

Abstract
Safe and effective adjuvants are needed for many vaccines with limited commercial appeal, such as vaccines to infrequent (orphan) diseases or to neglected and poverty-related diseases. Here we found that three nonproprietary liposome formulations containing monophosphoryl lipid A each induced 3-fold to 5-fold increased titers of binding and neutralizing antibodies to anthrax protective antigen compared to aluminum hydroxide-adsorbed antigen in monkeys. All vaccinated monkeys were protected against lethal challenge with aerosolized Ames strain spores.
Keywords: Adjuvants, Liposomes, Monophosphoryl lipid A, Bacteriophage T4, Anthrax, Non-human primates
1. Introduction
A major challenge in current vaccinology is to develop new adjuvants to enhance the ability of highly purified recombinant protein and peptide antigens to induce adaptive immunity in humans [1]. Numerous commercial adjuvants and “adjuvant systems” have been tested as constituents of vaccines against various diseases [2–4] but these are often not easily accessible for many vaccine developers due to commercial or proprietary reasons [5]. Adjuvants are a priority for vaccines against biothreat agents such as anthrax and plague, and for diseases associated with poverty such as malaria, HIV/AIDS, and tuberculosis [5]. Anthrax vaccine is an example of a limited use vaccine which, because of policy and safety issues and highly specialized use, is essentially an “orphan” vaccine, as defined by U.S. law and federal regulations [6]. Current approved anthrax vaccines have many real and perceived shortcomings [7], and new types of less reactogenic and more potent adjuvants are needed for larger scale deployment of an improved vaccine [8,9].
A further challenge in vaccinology pertains to those vaccines for which human efficacy studies are not ethical or feasible. Regulatory approval for marketing such vaccines allows for efficacy testing under certain conditions in non-human animal species, the so-called “animal rule” [10], but it has been suggested, based on comparative adjuvant studies, that the relative effects observed in many commonly used animal models, such as mice, rats, guinea pigs, or rabbits, do not reliably predict adjuvant efficacy in humans [2,11,12]. Non-human primates (NHPs) are thought to be a valid surrogate model to predict the efficacies of many vaccines in humans, including recombinant anthrax protective antigen (PA) adsorbed to aluminum hydroxide (AH) [8]. However, the question still remains whether NHPs are attractive as a primary alternative to rodents to select new and superior adjuvants. Approaches designed to address this question could lead to more rapid and reliable prediction of relative potencies of vaccine adjuvants for a variety of orphan and poverty-related diseases than by comparisons of adjuvant effects in smaller animals.
2. Materials and methods
2.1. Animals and reagents
Thirty-nine Indian-origin Rhesus macaques (5–7 years old) that were negative for simian retroviruses (SRV, SIV and STLV), herpes B virus, and pre-screened for antibodies to anthrax protective antigen to ensure no prior exposure, were obtained from the Walter Reed Army Institute of Research (WRAIR) primate pool. The study was conducted in compliance with the animal welfare act and adhered to the principles in the guide for care and use of laboratory animals. The investigators used facilities accredited by the Association for Assessment and Accreditation of Laboratory Animal Care International. The WRAIR Animal Care and Use Committee approved all animal experiments. Animals were housed under ABSL-2 conditions and transferred to BSL-3 facilities at the WRAIR one week prior to challenge with Bacillus anthracis Ames strain spores.
Lipid A detoxified (Salmonella minnesota R595) which contains predominantly monophosphoryl lipid A (MPLA), 1,2 dimyristoyl-sn-glycero-3-phosphocholine (DMPC), 1,2 dimyristoyl-sn-glycero-3-phosphoglycerol (DMPG), and cholesterol were purchased from Avanti Polar Lipids, Inc. Recombinant PA produced in B. anthracis strain BH445 and purified as described earlier [13] was a gift from Dr. Stephen Leppla (NIH). Escherichia coli heat-labile enterotoxin was a gift from Dr. John Clements (Tulane University, New Orleans, LA). Alhydrogel® (AH) was purchased from E.M. Sergeant Pulp and Chemical Co., Inc.
2.2. Preparation of seven anthrax protective antigen (PA)-vaccine adjuvant formulations
(Formulation 1) Recombinant PA was adsorbed to AH to give a final dose of 200 μg/ml of PA on AH having 2.4 mg of aluminum. (Formulation 2) For transcutaneous immunization (TCI) [14,15], PA (50 μg/dose) was mixed with E. coli heat-labile enterotoxin (100 μg/dose) just before application on the surface of the skin on the arms of the Rhesus macaques (PA + HLT).
(Formulations 3 and 4) PA was over-expressed in E. coli and purified as a fusion protein with bacteriophage T4 proteins, highly antigenic outer capsid protein (Hoc) and small outer capsid protein (Soc) and then displayed on the surface of hoc−soc− bacteriophage T4 nanoparticle (T4-PA) [16–18]. For each animal, about 1.2 × 1012 of purified hoc−soc− 4 phage particles were centrifuged at 15,000 rpm for 30 min using Lobind Eppendorf tubes. The phage pellet was then resuspended in 200 μl of PBS buffer, pH 7.4. A total of 1.15 mg PA-Hoc was first incubated with phage particles at 37 °C for 45 min in a total reaction volume of 1 ml. The reaction mixture was cooled to 8 °C followed by the addition of 0.57 mg of Soc-PA for 45 min. The phage particles loaded with PA-Hoc and Soc-PA were then sedimented by high-speed centrifugation at 15,000 rpm for 30 min. The supernatant containing the unbound PA-Hoc and Soc-PA antigens was discarded and the phage pellet was washed twice with excess PBS buffer to remove any remaining unbound antigen. The pellet was resuspended in 500 μl of PBS buffer and transferred to a new Eppendorf tube and stored at 4 °C. The copy number of displayed PA-Hoc and Soc-PA per capsid was quantified by SDS-PAGE and laser densitometry (PDSI, GE Healthcare) as described earlier [16,17]. The T4-PA antigen (+/– HLT) used for immunizations contained 155 copies of PA-Hoc and 200 copies of Soc-PA per phage capsid, which is equivalent to ~50 μg of PA/animal.
(Formulation 5) PA was encapsulated in liposomes containing monophosphoryl lipid A [L(PA + MPLA)] to give a final concentration of 125 mM phospholipids, 200 μg/ml PA, and 400 μg/ml of MPLA. Detailed procedures for the preparation of liposomes and liposomal emulsion were described previously [19]. Briefly, multilamellar liposomes composed of DMPC, cholesterol, and DMPG (9:7.5:1) with MPLA were prepared by dispersion of lyophilized mixtures of lipids at a phospholipid concentration of 125 mM in Dulbecco’s PBS either containing or lacking PA. (Formulation 6) Alternatively, PA was added to preformed liposomal MPLA [PA + L(MPLA)]. (Formulation 7) Liposome-stabilized oil-in-water emulsion was formulated with L(PA + MPLA) and 20% light mineral oil. The final phospholipid concentration of the emulsion [L(PA + MPLA)–emulsion] (also referred to as PA–emulsion) was 125 mM. The emulsion was formulated by mixing the liposomes and the oil just before use as previously described [20].
2.3. Immunization and challenge
Four Rhesus macaques were used for 6 of the 7 adjuvant groups while 8 animals were used with [L(PA + MPLA)] and 7 for the naive group. The immunization and analysis scheme is shown in Fig. 1A. Rhesus macaques were immunized under ABSL-2 conditions by the intramuscular (250 μl dose/animal) or transcutaneous routes (500 μl dose/animal) at weeks 0, 4, and 8 with PA-adjuvant formulations described above. Each group consisted of 4 animals, except the naive group which had 7 animals, and the [L(PA + MPLA)] group, which had 8 animals. Each animal received 50 μg of PA and 100 μg of MPLA or 100 μg of E. coli heat-labile enterotoxin. TCI (on the arm) was carried out as previously described [14,15]. T4-PA was injected by the intramuscular route and in one of the groups HLT (100 μg/animal) was applied on the surface of the arm at the site of injection.
Fig. 1.
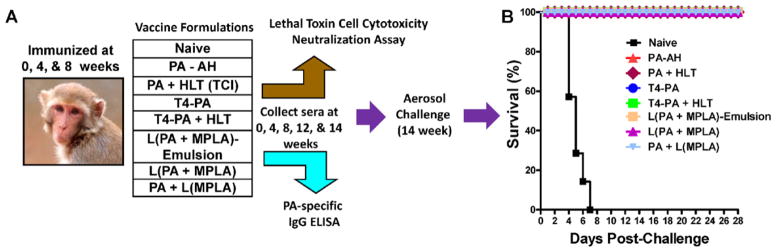
Immunization scheme and challenge with aerosolized Bacillus anthracis spores in Rhesus macaques. (A) Immunization scheme for various vaccine formulations. Each animal received 50 μg of PA/animal/immunization. (B) At week-14, animals were challenged with aerosolized B. anthracis Ames strain spores (9.6–86.9 LD50 , Table 1) and the data are shown as a Kaplan Meyer survival curve. All of the 7 naive animals succumbed to disease by day 7. All of the 32 immunized animals remained healthy and survived through the 28-day post-challenge period.
Naive and immunized animals were transferred to BSL-3 facilities at the Walter Reed Army Institute of Research on week-13 and challenged with aerosolized Bacillus anthracis Ames strain spores (9.6–86.9 LD50; see Table 1) on week-14 using a class III glove box under strict ABSL-3 conditions. The challenge consisted of 5.5–7 × 104 cfu delivered as an aerosol between 5 and 15 min using a head-only exposure system and the plethsymography data collected real-time using a Skornik plethsymography system with the animal contained in a sealed and head inside-only exposure chamber. The animals were observed at 4-h intervals daily following exposure for 7 days and then twice a day for 28 days for signs of illness or morbidity. Blood and fecal material were collected every second day after challenge for the first 7 days and then at weekly intervals until the end of the study. All surviving animals were humanely euthanized at the end of the study. All of the animals that either succumbed to the challenge or were euthanized at the end of the study were necropsied for both gross and fine histological changes. All organs were immersion fixed in neutral-buffered 10% formalin for at least 21 days before being removed from ABSL-3 containment for histology processing.
Table 1.
Summary of analyses related to the challenge of vaccinated Rhesus macaques.
| Formulation | Monkey no. | Antibodies (week-14, pre-challenge)
|
||||
|---|---|---|---|---|---|---|
| PA-specific IgG (μg/ml) | Neutralizing antibody titer (ED50 ) | Aerosol challenge dose (LD50 ) | Bacteremia blood (CFU/ml) | Bacteria in feces (CFU/g) | ||
| Naive | D88Z | <1 | 0 | 22 | 50 | 70 |
| DAIP | <1 | 0 | 19.9 | 130 | 124 | |
| DA83 | <1 | 0 | 15.9 | 2000 | 60 | |
| DA91 | <1 | 0 | 16.8 | 20 | 90 | |
| 03D085 | <1 | 0 | 26.1 | 24 | 0 | |
| 03D135 | <1 | 0 | 14.3 | No sample | No sample | |
| CL26 | <1 | 0 | 21.8 | 24 | 0 | |
| PA-AH | A0X74 | 360 | 859 | 22 | 0 | 0 |
| CJ3J | 842 | 2651 | 22.7 | 0 | 0 | |
| DA98 | 604a | 5121a | 17.9 | 0 | 0 | |
| DB10 | 299 | 1222 | 85.6 | 0 | 0 | |
| PA + HLT | D117 | 364 | 1459 | 19 | 0 | 6 |
| CX5G | 811 | 4353 | 25.3 | 0 | 0 | |
| DA2P | 641 | 2613 | 20.1 | 0 | 26 | |
| DA2W | 83 | 534 | 86.9 | 0 | 0 | |
| T4-PA + HLT | DB4T | 909 | 1604 | 15.9 | 0 | 0 |
| 04D256 | 1146 | 2809 | 21.3 | 0 | 0 | |
| 03D116 | 718 | 1439 | 9.6 | 0 | 0 | |
| 04D110 | 274 | 566 | 13.7 | 0 | 0 | |
| T4-PA | DA9V | 445 | 1354 | 85.3 | 0 | 0 |
| DA9H | 340 | 755 | 12.3 | 0 | 14 | |
| D151 | 317 | 1111 | 23.3 | 0 | 0 | |
| CL1T | 571a | 3404a | 18.3 | 0 | 20 | |
| L(PA + MPLA) | D060 | 599 | 4196 | 17.4 | 0 | 0 |
| DA1E | 1005 | 6081 | 15.3 | 0 | 6 | |
| CX5A | 671 | 1297 | 64.5 | 0 | 0 | |
| DA64 | 808 | 1930 | 53.2 | 0 | 0 | |
| 04C073 | 2918 | 6018 | 18.9 | 0 | 0 | |
| 04D145 | 1811 | 5323 | 18.7 | 0 | 0 | |
| 03D096 | 1604 | 5901 | 15.7 | 0 | 0 | |
| 04D233 | 2109 | 5344 | 15 | 0 | 0 | |
| PA + L(MPLA) | DB6V | 3653 | 10,463 | 17.6 | 0 | 0 |
| 04D109 | 1229 | 7497 | 18.8 | 0 | 0 | |
| M001 | 1375 | 6012 | 16.3 | 0 | 0 | |
| 04D085 | 3614 | 6155 | 12.5 | 0 | 0 | |
| L(PA + MPLA) - emulsion | 04D060 | 1881 | 8249 | 20.4 | 0 | 0 |
| 04D283 | 2075 | 4054 | 13.8 | 0 | 0 | |
| 04D081 | 1574 | 2099 | 9.7 | 0 | 0 | |
| DB98 | 1640 | 1448 | 14.7 | 0 | 0 | |
Values shown were obtained at week-12.
2.4. PA-specific IgG antibodies
PA-specific IgG antibodies were analyzed in individual NHP serum samples collected on weeks 0, 4, 8, 12, and 14 by ELISA as previously described [21]. Human reference serum AVR801 (pooled serum derived from AVA-vaccinated human donors, a gift from Dr. Conrad Quinn, CDC) was used to generate a standard curve, which was used to quantitate the PA-specific IgG antibodies (μg/ml).
2.5. Lethal toxin neutralizing antibodies
Lethal toxin (LTx) neutralizing antibody titers were determined by the ability of individual serum samples to neutralize the cytotoxicity of LTx in a J774A.1 macrophage cell line as described previously [15,22]. A 4-parametric sigmoid regression curve was used to determine the dilution of antiserum that resulted in 50% reduction in toxicity (ED50) of anthrax LTx. Each plate contained control wells that had either toxin alone (no antiserum) or cells alone (no toxin and no antiserum). Polyclonal rabbit anti-PA-specific antibody and human reference serum AVR801 were used as positive controls.
2.6. Statistical analysis
PA-specific IgG concentrations and LTx ED50 titers between the different PA-adjuvant vaccine formulations were analyzed using 2-way ANOVA with Bonferroni posttests.
3. Results
3.1. Challenge of immunized animals with aerosolized anthrax spores
Seven test adjuvant formulations were compared, each with anthrax PA as a model antigen, using the immunization scheme shown in Fig. 1A. The animals were challenged with 100 LD50 of Bacillus anthracis Ames strain spores at week-14, 6 weeks after the second boost (Table 1). All of the 7 naive animals died by day 7 (Fig. 1B and Table 1). However, all the animals in each of the adjuvant groups survived (Fig. 1B).
The observed gross and histopathologic lesions in naive animals, examined first and unblinded for comparative purposes, were consistent with documented lesions of inhalation anthrax infection in Rhesus macaques, although there were some slight variations in the lesions among the 7 naive animals (Table 1). Bacilli were noted in vessels and other vascular spaces in the following tissues: tongue, mandibular lymph node, mandibular salivary gland, aorta, trachea, lung, mediastinal lymph node, liver, heart, pancreas, stomach, duodenum, jejunum, ileum, cecum, colon, kidney, adrenal gland, urinary bladder, inguinal lymph node, testicle, epididymis, prostate gland, skeletal muscle, pituitary gland, brain, and eye. Six of the 7 naive animals were positive for bacteremia at day 4 (Table 1) and for one of the animals the sample was not available. Blinded examinations of all of the animals in the 7 adjuvant formulation groups revealed that, when compared with the unblinded naive animals, none of the immunized animals showed any bacteremia, bacilli in the tissues, or anthrax lesions (Table 1). This demonstrated that PA-AH and the other formulations were highly protective for preventing lethal effects of aerosolized anthrax, but the challenge results could not distinguish between the relative effects of the different adjuvants.
3.2. Comparative effects of adjuvants on induction of PA-specific immunity
In contrast to the pathologic findings after challenge, substantial differences were observed between adjuvants in the levels of PA-specific IgG antibody concentrations (Fig. 2A) and lethal toxin neutralizing antibody titers (Fig. 2B). When compared to PA-AH, PA - emulsion and [PA + L(MPLA)] each induced significantly higher levels of PA-specific IgG antibodies at weeks 8, 12, or 14, as indicated by asterisks in Fig. 2. PA–emulsion and [L(PA + MPLA)] also induced significantly higher levels of lethal toxin neutralizing antibodies. However, PA + L(MPLA) induced the highest levels of toxin neutralizing antibodies (p < 0.001), nearly 4-fold and 5-fold greater than PA-AH at weeks 12 and 14, respectively, the highest toxin neutralizing antibody titers to PA yet reported in NHPs [23–25]. Interestingly, after only a single injection of [PA + L(MPLA)], the PA-specific neutralization titer at week-4 was equivalent to that after injection of PA-AH at week-4.
Fig. 2.
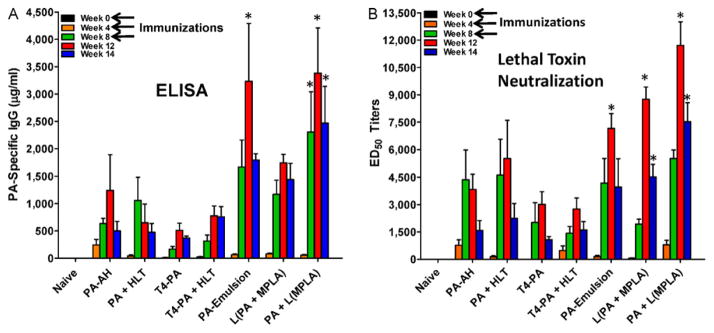
Immune responses to PA-adjuvant formulations in Rhesus macaques. (A) PA-specific IgG antibodies in sera of naive and immunized animals as determined by ELISA. Shown are the average concentrations of PA-specific IgG ± S.D. from each of the groups on weeks 0, 4, 8, 12, and 14, respectively. PA-emulsion induced significantly higher (p < 0.01) PA-specific IgG on week-12 compared to PA-AH. [PA + L(MPLA)] induced significantly higher PA-specific IgG on weeks 8 (p < 0.05), 12 (p < 0.001), and 14 (p < 0.01), respectively than PA-AH. (b) Lethal toxin neutralizing antibody titers (ED50 ) were determined in individual serum samples. At week 12, PA-emulsion immunized animals had significantly higher titers (p < 0.05) compared to PA-AH, while animals immunized with [L(PA + MPLA)] had significantly higher titers on weeks 12 (p < 0.001) and 14 (p < 0.05). The highest titers were induced in animals immunized with [PA + L(MPLA)] and ED50 titers were significantly higher than the titers induced by PA-AH at weeks 12 (p < 0.001) and 14 (p < 0.001).
4. Discussion
Although aluminum salt adjuvants are deemed relatively safe, they are sometimes associated with local reactions, including subcutaneous nodules, erythema, induration, and contact hypersensitivity [26], and new generic adjuvant formulations that exhibit a high level of safety, lower reactogenicity, but superior immunopotency would be preferred. The safety and lack of reactogenicity of liposomal MPLA has been previously demonstrated in human phase I trials with several candidate vaccine formulations for malaria, HIV, and cancer [11].
Our results with liposome formulations in NHPs are consistent with similar relative potencies observed in humans, but contrast with effects of similar formulations in mice and rabbits. For example, the best performing adjuvant system in this study, [L(MPLA)] mixed with PA, failed to show any adjuvant activity in mice when [L(MPLA)] was mixed with malaria antigen [27], and with HIV antigen in mice it induced antibody titers only 16% of those induced by antigen encapsulated in L(MPLA) [28]. Although [L(MPLA) + antigen] has never been tested in humans, it is a major constituent of both AS01b and AS15, which consist of empty liposome formulations that contain both MPLA and QS21 (AS01b), or MPLA, QS21, and CpG (AS15), that are simply mixed with antigen [3,29]. AS01b and AS15 are both in late stage (phase 3) clinical testing in humans [29]. Furthermore [L(PA + MPLA) – emulsion] had much less adjuvant activity than PA-AH in mice and rabbits (unpublished data), but the liposome-stabilized emulsion was the best performing adjuvant for inducing immune responses to prostate specific antigen in elderly and immunosuppressed late stage prostate cancer patients [11].
Our approach provides evidence that comparative adjuvant studies in NHPs may be a useful alternative approach to the use of other animal models for primary studies to identify safe, generic, easily manufactured, and potent adjuvants that could substitute for aluminum salts in candidate human vaccines, including anthrax vaccine. In the case of anthrax vaccine, the protective effects of PA adsorbed to AH are consistent with the presumed efficacies of licensed anthrax vaccines. However, because of perceptions and issues related to current vaccines [7], new generations of anthrax vaccine may have greater public acceptance. Although aluminum salts are relatively inexpensive, they are also sometimes weak adjuvants, and they do have a history of side effects [26]. The present results in macaques, that compare PA as a model antigen in combination with different 7 adjuvants and adjuvant systems, suggest that generic adjuvant systems containing liposomal MPLA might provide a safe and more potent alternative to AH for an anthrax vaccine.
The recent development of potentially effective malaria vaccines, which has been partly based on demonstration of preclinical effectiveness in macaques of novel strong and safe commercial adjuvant systems that yield better efficacies than those obtained with AH, further illustrates the potential usefulness of testing of new types of adjuvant systems in macaques [3]. Although PA was used as a model antigen in this study, encapsulation of a variety of antigens in liposomal adjuvant systems containing MPLA have demonstrated both safety and effectiveness in macaques and humans [3,11,29]. For vaccines to important poverty-related diseases, such as malaria, HIV/AIDS, tuberculosis, and many other difficult diseases that could benefit from strong and safe nonproprietary adjuvant systems, three generic liposome formulations containing MPLA are identified in this study that may have utility.
Acknowledgments
We gratefully acknowledge the assistance of Ms. Elaine Morrison for all the immunizations and sera collection; Ms. Sarah McCormack for performing the PA-specific IgG ELISAs; Ms. Stacy Banko, and PFC Allen J. Mueller for assistance with all the work conducted under BSL-3 conditions; and Ms. Zhihong Zhang for assistance with the preparation of bacteriophage T4 displayed antigens. We thank Drs. Stephen Leppla, John Clements, and Conrad Quinn for providing valuable reagents (recombinant PA, HLT, and AVR801 antibody). This research was supported by a cooperative agreement (W81XWH-07-2-0067) between the Henry M. Jackson Foundation for the Advancement of Military Medicine, Inc., and the U.S. Department of Defense. The research was funded, in part, by the U.S. National Institute of Allergy and Infectious Diseases (VBR), National Institutes of Health, NIAID Grant U01-AI056443.
Footnotes
The views expressed are those of the authors and should not be construed to represent the positions of the U.S. Department of Defense.
References
- 1.Guy B. The perfect mix: recent progress in adjuvant research. Nat Rev Microbiol. 2007;5:505–17. doi: 10.1038/nrmicro1681. [DOI] [PubMed] [Google Scholar]
- 2.Sesardic D, Rijpkema S, Patel BP. New adjuvants: EU regulatory developments. Expert Rev Vaccines. 2007;6:849–61. doi: 10.1586/14760584.6.5.849. [DOI] [PubMed] [Google Scholar]
- 3.Garçon N, Chomez P, Van Mechelen M. GlaxoSmithKline Adjuvant Systems in vaccines: concepts, achievements and perspectives. Exp Rev Vaccines. 2007;6:723–39. doi: 10.1586/14760584.6.5.723. [DOI] [PubMed] [Google Scholar]
- 4.Reed SG, Bertholet S, Coler RN, Friede M. New horizons in adjuvants for vaccine development. Trends Immunol. 2009;30:23–32. doi: 10.1016/j.it.2008.09.006. [DOI] [PubMed] [Google Scholar]
- 5.Harandi AM, Davies G, Olesen OF. Vaccine adjuvants: scientific challenges and strategic initiatives. Exp Rev Vaccines. 2009;8:293–8. doi: 10.1586/14760584.8.3.293. [DOI] [PubMed] [Google Scholar]
- 6.FDA. 21 CFR PART 316: Final rule. 1992 http://www.fda.gov/ForIndustry/DevelopingProductsforRareDiseasesConditions/HowtoapplyforOrphanProductDesignation/ucm124562.htm.
- 7.Grabenstein JD. Countering anthrax: vaccines and immunoglobulins. Clin Infect Dis. 2008;46:129–36. doi: 10.1086/523578. [DOI] [PubMed] [Google Scholar]
- 8.Friedlander AM, Little SF. Advances in the development of next-generation anthrax vaccines. Vaccine. 2009;27(Suppl 4):D28–32. doi: 10.1016/j.vaccine.2009.08.102. [DOI] [PubMed] [Google Scholar]
- 9.Cybulski RJ, Jr, Sanz P, O’Brien AD. Anthrax vaccination strategies. Mol Aspects Med. 2009;30:490–502. doi: 10.1016/j.mam.2009.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sullivan NJ, Martin JE, Graham BS, Nabel GJ. Correlates of protective immunity for Ebola vaccines: implications for regulatory approval by the animal rule. Nat Rev Microbiol. 2009;7:393–400. doi: 10.1038/nrmicro2129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Alving CR. Design and selection of vaccine adjuvants: animal models and human trials. Vaccine. 2002;20(Suppl 3):S56–64. doi: 10.1016/s0264-410x(02)00174-3. [DOI] [PubMed] [Google Scholar]
- 12.Kenney RT, Edelman R. Survey of human-use adjuvants. Exp Rev Vaccines. 2003;2:167–88. doi: 10.1586/14760584.2.2.167. [DOI] [PubMed] [Google Scholar]
- 13.Ramirez DM, Leppla SH, Schneerson R, Shiloach J. Production, recovery and immunogenicity of the protective antigen from a recombinant strain of Bacillus anthracis. J Ind Microbiol Biotechnol. 2002;28:232–8. doi: 10.1038/sj/jim/7000239. [DOI] [PubMed] [Google Scholar]
- 14.Peachman KK, McLean DM, Tong JC, Alving CR, Rao M. Ganglioside GM1 binding peptides: a potential adjuvant for transcutaneous immunization. Open Immunol J. 2009;2:94–102. [Google Scholar]
- 15.Peachman KK, Rao M, Alving CR, Burge R, Leppla SH, Rao VB, et al. Correlation between lethal toxin-neutralizing antibody titers and protection from intranasal challenge with Bacillus anthracis Ames strain spores in mice after transcutaneous immunization with recombinant anthrax protective antigen. Infect Immun. 2006;74:794–7. doi: 10.1128/IAI.74.1.794-797.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shivachandra SB, Rao M, Janosi L, Sathaliyawala T, Matyas GR, Alving CR, et al. In vitro binding of anthrax protective antigen on bacteriophage T4 capsid surface through Hoc–capsid interactions: a strategy for efficient display of large full-length proteins. Virology. 2006;345:190–8. doi: 10.1016/j.virol.2005.10.037. [DOI] [PubMed] [Google Scholar]
- 17.Li Q, Shivachandra SB, Zhang Z, Rao VB. Assembly of the small outer capsid protein, Soc, on bacteriophage T4: a novel system for high density display of multiple large anthrax toxins and foreign proteins on phage capsid. J Mol Biol. 2007;370:1006–19. doi: 10.1016/j.jmb.2007.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li Q, Fokine A, O’Donnell E, Rao VB, Rossmann MG. The structure of the small outer capsid protein, soc: a clamp for stabilizing capsids of T4-like phages. J Mol Biol. 2010;395:728–41. doi: 10.1016/j.jmb.2009.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Matyas GR, Muderhwa JM, Alving CR. Oil-in-water liposomal emulsions for vaccine delivery. Methods Enzymol. 2003;373:34–50. doi: 10.1016/S0076-6879(03)73003-1. [DOI] [PubMed] [Google Scholar]
- 20.Muderhwa JM, Matyas GR, Spitler LE, Alving CR. Oil-in-water liposomal emulsions: characterization and potential use in vaccine delivery. J Pharm Sci. 1999;88:1332–9. doi: 10.1021/js990011u. [DOI] [PubMed] [Google Scholar]
- 21.Li Q, Peachman KK, Sower L, Leppla SH, Shivachandra SB, Matyas GR, et al. Anthrax LFn-PA hybrid antigens: biochemistry, immunogenicity, and protection against lethal ames spore challenge in rabbits. Open Vaccine J. 2009;2:92–9. doi: 10.2174/1875035400902010092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hering D, Thompson W, Hewetson J, Little S, Norris S, Pace-Templeton J. Validation of the anthrax lethal toxin neutralization assay. Biologicals. 2004;32:17–27. doi: 10.1016/j.biologicals.2003.09.003. [DOI] [PubMed] [Google Scholar]
- 23.Darlow HM, Belton FC, Henderson DW. The use of anthrax antigen to immunize man and monkey. Lancet. 1956;271:476–9. doi: 10.1016/s0140-6736(56)91968-7. [DOI] [PubMed] [Google Scholar]
- 24.Ivins BE, Pitt ML, Fellows PF, Farchaus JW, Benner GE, Waag DM, et al. Comparative efficacy of experimental anthrax vaccine candidates against inhalation anthrax in rhesus macaques. Vaccine. 1998;16:1141–8. doi: 10.1016/s0264-410x(98)80112-6. [DOI] [PubMed] [Google Scholar]
- 25.Livingston BD, Little SF, Luxembourg A, Ellefsen B, Hannaman D. 2010 Comparative performance of a licensed anthrax vaccine versus electroporation based delivery of a PA encoding DNA vaccine in rhesus macaques. Vaccine. 2010;28:1056–61. doi: 10.1016/j.vaccine.2009.10.111. [DOI] [PubMed] [Google Scholar]
- 26.Baylor NW, Egan W, Richman P. Aluminum salts in vaccines—US perspective. Vaccine. 2002;20(Suppl 3):S18–23. doi: 10.1016/s0264-410x(02)00166-4. [DOI] [PubMed] [Google Scholar]
- 27.Verma JN, Rao M, Amselem S, Krzych U, Alving CR, Green SJ, et al. Adjuvant effects of liposomes containing lipid A: enhancement of liposomal antigen presentation and recruitment of macrophages. Infect Immun. 1992;60:2438–44. doi: 10.1128/iai.60.6.2438-2444.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rao M, Matyas GR, Vancott TC, Birx DL, Alving CR. Immunostimulatory CpG motifs induce CTL responses to HIV type I oligomeric gp140 envelope protein. Immunol Cell Biol. 2004;82:523–30. doi: 10.1111/j.0818-9641.2004.01283.x. [DOI] [PubMed] [Google Scholar]
- 29.Garçon N, Goldman M. Boosting vaccine power. Sci Am. 2009;301:72–9. doi: 10.1038/scientificamerican1009-72. [DOI] [PubMed] [Google Scholar]