for rs28498976 (appendix). The direction of effect was consistent across all cohorts and we noted no evidence of heterogeneity. Although achieving genome-wide significance only for the all-epilepsy phenotype, the PCDH7 signal seemed stronger in genetic generalised epilepsy than in focal epilepsy (appendix).
Figure 3.
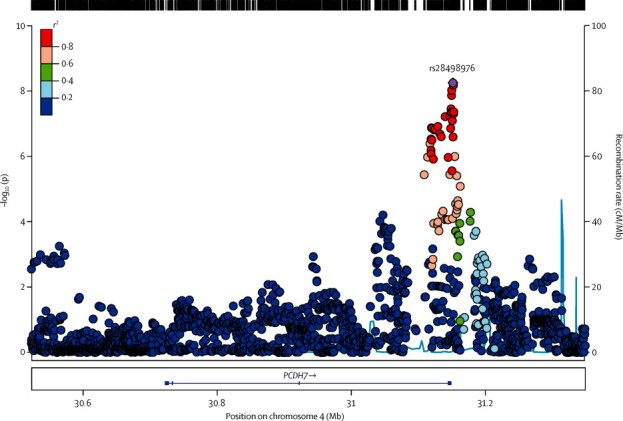
Genomic context of 4p15.1 signal from all-epilepsy analysis
Plot created with LocusZoom (version 1.1). Linkage disequilibrium data taken from the 1000 Genomes Project, HG19, March 2012.
PCDH7 encodes a calcium-dependent adhesion protein, not previously associated with epilepsy. It is a member of the cadherin gene family. The gene is expressed in the CNS, specifically in thalamocortical circuits and the hippocampus,30, 31 and expression of PCDH7 is controlled by MECP2,32 mutations in which cause Rett syndrome. The cytoplasmic domain of the PCDH7 protein binds to protein phosphatase 1α (PPP1CA), which is enriched in dendritic spines and is important in learning and memory,33 and to template activation factor 1 (TAF1), which along with PCDH7 is involved in neurite extension.34, 35
Suggestive signals of note (p<5 × 10−7) for the all-epilepsy phenotype were detected at 3q26.2 (p=4·42 × 10−7) and 4p12 (p=1·71 × 10−7; table 2). The 3q26.2 region contained the 5′ end of GOLIM4 (appendix). This gene encodes Golgi internal membrane protein 4, which is degraded when manganese increases above normal concentrations, suggesting a role for this protein in manganese homoeostasis.36 Almost all brain manganese is bound to glutamine synthetase, an enzyme playing a key part in production or degradation of the neurotransmitters glutamate, glutamine, and GABA. Decreased brain glutamine synthetase and manganese concentrations have been reported in epilepsy.37, 38 The 4p12 region contained the 3′ end of the GABA receptor, α2-subunit gene (GABRA2). Mutations in other GABA receptors have been reported to cause epilepsy.39
After quality control, we included 21 596 individuals (2606 cases and 18 990 controls) across eight cohorts in the meta-analysis of genetic generalised epilepsy (table 1), a subset of those included in the all-epilepsy analysis. Results from the genetic generalised epilepsy meta-analysis suggested an inflation factor of 1·05 (appendix).
A single signal achieved the threshold of genome-wide significance (figure 1). Located at 2p16.1, the interval contained genes encoding vaccinia-related kinase 2 (VRK2) and Fanconi anaemia, complementation group L (FANCL; figure 4). The most strongly associated variant in this region was the intergenic variant rs2947349 (p=9·99 × 10−9, OR 1·23, 95% CI 1·16–1·31; table 2). Logistic regression analysis supported the association with 2p16.1 (appendix). We noted no additional significant signals from 2p16.1 on conditioning for rs2947349 (appendix). The direction of effect was consistent across all cohorts, and the association seemed to be specific to genetic generalised epilepsy (appendix).
Figure 4.
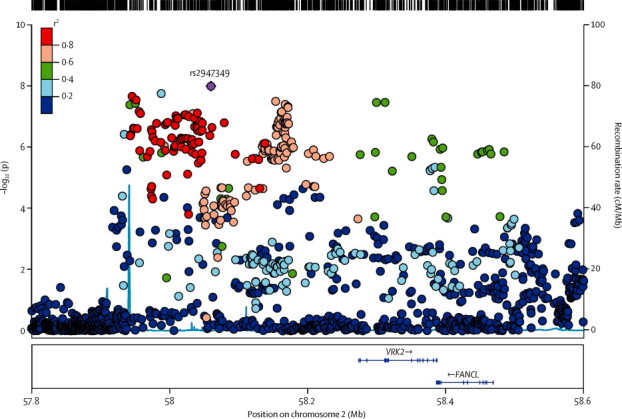
Genomic context of 2p16.1 signal from analysis of genetic generalised epilepsy
Plot created with LocusZoom (version 1.1). Linkage disequilibrium data taken from the 1000 Genomes Project, HG19, March 2012.
VRK2 is a serine-threonine protein kinase involved in signal transduction and apoptosis.40, 41 Variation in VRK2 has previously been suggested as a risk factor for epilepsy16 and schizophrenia.42, 43, 44 Indeed, the schizophrenia-associated risk variant (rs2312147)43 shows also a strong signal for genetic generalised epilepsy (p=2·3 × 10−6, OR 1·22, 95% CI 1·14–1·30) and is in high linkage disequilibrium with the strongest variant for genetic generalised epilepsy (r2=0·82), although the direction of the effect is opposite (ie, the protective variant for epilepsy raises risk for schizophrenia). The EPICURE cohort, in which 2p16.1 was originally proposed as a risk factor for genetic generalised epilepsy, was included in our meta-analysis. After exclusion of the EPICURE cohort, the top single nucleotide polymorphism from their study (rs13026414)16 remained nominally significant at p=7 × 10−3 here. These results provide further support to the suggestion that VRK2 is a risk locus for both epilepsy and schizophrenia. The other gene in the region, FANCL, codes for a RING-type E3 ubiquitin ligase of the Fanconi anaemia pathway. FANCL mono-ubiquitinates FANCD2 and FANCI, proteins involved in DNA repair and homologous recombination.45 FANCL has not been previously implicated in epilepsy or any seizure-related phenotype.
We detected suggestive evidence for association with genetic generalised epilepsy at 4p15.1 (p=1·87 × 10−7), 5q22.3 (p=6·34 × 10−8), and 11q22.2 (p=2·37 × 10−8; table 2). The 4p15.1 PCDH7 signal was the same as that with genome-wide significance for the all-epilepsy phenotype (figure 3, appendix). The 5q22.3 signal was intergenic (appendix). The 11q22.2 signal contained the 5′ end of the matrix metallopeptidase gene MMP8 (appendix). The direction of effect was consistent across all cohorts and seemed specific to genetic generalised epilepsy (appendix). With a p value of 2·37 × 10−8, the 11q22.2 signal reached the conventional threshold for genome-wide significance (p<5 × 10−8), but not our more stringent value (p<1·66 × 10−8). Matrix metallopeptidases are zinc-dependent endopeptidases involved in the breakdown of the extracellular matrix in physiological processes and in blood–brain inflammation.46 Increased expression of MMPs has been recorded in various neurological disease states,47 and epileptogenesis is decreased in MMP9 knockout mice but increased in transgenic rats overexpressing MMP9.48
After quality control, we included 28 916 individuals (5310 cases and 23 606 controls) from ten cohorts in our meta-analysis of focal epilepsy. No signal achieved genome-wide significance. Results from the focal meta-analysis suggested an inflation factor of 1·014 (appendix). We observed one notable subthreshold signal (rs12987787, p=1·45 × 10−7) from 2q24.3, the region containing SCN1A (appendix).
Targeted genotyping of the three GWAS-significant signals supported the accuracy of imputation, with a minimum correlation of 0·98 noted between experimentally determined and imputed genotypes (appendix).
Assessment of enrichment of gene ontology terms for regions containing variants with nominally significant p values (p<1 × 10−5) for each of the three phenotypes showed enrichment in several signalling pathways (appendix). Although none of these variants remained significant after correction for multiple testing, our results suggest pathways with biological plausibility.
Finally, we investigated whether any of the four susceptibility loci at nominal genome-wide significance (p<5 × 10−8) were associated with outcome of newly treated epilepsy with use of data from Speed and colleagues.21 We used both the index single nucleotide polymorphism (table 2) and single nucleotide polymorphisms within a 20 kilobase window around each of the five genes (SCN1A, PCDH7, VRK2/FANCL, and MMP8; appendix). The minimum p value of association with outcome of newly treated epilepsy for any susceptibility locus was 8·14 × 10−4 (MMP8). We noted no evidence for an association between SCN1A (the gene that codes for the target of sodium-channel-blocking class antiepileptic drugs) and epilepsy outcome.
Discussion
In this genome-wide association meta-analysis of epilepsy and its most common subtypes, we identified three loci with genome-wide significance, and our findings suggest that some loci might be specifically associated with an epilepsy type.
In the whole cohort consisting of all epilepsy, the region of the sodium channel subunit gene SCN1A was clearly associated with the disease. This gene is a well-established cause of genetic epilepsy with febrile seizures plus (GEFS+),28, 29 a generally mild, familial form of epilepsy, and with Dravet syndrome, a severe epileptic encephalopathy usually arising from de-novo mutations.7 SCN1A was associated with mesial temporal lobe epilepsy and hippocampal sclerosis with febrile seizures in a recent GWAS14 and in a meta-analysis of SCN1A rs3812718.49 SCN1A mutations have also been reported in a range of paroxysmal neurological disorders including familial hemiplegic migraine50 and, more rarely, in some focal epilepsies.51 Whether this robust association with all epilepsy is a true common variant association or a synthetic association due to tagged rare variants in cases with GEFS+ is therefore not clear. Although the cohorts might have included individuals from monogenic GEFS+ families with SCN1A mutations of large effect, review of the phenotyping data suggested that inclusion of more than a few such cases was unlikely; moreover, SCN1A variants have been reported only in about 10% of large GEFS+ families.52
Our all-epilepsy analysis identified a second locus (4p15.1) that satisfied our threshold for genome-wide significance. This locus is newly associated with epilepsy and implicates the gene PCDH7. This protocadherin gene is a plausible candidate for common forms of epilepsy, as mutations in another protocadherin gene, PCDH19, cause epilepsy and mental retardation in female patients.53
For the specific category of genetic generalised epilepsy, we noted the association at 2p16.1 that was previously reported in the EPICURE cohort;16 this cohort provided about half of our sample for the meta-analysis of this subtype (table 1). The association maintained nominal significance after removal of EPICURE cases for this locus, where the genes VRK2 and FANCL are within close proximity. With our additional samples, we did not note significance for the 17q21 locus reported by EPICURE investigators for genetic generalised epilepsy (appendix).
For the subcategory of focal epilepsy, we did not note any locus with genome-wide significance, consistent with negative findings from the EPIGEN study of focal epilepsy (samples from which were included in our analysis).13 However, a signal at 2q24.3 (containing SCN1A) in focal epilepsy approached but did not achieve significance (appendix). This signal in focal epilepsy was in high linkage disequilibrium with that noted for all epilepsy (r2=0·85). Importantly, the 2q24.3 signal for focal epilepsy that we recorded differed to that reported in a recent study of the narrow focal epilepsy phenotype of mesial temporal lobe epilepsy and hippocampal sclerosis with febrile seizures.14 rs7587026 (the previously reported variant) was not significant in our analysis of a broader focal epilepsy phenotype consisting of all focal epilepsies (p=0·01; appendix). We also did not note the association at 1q32.1 (implicating CAMSAP1L1) that was previously reported in the Hong Kong cohort,15 which was included in our sample (appendix). Most patients in this cohort had focal epilepsy due to known lesions.
Consistent with experience of GWAS in other neuropsychiatric disorders, and common disorders in general, this study reinforces the value of large sample sizes. In the epilepsies, electroclinical and imaging data allow the identification of clinical syndromes that share common clinical features. Our study findings suggest that an experimental design that includes fractionation of samples into clinical subtypes can reveal syndrome-specific risk alleles, but the identification of these alleles will be assisted by the collection and genotyping of larger sample sizes. Although this lumping versus splitting debate in genetic analyses is not unique to the epilepsies, there has been long-standing controversy about it in clinical epileptology,54 which genetics will help to inform.
Limitations of our study include sample size; although ours is large, even larger samples have yielded more findings in other disorders.55, 56, 57 Larger samples would enable further analysis of epilepsy subtypes, and the International League Against Epilepsy Consortium on Complex Epilepsies now provides a useful vehicle for future efforts. Second, our meta-analysis relied on genotypes generated separately on various platforms, an issue common to most meta-analyses. Third, extension of the phenotyping data to include treatment outcome would be ideal, but in a cross-sectional cohort this approach has methodological difficulties. Finally, we did not have an independent replication sample. However, stringent criteria for statistical significance were set a priori, and for loci achieving our threshold of genome-wide significance the direction of effects were largely consistent across the cohorts, and extended over multiple variants in high linkage disequilibrium.
Taken together, these data show that, with sufficient sample size, susceptibility loci for common epilepsies can be identified through the analysis of common variation. The role of rare variants of large effect is also well established, particularly in rarer Mendelian epilepsies.3, 4, 5, 6, 7 The role of rare variants in the common epilepsies is at present under exploration by deep-sequencing approaches.11, 58, 59 A dual approach of identification of both rare and common variation will result in improved understanding of the genetic architecture for the overall population of people with epilepsy, necessary for precision medicine. Although our findings will not be of immediate clinical usefulness, they are an important first step to understand the genetic architecture of the epilepsies, which could lead to clinically relevant markers of prognosis and outcome.
Acknowledgments
Acknowledgments
We are grateful to the patients and volunteers who participated in this research. We thank the following clinicians and research scientists for their contribution through sample collection (cases and controls), data analysis, and project support: Wim Van Paesschen, Benjamin Legros, Patrick Tugendhaft, Kevin Shianna, Edouard Louis, Michel Georges, William Gallentine, Aatif Husain, Mohamad Mikati, Saurabh Sinha, Raju Yerra, Chris French, Zelko Matkovic, Steven J Howell, Paul Cooper, Mark Kellett, Brendan McLean, Marcus Reuber, Peter Cleland, Kathleen White, Peter Goulding, Richard E Appleton, Mark Lawden, Basil Sharrack, Guiliano Avanzini, Ditte B Kjelgaard, Oebele Brouwer, Floor Jansen, Kees Braun, Hans Carpay, Willem Frans Arts, Paul Boon, Lenora Lehwald, Jorge Vidaurre, Pedro Weisleder, Chang-Yong Tsao, Annie Kung Wai-Chee, Monica Islam, Emily de los Reyes, Jennifer McKinney, Laurel Slaughter, Bethanie Morgan-Followell, Lori Hamiwka, Deborah Terry, Molly Taylor, Sally Steward, Mary Karn, Jo Ellen Lee, Donna Kring, Sarah Borror, Karen Carter, Cathy Schumer, Guy Rouleau, Micheline Gravel, Virginia Wong, Colin H T Lui, Sin Ngai Chuen, Tak-Hong Tsoi, Rhian Gwilliam, and all contributing clinicians from the Department of Clinical and Experimental Epilepsy at the National Hospital for Neurology and Neurosurgery, UCL Institute of Neurology, and the Wellcome Trust Case Control Consortium (WTCCC). The International League Against Epilepsy facilitated the Consortium through the Commission on Genetics and by financial support; however, the opinions expressed in the paper are not necessarily those of the International League Against Epilepsy.
Funding
This work was in part supported by an award (2009/001) from Brainwave—the Irish Epilepsy Association; by the Medical Research Charities Group of Ireland, the Health Research Board and by a Translational Research Scholars award from the Health Research Board of Ireland (CW). Further funding sources include the Wellcome Trust (grant 084730), NIHR (08-08-SCC), GIHE (NIH R01-NS-49306-01; RJB), GSCFE (NIH R01NS064154-01; RJB and HH), NIH (UL1TR001070), Development Fund from The Children's Hospital of Philadelphia (HH), NHMRC Program Grant (ID 628952; SFB, IES, KLO); The Royal Melbourne Hospital Foundation Lottery Grant (SP), The RMH Neuroscience Foundation (TJO'B), European Union's Seventh Framework Programme (FP7/2007-2013) under grant agreement number 279062 and 602102, Department of Health's NIHR Biomedical Research Centers funding scheme, European Community (EC, FP6 project; EPICURE, LSHM-CT-2006-037315); German Research Foundation (DFG, SA434/4-1/4-2), EuroEPINOMICS Consortium (European Science Foundation/DFG: SA434/5-1, NU50/8-1, LE1030/11-1, HE5415/3-1); the German Federal Ministry of Education and Research, National Genome Research Network (NGFNplus/EMINet: 01GS08120, and 01GS08123; IntenC, TUR 09/I10); The Netherlands National Epilepsy Fund (grant 04-08); EC (FP7 project EpiPGX 279062); Research Grants Council of the Hong Kong Special Administrative Region, China project numbers HKU7623/08M (SSC, PK, LWB, PCS), HKU7747/07M (SSC, PCS) and CUHK4466/06M (PK, LWB). Collection of Belgian cases was supported by the Fonds National de la Recherche Scientifique, Fondation Erasme, Université Libre de Bruxelles. GlaxoSmithKline funded the recruitment and data collection for the GenEpA Consortium samples. We acknowledge the support of Nationwide Children's hospital in Columbus, Ohio, USA. The Wellcome Trust (WT066056) and The NIHR Biomedical Research Centres Scheme (P31753) supported UK contributions. Further support was received through the Intramural Research Program of the Eunice Kennedy Shriver National Institute of Child Health and Human Development (Contract N01HD33348). The project was also supported by the popgen 2.0 network through a grant from the German Ministry for Education and Research (01EY1103). The KORA research platform (KORA, Cooperative Research in the Region of Augsburg) was initiated and financed by the Helmholtz Zentrum München—German Research Center for Environmental Health, which is funded by the German Federal Ministry of Education and Research and by the State of Bavaria. Furthermore, KORA research was supported within the Munich Center of Health Sciences (MC Health), Ludwig-Maximilians-Universität, as part of LMUinnovativ. A complete list of p values for all SNPs analysed can be found at the Epilepsy Genetic Association Database.
International League Against Epilepsy Consortium on Complex Epilepsies
Richard J L Anney, Andreja Avbersek, David Balding, Larry Baum, Felicitas Becker, Samuel F Berkovic, Jonathan P Bradfield, Lawrence C Brody, Russell J Buono, Claudia B Catarino, Gianpiero L Cavalleri, Stacey S Cherny, Krishna Chinthapalli, Alison J Coffey, Alastair Compston, Patrick Cossette, Gerrit-Jan de Haan, Peter De Jonghe, Carolien G F de Kovel, Norman Delanty, Chantal Depondt, Dennis J Dlugos, Colin P Doherty, Christian E Elger, Thomas N Ferraro, Martha Feucht, Andre Franke, Jacqueline French, Verena Gaus, David B Goldstein, Hongsheng Gui, Youling Guo, Hakon Hakonarson, Kerstin Hallmann, Erin L Heinzen, Ingo Helbig, Helle Hjalgrim, Margaret Jackson, Jennifer Jamnadas-Khoda, Dieter Janz, Michael R Johnson, Reetta Kälviäinen, Anne-Mari Kantanen, Dalia Kasperavičiūte, Dorothee Kasteleijn-Nolst Trenite, Bobby P C Koeleman, Wolfram S Kunz, Patrick Kwan, Yu Lung Lau, Anna-Elina Lehesjoki, Holger Lerche, Costin Leu, Wolfgang Lieb, Dick Lindhout, Warren Lo, Daniel H Lowenstein, Alberto Malovini, Anthony G Marson, Mark McCormack, James L Mills, Martina Moerzinger, Rikke S Møller, Anne M Molloy, Hiltrud Muhle, Mark Newton, Ping-Wing Ng, Markus M Nöthen, Peter Nürnberg, Terence J O'Brien, Karen L Oliver, Aarno Palotie, Faith Pangilinan, Katharina Pernhorst, Slave Petrovski, Michael Privitera, Rodney Radtke, Philipp S Reif, Felix Rosenow, Ann-Kathrin Ruppert, Thomas Sander, Theresa Scattergood, Steven Schachter, Christoph Schankin, Ingrid E Scheffer, Bettina Schmitz, Susanne Schoch, Pak C Sham, Sanjay Sisodiya, David F Smith, Philip E Smith, Doug Speed, Michael R Sperling, Michael Steffens, Ulrich Stephani, Pasquale Striano, Hans Stroink, Rainer Surges, K Meng Tan, The KORA study group, G Neil Thomas, Marian Todaro, Anna Tostevin, Rossana Tozzi, Holger Trucks, Frank Visscher, Sarah von Spiczak, Nicole M Walley, Yvonne G Weber, Zhi Wei, Christopher Whelan, Wanling Yang, Federico Zara, and Fritz Zimprich. Affiliations listed in appendix (p 2).
Analysis committee
JPB, SSC, CGFdK, HG, CL, DS, ZW, and CW (association analysis); GLC (coordination); JPB, SSC, CGFdK, HG, DS, and CW (imputation); and JPB, GLC, CL, DS, and ZW (protocol development).
Phenotyping committee
CD, DJD, WSK, PK, DHL, AGM, MRS, and PS.
Strategy committee
LB, SFB, RJB, HHak, ELH, MRJ, BPCK, PK, HL, TJO'B, KLO (ex officio), and SSis.
Governance committee
SFB, AC, A-EL, and DHL.
Writing committee
SFB, GLC, and MRJ.
Patient recruitment and phenotyping
AA, FB, RJB, CBC, KC, PC, G-JdH, PDJ, ND, CD, DJD, CPD, CEE, TNF, MF, JF, VG, HHja, IH, MJ, JJ-K, DJ, MRJ, RK, A-MK, DK-NT, WSK, PK, HL, DL, WLo, AGM, MMoe, RSM, HM, MN, P-WN, TJO'B, MP, RR, PSR, FR, TSan, TSca, StSc, SuSc, SSis, CS, IES, BS, DFS, PES, MRS, US, PS, HS, RS, KMT, MT, AT, RT, FV, SV, NMW, YGW, CW, and FZim.
Genotyping and bioinformatics
RJLA, DB, LB, JPB, GLC, SSC, AJC, CGFdK, DBG, HG, YG, HHak, ELH, IH, DK, CL, AM, MMcC, PN, SP, A-KR, TSan, PCS, DS, MS, HT, ZW, CW, and FZar.
Control cohorts
LCB, AF, HHak, Y-LL, WLi, JLM, AMM, MMN, AP, FP, HS, GNT, the KORA study group, and WY.
Declaration of interests
SFB reports grants from the National Health and Medical Research Council during the conduct of the study, and grants from UCB Pharma, Sanofi-Aventis, Jansen Cilag, and SciGen outside the submitted work. SFB has a patent for SCN1A testing held by Bionomics Inc and licensed to various diagnostic companies (no financial return) and a patent for PCDH19 testing through SA Pathology/Univ Melbourne pending. GLC and MRJ report grants from UCB Pharma outside the submitted work.
Supplementary Material
References
- 1.Hesdorffer DC, Logroscino G, Benn EK, Katri N, Cascino G, Hauser WA. Estimating risk for developing epilepsy: a population-based study in Rochester, Minnesota. Neurology. 2011;76:23–27. doi: 10.1212/WNL.0b013e318204a36a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Berg AT, Berkovic SF, Brodie MJ. Revised terminology and concepts for organization of seizures and epilepsies: report of the ILAE Commission on Classification and Terminology, 2005–2009. Epilepsia. 2010;51:676–685. doi: 10.1111/j.1528-1167.2010.02522.x. [DOI] [PubMed] [Google Scholar]
- 3.Helbig I, Scheffer IE, Mulley JC, Berkovic SF. Navigating the channels and beyond: unravelling the genetics of the epilepsies. Lancet Neurol. 2008;7:231–245. doi: 10.1016/S1474-4422(08)70039-5. [DOI] [PubMed] [Google Scholar]
- 4.Poduri A, Lowenstein D. Epilepsy genetics—past, present, and future. Curr Opin Genet Dev. 2011;21:325–332. doi: 10.1016/j.gde.2011.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Epi4K Consortium. Epilepsy Phenome/Genome Project. Allen AS. De novo mutations in epileptic encephalopathies. Nature. 2013;501:217–221. doi: 10.1038/nature12439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Carvill GL, Heavin SB, Yendle SC. Targeted resequencing in epileptic encephalopathies identifies de novo mutations in CHD2 and SYNGAP1. Nat Genet. 2013;45:825–830. doi: 10.1038/ng.2646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Claes L, Del-Favero J, Ceulemans B, Lagae L, Van Broeckhoven C, De Jonghe P. De novo mutations in the sodium-channel gene SCN1A cause severe myoclonic epilepsy of infancy. Am J Hum Genet. 2001;68:1327–1332. doi: 10.1086/320609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chen Y, Lu J, Pan H. Association between genetic variation of CACNA1H and childhood absence epilepsy. Ann Neurol. 2003;54:239–243. doi: 10.1002/ana.10607. [DOI] [PubMed] [Google Scholar]
- 9.Heinzen EL, Radtke RA, Urban TJ. Rare deletions at 16p13.11 predispose to a diverse spectrum of sporadic epilepsy syndromes. Am J Hum Genet. 2010;86:707–718. doi: 10.1016/j.ajhg.2010.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Helbig I, Mefford HC, Sharp AJ. 15q13.3 microdeletions increase risk of idiopathic generalized epilepsy. Nat Genet. 2009;41:160–162. doi: 10.1038/ng.292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Epi4K Consortium Epi4K: gene discovery in 4,000 genomes. Epilepsia. 2012;53:1457–1467. doi: 10.1111/j.1528-1167.2012.03511.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dibbens LM, Heron SE, Mulley JC. A polygenic heterogeneity model for common epilepsies with complex genetics. Genes Brain Behav. 2007;6:593–597. doi: 10.1111/j.1601-183X.2007.00333.x. [DOI] [PubMed] [Google Scholar]
- 13.Kasperaviciute D, Catarino CB, Heinzen EL. Common genetic variation and susceptibility to partial epilepsies: a genome-wide association study. Brain. 2010;133:2136–2147. doi: 10.1093/brain/awq130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kasperaviciute D, Catarino CB, Matarin M. Epilepsy, hippocampal sclerosis and febrile seizures linked by common genetic variation around SCN1A. Brain. 2013;136:3140–3150. doi: 10.1093/brain/awt233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Guo Y, Baum LW, Sham PC. Two-stage genome-wide association study identifies variants in CAMSAP1L1 as susceptibility loci for epilepsy in Chinese. Hum Mol Genet. 2012;21:1184–1189. doi: 10.1093/hmg/ddr550. [DOI] [PubMed] [Google Scholar]
- 16.EPICURE Consortium. EMINet Consortium. Steffens M. Genome-wide association analysis of genetic generalized epilepsies implicates susceptibility loci at 1q43, 2p16.1, 2q22.3, and 17q21.32. Hum Mol Genet. 2012;21:5359–5372. doi: 10.1093/hmg/dds373. [DOI] [PubMed] [Google Scholar]
- 17.Berkovic SF, Howell RA, Hay DA, Hopper JL. Epilepsies in twins: genetics of the major epilepsy syndromes. Ann Neurol. 1998;43:435–445. doi: 10.1002/ana.410430405. [DOI] [PubMed] [Google Scholar]
- 18.Ottman R, Lee JH, Hauser WA, Risch N. Are generalized and localization-related epilepsies genetically distinct? Arch Neurol. 1998;55:339–344. doi: 10.1001/archneur.55.3.339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Peljto AL, Barker-Cummings C, Vasoli VM. Familial risk of epilepsy: a population-based study. Brain. 2014;137:795–805. doi: 10.1093/brain/awt368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Commission on Classification and Terminology of the International League Against Epilepsy Proposal for revised classification of epilepsies and epileptic syndromes. Epilepsia. 1989;30:389–399. doi: 10.1111/j.1528-1157.1989.tb05316.x. [DOI] [PubMed] [Google Scholar]
- 21.Speed D, Hoggart C, Petrovski S. A genome-wide association study and biological pathway analysis of epilepsy prognosis in a prospective cohort of newly treated epilepsy. Hum Mol Genet. 2014;23:247–258. doi: 10.1093/hmg/ddt403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lippert C, Listgarten J, Liu Y, Kadie CM, Davidson RI, Heckerman D. FaST linear mixed models for genome-wide association studies. Nat Methods. 2011;8:833–835. doi: 10.1038/nmeth.1681. [DOI] [PubMed] [Google Scholar]
- 23.Willer CJ, Li Y, Abecasis GR. METAL: fast and efficient meta-analysis of genomewide association scans. Bioinformatics. 2010;26:2190–2191. doi: 10.1093/bioinformatics/btq340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dempster ER, Lerner IM. Heritability of Threshold Characters. Genetics. 1950;35:212–236. doi: 10.1093/genetics/35.2.212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Banerjee PN, Filippi D, Allen Hauser W. The descriptive epidemiology of epilepsy—a review. Epilepsy Res. 2009;85:31–45. doi: 10.1016/j.eplepsyres.2009.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Purcell S, Neale B, Todd-Brown K. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007;81:559–575. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lee PH, O'Dushlaine C, Thomas B, Purcell SM. INRICH: interval-based enrichment analysis for genome-wide association studies. Bioinformatics. 2012;28:1797–1799. doi: 10.1093/bioinformatics/bts191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Escayg A, MacDonald BT, Meisler MH. Mutations of SCN1A, encoding a neuronal sodium channel, in two families with GEFS+2. Nat Genet. 2000;24:343–345. doi: 10.1038/74159. [DOI] [PubMed] [Google Scholar]
- 29.Wallace RH, Scheffer IE, Barnett S. Neuronal sodium-channel α1-subunit mutations in generalized epilepsy with febrile seizures plus. Am J Hum Genet. 2001;68:859–865. doi: 10.1086/319516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kim SY, Chung HS, Sun W, Kim H. Spatiotemporal expression pattern of non-clustered protocadherin family members in the developing rat brain. Neuroscience. 2007;147:996–1021. doi: 10.1016/j.neuroscience.2007.03.052. [DOI] [PubMed] [Google Scholar]
- 31.Kim SY, Mo JW, Han S. The expression of non-clustered protocadherins in adult rat hippocampal formation and the connecting brain regions. Neuroscience. 2010;170:189–199. doi: 10.1016/j.neuroscience.2010.05.027. [DOI] [PubMed] [Google Scholar]
- 32.Miyake K, Hirasawa T, Soutome M. The protocadherins, PCDHB1 and PCDH7, are regulated by MeCP2 in neuronal cells and brain tissues: implication for pathogenesis of Rett syndrome. BMC Neurosci. 2011;12:81. doi: 10.1186/1471-2202-12-81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Braithwaite SP, Stock JB, Lombroso PJ, Nairn AC. Protein phosphatases and Alzheimer's disease. Prog Mol Biol Transl Sci. 2012;106:343–379. doi: 10.1016/B978-0-12-396456-4.00012-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kim SY, Yasuda S, Tanaka H, Yamagata K, Kim H. Non-clustered protocadherin. Cell Adh Migr. 2011;5:97–105. doi: 10.4161/cam.5.2.14374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Piper M, Dwivedy A, Leung L, Bradley RS, Holt CE. NF-protocadherin and TAF1 regulate retinal axon initiation and elongation in vivo. J Neurosci. 2008;28:100–105. doi: 10.1523/JNEUROSCI.4490-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Masuda M, Braun-Sommargren M, Crooks D, Smith DR. Golgi phosphoprotein 4 (GPP130) is a sensitive and selective cellular target of manganese exposure. Synapse. 2013;67:205–215. doi: 10.1002/syn.21632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Eid T, Tu N, Lee TS, Lai JC. Regulation of astrocyte glutamine synthetase in epilepsy. Neurochem Int. 2013;63:670–681. doi: 10.1016/j.neuint.2013.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gonzalez-Reyes RE, Gutierrez-Alvarez AM, Moreno CB. Manganese and epilepsy: a systematic review of the literature. Brain Res Rev. 2007;53:332–336. doi: 10.1016/j.brainresrev.2006.10.002. [DOI] [PubMed] [Google Scholar]
- 39.Lerche H, Shah M, Beck H, Noebels J, Johnston D, Vincent A. Ion channels in genetic and acquired forms of epilepsy. J Physiol. 2013;591:753–764. doi: 10.1113/jphysiol.2012.240606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Fernandez IF, Perez-Rivas LG, Blanco S, Castillo-Dominguez AA, Lozano J, Lazo PA. VRK2 anchors KSR1-MEK1 to endoplasmic reticulum forming a macromolecular complex that compartmentalizes MAPK signaling. Cell Mol Life Sci. 2012;69:3881–3893. doi: 10.1007/s00018-012-1056-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Monsalve DM, Merced T, Fernandez IF, Blanco S, Vazquez-Cedeira M, Lazo PA. Human VRK2 modulates apoptosis by interaction with Bcl-xL and regulation of BAX gene expression. Cell Death Dis. 2013;4:e513. doi: 10.1038/cddis.2013.40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Irish Schizophrenia Genomics Consortium and the Wellcome Trust Case Control Consortium 2 Genome-wide association study implicates HLA-C*01:02 as a risk factor at the major histocompatibility complex locus in schizophrenia. Biol Psychiatry. 2012;72:620–628. doi: 10.1016/j.biopsych.2012.05.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Li M, Wang Y, Zheng XB. Meta-analysis and brain imaging data support the involvement of VRK2 (rs2312147) in schizophrenia susceptibility. Schizophr Res. 2012;142:200–205. doi: 10.1016/j.schres.2012.10.008. [DOI] [PubMed] [Google Scholar]
- 44.Steinberg S, de Jong S, Andreassen OA. Common variants at VRK2 and TCF4 conferring risk of schizophrenia. Hum Mol Genet. 2011;20:4076–4081. doi: 10.1093/hmg/ddr325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Dao KH, Rotelli MD, Brown BR. The PI3K/Akt1 pathway enhances steady-state levels of FANCL. Mol Biol Cell. 2013;24:2582–2592. doi: 10.1091/mbc.E13-03-0144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Vandenbroucke RE, Dejonckheere E, Van Lint P. Matrix metalloprotease 8-dependent extracellular matrix cleavage at the blood-CSF barrier contributes to lethality during systemic inflammatory diseases. J Neurosci. 2012;32:9805–9816. doi: 10.1523/JNEUROSCI.0967-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yong VW. Metalloproteinases: mediators of pathology and regeneration in the CNS. Nat Rev Neurosci. 2005;6:931–944. doi: 10.1038/nrn1807. [DOI] [PubMed] [Google Scholar]
- 48.Wilczynski GM, Konopacki FA, Wilczek E. Important role of matrix metalloproteinase 9 in epileptogenesis. J Cell Biol. 2008;180:1021–1035. doi: 10.1083/jcb.200708213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Baum L, Haerian BS, Ng HK. Case-control association study of polymorphisms in the voltage-gated sodium channel genes SCN1A, SCN2A, SCN3A, SCN1B, and SCN2B and epilepsy. Hum Genet. 2014;133:651–659. doi: 10.1007/s00439-013-1405-1. [DOI] [PubMed] [Google Scholar]
- 50.Dichgans M, Freilinger T, Eckstein G. Mutation in the neuronal voltage-gated sodium channel SCN1A in familial hemiplegic migraine. Lancet. 2005;366:371–377. doi: 10.1016/S0140-6736(05)66786-4. [DOI] [PubMed] [Google Scholar]
- 51.Abou-Khalil B, Ge Q, Desai R. Partial and generalized epilepsy with febrile seizures plus and a novel SCN1A mutation. Neurology. 2001;57:2265–2272. doi: 10.1212/wnl.57.12.2265. [DOI] [PubMed] [Google Scholar]
- 52.Hirose S, Scheffer IE, Marini C. SCN1A testing for epilepsy: application in clinical practice. Epilepsia. 2013;54:946–952. doi: 10.1111/epi.12168. [DOI] [PubMed] [Google Scholar]
- 53.Dibbens LM, Tarpey PS, Hynes K. X-linked protocadherin 19 mutations cause female-limited epilepsy and cognitive impairment. Nat Genet. 2008;40:776–781. doi: 10.1038/ng.149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Berg AT, Blackstone NW. Of cabbages and kings: Perspectives on classification from the field of systematics. Epilepsia. 2003;44:8–12. [Google Scholar]
- 55.International Multiple Sclerosis Genetics Consortium. Beecham AH, Patsopoulos NA. Analysis of immune-related loci identifies 48 new susceptibility variants for multiple sclerosis. Nat Genet. 2013;45:1353–1360. doi: 10.1038/ng.2770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.International Multiple Sclerosis Genetics Consortium. Wellcome Trust Case Control Consortium 2. Sawcer S. Genetic risk and a primary role for cell-mediated immune mechanisms in multiple sclerosis. Nature. 2011;476:214–219. doi: 10.1038/nature10251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ripke S, O'Dushlaine C, Chambert K. Genome-wide association analysis identifies 13 new risk loci for schizophrenia. Nat Genet. 2013;45:1150–1159. doi: 10.1038/ng.2742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Heinzen EL, Depondt C, Cavalleri GL. Exome sequencing followed by large-scale genotyping fails to identify single rare variants of large effect in idiopathic generalized epilepsy. Am J Hum Genet. 2012;91:293–302. doi: 10.1016/j.ajhg.2012.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Klassen T, Davis C, Goldman A. Exome sequencing of ion channel genes reveals complex profiles confounding personal risk assessment in epilepsy. Cell. 2011;145:1036–1048. doi: 10.1016/j.cell.2011.05.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.