

Abstract
While the three-dimensional structures of heme- and flavin-binding domains of the NOS isoforms have been determined, the structures of the holoenzymes remained elusive. Application of electron cryo-microscopy and structural modeling of the bovine endothelial nitric oxide synthase (eNOS) holoenzyme produced detailed models of the intact holoenzyme in the presence and absence of Ca2+/calmodulin (CaM). These models accommodate the cross-electron transfer from the reductase in one monomer to the heme in the opposite monomer. The heme domain acts as the anchoring dimeric structure for the entire enzyme molecule, while the FMN domain is activated by CaM to move flexibly to bridge the distance between the reductase and oxygenase domains. Our results indicate that the key regulatory role of CaM involves the stabilization of structural intermediates and precise positioning of the pivot for the FMN domain tethered shuttling motion to accommodate efficient and rapid electron transfer in the homodimer of eNOS.
Keywords: Calmodulin, Electron cryomicroscopy, Three-dimensional reconstruction, Nitric oxide synthase, Electron transfer, Image processing
Introduction
Nitric oxide (NO) produced in biological systems is necessary for signaling events in vascular hemodynamics, neural transmission and cellular defenses, but must be regulated quite tightly to maintain a homeostatic state in which the appropriate amount is being synthesized, as excessive amounts may result in low blood pressure or adverse cytotoxic events. Nitric oxide synthases (NOSs) are mono-oxygenases that produce NO and L-citrulline from L-arginine (L-Arg) through two sequential oxygenation steps. Three isoforms exist, encoded by different genes: neuronal (nNOS; NOSI), endothelial (eNOS; NOSIII), and inducible (iNOS; NOSII). Both nNOS and eNOS are constitutively expressed, while iNOS is inducible at the transcriptional level. NO exerts its physiological effects in accordance with the NOS isoform producing it and the cell type in which the isoform is expressed (for reviews, see Alderton et al., 2001; Roman et al., 2002; Stuehr et al., 2009; Feng, 2012). Isoform specificity for the NOS is achieved through subcellular localization, direct binding of other proteins and/or phosphorylation, nitration, nitrosation in neurons, skeletal muscle or endothelial cells (Villanueva and Giulivi, 2010).
The NOSs are functional dimers, with each monomer consisting of an N-terminal oxygenase domain containing binding sites for L-Arg, heme, and tetrahydrobiopterin (H4B), attached within the polypeptide chain to a reductase domain, which contains binding sites for FAD, FMN, and NADPH (the source of electrons), as well as several regulatory sequences. These domains are connected by a Ca2+/calmodulin (CaM) binding site, which is occupied upon agonist-induced elevation of intracellular Ca2+, thereby activating nNOS and eNOS, the constitutive isoforms. Despite their different sizes (ranging from ~130–160 kDa per monomer), NOSs share 50–60% overall amino acid sequence homology (Bredt and Snyder, 1994). NOS homodimers are joined by an extensive protein-protein surface interface (~3000Å2), as well as a tetrahedrally coordinated zinc atom, in the oxygenase domain dimer interface (Raman et al., 1998). In addition to the CaM-binding site, at least three CaM-responsive sequences reside in the reductase domains – an autoregulatory region (AR), originally identified as a ~40 residue sequence in the FMN-binding subdomain (Salerno et al., 1997; Daff et al., 1999; Lane and Gross, 2000; Montgomery et al., 2000), a C-terminal tail sequence (CT), which differs in length among the three isoforms (Roman et al., 2000; Roman and Masters, 2006), and a small insertion (β-finger) in the hinge region (Zhang et al., 2001; Knudsen et al., 2003; Jones et al., 2004).
It is clear from the partial crystal structure obtained for the nNOS reductase domain (Garcin et al., 2004) and by homology to cytochrome P450 reductase that electron transfer between flavin moieties and then from the FMN to heme must require significant conformational changes to occur. Ghosh and Salerno (Ghosh and Salerno, 2003) proposed a “tethered shuttle” model in which the FMN-binding domain is in close proximity to the FAD/NADPH-binding domain to receive electrons from the FAD (the closed or input state), but must reorient to supply electrons to the oxygenase domain (the open or output state). In the absence of CaM, the enzyme is in the closed conformation, but is unlocked and forms the open conformation in its presence. The extreme flexibility and mobility of these protein domains required to alternate between these conformations makes crystallization of the holo-enzyme very challenging and, to date, no structural data of any holo-NOS isoform has been possible. These conformational rearrangements, including the CaM-responsive elements AR and CT, must occur in a regulated manner for enzymatic activity to ensue. Roman and Masters (Roman and Masters, 2006) proposed a concerted model of NOS regulation in which the AR in the constitutive isoforms, in combination with the CT, stabilizes either a closed or an open form of the enzyme, depending on the binding of CaM and/or NADP+/NADPH. Isoform specificity for the NOS is achieved through subcellular localization, direct binding of other proteins and/or phosphorylation, nitration, nitrosation in neurons, skeletal muscle or endothelial cells (Förstermann and Sessa, 2012).
To discover the structural conformation(s) that permit the transfer of electrons from the reductase domain into the oxygenase domain within the NOS isoform homodimers and, thus, to determine how the individual isoform structures contribute to the differences in reaction rates and stoichiometry of product formation among the NOS isoforms constitute a search for a Holy Grail. Endothelial NOS is the most tightly coupled isoform, with >90% of the NADPH-donated electrons producing NO (Gao et al., 2007a; Gao et al., 2007b), required for maintaining homeostasis in vascular endothelial cells. Although several structures of isolated domains of the NOS isoforms have been determined (Li et al., 2001; Raman et al., 1998; Zhang et al., 2001; Crane et al., 1997; Crane et al., 1998; Garcin et al., 2004), currently there is no full-length atomic structure available for any of the NOS isoforms, as multiple attempts to determine the X-ray structures have met with failure. A recent electron microscopy study employing negative staining of chemically cross-linked nNOS holoenzymes (Yokom et al., 2014) is not only of a different isoform, it also suffers from potential sample preparation artifacts and an electron cryo-microscopy study of eNOS holoenzymes only shows incomplete density that only accommodates the oxygenase domains (Persechini et al., 2013), leaving the conformations of the eNOS holoenzyme an open question.
The following report describes the structure of full-length eNOS, as determined by combining the techniques of electron cryo-electron microscopy, 3D reconstruction, and fitting of high-resolution domain structures, to determine near-atomic resolution structures of the conformations of the complete CaM-bound and CaM-free forms of eNOS in their native environment and without chemical modifications. The reconstructions presented herein enable accurate placement of both the oxygenase and the reductase domains, and allow unequivocal assignment of CaM through difference mapping. Our results indicate that a key regulatory role of CaM is allosteric in nature, stabilizing a conformation of eNOS that enables accurate positioning of the pivot for the FMN domain tethered shuttling motion to accommodate efficient electron transfer in the homodimer.
Materials and Methods
Protein Expression and Purification
eNOS was expressed and purified as previously described (Martasek et al., 1996), with a few modifications. The cultures were grown for 48 hours at room temperature in 500 ml TB medium in Fernbach flasks. Delta-aminolevulinic acid (60mg/l) was added to the culture at the time of IPTG induction, as well as after 24 hours. After sonication, the lysate was applied to a 30-ml 2′5′-ADP Sepharose column equilibrated in 20 mM Tris-HCl, pH7.4, 400 mM NaCl, 0.5 mM L-Arginine, 0.1 mM EDTA, 1.0 mM β-mercaptoethanol, and 10% glycerol (Buffer B). The column was washed with 10x volume of Buffer B and the protein eluted with 50 ml Buffer B containing 500 mM NaCl and 5 mM 2′-AMP. Fractions showing a spectral maximum at 397nm were concentrated, and 500 μM H4B was added. Samples were then loaded onto a Sepharose 6 gel filtration column (GE Healthcare Biosciences, Pittsburgh, PA) equilibrated in buffer B containing 5% glycerol, and fractions containing eNOS dimers were collected and concentrated to approximately 10–12 mg/mL. All samples demonstrated spectra with maxima at 397 nm and 280/397 nm ratios of 2.5–2.8. NO synthesis activity, as measured by hemoglobin capture varied from 15–30 min−1. Where described, CaM was added in 10x molar excess; unbound CaM was removed by concentration/dilution using 50 kD Centricon protein concentrators (Millipore Corporation, Billerica, MA). The presence of CaM was verified by PAGE. CaM was prepared by the method of Zhang and Vogel (Zhang and Vogel, 1994). (6R)-5,6,7,8-tetrahydrobiopterin was from Schircks Laboratories (Switzerland). All other chemicals were from Sigma-Aldrich (St. Louis, MO) and were of the highest grade available.
Activity Measurements
Nitric oxide formation (hemoglobin capture assay) was measured at 23° C as described (Hevel and Marletta, 1994), with the exception that the assay was performed in a buffer containing 50 mM Tris-HCl, pH 7.4, 100 mM NaCl, and 200 μM CaCl2. The rate of NO synthesis was determined using an extinction coefficient of 60 mM−1cm−1 at 401 nm.
Electron Cryo-microscopy Sample Preparation and Data Collection
Three independent eNOS preparations were used for the electron cryo-microscopy studies described here. Fresh and/or freshly thawed eNOS samples concentrations were between ~60 to 250nM. The enzyme was diluted into eNOS dilution buffer (50 mM Tris-HCL, pH 7.4, 100 mM NaCl, 0.1 mM EDTA, 0.1 mM BME, 0.5 mM L-Arg) with or without a 5-fold molar excess of CaM in the presence of 1mM CaCl2. Following a 10-min incubation in a humidified chamber, a drop of 4 μl reaction sample was then applied to glow-discharged, plasma-cleaned (Solaris, Gatan Inc) Quantifoil 1.2/1.3 holey carbon-coated 400 mesh electron microscopy grids (Quantifoil Micro Tools GmbH). Excess liquid was blotted and the grids were then frozen in liquid N2-cooled ethane, either manually or with Vitrobot IV (FEI company). Images of all samples suspended over holes were acquired under low-dose conditions using either a Tecnai F20 G2 Twin transmission electron microscope (FEI company), equipped with an field emission gun (FEG) operated at 200kV and a nominal magnification of 67,000x, or a Titan Krios Twin transmission electron microscope (FEI company), equipped with an FEG operated at 300kV and a nominal 100,000x magnification. Micrographs (F20) were recorded on Kodak ISO-163 plates (Eastman Kodak Co) with a total dose of 20–50e−/Å2 and under defocus ranging between 1.5 μm and 2.5 μm. The micrographs were developed in full-strength Kodak D19 developer, digitized using a SCAI scanner (Integraph) at 7-μm raster and binned to a final pixel size of 0.67 nm. Digital images (Titan) were obtained, using an Ultrascan 4kx4k charge coupled device (CCD) model 895 (Gatan Inc), acquiring defocus pairs with 4–8 μm with pixel size of 0.36 nm.
Image Processing and Three-Dimensional Reconstructions
A total of 21,000 particles were interactively selected from 316 micrographs for the CaM-free eNOS data and 10,000 particles were selected for the CaM-bound eNOS data from 245 micrographs. The particles were processed and analyzed using the EMAN (Ludtke et al., 1999), SPARX (Hohn et al., 2007) and CoAn (Volkmann and Hanein, 1999) software packages. Phase correction for the contrast transfer function was applied for all images using EMAN. The independent experimental data sets was initially independently reconstructed using starting models produced directly from the respective data using common lines (Ludtke et al., 1999). In later runs, each data set was was iteratively sorted into separate classes following a protocol that explicitly accounts for reference bias and was previously described elsewhere (Xu et al., 2011; Janssen et al., 2012; Xu et al., 2013). We used this procedure with the aim to sort out and reconstruct the most prominent conformation of each condition. For all data sets, convergence for the sorting was achieved within 8–25 iterations. Alteration of alignment and classification strategies yielded similar sorting results. Cross-validation for the most prominent conformation within the respective condition (with or without CaM) was performed by comparing the individual reconstructions of the most prominent classes derived from the independent data sets (two or three per condition). The cross-validation test was considered passed when the Fourier Shell Correlation (FSC) remained above 0.5 for all frequencies smaller than 1/4 nm−1. Consensus reconstructions were then generated by averaging the respective cross-validated reconstructions of the most prominent classes of each independent data set, separately for the two condition.
In order to improve the accuracy and quality of the initial sorting and reconstruction, iterative reference-based sorting and refinement was performed using the consensus reconstruction from the previous step as reference with the combined, complete data sets for each condition. A final test for the absence of over-fitting was performed by using the final sorted data and swapping out the reference with the respective initial common-line based models from the unsorted data (Xu et al., 2011). No statistically significant difference between the reconstructions based on the initial consensus reference as a starting model and the reconstruction with the swapped reference as a starting model was observed.
Because the iteration of the sorting procedure inherently breaks the assumption of complete independence for the refinement, the ‘gold-standard’ criterion for resolution determination, which relies on independence, is not an adequate choice for this study. Instead we limited the data during refinement to 1/2 nm−1, an alternative approach to avoid model bias in resolution determination that yields similarly unbiased estimates as the ‘gold standard’ (Scheres and Chen, 2012). For this procedure, resolution is defined as the frequency were the FSC drops below 0.5. Differences between reconstructions were considered significant when they corresponded to peaks in a difference map that were at least three standard deviations larger in magnitude than the mean of the difference map.
Structural Modeling
Models of the entire eNOS holoenzyme were constructed in the following way: A homology model based on the crystal structure of nNOS reductase fragment, PDB ID 1TLL (Garcin et al., 2004) was generated using the HOMER homology modeling server (Tosatto, 2005). A homology model based on the crystal structure of an iNOS FMN/CaM-binding domain fragment (including the bound CaM), PDB ID 3HR4 (Xia et al., 2009) was generated using HOMER. The common residues of the two homology models were aligned with Chimera (Pettersen et al., 2004) and the two models were merged. Clashes in the merged homology model were relieved using the idealization routine in REFMAC5 (Murshudov et al., 2011). The reconstructions were segmented into compartments using the watershed transform (Volkmann, 2002). A crystal structure of the eNOS oxygenase dimer was fitted into the corresponding segment using the statistics-based fitting program CoAn (Volkmann and Hanein, 1999). The statistics were calculated as described (Volkmann, 2009). The combined reductase/CaM-binding domain/CaM homology model was fitted into the reductase segments using CoAn. The segments for the reductase and oxygenase portions of the reconstructions were iteratively refined during the fitting procedure as described (Volkmann, 2009; Xu et al., 2011). A few rounds of normal-mode based refinement (Tama et al., 2004) as implemented in CoAn were applied. The statistical analysis (Volkmann, 2009) indicates that the fit is globally unique with an uncertainty in domain placements of ~4 Å root mean square deviation. After the final fits were obtained, the linker connecting the oxygenase domain and the CaM binding domain was modeled de-novo using Pymol (DeLano, 2002), Chimera, REFMAC5, and CoAn. Modeling of the open conformation was achieved using the same set of programs. Electrostatic potentials were calculated within Chimera. Figures were generated with Chimera.
Results
eNOS holoenzyme particles exhibit two-fold symmetry
Electron cryo-microscopy and three-dimensional (3D) image reconstruction techniques were employed in conjunction with molecular modeling to elucidate the structure of the bovine eNOS holoenzyme in the presence and absence of CaM. For both samples, well-defined protein particles were distributed with random orientations, while suspended in buffer over holes under fully hydrated conditions (Fig. 1A), allowing the use of single-particle reconstruction techniques to determine the 3D structure. For both eNOS in the presence and in the absence of CaM, initial independently derived reconstructions clearly showed signs of two-fold symmetry, consistent with the dimeric state of the holoenzyme. Two-fold symmetry was enforced for the remainder of the analysis.
Figure 1. Cryo-EM data quality indicators and initial reconstruction.

A. Typical electron cryo micrograph with boxed eNOS particles in the presence of CaM. Micrographs of eNOS in the absence of CaM look similar.
B. Projections of the final model (column ‘M’) for the most prominent conformation of eNOS in the absence (lower panel) and presence of CaM (upper panel) and refernece-free class averages from the unsorted data (columns ‘R’) show excellent agreement. The dimensions of the boxes in A and B are 110 Å by 110 Å.
C. Two orthogonal views of the initial reconstruction of the eNOS holoenzyme in the presence of CaM obtained without sorting the data. Two contour levels are shown. The contour level represented by the solid surface is chosen so that the ‘head’ contains the volume expected for the oxygenase dimer. At this contour level, the two ‘legs’ are not connected to the ‘head’ of the reconstruction and are also separate from each other, indicating high mobility of the underlying structural elements. The contour level represented by the wire mesh shows that, at lower density levels, the legs indeed connect to the head and are not separate entities. The eNOS oxygenase dimer crystal structure is overlaid onto the density of the ‘head’ for reference. The bar represents 25 Å.
D. Fourier shell correlation from two halves of frequency limited sorted data for eNOS in the absence (red) and presence (cyan) of CaM. The curves drop below 0.5 (indicated by grey lines) at 28 Å (without CaM) and 25 Å (with CaM), respectively.
eNOS holoenzymes exist in conformational equilibria
Both reconstructions had very similar appearance, with a well-defined ‘head’ and two ‘legs’ at an approximately 40° angle to the head axis (Fig 1C). Despite the use of up to 21,000 particles for the reconstructions, the resolution for both these initial reconstructions was restricted to ~45 Å, strongly indicating the presence of conformational mixtures in the samples. Within the reconstructions, the ‘head’ was roughly comparable in shape and in volume with the crystal structures of the eNOS oxygenase dimer (Fig. 1D), but the volume of the individual ‘legs’ was too small to accommodate the reductase domain. We concluded that the conformational variability within the samples is caused by flexibility in the connection regions between the oxygenase dimer and the individual reductase domains. At this stage and at 45 Å resolution, there was no statistically significant difference (three sigma level) detectable between these reconstructions in the presence and in the absence of CaM.
CaM is located between the oxygenase and reductase domains
Next, multi-reference sorting protocols (Spahn and Penczek, 2009; Xu et al., 2011) were employed to extract the most dominant conformations independently for the two samples. The resolution for the resulting reconstructions was dramatically improved to ~25 Å (Fig. 1D). Interestingly, the percentage of particles contributing to the most dominant conformation in the absence of CaM was only 35%, whereas a total of 50% of the particles contributed in its presence. This result indicates that the presence of CaM locks a significantly larger percentage of the holoenzymes into a well-defined conformation. Both reconstructions retained the head-legs configuration of the mixed states, but this time the volumes of the legs do accommodate that of the reductase domains (Fig. 2A, B). In addition, a link between the two legs is clearly evident, suggesting an additional dimer contact in this region that stabilizes the dominant conformation. The crystal structure of the eNOS oxygenase dimer can be docked unequivocally into the ‘head’ of the reconstruction (Fig. 3). The primary difference (three sigma level) between the reconstructions in the presence and absence of CaM is some extra density close to the connections between the oxygenase dimer and the legs when CaM is present (arrows in Fig. 2). This extra density can thus be attributed to bound CaM. This is consistent with the fact that the docking of the reductase domain univocally places the CaM binding helix near this extra density.
Figure 2. Three-dimensional reconstructions of human eNOS holoenzyme in the presence and absence of CaM.
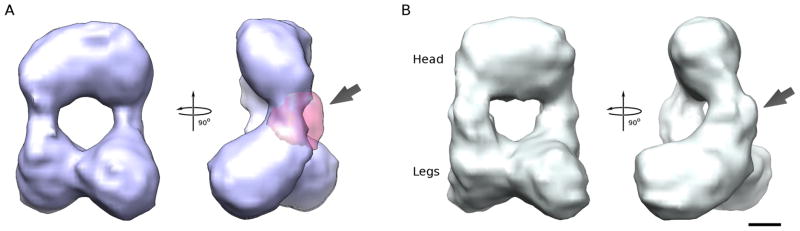
A. Reconstruction of eNOS in the absence of CaM. Two orthogonal views around the vertical axis are shown. The main difference between this reconstruction and the reconstruction of eNOS in the presence of CaM is shown as a pink transparent surface for one of the two monomers in the second view(arrow).
B. Reconstruction of eNOS in the presence of CaM. The arrow in B matches the location of the arrow in A. The bar represents 25 Å.
Figure 3. Fitting of eNOS homology model into the reconstructions human eNOS holoenzyme.
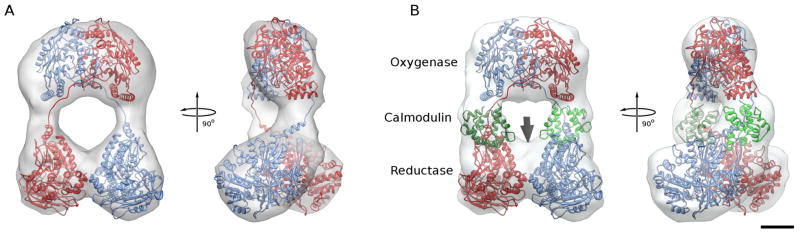
A. Fit of final model of the eNOS homo dimer into the reconstruction of eNOS in the absence of CaM. One monomer is shown in red, the other in blue. Same views as in Figure 2A.
B. Fit of final model of the eNOS homo dimer with bound CaM into the reconstruction of eNOS in the presence of CaM. The two CaMs are shown in different shades of green and occupy the extra density seen in Figure 2B. There is some unoccupied density near the place where the C-termini of the reductase domains interact (arrow), indicating that the C-terminal residues disordered in the crystal structure are more ordered in this conformation and possibly contribute to the stabilization of the reductase domain dimer interface.
Topology of the eNOS holoenzyme
A homology model of the eNOS reductase domain with bound CaM was built, based on the crystal structures of the nNOS reductase domain (Garcin et al., 2004) and a CaM-bound iNOS reductase fragment (Xia et al., 2009). The resulting model docks readily and without ambiguity as a rigid body into the ‘leg’ portion of the reconstruction of eNOS in the presence of CaM, with the CaM ending up inside the extra density, the FMN domain on the side closest to the oxygenase domain, and the resolved portion of the C-terminal end at the interface between the two ‘legs’. This docking solution is significantly better than any other solution at a confidence level of 99.99% (Volkmann and Hanein, 1999; Volkmann and Hanein, 2009). Constraints to bias the solution towards the positioning of the CaM derived from the difference between the reconstructions were not used. Normal mode-based refinement of the fit slightly repositioned the CaM and its binding domain with respect to the rest of the reductase domain, moving it closer to the oxygenase dimer. The final model provides an excellent fit to both the reconstruction in the presence of CaM (Fig. 3A) and, with CaM omitted from the model, with the reconstruction in the absence of CaM (Fig. 3B).
The C-termini of the reductase domains form a stabilizing dimer interface
The docking of the reductase homology model brings the C-termini of the individual reductase domains into close apposition in the region where the two legs of the reconstructions connect. The interface between the reductase domains appears to be relatively small, potentially involving eight residues (Glu-883, Arg-885, Gln-888, Glu-889, Arg-898, Arg-899, Glu-902, Phe-1178), including the most C-terminal residue (Phe-1178) resolved in the model. Charged residues with several potential salt bridges dominate this interface. There is some unoccupied density near the place where the C-termini of the reductase domains interact (arrow Fig. 3B), indicating that the C-terminal residues disordered in the crystal structure are more ordered in this conformation, thus raising the possibility that the residues at the C-terminus contribute to the stabilization of the reductase domain dimer interface.
The oxygenase and reductase domains connect cross-wise
The oxygenase and reductase domains are covalently linked by a flexible 25-residue linker. With moderate modifications at the resolved ends of the two domain structures, the linker can be readily modeled to connect either the two neighboring domains or two cross domains. The distance between the two resolved ends of the domain structures is slightly shorter for the nearest neighbor configuration, but this connection necessitates leading the chain from one side of the complex to the other, introducing two backwards kinks. The connection from one oxygenase domain to the reductase domain across from it is slightly more taut but connects on the same side without kinks (Fig. 3), consistent with data favoring trans-dimer electron transfer (Siddhanta et al., 1996).
Structural Modeling studies indicates a large shuttling motion of the FMN domain
The distance between the flavin and heme cofactors in our structure does not support electron transport (Fig. 4). Pivoting the FMN domain using a simple rigid hinge at residue Ser-517 (Li et al., 2012; Xia et al., 2009), can bring the closest distance between the aromatic rings of the heme and flavin cofactors into the 25 Å range, but this distance is still too large to support electron transport at reasonable rates. However, the introduction of some flexibility in the hinge region brings the edge-to-edge distance between the aromatic rings to 13.5 Å (Fig. 4C). The log electron transfer rate in this configuration is 3.2 ± 1.2 in log(s−1), yielding a rate of about 1600 s−1 (Page et al., 1999; Bartberger et al., 2002). Thus, these structural and modeling studies suggest that electron transfer in the eNOS holoenzyme involves shuttling of the electrons from the reductase domain to the oxygenase domain via a fairly large-scale, tethered movement of the FMN domain.
Figure 4. Model of electron transfer in the eNOS holoenzyme.

A. Conformation of eNOS with bound CaM as visualized by cryo-EM. For clarity only one oxygenase domain (red) and the reductase domain of the other monomer (blue) with bound CaM (green) are shown. The orientation is identical to that shown in the right panel of Figure 2B. The FMN domain is shown in yellow. In order to transfer electrons to the heme in the oxygenase domain, the FMN domain needs to swing from its position towards the oxygenase (curved yellow arrow).
B. A model of the conformation in which electrons are transferred from the FMN domain to the heme in the presence of NADPH. This conformational change can be achieved by a rigid body swing of the FMN domain, pivoted at the CaM binding helix (transition between yellow and blue) and tethered to the rest of the reductase domain at the C-terminus of the FMN domain. To target this movement, CaM is necessary to lock the eNOS CaM-binding domain into place to provide a stable pivot.
C. Close-up of the contact area between the modeled FMN domain and the oxygenase domains. In this geometry the edge-to-edge distance between the aromatic rings of the flavin and the heme is 13.5 Å.
D. View of the reductase dimer interface from the ‘bottom’ in relation to Figures A–C, along the two-fold symmetry axis. The cofactors are shown in green (NADPH), yellow (FAD) and orange (FMN). The two reductase domains are shown in red and blue. The CaMs are just visible in pale green blending into the background (labeled ‘CaM’). Note that the FMN domain is located in between the bulk of the reductase domain and the CaM. As a consequence, the FMN and the FMN domain are largely obstructed by the rest of the reductase domain. Several of the potentially interacting residues are shown and labeled, including the most C-terminal residue resolved in the reductase crystal structure (labeled ‘F1178 (C)’) and many oppositely charged residues.
Discussion
Here we presented reconstructions of fully hydrated full-length eNOS holoenzymes in the presence and absence of CaM. The electron density maps accounts for the entire holoenzyme. The spatial relationship between the FMN domain and the CaM/CaM-binding domains in the holoenzyme structures is very similar to that observed in the crystallographic iNOS structure of the isolated FMN/CaM/CaM-binding domain structure (Xia et al., 2009). The conformations of the oxygenase and reductase domains are also quite similar to the respective crystallographic structures (Raman et al., 1998; Garcin et al., 2004). However, the extensive crystallographic interface of the nNOS reductase domain dimer structure (Garcin et al., 2004) does not occur in the cryoEM full-length holoenzyme reconstructions reported herein.
In our reconstructions, the oxygenase and reductase domain structures are fully accommodated in the cryo-EM density in the context of the holo-enzyme. Both structural elements can be univocally docked as rigid bodies into the density with an accuracy of ~0.4 nm using objective, statistics-based docking algorithms (Volkmann and Hanein, 1999; Volkmann, 2009). CaM can be unequivocally placed into the density through difference mapping between the reconstruction of the CaM-free sample and the sample in the presence of CaM. The CaM site from the difference mapping coincides with the location of CaM from the docking results (Fig. 3).
A previous cryo-EM study of eNOS (Persechini et al., 2013), which - in contrast to the study described here - did not employ sorting strategies, only showed density for the oxygenase domain and no density for the reductase domain. A small amount of additional density was observed when CaM was present. The authors interpret the additional density as remnants of the bound CaM and predict its binding mode. In light of our complete structure, their conclusions are not supported. It is likely that the extra density they see in their CaM-bound eNOS reconstruction is primarily caused by the stabilization of the reductase domain rather than by the bound CaM directly.
Random conical tilt reconstructions of negatively stained, chemically cross-linked nNOS holoenzymes in the presence of CaM (Yokom et al., 2014) show a roughly similar shape to our cryo-EM reconstructions. However, the manual docking of the reductase domain presented in the negative stain study is completely incompatible with the unique fit we obtain from objective docking into our cryo-EM reconstructions by being about 180 degrees rotated around the long axis of the domain. It is plausible that nNOS and eNOS follow different structural pathways but it is more likely that one or more steps of the sample preparation and reconstruction process during the nNOS study led to distortions that hampered the accuracy of the subjective manual docking process.
Interestingly, our results indicate that CaM binding does not induce dramatic conformational changes in eNOS but primarily results in a shift of the conformational equilibrium towards a conformation in which the two reductase domains interact near the C-terminal region. In this conformation, CaM stabilizes the interface between the oxygenase and reductase domains, thus holding the reductase domains in place to form contacts near the C-terminus. This interaction involves only a relatively small area that includes several potential salt bridges. In the crystal structure of the nNOS reductase domain (Garcin et al., 2004), the resolved portion of the C-terminal region provides extra contacts between the FAD and FMN domains, presumably inhibiting hinge opening.
The last resolved residue of the C-terminus, Phe1178 in eNOS, is directly involved in the reductase dimer interactions (Fig. 4D), likely causing conformational changes further along in the sequence towards the N-terminus, including Phe1160 (Phe1395 in nNOS), which stacks with the FAD isoalloxazine ring. This residue must move considerably during catalysis in order for NADPH to transfer a hydride to the FAD (Welland and Daff, 2010). The nearby residue Arg1165 (Arg1400 in nNOS) was implicated in NADPH-mediated regulation, as well (Tiso et al., 2005). The 25 residues, unresolved in the crystal structure, are most likely ordered in the CaM-stabilized reconstructions evidenced by the unoccupied density present in this region that can account for all of the missing residues. Another structural element clearly affected by CaM binding is the β-finger (or SI, small insertion, residues 831–844) in the hinge region (Zhang et al., 2001; Knudsen et al., 2003; Jones et al., 2004), possibly weakening contacts within the FMN domain. Thus, the effect of CaM binding is allosteric in nature, stabilizing a conformation of eNOS that primes the release of the FMN domain from the FAD-binding sub-domain by weakening autoinhibitory interactions (Salerno et al., 1997; Roman et al., 2000a; Roman and Masters, 2006; Salerno et al., 2013).
The AR region appears to be less likely to be directly affected by CaM binding in this closed conformation because it is neither near the reductase interface nor near the β-finger or the bound CaM itself. It is possible and even likely, however, that the AR region interacts with the C-terminus of eNOS, which is the longest of the three isoforms (Roman et al., 2000b). The disordered portion in the isolated nNOS reductase crystal structure (Garcin et al., 2004) appears to be disordered in the CaM-stabilized conformation as well, as no extra density is present in the vicinity of AR in this CaM-bound form. It is noteworthy to mention that we cannot rule out that the AR region becomes ordered and/or that interactions between AR and CaM or other regions occur in the open conformation of eNOS, because the structure of this conformation was not determined in this study.
The structures resolved in this study are of the most predominant, stable conformations present under the experimental conditions. They represent the putative “closed” conformation, in which electrons are passed from the FAD to FMN. A predicted “open” conformation would be expected only with CaM and NADP+ bound to the enzyme (Craig et al., 2002). These studies were not performed in the presence of NADP+, so the open conformation would not be captured. In this closed conformation, the distance between the flavin and heme cofactors in the cryo-EM structure of eNOS in the presence of CaM does not support electron transport. Pivoting at residue Ser517 (Xia et al., 2009) with some limited flexibility shortens the edge-to-edge distance between the aromatic rings to 13.5 Å (Fig 4B), compatible with recent pulsed EPR measurements (Astashkin et al., 2010). Thus, our structural and modeling studies support the tethered shuttle model of electron transfer in the eNOS holoenzyme, involving the transfer of the electrons from the reductase domain to the oxygenase domain via a fairly large-scale, concerted movement of the FMN domain. The tethered shuttle model is also strongly supported by kinetic studies (Feng et al., 2007; Haque et al., 2007) and a similar large-scale rearrangement of an FMN domain to enable electron transfer was observed in yeast-human NADPH-cytochrome P450 reductase (Aigrain et al., 2009).
The oxygenase/reductase interface resulting from the modeling (Fig. 5) is near the cysteine tetracoordinated Zn center of the oxygenase domain and is quite similar to the site proposed previously on grounds of charge complementarity (Raman et al., 1998). An alternative interface proposed (Garcin et al., 2004), which is based on a shuttle movement from a different starting point, is incompatible with the CaM-bound conformation without major conformational readjustments. In addition, one consequence of the arrangement proposed here is that only one FMN domain at a time can dock onto the oxygenase dimer, the second FMN domain would be sterically hindered by the first one.
Figure 5. Potential surface patches interacting in the open conformation.
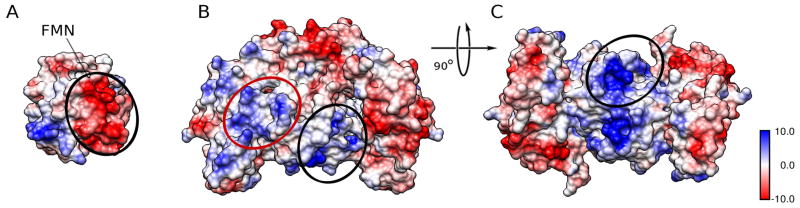
A. Coloumbic potential mapped onto the surface of the FMN domain. This view is rotated 90 degrees counter clockwise form the view in Figure 5B and C. The oval marks the most negatively charged patch of the domain, which is buried in the closed conformation in the interface with the rest of the reductase domain. The flavin location is marked as well
B. Electrostatic potential mapped onto the surface of the oxygenase dimer. This view is rotated 90 degrees clockwise from the view in Figure 4B and C so this view faces the FMN surface depicted in (A). The positively charged interaction surface recently proposed based on the partial nNOS reductase crystal structure (Garcin et al., 2004) is marked by a red oval. This surface is not accessible by the FMN domain without major re-arrangements in the closed structure observed here. The alternative interaction surface originally proposed on grounds of electrostatics (Raman et al., 1998) is mapped with a black oval and corresponds well with the interaction area in our model for the open conformation.
C. View of oxygenase dimer rotated by 90 degrees around the horizontal axis from (B) to show the continuation of the potential interaction surface (black oval) shown in (B). Note that the overall charge in the region marked by the black ovals is more positive than that in the red oval. The bar shows the coloumbic electrostatic potential distribution in kcal/(mol * e−).
Our results indicate that the primary role of CaM binding appears to be the stabilization and accurate positioning of the pivot for the tethered FMN domain shuttling motion, which is at the end of the CaM binding region. In the absence of CaM, this region only loosely connects between the oxygenase and reductase domains, leading to high flexibility, preventing the pivot from adopting a defined position necessary to support the shuttling of electrons from the FMN (Fig. 6). Once CaM binds, the pivot is locked in place, further stabilized by the reductase-reductase interaction in the C-terminal region. In addition, CaM binding also allosterically affects other regulatory elements in the holoenzyme that, for example, lock the FMN domain in its place in the closed conformation. Once NADPH+ binds to the eNOS-CaM complex, the FMN domain can be released and positioned accurately for electron transfer by a large-scale shuttle movement.
Figure 6. Schematic drawing showing how CaM stabilizes the conformation observed here and how CaM positions the pivot for the FMN movement.
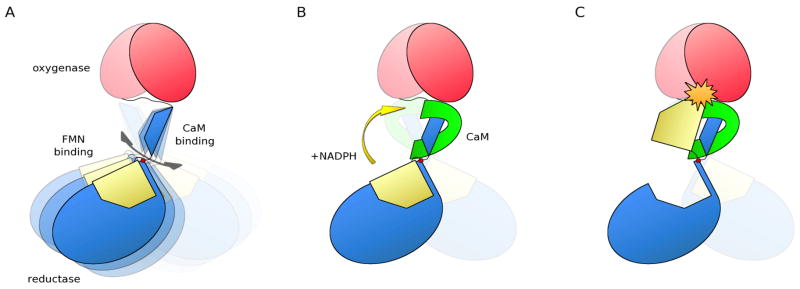
A. Without CaM, the reductase domains (blue, FMN domain yellow) and the oxygenase dimer (red) are only loosely coupled through the CaM binding domain (blue trapeze) and its connecting loops (dark grey). As a consequence, the location of the reductase domains as well as the pivot for the FMN-domain shuttling motion are not well defined in relation to the oxygenase dimer (double arrow) and if electron transfer occurs at all, it will be very inefficient.
B. Once CaM (green) binds, the CaM binding domain is locked into place and the pivot position and reductase location are well defined in relation to the oxygenase dimer, priming the FMN-domain to swing over to the oxygenase domain for electron transfer. This configuration is further stabilized by the reductase-reductase domain interactions (Fig. 4D).
C. With a stable pivot point, the FMN-domain swings into the precise position necessary for efficient electron transfer between the FMN and the heme when NADPH is present.
Conclusion
The elucidation of a full-length structure of the eNOS holoenzyme, which is fully functional, is essential for understanding the mechanism. Here the structural approach used has permitted the establishment of the heme dimer as the stabilizing element of the functional holoenzyme dimer and the unequivocal spatial location of the CaM binding site and the reductase domains in relation to the oxygenase domain. Given the CaM-bound and CaM-free eNOS structural models, we modeled the conformational changes required for electron transfer from the reductase domain of one monomer to the oxygenase domain of the opposite monomer in the functional dimer (Siddhanta et al., 1996) incorporating predicted flexibilities within the NOS structures (Daff et al., 1999; Feng, 2012; Roman and Masters, 2006; Salerno et al., 1997; Salerno et al., 2013; Tiso et al., 2005). The results in the present report lead to unique insights into the structural properties of eNOS that both permit its interaction with its obligate activating protein partner, CaM, and directly demonstrate the stabilization of the most frequently occurring conformations by CaM binding. Our results indicate that a key regulatory role of Ca+2/CaM is allosteric in nature, stabilizing structural intermediates of eNOS and enabling accurate positioning of the pivot for the FMN tethered shuttling motion to accommodate efficient electron transfer in the homodimer of eNOS.
Acknowledgments
DH thanks Myint Aung, Chung-Yueh (Greta) Ouyang and Larnele Hazelwood for cryo-EM sample preparations. The Titan Krios TEM (FEI Company) used in this study is part of Cryo-Electron Microscope Facility at the Centre for Bioimaging Science at National University of Singapore (P. Matsudaira). The studies presented here were supported by NIH grant R01 GM052419 to BSM, LJR and DH. BSM is the Robert A. Welch Distinguished Chair in Chemistry (AQ-0012). PM was also supported by Charles University in Prague institutional programs PRVOUK-P24/LF1/3 and by the European Regional Development Fund (CZ.1.05/1.1.00/02.0109). The image analysis and modeling were supported by NIH grant R01 CA179087 to NV.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Aigrain L, Pompon D, Moréra S, Truan G. Structure of the open conformation of a functional chimeric NADPH cytochrome P450 reductase. EMBO Rep. 2009;10:742–747. doi: 10.1038/embor.2009.82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alderton WK, Cooper CE, Knowles RG. Nitric oxide synthases: structure, function and inhibition. Biochemical Journal. 2001;357:593–615. doi: 10.1042/0264-6021:3570593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Astashkin AV, Elmore BO, Fan W, Guillemette JG, Feng C. Pulsed EPR determination of the distance between heme iron and FMN centers in a human inducible nitric oxide synthase. J Am Chem Soc. 2010;132:12059–12067. doi: 10.1021/ja104461p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartberger MD, Liu W, Ford E, Miranda KM, Switzer C, Fukuto JM, Farmer PJ, Wink DA, Houk KN. The reduction potential of nitric oxide (NO) and its importance to NO biochemistry. Proc Natl Acad Sci U S A. 2002;99:10958–10963. doi: 10.1073/pnas.162095599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bredt DS, Snyder SH. Nitric oxide: a physiologic messenger molecule. Annu Rev Biochem. 1994;63:175–195. doi: 10.1146/annurev.bi.63.070194.001135. [DOI] [PubMed] [Google Scholar]
- Craig DH, Chapman SK, Daff S. Calmodulin activates electron transfer through neuronal nitric-oxide synthase reductase domain by releasing an NADPH-dependent conformational lock. J Biol Chem. 2002;277:33987–33994. doi: 10.1074/jbc.M203118200. [DOI] [PubMed] [Google Scholar]
- Crane BR, Arvai AS, Gachhui R, Wu C, Ghosh DK, Getzoff ED, Stuehr DJ, Tainer JA. The structure of nitric oxide synthase oxygenase domain and inhibitor complexes. Science. 1997;278:425–431. doi: 10.1126/science.278.5337.425. [DOI] [PubMed] [Google Scholar]
- Crane BR, Arvai AS, Ghosh DK, Wu C, Getzoff ED, Stuehr DJ, Tainer JA. Structure of nitric oxide synthase oxygenase dimer with pterin and substrate. Science. 1998;279:2121–2126. doi: 10.1126/science.279.5359.2121. [DOI] [PubMed] [Google Scholar]
- Daff S, Sagami I, Shimizu T. The 42-amino acid insert in the FMN domain of neuronal nitric-oxide synthase exerts control over Ca(2+)/calmodulin-dependent electron transfer. J Biol Chem. 1999;274:30589–30595. doi: 10.1074/jbc.274.43.30589. [DOI] [PubMed] [Google Scholar]
- DeLano WL. The PyMOL molecular graphics system. DeLano Scientific; Palo Alto, CA, USA: 2002. [Google Scholar]
- Feng C. Mechanism of Nitric Oxide Synthase Regulation: Electron Transfer and Interdomain Interactions. Coord Chem Rev. 2012;256:393–411. doi: 10.1016/j.ccr.2011.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng C, Tollin G, Hazzard JT, Nahm NJ, Guillemette JG, Salerno JC, Ghosh DK. Direct measurement by laser flash photolysis of intraprotein electron transfer in a rat neuronal nitric oxide synthase. J Am Chem Soc. 2007;129:5621–5629. doi: 10.1021/ja068685b. [DOI] [PubMed] [Google Scholar]
- Förstermann U, Sessa WC. Nitric oxide synthases: regulation and function. Eur Heart J. 2012;33:829–37. 837a–837d. doi: 10.1093/eurheartj/ehr304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao YT, Panda SP, Roman LJ, Martasek P, Ishimura Y, Masters BSS. Oxygen Metabolism by Neuronal Nitric-oxide Synthase. J Biol Chem. 2007a;282:7921–7929. doi: 10.1074/jbc.M609814200. [DOI] [PubMed] [Google Scholar]
- Gao YT, Roman LJ, Martasek P, Panda SP, Ishimura Y, Masters BSS. Oxygen Metabolism by Endothelial Nitric-oxide Synthase. J Biol Chem. 2007b;282:28557–28565. doi: 10.1074/jbc.M704890200. [DOI] [PubMed] [Google Scholar]
- Garcin ED, Bruns CM, Lloyd SJ, Hosfield DJ, Tiso M, Gachhui R, Stuehr DJ, Tainer JA, Getzoff ED. Structural basis for isozyme-specific regulation of electron transfer in nitric-oxide synthase. J Biol Chem. 2004;279:37918–37927. doi: 10.1074/jbc.M406204200. [DOI] [PubMed] [Google Scholar]
- Ghosh DK, Salerno JC. Nitric oxide synthases: domain structure and alignment in enzyme function and control. Front Biosci. 2003;8:d193–d209. doi: 10.2741/959. [DOI] [PubMed] [Google Scholar]
- Haque MM, Panda K, Tejero J, Aulak KS, Fadlalla MA, Mustovich AT, Stuehr DJ. A connecting hinge represses the activity of endothelial nitric oxide synthase. Proc Natl Acad Sci U S A. 2007;104:9254–9259. doi: 10.1073/pnas.0700332104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hevel JM, Marletta MA. Nitric-oxide synthase assays. Methods Enzymol. 1994;233:250–258. doi: 10.1016/s0076-6879(94)33028-x. [DOI] [PubMed] [Google Scholar]
- Hohn M, Tang G, Goodyear G, Baldwin PR, Huang Z, Penczek PA, Yang C, Glaeser RM, Adams PD, Ludtke SJ. SPARX, a new environment for Cryo-EM image processing. J Struct Biol. 2007;157:47–55. doi: 10.1016/j.jsb.2006.07.003. [DOI] [PubMed] [Google Scholar]
- Janssen ME, Liu H, Volkmann N, Hanein D. The C-terminal tail domain of metavinculin, vinculin’s splice variant, severs actin filaments. J Cell Biol. 2012;197:585–593. doi: 10.1083/jcb.201111046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones RJ, Smith SM, Gao YT, DeMay BS, Mann KJ, Salerno KM, Salerno JC. The function of the small insertion in the hinge subdomain in the control of constitutive mammalian nitric-oxide synthases. J Biol Chem. 2004;279:36876–36883. doi: 10.1074/jbc.M402808200. [DOI] [PubMed] [Google Scholar]
- Knudsen GM, Nishida CR, Mooney SD, Ortiz de Montellano PR. Nitric-oxide Synthase (NOS) Reductase Domain Models Suggest a New Control Element in Endothelial NOS That Attenuates Calmodulin-dependent Activity. J Biol Chem. 2003;278:31814–31824. doi: 10.1074/jbc.M303267200. [DOI] [PubMed] [Google Scholar]
- Lane P, Gross SS. The autoinhibitory control element and calmodulin conspire to provide physiological modulation of endothelial and neuronal nitric oxide synthase activity. Acta Physiol Scand. 2000;168:53–63. doi: 10.1046/j.1365-201x.2000.00654.x. [DOI] [PubMed] [Google Scholar]
- Li H, Raman CS, Martasek P, Masters BS, Poulos TL. Crystallographic studies on endothelial nitric oxide synthase complexed with nitric oxide and mechanism-based inhibitors. Biochemistry. 2001;40:5399–5406. doi: 10.1021/bi002658v. [DOI] [PubMed] [Google Scholar]
- Li W, Fan W, Chen L, Elmore BO, Piazza M, Guillemette JG, Feng C. Role of an isoform-specific serine residue in FMN-heme electron transfer in inducible nitric oxide synthase. J Biol Inorg Chem. 2012;17:675–685. doi: 10.1007/s00775-012-0887-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ludtke SJ, Baldwin PR, Chiu W. EMAN: semiautomated software for high-resolution single-particle reconstructions. J Struct Biol. 1999;128:82–97. doi: 10.1006/jsbi.1999.4174. [DOI] [PubMed] [Google Scholar]
- Martasek P, Liu Q, Liu J, Roman LJ, Gross SS, Sessa WC, Masters BS. Characterization of bovine endothelial nitric oxide synthase expressed in E. coli. Biochem Biophys Res Commun. 1996;219:359–365. doi: 10.1006/bbrc.1996.0238. [DOI] [PubMed] [Google Scholar]
- Montgomery HJ, Romanov V, Guillemette JG. Removal of a putative inhibitory element reduces the calcium-dependent calmodulin activation of neuronal nitric-oxide synthase. J Biol Chem. 2000;275:5052–5058. doi: 10.1074/jbc.275.7.5052. [DOI] [PubMed] [Google Scholar]
- Murshudov GN, Skubák P, Lebedev AA, Pannu NS, Steiner RA, Nicholls RA, Winn MD, Long F, Vagin AA. REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystallogr D Biol Crystallogr. 2011;67:355–367. doi: 10.1107/S0907444911001314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Page CC, Moser CC, Chen X, Dutton PL. Natural engineering principles of electron tunnelling in biological oxidation-reduction. Nature. 1999;402:47–52. doi: 10.1038/46972. [DOI] [PubMed] [Google Scholar]
- Persechini A, Tran QK, Black DJ, Gogol EP. Calmodulin-induced structural changes in endothelial nitric oxide synthase. FEBS Lett. 2013;587:297–301. doi: 10.1016/j.febslet.2012.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. UCSF Chimera--a visualization system for exploratory research and analysis. J Comput Chem. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- Raman CS, Li H, Martásek P, Král V, Masters BS, Poulos TL. Crystal structure of constitutive endothelial nitric oxide synthase: a paradigm for pterin function involving a novel metal center. Cell. 1998;95:939–950. doi: 10.1016/s0092-8674(00)81718-3. [DOI] [PubMed] [Google Scholar]
- Roman LJ, Martasek P, Masters BS. Intrinsic and extrinsic modulation of nitric oxide synthase activity. Chem Rev. 2002;102:1179–1190. doi: 10.1021/cr000661e. [DOI] [PubMed] [Google Scholar]
- Roman LJ, Martásek P, Miller RT, Harris DE, de La Garza MA, Shea TM, Kim JJ, Masters BS. The C termini of constitutive nitric-oxide synthases control electron flow through the flavin and heme domains and affect modulation by calmodulin. J Biol Chem. 2000a;275:29225–29232. doi: 10.1074/jbc.M004766200. [DOI] [PubMed] [Google Scholar]
- Roman LJ, Masters BS. Electron transfer by neuronal nitric-oxide synthase is regulated by concerted interaction of calmodulin and two intrinsic regulatory elements. J Biol Chem. 2006;281:23111–23118. doi: 10.1074/jbc.M603671200. [DOI] [PubMed] [Google Scholar]
- Roman LJ, Miller RT, de La Garza MA, Kim JJ, Siler Masters BS. The C terminus of mouse macrophage inducible nitric-oxide synthase attenuates electron flow through the flavin domain. J Biol Chem. 2000b;275:21914–21919. doi: 10.1074/jbc.M002449200. [DOI] [PubMed] [Google Scholar]
- Salerno JC, Harris DE, Irizarry K, Patel B, Morales AJ, Smith SM, Martásek P, Roman LJ, Masters BSS, Jones CL, Weissman BA, Lane P, Liu Q, Gross SS. An autoinhibitory control element defines calcium-regulated isoforms of nitric oxide synthase. J Biol Chem. 1997;272:29769–29777. doi: 10.1074/jbc.272.47.29769. [DOI] [PubMed] [Google Scholar]
- Salerno JC, Ray K, Poulos T, Li H, Ghosh DK. Calmodulin activates neuronal nitric oxide synthase by enabling transitions between conformational states. FEBS Lett. 2013;587:44–47. doi: 10.1016/j.febslet.2012.10.039. [DOI] [PubMed] [Google Scholar]
- Scheres SH, Chen S. Prevention of overfitting in cryo-EM structure determination. Nat Meth. 2012;9:853–854. doi: 10.1038/nmeth.2115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siddhanta U, Wu C, Abu-Soud HM, Zhang J, Ghosh DK, Stuehr DJ. Heme iron reduction and catalysis by a nitric oxide synthase heterodimer containing one reductase and two oxygenase domains. J Biol Chem. 1996;271:7309–7312. doi: 10.1074/jbc.271.13.7309. [DOI] [PubMed] [Google Scholar]
- Spahn CM, Penczek PA. Exploring conformational modes of macromolecular assemblies by multiparticle cryo-EM. Curr Opin Struct Biol. 2009;19:623–631. doi: 10.1016/j.sbi.2009.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stuehr DJ, Tejero J, Haque MM. Structural and mechanistic aspects of flavoproteins: electron transfer through the nitric oxide synthase flavoprotein domain. FEBS J. 2009;276:3959–3974. doi: 10.1111/j.1742-4658.2009.07120.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tama F, Miyashita O, Brooks CL3. Flexible multi-scale fitting of atomic structures into low-resolution electron density maps with elastic network normal mode analysis. J Mol Biol. 2004;337:985–999. doi: 10.1016/j.jmb.2004.01.048. [DOI] [PubMed] [Google Scholar]
- Tiso M, Konas DW, Panda K, Garcin ED, Sharma M, Getzoff ED, Stuehr DJ. C-terminal tail residue Arg1400 enables NADPH to regulate electron transfer in neuronal nitric-oxide synthase. J Biol Chem. 2005;280:39208–39219. doi: 10.1074/jbc.M507775200. [DOI] [PubMed] [Google Scholar]
- Tosatto SC. The victor/FRST function for model quality estimation. J Comput Biol. 2005;12:1316–1327. doi: 10.1089/cmb.2005.12.1316. [DOI] [PubMed] [Google Scholar]
- Villanueva C, Giulivi C. Subcellular and cellular locations of nitric oxide synthase isoforms as determinants of health and disease. Free Radic Biol Med. 2010;49:307–316. doi: 10.1016/j.freeradbiomed.2010.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volkmann N. A novel three-dimensional variant of the watershed transform for segmentation of electron density maps. J Struct Biol. 2002;138:123–129. doi: 10.1016/s1047-8477(02)00009-6. [DOI] [PubMed] [Google Scholar]
- Volkmann N. Confidence intervals for fitting of atomic models into low-resolution densities. Acta Crystallogr D Biol Crystallogr. 2009;65:679–689. doi: 10.1107/S0907444909012876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volkmann N, Hanein D. Quantitative fitting of atomic models into observed densities derived by electron microscopy. J Struc Biol. 1999;125:176–184. doi: 10.1006/jsbi.1998.4074. [DOI] [PubMed] [Google Scholar]
- Volkmann N, Hanein D. Electron microscopy in the context of systems biology. In: Gu J, Bourne PE, editors. Structural Bioinformatics. Wiley-Blackwell; New York: 2009. pp. 143–170. [Google Scholar]
- Welland A, Daff S. Conformation-dependent hydride transfer in neuronal nitric oxide synthase reductase domain. FEBS J. 2010;277:3833–3843. doi: 10.1111/j.1742-4658.2010.07787.x. [DOI] [PubMed] [Google Scholar]
- Xia C, Misra I, Iyanagi T, Kim JJ. Regulation of interdomain interactions by calmodulin in inducible nitric-oxide synthase. J Biol Chem. 2009;284:30708–30717. doi: 10.1074/jbc.M109.031682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu XP, Rouiller I, Slaughter BD, Egile C, Kim E, Unruh JR, Fan X, Pollard TD, Li R, Hanein D, Volkmann N. Three-dimensional reconstructions of Arp2/3 complex with bound nucleation promoting factors. EMBO J. 2011;31:236–247. doi: 10.1038/emboj.2011.343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu XP, Slaughter BD, Volkmann N. Probabilistic determination of probe locations from distance data. J Struc Biol. 2013;184:78–82. doi: 10.1016/j.jsb.2013.05.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yokom AL, Morishima Y, Lau M, Su M, Glukhova A, Osawa Y, Southworth DR. Architecture of the Nitric Oxide Synthase Holoenzyme Reveals Large Conformational Changes and a Calmodulin-Driven Release of the FMN Domain. J Biol Chem. 2014 doi: 10.1074/jbc.M114.564005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Martàsek P, Paschke R, Shea T, Siler Masters BS, Kim JJ. Crystal structure of the FAD/NADPH-binding domain of rat neuronal nitric-oxide synthase. Comparisons with NADPH-cytochrome P450 oxidoreductase. J Biol Chem. 2001;276:37506–37513. doi: 10.1074/jbc.M105503200. [DOI] [PubMed] [Google Scholar]
- Zhang M, Vogel HJ. Characterization of the calmodulin-binding domain of rat cerebellar nitric oxide synthase. J Biol Chem. 1994;269:981–985. [PubMed] [Google Scholar]