

Summary
Promoter proximal pausing of RNA polymerase II (Pol II) is a major checkpoint in transcription. An unbiased search for novel human proteins that could regulate paused Pol II at the HSPA1B gene identified TRIM28. In vitro analyses indicated HSF1-dependent attenuation of Pol II pausing upon TRIM28 depletion, whereas in vivo data revealed de novo expression of HSPA1B and other known genes regulated by paused Pol II upon TRIM28 knockdown. These results were supported by genome-wide ChIP-sequencing analyses of Pol II occupancy that revealed a global role for TRIM28 in regulating Pol II pausing and pause release. Furthermore, in vivo and in vitro mechanistic studies suggest that transcription-coupled phosphorylation regulates Pol II pause release by TRIM28. Collectively, our findings identify TRIM28 as a novel factor that modulates Pol II pausing and transcriptional elongation at a large number of mammalian genes.
Promoter proximal pausing of RNA polymerase II (Pol II) represents a major checkpoint in transcription. Typically, Pol II enzymes pause at around +30–100 relative to the transcriptional start site (TSS) until activating cellular signals induce elongation1. Although promoter proximal pausing was discovered over two decades ago, it was initially thought to occur at only a limited set of genes2. Recently however, genome-wide analyses such as Chromatin Immuno- Precipitation followed by sequencing (ChIP-seq) and Global Run-On Sequencing (GRO-seq) have shown that promoter-proximal pausing is widespread1,3–7. For instance, approximately 30% of coding genes and over 70% of developmental or inducible genes harbor Pol II paused at promoter-proximal sites8,9. Thus, promoter-proximal pausing is considered a major cellular mechanism to regulate gene expression.
Although the mechanisms of Pol II pausing and pause release are incompletely understood, several transcription factors are known to regulate these processes. DSIF and NELF induce and stabilize pausing10 while TFIIS3, Myc, and P-TEFb help release Pol II from the pausing site1. P- TEFb phosphorylates DSIF, NELF, and the C-terminal domain of Pol II (Pol II CTD), which correlates with pause release11. HSP70 is a model gene appropriate for study of promoter- proximal pausing. Especially in Drosophila, Pol II pausing has been extensively studied at HSP70: Pol II pausing occurs around +3012 relative to the TSS and factors such as NELF and DSIF10 regulate pausing. However, less is known regarding pausing at human genes including HSP70. Although the fundamental mechanisms of promoter-proximal pausing might be evolutionarily conserved, the degree of conservation is uncertain. For example, HSF1, a master HSP70 activator, is conserved in the DNA binding and trimerization domains but is distinctive in its regulatory and activation domains between Drosophila and human13. Spt5 (a subunit of DSIF) knock-down (KD) largely controls pausing, but NelfA KD is less effective in mouse embryonic stem (mES) cells1, and NELF KD unexpectedly decreases Pol II density at NELF-regulated genes in humans14. NELF is less conserved in eukaryotes, and is lacking in C. elegans and S. cerevisiae. In addition, elongation factors such as Gdown115 and ELL16 have been reported to regulate Pol II promoter-proximal pausing in humans yet orthologs for these factors are unknown in Drosophila. These observations imply diversity and complexity of Pol II promoter proximal pause regulatory mechanisms in mammals.
To further probe the factors and mechanisms that regulate Pol II pausing, we initiated an unbiased approach to screen for factors that could selectively bind the non-template strand of the well-studied human HSPA1B (HSP70–2) gene. These experiments were based upon the concept of nucleic acid aptamers, short single-stranded nucleic acid polymers that can bind protein factors with high affinity and selectivity17. We reasoned that, for a paused Pol II enzyme, a non- template single-stranded DNA of about 20 bases around the transcription bubble could be available for protein factors to bind. Genes regulated by paused Pol II typically contain GC-rich sequences, such as GAGA motifs or the pause button motif, at their promoters18,19. The high GC content at the TSS of HSPA1B could facilitate formation of secondary structures that might form a binding motif for a specific protein factor (Supplementary Fig. 1a).
Using single-stranded DNA (ssDNA) oligo-protein binding assays and mass spectrometry, we identified the TRIM28 protein as a factor that bound specifically and selectively to the non- template strand of the HSPA1B promoter. TRIM28 is a multidomain transcriptional regulator linked to activation and repression of a subset of genes20. Its abnormal expression is implicated in disorders related to cell growth21, development22, and differentiation23,24. To understand the function of TRIM28 bound at the promoter-proximal pausing site, we performed in vitro transcription assays and in vivo KD experiments. Genome-wide ChIP-seq analyses of Pol II comparing WT and TRIM28 KD mES cells suggest a global function of TRIM28 in controlling Pol II pause release. In addition, a rapid phosphorylation event in TRIM28 was identified upon HSPA1B activation in vivo. We thus propose that a novel pausing factor, TRIM28, regulates Pol II promoter-proximal pausing and progression into processive elongation at a large number of genes in mammals, and regulation of its activity involves post-translational modification.
Results
TRIM28 binds to the HSPA1B promoter-proximal pausing site
Firstly, using protein pull-down with the immobilized HSPA1B ssDNA non-template strand, followed by mass spectrometry (MS), we screened for potential unidentified pausing factors. Considering the structural flexibility of non-template ssDNA and its likely accessibility outside the Pol II cleft25, we hypothesized that non-template DNA might be recognized by proteins that regulate pausing. The non-template ssDNA oligos of HSPA1B, +1 to +50 and +1 to +80 along with +1 to +50 of GREB1 (used as a non-pausing control gene; see Supplementary Fig. 1a)26 were subjected to pull-down assays with HeLa NE. Stringent wash conditions were applied to screen proteins bound to each oligo with high affinity. Only a few proteins were identified with high confidence through such wash conditions by MS (Fig. 1a, Table 1 and Supplementary Fig. 1b). TRIM28 was associated with the +1 to +80 sequence but not with +1 to +50 sequences of HSPA1B or GREB1 (Table 1 and Fig. 1b). Immunoblotting confirmed that TRIM28 specifically bound to the +1 to +80 sequence but not to +1 to +50 of HSPA1B or GREB1 (Fig. 1b). These results suggested that TRIM28 binds to the non-template DNA of HSPA1B, in particular the segment between +50 and +80. Although TRIM28 is not known as a single- stranded DNA binding protein (e.g. unlike PURb, Fig. 1b), we identified TRIM28 binding motifs, GCCGCG23, at +60 to +65 and +62 to +67 of the HSPA1B promoter, both with an 83% homology to consensus (Fig. 1c). These data prompted further examination to determine whether TRIM28 might function in Pol II promoter-proximal pause regulation.
Figure 1. Identification of TRIM28 bound at the HSPA1B promoter-proximal site and in vitro visualization of promoter-proximal pausing in the native human HSPA1B gene.


(a) Proteins bound to non-template ssDNAs (silver stain). SM, Size Marker; CTRL, non- template (NT) DNA of GREB1 +1 to +50; HSP1-50, NT DNA of HSPA1B +1 to +50; HSP1-80, NT DNA of HSPA1B +1 to +80. (b) Immunoblots showing specific binding of TRIM28 to +1 to +80 of the HSPA1B NT DNA. Oligo, the NT DNAs used for the pull-down assay. Uncropped images are shown in Supplementary Data Set 1a (c) The sequence of the HSPA1B promoter- proximal site. TRIM28 Binding Motifs (TRIM28*) are shown in italics and underlined. (d) The immobilized HSPA1B DNA template used in this study. MBS, Myc Binding Site; HSE, Heat Shock Element (HSF1 binding site); TATA, TBP binding motif. (e) A schematic of in vitro transcription assays used in this study. (f) In vitro transcription assay showing a stable promoter- proximal Pol II pause around +70 from TSS of human HSPA1B. SM, Size Marker; Run-off, full- length transcripts; Paused, RNA transcripts generated by paused Pol II; Aborted, short abortive RNA products.
Table 1. Summary of MS data from the ssDNA pull-down assay.
Proteins commonly identified from the control (GREB1) and HSPA1B oligos (HSPA1B 1-50 and HSPA1B 1-80) are marked in italics. Seven proteins in bold were confirmed through immunoblotting. The MOWSE score and number of peptide matches are listed in a parenthesis.
| Control (GREB1) | HSPA1B 1-50 | HSPA1B 1-80 |
|---|---|---|
|
| ||
| CSDA (374;13) | CSDA (616;23) | CSDA (422;21) |
| YBX1 (262;7) | YBX1 (601;23) | TRIM28 (232;2) |
| U2AF2 (72;1) | HLTF (93;4) | YBX1 (193;12) |
| MSH6 (58;1) | MSH2 (73;3) | PURb (112;2) |
| FEN1 (53;1) | ACACA (71;1) | |
| PCMT1 (63;2) | ||
| SSBP (62;1) | ||
| PC4 (52;1) | ||
| DHX36 (50;1) | ||
Pol II pausing at human HSPA1B in vitro
A biotinylated fragment of the native human HSPA1B gene (−467 to +216) was immobilized onto streptavidin beads. This template was used to assess transcription in vitro using HeLa NE (Fig. 1d,e and Supplementary Fig. 2b). Importantly, our transcription system visualized all species of de novo transcripts, enabling us to monitor transcription activities ranging from abortive initiation to processive elongation. Through this method, a Pol II pausing site was visualized on human HSPA1B. Upon titrating NE, a stable and prominent RNA transcript was observed at around +70 without detectable run-off or extended transcripts, suggesting a native tendency of the HSPA1B template for Pol II pausing (Fig. 1f). We also noted that Pol II pausing occurred near the TRIM28 binding motif (Fig. 1c). These data suggested that our in vitro system could accurately recapitulate stable Pol II pausing at the HSPA1B promoter, with stable pausing occurring at around +70, near the TRIM28 binding site.
TRIM28 attenuates HSF1-mediated pause release in vitro
We hypothesized that TRIM28 might repress HSPA1B transcription by augmenting promoter- proximal pausing. To assess this possibility, transcription assays were performed as described (Fig. 1e), using TRIM28-immunodepleted HeLa NE (TRIM28ΔNE) (Fig. 2a and Supplementary Fig. 2e). TRIM28ΔNE displayed transcription potential comparable to WT NE (Fig. 2b), indicating that TRIM28 depletion does not affect the general transcription efficiency at the HSPA1B promoter under these conditions.
Figure 2. TRIM28 regulates Pol II promoter-proximal pausing upon HSPA1B activation.
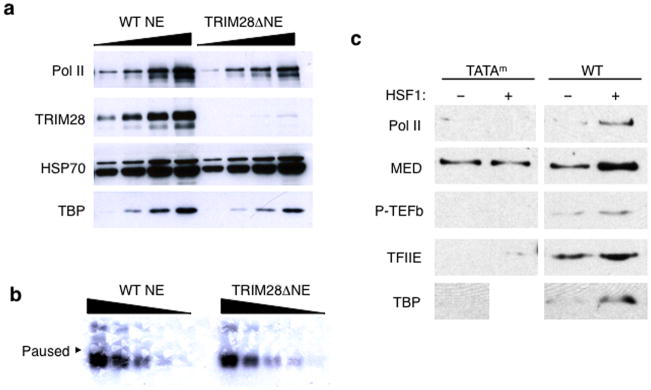

(a) Immunoblots showing TRIM28 immuno-depletion from HeLa NE. Uncropped images are shown in Supplementary Data Set 1b. (b) In vitro transcription assay with the HSPA1B template displaying comparable transcription capability between WT and TRIM28ΔNE. An uncropped image is shown in Supplementary Data Set 1c (c) Immobilized template assay showing HSF1 stimulates PIC assembly at HSPA1B. TATAm, TATA box mutant template; WT, wild type template. Uncropped images are shown in Supplementary Data Set 1d. (d) In vitro transcription assay showing HSF1-mediated transcription activation in WT and TRIM28ΔNE. t0, HSF1 added during PIC assembly. An uncropped image is shown in Supplementary Data Set 1e. (e) In vitro transcription assay demonstrating the function of TRIM28 in Pol II promoter- proximal pausing. t1, HSF1 supplied to PIC, immediately before NTP addition; t2, HSF1 added after Pol II pausing. An uncropped image is shown in Supplementary Data Set 1f. (f) In-gel digestion of gel pieces (Gel I, Gel II, and Gel III) followed by MS identifying the proteins co- immunodepleted with TRIM28. Peptide number identified with each protein is marked in parentheses. (g) In vitro transcription assay showing increased accumulation of paused RNA transcripts with addition of full-length TRIM28 (FL-T) to TRIM28ΔNE. An uncropped image is shown in Supplementary Data Set 1g.
In vivo, HSF1 is rapidly recruited to HSP70 promoters upon heat-shock to activate transcription27. In our immobilized template assays, HSF1 enhanced recruitment of Pre-Initiation Complex (PIC) factors and P-TEFb proteins to the HSPA1B template DNA (Fig. 2c). Therefore, we included HSF1 to recapitulate HSPA1B activation in vitro. Native PIC and naturally associating proteins were assembled on the template using NE. Proteins unbound or bound loosely were removed by washing with transcription buffer; NTPs, including radioactively labeled CTP, were then added to initiate transcription and to allow visualization of nascent RNA products (Fig. 1e).
HSF1 was introduced to the transcription reaction at different time points in order to stimulate HSPA1B at distinct transcriptional stages: t0, during PIC formation; t1, immediately after PIC formation; and t2, after Pol II paused at the promoter-proximal site (Fig. 1e). Evidence for Pol II pausing was observed in the absence of HSF1 in both WT and TRIM28ΔNE with little evidence of run-off transcripts (Fig. 2d, lanes 1 & 3). This suggested that Pol II pausing occurred in uninduced HSPA1B with or without TRIM28. When HSF1 was added at t0, transcription was stimulated in both WT and TRIM28ΔNEs (Fig. 2d, lanes 2 & 4), perhaps due to enhanced recruitment of PIC proteins by HSF1 (Fig. 2c). Importantly, pausing was dramatically diminished in TRIM28ΔNE, but not in WT NE, when HSF1 was introduced to the established PIC (t1) or the paused complex (t2; Fig. 2e). Thus, HSF1 added at t1 and t2 appeared to release paused Pol II more effectively in the absence of TRIM28. Addition of HSF1 at t1 (after PIC assembly) or t2 (after formation of paused Pol II) mimics HSF1-mediated HSPA1B activation in vivo in which Pol II has assembled into the PIC or has paused near the promoter. Because paused transcripts (+70) were diminished and run-off transcript levels increased in TRIM28ΔNE, these results suggested that TRIM28 could be functioning to stabilize paused Pol II in the presence of HSF1.
To verify that other proteins immunodepleted along with TRIM28 were not contributing to the difference in TRIM28ΔNE activity, we identified proteins bound to the anti-TRIM28 antibody by MS (Fig. 2f and Supplementary Data Source 1,2). Based on their known functions, the proteins that co-immunoprecipitated appeared unlikely to function in promoter-proximal pausing. More importantly, purified TRIM28 re-supplied to TRIM28ΔNE in the transcription assay restored Pol II pausing, validating that TRIM28 was indeed responsible for the formation of stably paused Pol II complexes (Fig. 2g and Supplementary Fig. 2c).
TRIM28 controls expression of known paused genes in vivo
Because TRIM28 was shown to affect HSPA1B transcription in vitro, we next examined whether TRIM28 KD would alter expression of HSPA1B in cells. Since TRIM28 depletion facilitated Pol II pause release in vitro, we hypothesized that Pol II pausing may become attenuated in TRIM28 KD cells, and increase the basal levels of HSPA1B mRNA expression in vivo. Using two short- hairpin RNA (KD1 and KD2) species targeting TRIM28, the factor was knocked-down in HEK293 cells. As shown in Fig. 3a, the mRNA and protein levels of HSPA1B, measured by quantitative PCR (qPCR) and immunoblotting, were increased in TRIM28 KD cells but not in controls with scrambled shRNA species. Therefore, as expected, Pol II pausing appeared attenuated in TRIM28 KD cells, permitting Pol II progression from the pause site into the ORF of HSPA1B.
Figure 3. TRIM28 KD increases the expression of paused genes in vivo.

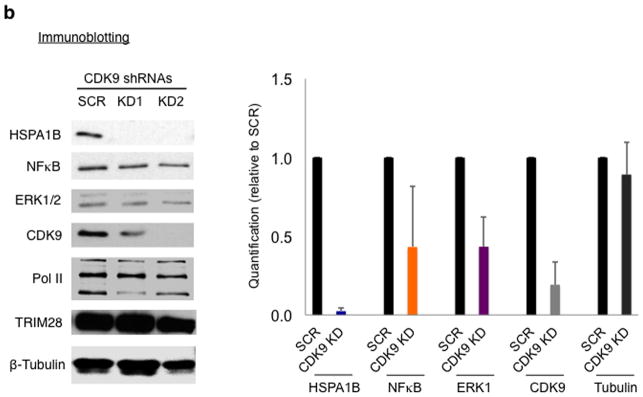

(a) HSPA1B mRNA and primer sets for qPCR (arrows, upper left). qPCR data (left and bottom right panel, n= 2 cell cultures) and western blot (upper right panel) showing increased expression of HSPA1B in TRIM28 KD cells. SCR, HEK293 cells transfected with a scrambled shRNA; KD1 and KD2, TRIM28 KD cells with two different shRNA species targeting TRIM28. Uncropped images are shown in Supplementary Data Set 1h,i,k. (b) Western blot data in CDK9 KD HEK293 cells. SCR, HEK293 cells transfected with a scrambled shRNA; KD1 and KD2, CDK9 KD cells by two different shRNAs. Error bars represent standard deviations (n= 3 cell cultures) (c) qPCR (n= 2 cell cultures) and immunoblotting of NFκb and ERK1, showing increased mRNA (left and upper right panel) and protein (bottom right panel) levels upon TRIM28 KD. Uncropped images are shown in Supplementary Data Set 1j,k.
Recently, it has been reported that genes in immune responsive and signal transduction pathways can be highly paused28. Because TRIM28 is known to regulate immune responses and cell proliferation21,24, we examined NFκB and ERK1. These genes broadly regulate immune response and proliferation and were shown to have paused Pol II at their promoters28. Consistent with this, CDK9 KD reduced the expression of HSPA1B, NFκB, and ERK1, suggesting these genes are dependent on P-TEFb and thus regulated by Pol II pausing in HEK293 cells (Fig. 3b). We also noted that NFκB and ERK1 possess TRIM28 binding motifs within their promoter regions (+47 to +52 for ERK1 and +7 to +12 for NFκB with 100% consensus). Immunoblotting and qPCR indicated up-regulation of NFκB and ERK1 in TRIM28 KD cells (Fig. 3c).
TRIM28 regulates Pol II pausing genome-wide
Existing ChIP-chip data indicate that TRIM28 is frequently located near the TSS of many protein-coding genes23. Based upon our cellular, biochemical, and in vitro data, we hypothesized that TRIM28 may function in Pol II promoter proximal pausing genome-wide. ChIP-seq was therefore performed to investigate the global impact of TRIM28 on Pol II occupancy. Given the overall coding conservation between mouse and human29, murine ES (mES) cells provided a useful model system for this investigation. Specifically, the pausing genes evaluated in this study such as EGR1, JUN, ERK1, and HSPA1B are highly conserved between human and mouse with over 92% coding similarity.
TSS and TTS-proximal windows were defined as start or end + or −250 respectively. Gene body windows were defined as +500 to +2500; if the size of a gene was smaller than 2500 bp, the gene body window was defined as +500 to the gene end. When TRIM28 was knocked-down using siRNA in mES cells (Fig. 4a), the pausing index (Pol II TSS occupancy/gene body occupancy) was either increased or decreased by over twofold at 41.3% of mappable protein coding genes, compared to WT. As expected, the pausing index of established paused genes such as JUN and EGR1 was decreased in TRIM28 KD (Fig. 4b). The pausing index of HSPA1B and ERK1 was decreased consistent with the in vivo KD and gene expression results (Fig. 3 and 4b).
Figure 4. TRIM28 regulates Pol II pause release.


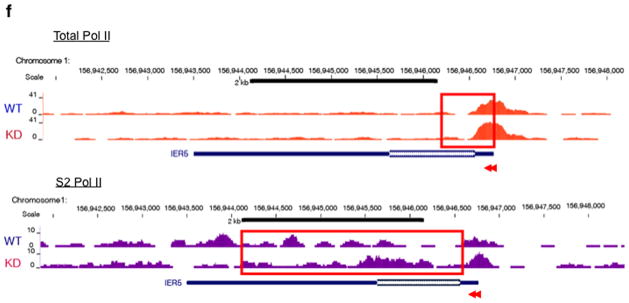
(a) TRIM28 KD in mES cells using two different siRNAs (si2 and si3). Immunoblotting (upper panel) and cell morphology (bottom panel) are shown, compared with control (Mock). si3 was used for ChIP-seq. (b) Pausing index change at individual genes upon TRIM28 KD. (c) Box plots showing change in gene body Pol II occupancy at paused genes, indicating a modest increase in gene body Pol II for TRIM28 bound genes upon TRIM28 KD (p-value based on Wilcoxon Rank Sum test). Center line: median; Box limits: 25th and 75th percentile (interquartile range); Upper Whisker: largest observation less than or equal to 75th percentile plus 1.5x the interquartile range; Lower Whisker: smallest observation greater than or equal to 25th percentile minus 1.5x the interquartile range. (d) Heat maps of total Pol II sorted by WT gene body Pol II occupancy (N=32,729). Normalized, input adjusted total Pol II ChIP-seq reads from WT mES cells (WT) and TRIM28 KD (KD), and changes upon knock down (KD-WT) are shown. (e) Metagene analysis from Ser2 phospho-Pol II ChIP-seq comparing 15,917 coding genes. (f) Chromosome views of total Pol II and Ser2-Phospho-Pol II (S2 Pol II) in IER5 (Immediate Early Response 5), illustrating facilitated Pol II entry into the downstream of the promoter-proximal pausing site in TRIM28 KD. TSS and orientation of IER5 are marked with arrowheads and the regions with noticeable increase in Pol II occupancy in KD are embraced in red boxes.
We also report a series of genes in different signaling pathways whose pausing index was notably altered by TRIM28 KD (Table 2). In agreement with our results, a metagene analysis based on previous TRIM28 ChIP-chip data23 indicate that TRIM28 bound genes increased the mean gene body Pol II occupancy upon TRIM28 KD (Fig. 4c and Supplementary Fig. 3a,b). Importantly, TRIM28 KD led to elevation of Pol II occupancy within the gene body (between +250 and +999 from the TSS) at a number of coding genes (Fig. 4d and Supplementary Fig. 3c–e and 4a–d). TRIM28 KD also increased Ser2-phospho Pol II occupancy within the gene body, between +250 and +999 from the TSS, as shown by metagene analysis of 15,917 coding genes (Fig. 4e). Chromosome views of individual loci (IER5 and DQ072391) further illustrate a role for TRIM28 in Pol II progression (Fig. 4f and Supplementary Fig. 6). At IER5, Pol II occupancy increases downstream of TSS and in the gene body as assessed by total Pol II (Fig. 4f, upper panel) and Ser2-phospho Pol II (Fig. 4f, bottom panel) upon TRIM28 KD. Overall, these results support a novel function for TRIM28 in regulating Pol II promoter-proximal pausing and pause release.
Table 2.
Genes with altered pausing index in TRIM28 KD mES cells
| Apoptotic pathway
| |
|---|---|
| Gene | Pausing index (% of WT) |
|
| |
| Casp3 | 9.5 |
| Casp7 | 19.3 |
| Casp2 | 0.7 |
| Jun | 0.1 |
| Bat3 | 6.5 |
| Bid | 14.4 |
| Bak1 | 26 |
| Fastk | 22.6 |
| Fadd | 16.7 |
|
| |
|
Cell growth and tumorigenesis
| |
| Gene | Pausing index (% of WT) |
|
| |
| Gmnn | 24.3 |
| Rbbp8 | 14.2 |
| Sipa1 | 3.9 |
| FGFR1 | 23.6 |
| Rb1 | 1.34 |
|
| |
|
Immune response
| |
| Gene | Pausing index (% of WT) |
|
| |
| NFκB | 2179.3 |
| Bcl3 | 21.3 |
| Irak1 | 22.3 |
| Traf3 | 16 |
| Il17b | 0.03 |
| Jab | 9.7 |
| Mal | 13.3 |
|
| |
|
MAPK pathway
| |
| Gene | Pausing index (% of WT) |
|
| |
| Mapk3 | 36.7 |
| Mapk7 | 39.9 |
| Tirap | 13.3 |
| Map3k4 | 10.7 |
| Map3k11 | 11.2 |
| Braf | 26 |
Rapid phosphorylation of TRIM28 upon HSPA1B activation
We next attempted to identify mechanisms through which TRIM28 inhibition of Pol II pause release might be regulated. We initially asked whether TRIM28 was released from the HSPA1B promoter upon activation of HSPA1B transcription. Thus HEK293 cells were heat-shocked over a time course of 0, 0.5, 2, and 5 minutes and TRIM28 occupancy at HSPA1B was monitored by ChIP followed by qPCR with primer sets to amplify the HSPA1B promoter and TSS (Fig. 5a). As reported for Drosophila HSP7027, HSF1 was rapidly recruited to the HSPA1B promoter within 30 seconds of heat-shock (Fig. 5b). Notably, TRIM28 did not dissociate from the TSS of HSPA1B at these time points (Fig. 5c).
Figure 5. Release of TRIM28-mediated transcriptional repression involves TRIM28 phosphorylation at S824.




(a) HSPA1B promoter and TSS with marked primers (arrowheads) used in ChIP-qPCR; HSE, Heat Shock Element (HSF1 binding site). (b) ChIP-qPCR showing rapid association of HSF1 with the HSPA1B promoter upon heat-shock. (c) ChIP-qPCR showing TRIM28 remaining at HSPA1B TSS upon transcriptional activation. (d) ChIP-qPCR showing TRIM28 (S824) phosphorylation upon transcriptional activation. (e) ChIP-qPCR displaying that DNA-PK and ATM inhibitors interfere TRIM28 S824 phosphorylation and Pol II progression into the HSPA1B gene body upon heat-shock. Uncropped images for Fig. 5b-e are shown in Supplementary Data Set 1l,m. (f) In vitro transcription assay showing that TRIM28ΔCTD omitting S824 stabilizes Pol II pausing. (g) In vitro transcription assay validating that phosphorylation of TRIM28 S824 regulates Pol II pause release at HSPA1B. An uncropped image is shown in Supplementary Fig. 8b. (h) The efficiency of WT, S824A or S824D TRIM28 in stabilizing Pol II pausing in vitro was quantified (error bars, s.d., n= 3 technical replicates). (i) Model of TRIM28-mediated pausing regulation. Top: TRIM28 bound near the TSS represses transcriptional elongation by stabilizing Pol II promoter-proximal pausing. Bottom: a gene-activating signal modulates TRIM28 to suppress its repressive function and Pol II is released from the pausing site into the gene body. At HSPA1B, TRIM28 remains at the TSS but is phosphorylated at S824 by DNA-PK and ATM upon heat-shock. This post-translational modification correlates with Pol II pause release at HSPA1B.
TRIM28 is reportedly phosphorylated at S824 under specific circumstances, which has been associated with chromatin structural changes30–32. In addition, a study showed that TRIM28 was phosphorylated at S824 when p21 (CDKN1A), a TRIM28 regulated gene, was transcriptionally activated33. Therefore, we inspected TRIM28 S824 phosphorylation at HSPA1B upon heat-shock induction. HSPA1B expression in HEK293 cells was induced by heat-shock over a time course of 0, 0.5, 2, and 5 minutes, and phospho-TRIM28 (S824) occupancy at HSPA1B was quantified using ChIP followed by qPCR. It was observed that TRIM28 became rapidly phosphorylated at S824, an effect apparent within 30 seconds of heat-shock, and dramatically increased in an activating signal dependent manner (Fig. 5d).
To search for the kinase that phosphorylates TRIM28 at S824, we utilized immunoprecipitation followed by MS. Because protein kinases were not among the few proteins that co- immunoprecipitated with TRIM28 under stringent wash conditions, we initiated a set of TRIM28 IP experiments that utilized less-stringent washes followed by medium salt elutions (Supplementary Fig. 7b). Through this method, DNA-dependent Protein Kinase (DNA-PK) was identified by MS to co-immunoprecipitate with TRIM28 (See Supplementary Data for list of identified proteins). In addition, Ataxia Telangiectasia Mutated (ATM), another member of the PI-3 kinase family, is known to phosphorylate TRIM28 at S824 in the DNA repair process32,33. Therefore, we investigated whether DNA-PK or ATM were required for TRIM28 phosphorylation at S824 during HSPA1B activation by using specific inhibitors for each kinase, NU744134 and KU5593334. In addition, an HSF1 inhibitor, KRIBB1135 which allows HSF binding to the promoter but interferes with HSF-mediated P-TEFb recruitment, was included to test whether HSF1-P-TEFb interaction was responsible for TRIM28 phosphorylation at S824. Each kinase inhibitor (NU7441, 2 μM; KU55933, 10 μM; KRIBB11, 10 μM in 0.1% DMSO as a final concentration) was added to HEK293 cells an hour prior to 5-minute heat-shock (or non- heat shock control). The results showed that both DNA-PK and ATM inhibitors effectively reduced heat-shock induced TRIM28 S824 phosphorylation at the TSS of HSPA1B upon heat- shock (Fig. 5e). By contrast, the HSF1-P-TEFb inhibitor, KRIBB11 did not affect this phosphorylation, suggesting that TRIM28 S824 phosphorylation is not mediated by P-TEFb. Under similar experimental conditions, we measured Pol II occupancy at the TSS and a distal site of HSPA1B. Total Pol II occupancy at the TSS was increased with or without each of the inhibitors upon heat-shock (Fig. 5e), which implied that these inhibitors did not markedly influence Pol II recruitment to the TSS upon activation.
Importantly, DNA-PK and ATM inhibitors abolished the increase of Pol II occupancy at the 3″ end of HSPA1B upon heat-shock, contrasting with DMSO controls, implying reduced Pol II elongation (Fig. 5e). The inhibitor of the HSF1-P-TEFb interaction, KRIBB11, also reduced Pol II occupancy at the 3″ end of HSPA1B, probably due to reduced P-TEFb recruitment. Thus TRIM28 S824 phosphorylation appeared to be regulated by DNA-PK and ATM and correlated with Pol II progression toward the 3″ end and transcriptional elongation at HSPA1B.
Since a post-translational modification at the C-terminal domain of TRIM28 was involved in release of its repressive function, a C-terminal deletion mutant of TRIM28 (residues 1–617, TRIM28ΔCTD, Supplementary Fig. 2c) was tested for its effect on Pol II promoter-proximal pausing in vitro. When TRIM28ΔCTD was titrated to TRIM28ΔNE, Pol II pausing increased in a dose dependent manner upon HSF1 induction (Fig. 5f). This suggested that TRIM28ΔCTD could stabilize Pol II pausing.
To further test whether TRIM28 phosphorylation at S824 was critical to regulate Pol II pausing, TRIM28 S824A and S824D mutants were compared with WT TRIM28 in the in vitro transcription assay. Purified TRIM28 (WT, S824A, or S824D) was supplemented with WT or TRIM28ΔNE during PIC formation on HSPA1B template DNA (Supplementary Fig. 7a,c). As shown in Fig. 5g,h and Supplementary Fig. 8a,b, WT TRIM28 and the S824A mutant augmented Pol II pausing. By contrast, the TRIM28 S824D mutation (which mimics phosphorylation) destabilizes Pol II pausing in vitro. Together with the in vivo data (Fig. 5d,e), these results implicate TRIM28 S824 phosphorylation as a means to regulate Pol II pausing at HSPA1B. In particular, these data suggest that non-phosphorylated TRIM28 at S824 functions in stabilizing Pol II pausing whereas S824-phosphorylated TRIM28 facilitates Pol II pause release at HSPA1B.
Discussion
Using an unbiased approach, we have identified TRIM28 as a novel pausing factor that binds to the non-template DNA near the Pol II pausing site of HSPA1B. Our results suggest that TRIM28 can stabilize paused Pol II and that TRIM28 helps regulate release of paused Pol II complexes. TRIM28 depletion in vitro facilitated Pol II pause release by HSF1. In vivo, TRIM28 contributes to Pol II promoter proximal pausing genome wide, based upon experiments that showed increased Pol II occupancy downstream from the TSS (> +250) at a wide spectrum of genes following TRIM28 knockdown. In addition, we have shown that TRIM28 was phosphorylated at S824 upon HSPA1B activation, dependent on DNA-PK and ATM. Our in vivo and in vitro data suggest that TRIM28 phosphorylation at S824 regulates Pol II pause release.
TRIM28 has been shown to be a powerful suppressor of transcription through the formation of complexes with ZNFs and HP1 proteins36, and TRIM28 interacts with the repressive NuRD complex and SETDB137. Previous studies have shown that TRIM28 phosphorylation at S824 can promote chromatin relaxation30,32; similar chromatin-based changes could facilitate Pol II release from the pausing site of HSPA1B. It will be of future interest to determine whether this phosphorylation event regulates TRIM28 interaction with histone modifiers such as SETDB1 and the NuRD complex at HSPA1B. Notably, TRIM28 interaction with SETDB1 and the NuRD complex requires sumoylation on TRIM28, and this modification is inhibited by phosphorylation of TRIM28 at S82433,37. Furthermore, nucleosome occupancy is enriched at many genes immediately downstream of the Pol II pausing site at the +1 nucleosome38 and TRIM28 may thus function in stabilizing paused Pol II through interaction with such structures.
Our observation that TRIM28 regulates Pol II pausing and pause release at a broad array of protein-coding genes suggests that diverse regulatory mechanisms and signaling pathways might converge upon TRIM28 to regulate Pol II activity. TRIM28 is known to locate at the TSS of genes that regulate the cell cycle and growth, and deregulation of TRIM28 expression is closely linked to development of different cancers23,39. Consistently, our Pol II ChIP-seq data indicated that TRIM28 KD alters the pausing index of genes involved in cell growth and apoptotic pathways. It is plausible that disruption of TRIM28 function or expression de-regulates expression of key growth or tumor suppressor genes by altering the activity of paused pol II complexes. Thus, TRIM28 and the signaling pathways that control its phosphorylation state may provide a means to selectively control this key regulatory checkpoint, which could ultimately yield new therapeutic approaches for a subset of cancers.
Online Methods
Non-template DNA-Pulldown assay
Avidin beads were suspended in 0.1 M KCl HEMG. Forty μl of the beads were incubated with 1.4 nmoles of biotinylated oligos for 30 minutes at room temperature (RT). The oligo amount per reaction was determined by the concentration provided by the manufacturer and also quantified by denaturing PAGE (Fig. 1b). After spin- down, the supernatant was discarded and the beads were incubated with 1.6 mg of the nuclear extracts for 2 hours at 4 °C. Unbound proteins were removed by discarding the supernatant. The beads were washed with 0.5 M KCl HEMG (20 mM HEPES, pH 7.6, 10% glycerol, 500 mM KCl, 0.1 mM EDTA 7 mM MgCl2) including 0.1% NP40, followed by a final wash with 0.1 M KCl HEMG (7 mM MgCl2, 0.02% NP40). After washes, the bound proteins were eluted with the elution buffer (2% Sarkosyl, 0.02% NP40, 0.1 M HEMG). The elution was analyzed by the SDS- PAGE followed by silver-staining, MS, and WB.
Mass Spectrometry
SDS-PAGE gel slices were processed for LC/MS/MS based protein identification using the following method. Gel slices were washed 3X in 50% (v/v) acetonitrile. Proteins in each gel slice were then reduced by incubation in 100 mM NH4CO3, 10 mM DTT at o 56°C for 45 minutes. The DTT was washed away and reduced disulfide bonds were blocked by incubation in a solution of 100 mM NH4CO3, 55 mM iodoacetamide (IAA). The gel slice was then washed 3X to remove the IAA. Proteins were digested by incubating in 100 mM NH4CO3 containing 0.05 mg/ml sequencing grade bovine trypsin (ABSciex) overnight at 37°C. Generated peptides were extracted from gel slices by washing 3X in 40% (v/v) acetonitrile, 1% TFA, then collecting and pooling the supernatant. The volume of extracted peptides was reduced using vacuum centrifugation. Peptides from each gel slice were then run over a reverse phase column (C18 PepMap, Dionex) using nanoflow LC (Ulitmate Plus, Dionex) and printed to 4800 MALDI target plates by an in-line printing robot (Probot, Dionex) that mixed in CHCA (5.0 mg/ml stock solution) as the matrix. Peptides were identified by MALDI-TOF/TOF mass spectrometry on an ABSciex 4800 Plus instrument. Protein Pilot 3.0 software was used for peptide and protein identification.
Immobilized template assay and transcription assay
Dynabeads M-280 Streptavidin (Invitrogen) was prepared with 2X B&W buffer (10 mM Tris-HCl, pH 7.5, 1 mM EDTA, 2 M NaCl) and incubated with the biotin-conjugated HSPA1B template DNA (−467 to +216) at 2 fmol DNA/μg beads. The template-conjugated beads were washed with 1X B&W buffer and 0.1 M Buffer D (20% Glycerol, 20 mM HEPES, pH 7.6, pH 7.9, 0.1 mM EDTA, 100 mM KCl). Twenty five μg of beads-DNA complex was mixed with TF buffer (12.5 ng/μl dI-dC, 0.075% NP40, 5 mM MgCl2, 250 ng/μl BSA, 12.5 % Glycerol, 100 mM KCl, 12.5 mM HEPES, pH 7.6, 62.5 μM EDTA, 10 μM ZnCl2) for pre-incubation with transcription factors when indicated as t0. The resultant template-protein complex was pulled-down using a magnet stand (Invitrogen) and resuspended in NE buffer (17.5 ng/μl dI-dC, 0.1% NP40, 7.5 mM MgCl2, 1.25 μg/μl BSA, 8.7% Glycerol, 8.7 mM HEPES, pH 7.6, 44 μM EDTA, 130 mM KCl, 10 μM ZnCl2). HeLa NE was added at 160 μg/reaction and incubated for 30 minutes at room temperature (RT). The loosely or unbound proteins were removed by multiple washes with Wash buffer (0.0125% NP40, 833 nM MgCl2, 12.5% Glycerol, 12.5 mM HEPES, pH 7.6, 62.5 μM EDTA, 500 mM KCl). The proteins stably bound to the template were eluted by 2% Sarkosyl for Western blotting. For transcription assay, the steps are identical described above until the HeLa NE incubation except that 100 ng of the template DNA and 160 μg of HeLa NE per a reaction were used to assemble PIC. The template-protein complex was washed briefly with a 10 beads volume of TW buffer (13 mM HEPES, pH 7.6, 13% Glycerol, 60 mM KCl, 7 mM MgCl2, 7 mM DTT, 100 μM EDTA, 0.0125% NP40, 10 μM ZnCl2) and then resuspended in Transcription Buffer I (13 mM HEPES, pH 7.6, 13% Glycerol, 60 mM KCl, 7 mM MgCl2, 10 μM ZnCl2, 7 mM DTT, 100 μM EDTA, 15 ng/μl dI-dC, 10 mM Creatine phosphate). When indicated as t1, a transcription factor(s) was added immediately after supplying Transcription Buffer I. A mixture of NTP in final concentrations of 250 μM A/G/U and 10 μM C and 5–10 μCi CTP was added to initiate Pol II to polymerize mRNA molecules. When indicated as t2, a transcription factor(s) was introduced after 10–15 minutes of NTP addition. The reaction was allowed for 30 or 45 minutes as a total incubation time and collapsed with 5 volumes of 1.2 X Stop buffer (0.6 M Tris-HCl, pH 8.0, 12 mM EDTA, 100 μg/mL tRNA). The mixture was treated with an equal volume of Phenol: Chloroform: Isoamyl alcohol (25:24:1) to extract proteins and then the soluble phase was precipitated with 2.6 volumes of 100% Ethanol. RNA transcripts were separated in denaturing polyacrylamide gels and exposed to X-ray film. In order to evaluate the transcriptional potential of nuclear extracts or to characterize the HSPA1B template DNA, titrating amounts of WT or TRIM28ΔNE between 160 ng and 1.6 μg were mixed with 50 ng of the HSPA1B template DNA in Transcription buffer II (13 mM HEPES, pH 7.6, 13% Glycerol, 60 mM KCl, 7 mM MgCl2, 1 mM DTT, 100 nM EDTA, 200 ng/μl BSA, 1 μM ZnSO4), followed by incubation for 10 minutes. Transcription was initiated by NTP and allowed for 30 minutes before stopping the reaction and collecting RNA molecules as described above. In the TRIM28 replenishment experiment, approximately 50 ng of purified full-length TRIM28 was added in 160 μg of WT or TRIM28ΔNE and incubated for 10–20 minutes at RT before incubating the mixture with the DNA template. The full-length TRIM28 was assayed in 100 μM instead of routine 10 μM of ZnCl2 in the buffers described above. For the replenishment and titration experiment of TRIM28ΔCTD, 16 μg of NE or TRIM28ΔNE pre-supplemented with purified TRIM28ΔCTD was incubated with 50 ng of the HSPA1B template DNA in Transcription buffer II for 10–20 minutes at RT. Then, ATP was added in a 500 μM final concentration and the mixture was incubated for 10 minutes before addition of NTP (500 μM A, G, and U; 20 μM C in the final concentration) to initiate transcription. HSF1 was added in 10 minutes after NTP. All transcription reactions were done in 25 μl in a final volume per a reaction. TF, NE, TW, and Transcription buffer I described above included fresh protease inhibitors of 1 mM Benzamidine, 0.25 mM PMSF, aprotinin, sigma A6279, in 1:1000, and 1 mM Na metabisulfite.
ChIP-seq
Replicate libraries from input DNA as well as both S2 and total Pol II ChIP-seq from wild type and TRIM28 KD mES cells were examined. Sequence data from all 12 libraries were individually quality filtered using a mean base quality score <Q20 threshold. Filtered reads were then aligned as single end 36mers to the mouse mm9 reference genome index using the Bowtie short-read alignment program (v0.12.8 employing parameters -v2, -m1) retaining reads mapped to unique genomic locations with at most 2 mismatches. Aligned reads were then de-duplicated using Picard tools for subsequent analyses and the generation of coverage tracks. DNA fragment sizes were estimated to be approximately 150nt using HOMER 4.1 ChIP-Seq analysis tool. Reads were thus center shifted half the fragment length 75nt for downstream analyses. Gene model annotations were downloaded from the mm9 UCSC Known Gene table on March 18th 2013. Genes annotated to chrM as well as random chromosomes were excluded from downstream analyses. Additionally, in order to differentiate TSS-proximal and gene body search windows, genes smaller than 1000nt were excluded. Genes with multiple annotations initiating from the identical TSS were limited to a single entry to minimize redundant search spaces. For comparative analyses, replicates were combined and normalized by uniquely mappable depth, based on the assumption that global PolII occupancy is the same in WT and KD samples. Normalized DNA input reads were then subtracted for each window examined (search windows with negative read counts were set to zero). TSS-proximal windows were defined as start + or − 250; gene body windows were defined as +500 to +2500 or gene end if smaller than 2500, and the unique mappability of single end 36mers was determined for all search windows. Pausing indexes were defined as the ratio of reads per uniquely mappable base in the TSS-proximal window to the gene body window; gene models in which the TSS-proximal or gene body window was less than 50% mappable were excluded from pausing index-based analyses. Heat maps were generated using Partek Genomics Suite 6.5 and depict total Pol II ChIP-seq normalized and input-subtracted read counts in 50 nt bins, tiling the −500 to +999 region about each TSS. All heat maps are sorted by WT gene body Pol II occupancy, defined as the total normalized, input-subtracted read count in bins from +250 to +999. Box plots were generated using R 3.0.1, and depict change in gene body PolII ChIP-seq reads per kilobase. Genes were defined as bound by TRIM28 as in the original publication23, while CpG island intersection was based on the TSS + or −250 region and the UCSC Browser CpG Islands track, downloaded November 2013. Genomic data described in this study have been deposited in the Gene Expression Omnibus under accession number GSE48253.
DNA templates, plasmids, protein purification, and cells and standard methods (Western blot and immunoprecipitation, qPCR, and ChIP) are described in Supplementary Note. Original images of gels, autoradiographs and blots used in this study can be found in Supplementary Data Set 1.
Supplementary Material
Highlights.
TRIM28 binds specifically to the non-template DNA of human HSPA1B promoter
TRIM28 controls Pol II pausing and pause release at HSPA1B
TRIM28 modulates Pol II promoter-proximal pausing and elongation genome-wide
TRIM28 phosphorylation at S824 releases Pol II pausing and occurs upon HSPA1B transcriptional activation
Acknowledgments
We appreciate S. Elledge at Harvard Medical School for mediating collaborations and R. Young and D. Orlando at Massachusetts Institute of Technology (MIT) for providing with helpful comments and perspectives for the manuscript. We thank A. York, A. Schubert, M. Knuesel, T. Westerling, and J. Stinchfield for technical support and thank C. Wu in US National Cancer Institute (NCI) for HSF1 vector, R. Kelm Jr. at the University of Vermont for PURb antibody, and F. Zhu at the Florida State University for GST-TRIM28 vectors. We appreciate the Alper, the Pollak, and the Brown labs at Beth Israel Deaconess Medical Center for technical assistance, equipment, and discussions. First author thanks M. Cross and M. A. Stevenson at Beth Israel Deaconess Medical Center for administrative supports and J. Park and D. Bunch for loving encouragement throughout the work. This study was supported by grants from US National Institutes of Health (RO-1CA047407) and The Harvard JCRT to SKC and HB, by the Koch Institute for Integrative Cancer Research at MIT (P30-CA14051) and the MIT Center for Environmental Health Sciences (P30-ES002109) to SM and SL, and also by the NCI (R01 CA127364) and the American Cancer Society (RSG 0927401DMC) to DJT. This research was supported in part by the Intramural Research Program of the National Institutes of Health, National Institute of Environmental Health Sciences (1ZIAES102745-02) to GH and XZ and to AB and DF.
Footnotes
Accession Code
ChIP-seq genomic data described in this study have been deposited in the Gene Expression Omnibus under accession number GSE48253.
Author Contributions:
XZ and GH generated WT and TRIM28 KD mES cell extracts for ChIP-seq and ChIP-qPCR. SM and SL processed ChIP-seq. AB, GH, and DF performed Bioinformatics. STD and CE carried out MS. GB constructed TRIM28 plasmids. HB, DJT, and SKC designed the experiments and wrote the manuscript.
Competing Financial Interests
The authors declare no competing financial interests.
References
- 1.Rahl PB, et al. c-Myc regulates transcriptional pause release. Cell. 2010;141:432–45. doi: 10.1016/j.cell.2010.03.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rasmussen EB, Lis JT. In vivo transcriptional pausing and cap formation on three Drosophila heat shock genes. Proc Natl Acad Sci U S A. 1993;90:7923–7. doi: 10.1073/pnas.90.17.7923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nechaev S, et al. Global analysis of short RNAs reveals widespread promoter- proximal stalling and arrest of Pol II in Drosophila. Science. 2010;327:335–8. doi: 10.1126/science.1181421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Muse GW, et al. RNA polymerase is poised for activation across the genome. Nat Genet. 2007;39:1507–11. doi: 10.1038/ng.2007.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Core LJ, Waterfall JJ, Lis JT. Nascent RNA sequencing reveals widespread pausing and divergent initiation at human promoters. Science. 2008;322:1845–8. doi: 10.1126/science.1162228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zeitlinger J, et al. Whole-genome ChIP-chip analysis of Dorsal, Twist, and Snail suggests integration of diverse patterning processes in the Drosophila embryo. Genes Dev. 2007;21:385–90. doi: 10.1101/gad.1509607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Seila AC, et al. Divergent transcription from active promoters. Science. 2008;322:1849– 51. doi: 10.1126/science.1162253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Adelman K, Lis JT. Promoter-proximal pausing of RNA polymerase II: emerging roles in metazoans. Nat Rev Genet. 2012;13:720–31. doi: 10.1038/nrg3293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gaertner B, et al. Poised RNA polymerase II changes over developmental time and prepares genes for future expression. Cell Rep. 2012;2:1670–83. doi: 10.1016/j.celrep.2012.11.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wu CH, et al. NELF and DSIF cause promoter proximal pausing on the hsp70 promoter in Drosophila. Genes Dev. 2003;17:1402–14. doi: 10.1101/gad.1091403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Peterlin BM, Price DH. Controlling the elongation phase of transcription with P-TEFb. Mol Cell. 2006;23:297–305. doi: 10.1016/j.molcel.2006.06.014. [DOI] [PubMed] [Google Scholar]
- 12.Gilchrist DA, et al. NELF-mediated stalling of Pol II can enhance gene expression by blocking promoter-proximal nucleosome assembly. Genes Dev. 2008;22:1921–33. doi: 10.1101/gad.1643208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rabindran SK, Giorgi G, Clos J, Wu C. Molecular cloning and expression of a human heat shock factor, HSF1. Proc Natl Acad Sci U S A. 1991;88:6906–10. doi: 10.1073/pnas.88.16.6906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sun J, Li R. Human negative elongation factor activates transcription and regulates alternative transcription initiation. J Biol Chem. 2010;285:6443–52. doi: 10.1074/jbc.M109.084285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cheng B, et al. Functional association of Gdown1 with RNA polymerase II poised on human genes. Mol Cell. 2012;45:38–50. doi: 10.1016/j.molcel.2011.10.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Byun JS, et al. ELL facilitates RNA polymerase II pause site entry and release. Nat Commun. 2012;3:633. doi: 10.1038/ncomms1652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ulrich H. DNA and RNA aptamers as modulators of protein function. Med Chem. 2005;1:199–208. doi: 10.2174/1573406053175274. [DOI] [PubMed] [Google Scholar]
- 18.Hendrix DA, Hong JW, Zeitlinger J, Rokhsar DS, Levine MS. Promoter elements associated with RNA Pol II stalling in the Drosophila embryo. Proc Natl Acad Sci U S A. 2008;105:7762–7. doi: 10.1073/pnas.0802406105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lee C, et al. NELF and GAGA factor are linked to promoter-proximal pausing at many genes in Drosophila. Mol Cell Biol. 2008;28:3290–300. doi: 10.1128/MCB.02224-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Iyengar S, Ivanov AV, Jin VX, Rauscher FJ, 3rd, Farnham PJ. Functional analysis of KAP1 genomic recruitment. Mol Cell Biol. 2011;31:1833–47. doi: 10.1128/MCB.01331-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chen L, et al. Tripartite motif containing 28 (Trim28) can regulate cell proliferation by bridging HDAC1/E2F interactions. J Biol Chem. 2012;287:40106–18. doi: 10.1074/jbc.M112.380865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Messerschmidt DM, et al. Trim28 is required for epigenetic stability during mouse oocyte to embryo transition. Science. 2012;335:1499–502. doi: 10.1126/science.1216154. [DOI] [PubMed] [Google Scholar]
- 23.Hu G, et al. A genome-wide RNAi screen identifies a new transcriptional module required for self-renewal. Genes Dev. 2009;23:837–48. doi: 10.1101/gad.1769609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chikuma S, Suita N, Okazaki IM, Shibayama S, Honjo T. TRIM28 prevents autoinflammatory T cell development in vivo. Nat Immunol. 2012;13:596–603. doi: 10.1038/ni.2293. [DOI] [PubMed] [Google Scholar]
- 25.Vassylyev DG, et al. Structural basis for substrate loading in bacterial RNA polymerase. Nature. 2007;448:163–8. doi: 10.1038/nature05931. [DOI] [PubMed] [Google Scholar]
- 26.Kininis M, Isaacs GD, Core LJ, Hah N, Kraus WL. Postrecruitment regulation of RNA polymerase II directs rapid signaling responses at the promoters of estrogen target genes. Mol Cell Biol. 2009;29:1123–33. doi: 10.1128/MCB.00841-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zobeck KL, Buckley MS, Zipfel WR, Lis JT. Recruitment timing and dynamics of transcription factors at the Hsp70 loci in living cells. Mol Cell. 2010;40:965– 75. doi: 10.1016/j.molcel.2010.11.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gilchrist DA, et al. Regulating the regulators: the pervasive effects of Pol II pausing on stimulus-responsive gene networks. Genes Dev. 2012;26:933–44. doi: 10.1101/gad.187781.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Makalowski W, Boguski MS. Evolutionary parameters of the transcribed mammalian genome: an analysis of 2,820 orthologous rodent and human sequences. Proc Natl Acad Sci U S A. 1998;95:9407–12. doi: 10.1073/pnas.95.16.9407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ziv Y, et al. Chromatin relaxation in response to DNA double-strand breaks is modulated by a novel ATM- and KAP-1 dependent pathway. Nat Cell Biol. 2006;8:870–6. doi: 10.1038/ncb1446. [DOI] [PubMed] [Google Scholar]
- 31.White D, et al. The ATM substrate KAP1 controls DNA repair in heterochromatin: regulation by HP1 proteins and serine 473/824 phosphorylation. Mol Cancer Res. 2012;10:401–14. doi: 10.1158/1541-7786.MCR-11-0134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lee DH, et al. Phosphoproteomic analysis reveals that PP4 dephosphorylates KAP- 1 impacting the DNA damage response. EMBO J. 2012;31:2403–15. doi: 10.1038/emboj.2012.86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Li X, et al. Role for KAP1 serine 824 phosphorylation and sumoylation/desumoylation switch in regulating KAP1-mediated transcriptional repression. J Biol Chem. 2007;282:36177–89. doi: 10.1074/jbc.M706912200. [DOI] [PubMed] [Google Scholar]
- 34.Chen BP, et al. Ataxia telangiectasia mutated (ATM) is essential for DNA-PKcs phosphorylations at the Thr-2609 cluster upon DNA double strand break. J Biol Chem. 2007;282:6582–7. doi: 10.1074/jbc.M611605200. [DOI] [PubMed] [Google Scholar]
- 35.Yoon YJ, et al. KRIBB11 inhibits HSP70 synthesis through inhibition of heat shock factor 1 function by impairing the recruitment of positive transcription elongation factor b to the hsp70 promoter. J Biol Chem. 2011;286:1737–47. doi: 10.1074/jbc.M110.179440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wolf D, Goff SP. TRIM28 mediates primer binding site-targeted silencing of murine leukemia virus in embryonic cells. Cell. 2007;131:46–57. doi: 10.1016/j.cell.2007.07.026. [DOI] [PubMed] [Google Scholar]
- 37.Ivanov AV, et al. PHD domain-mediated E3 ligase activity directs intramolecular sumoylation of an adjacent bromodomain required for gene silencing. Mol Cell. 2007;28:823–37. doi: 10.1016/j.molcel.2007.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lee W, et al. A high-resolution atlas of nucleosome occupancy in yeast. Nat Genet. 2007;39:1235–44. doi: 10.1038/ng2117. [DOI] [PubMed] [Google Scholar]
- 39.Herquel B, et al. Transcription cofactors TRIM24, TRIM28, and TRIM33 associate to form regulatory complexes that suppress murine hepatocellular carcinoma. Proc Natl Acad Sci U S A. 2011;108:8212–7. doi: 10.1073/pnas.1101544108. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.