

Abstract
It is widely recognized that severed axons in the adult central nervous system (CNS) have limited capacity to regenerate. However, mounting evidence from studies of CNS injury response and repair is challenging the prevalent view that the adult mammalian CNS is incapable of structural reorganization to adapt to an altered environment. Animal studies demonstrate the potential to achieve significant anatomical repair and functional recovery following CNS injury by manipulating axon growth regulators alone or in combination with activity-dependent strategies. With a growing understanding in the cellular and molecular mechanisms regulating axon plasticity and the availability of new experimental tools to map detour circuits of functional importance, the future of directing circuit rewiring to achieve functional recovery may be in sight.
Axon plasticity following injury
The adult mammalian central nervous system (CNS) is commonly perceived as a rigid network resistant to change. This is in part true as demonstrated by the detrimental and often permanent effects of CNS injury that result from its lack of regenerative ability. However, accumulating evidence show that genetic and pharmacological manipulations can induce regeneration of severed axons and, in addition, extensive axonal sprouting occurs spontaneously in a number of mammalian species, including primates, following spinal cord injury (SCI). Axon plasticity is defined here as the ability of axons to undergo structural changes to adapt to an altered environment. It occurs on the levels of axon regeneration and sprouting, the modulation of which has the potential to restore functions in patients with spinal injuries. While axon regeneration is naturally repressed in the CNS by a combination of neuron-extrinsic inhibitors and a lack of neuron-intrinsic growth capacity, axon sprouting occurs spontaneously and can restore limited function in rodent models of incomplete SCI. Although sprouting is deemed a form of spontaneous plasticity that can be exploited for therapeutic gain, surprisingly little is known about its regulation and anatomical organization.
In this review, we will discuss: 1) molecular regulators of axon growth and reorganization, primarily in the context of rodent spinal cord injury models, as the use of mouse genetics is becoming prevalent in examining molecular mechanisms of the regenerative response; 2) injury-induced circuit remodeling by spontaneous sprouting; 3) therapeutic potential of combining rehabilitation with growth-enhancing strategies to achieve functional recovery; and 4) future directions in neural regeneration research.
Regeneration of lesioned axons at and around the injury site
The intuitive approach to repairing axonal injury is to promote regeneration of lesioned axons across the injury site. That is, to reconnect severed tracts with their original targets. Spurred by the seminal finding that injured CNS axons can grow into the growth-permissive environment of a peripheral nerve graft [1], early efforts in this area focused mainly on identifying inhibitory molecules in the CNS milieu after injury. Following genetic studies that showed modest effects of deleting various extrinsic inhibitors on axon regeneration (references in [2]), attention was then turned to promoting the neuron-intrinsic ability to regrow axons. The importance of neuron-intrinsic contribution to axon regeneration was first demonstrated by the conditioning effect of a prior peripheral nerve injury that boosts regeneration of the central branches of sensory axons in the absence of any modification to the CNS environment [3, 4]. Although the regenerative potential of CNS neurons declines with age, injured adult CNS axons can be coaxed to grow by activating neuron-intrinsic signaling pathways [5, 6]. While a general distinction is made between extrinsic and intrinsic factors, these programs interact, as extrinsic factors converge on neuronal intracellular signaling pathways.
Axon regeneration: extrinsic regulators
Comparative studies of the growth-permissive environment of the peripheral nervous system (PNS) and the growth-inhibitory environment of the CNS after injury identified prolonged exposure to CNS myelin-derived inhibitors and the formation of the glial scar as two major factors contributing to the regenerative failure of the CNS [7]. Axotomy generates cellular breakdowns at locations proximal and distal to the injury site in both the PNS and CNS. Whereas myelin debris is rapidly cleared in the PNS by Schwann cells, macrophages, and endogenous antibodies to allow for axon regeneration, it persists in the CNS due to the lack of Schwann cells and restricted access of anti-myelin antibodies [8–10]. In addition, astrocytes in the CNS form a glial scar that presents a physical barrier to regenerating axons and expresses additional inhibitors of axon growth [7, 11]. Below, we discuss the biological activities of these inhibitory components as well as strategies to overcome them. Not included in this review are repulsive axon guidance cues that limit axon growth [12] as well as the immune response that also features prominently in the survival and regeneration of axons [13].
Myelin-derived inhibitors
The three prototypical myelin derived inhibitory molecules are: myelin associated glycoprotein (MAG), Nogo, and oligodendrocyte-myelin glycoprotein (OMgp), the last two of which are specifically expressed by oligodendrocytes in the CNS but not by Schwann cells in the PNS. Detailed properties of these molecules and their axonal surface receptors (PirB, NgRs, and co-receptors p75NTR or TROY, and LINGO-1) were recently reviewed [14, 15]. Extensive and redundant crosstalk occurs among myelin inhibitory ligands and receptors, converging on the intracellular activation of the Ras homolog gene family member A and Rho-associated kinase (RhoA-ROCK) cascade that ultimately leads to growth cone collapse [16–19].
Pharmacological and genetic approaches have been used to determine whether blocking particular myelin inhibitors promotes axon regeneration. Developed even before the discovery of its antigen as Nogo-A, antibodies against the inhibitor of neurites-1 (IN-1) neutralize the growth-restrictive effects of CNS myelin [20]. Early studies supplying IN-1 antibody in the cerebrospinal fluid by cortical implantation of IN-1-secreting hybridoma cells significantly increased the maximum distance of growth from limited numbers of corticospinal tract (CST) axons past the mid-thoracic lesion site in rats [21, 22]. The fine CST axons observed below the injury site in IN-1 treated but not in control animals can be traced back to the site of injury through tortuous courses, indicating that they were newly grown, not spared, fibers. In retrospect, however, these newly grown axons would be consistent with either sprouting from uninjured axons or regeneration of injured axons (see more below). Regardless, the majority of CST-lesioned rats that received such IN-1 treatment recovered contact-placing response, a CST-dependent reflex, compared to no recovery in controls [22]. Subsequent studies intrathecally infusing various purified forms of Nogo or NgR inhibitors with improved target specificity also showed similar results [23, 24]. These pharmacological studies support the axon growth-promoting effect of anti-Nogo treatment that presumably contributed to the observed functional improvements. Again, whether it was the regeneration of damaged axonal pathways, or the sprouting from undamaged pathways, that contributed most to any improved recovery remained an open question (see below “Circuit rewiring: detour formation by sprouting”). Phase II clinical trial of anti-human Nogo-A antibody in acutely injured paraplegic and tetraplegic patients is now in preparation.
In contrast to the repeatedly reported pro-regenerative effect of the IN-1 antibody, single or combined genetic deletion of myelin-derived inhibitors/receptors induced sprouting but yielded mixed results on regeneration of the corticospinal tract [2]. Alternative explanations for the divergent results of the IN-1 antibody and genetic deletions are: 1) compensatory activation of currently unknown inhibitors following genetic deletions; 2) emerging dichotomous functions of prototypical myelin inhibitors in both axon protection/growth promotion and growth inhibition; and 3) beneficial effects of the IN-1 antibody in addition to blocking Nogo-A that are currently underexplored, such as a possible function in antibody-facilitated clearance of myelin debris after injury [10]. In support of the second possibility, MAG is required for normal axon maintenance and protects axons from degeneration under stress conditions such as CNS inflammation [25–27]. Furthermore, in contrast to its expected growth inhibitory role, genetically deleting MAG reduced CST sprouting after injury [28].
RhoA-ROCK are also appealing therapeutic targets as they are common intracellular targets of myelin and non-myelin inhibitors [7]. In rodent models, both the ROCK inhibitor (Y27632) and the selective RhoA inhibitor (C3 transferase) increased optic nerve and CST regeneration [16, 29, 30]. Additionally, Rho inhibition also provides neuroprotection [31, 32]. Clinical trial of a RhoA inhibitor, trademarked as Cethrin, suggests beneficial effects of treatment in patients with cervical injuries [33]. However, it is difficult to determine the extent to which Cethrin contributes to the observed neurological repair, as patients with incomplete SCI often exhibit spontaneous recovery [34].
Glial scar and CSPGs
In response to CNS injury, astrocytes become “reactive”, undergoing hypertrophy and proliferation to form a glial scar at the lesion site with meningeal fibroblasts, microglia, and oligodendrocyte precursors [35, 36]. In addition to the scar acting as a physical barrier to axon growth, reactive astrocytes secrete chondroitin sulfate proteoglycans (CSPGs), molecules that further impede axon elongation. CSPGs are extracellular matrix proteins decorated with sulfated glycosaminoglycan (GAG) chains that largely mediate the inhibitory effect of CSPGs. They interfere with axon regeneration by 1) blocking access of axons to the growth-promoting adhesion molecules laminin and integrins [37, 38]; and 2) triggering growth inhibitory signals through interaction with receptor protein tyrosine phosphatase sigma (RPTPσ), leukocyte common antigen-related (LAR) phosphatase, NgR1, and NgR3 on the axonal surface [39–42]. RPTPσ is another bimodal “inhibitory” molecule that can also promote axon extension depending on the ligand: binding to heparan sulfate proteoglycans (HSPGs) supports axon growth, whereas binding to CSPGs does the opposite [43].
A current approach to antagonizing the inhibitory effect of CSPGs on axon elongation is enzymatic digestion of GAG chains by the bacterial enzyme chondroitinase ABC (ChABC). This has been shown to lead to regeneration of lesioned axons and significant functional recovery [44–47]. Again, the distinction between the regeneration of injured axons and sprouting of uninjured axons was not always emphasized, except perhaps when there was a PNS graft. Intraspinal delivery of lentivirus expressing ChABC achieved stable and widespread digest of CSPGs that conferred neuroprotection following injury, possibly as a result of modulating the early inflammatory response to favor induction of the M2 macrophages that promote tissue repair [48]. Host immune reaction following prolonged exposure to bacterial ChABC, however, remains to be evaluated. Intriguingly, an alternative strategy that targets the synthesis, rather than the degradation, of CSPGs may be more effective than ChABC in promoting post-injury axon growth. In a proof of principle study of this novel approach, mice with genetic deletion of the enzyme N-acetylgalactosaminyltransferase-1 (T1) involved in the second step of chondroitin sulfate (CS) synthesis had reduced glial scar formation, increased number of serotonergic axon terminals caudal to the injury site, and better motor performance after SCI compared to their ChABC-treated counterparts [49]. Surprisingly, such beneficial effects of T1 deletion were primarily attributed to increased neuronal production of heparan sulfate (HS), the aforementioned growth promoting ligand of RPTPσ, in addition to decreased CS production [49]. This inverse co-regulation of HS and CS synthesis may be exploited therapeutically to augment axon growth potential.
Given that reactive astrocytes are the source of both CSPGs and the glial scar, selective depletion of proliferating reactive astrocytes may facilitate repair of the injured CNS. Although such depletion did lead to disoriented sprouting of local nerve fibers surrounding the lesion site, this effect was outweighed by many detrimental outcomes such as the failure to repair the blood-brain barrier, exacerbated infiltration of inflammatory cells, increased death of local neurons and oligodendrocytes, and worsened tissue degeneration and motor impairment [50, 51]. These findings indicate scar formation is a part of wound repair that serves to reduce inflammation and contain tissue damage, with the side effect of inhibiting axon growth. The balance between maintaining such damage control mechanisms and clearing a path for axon growth needs to be carefully calibrated to achieve optimal therapeutic outcomes.
Axon regeneration: intrinsic regulators
The search for neuron-intrinsic regulators of axon regeneration has benefitted from developments in cancer biology, developmental neurobiology, and the study of lower model organisms. Several intracellular signaling proteins that regulate cell proliferation were discovered to play analogous roles in regulating CNS axon regeneration. In parallel, genetic studies of axon biology in Drosophila melanogaster and Caenorhabditis elegans revealed candidate regulators of mammalian axon regeneration.
Optic nerve and CST lesions are two injury models commonly used to study neuron-intrinsic regulation of axon regeneration in the mammalian CNS. In the optic nerve crush model, axotomy of retinal ganglion cells (RGCs) leads to apoptosis of RGCs [52] and consequent optic nerve degeneration, underscoring cell survival as a prerequisite for axon regeneration in this system. Indeed, some intrinsic regulators promote both cell survival and axon growth in RGCs [53, 54]. However, supporting cell survival alone may not be sufficient to stimulate axon regeneration [53]. Although CST neurons, unlike RGCs, may not undergo substantial cell death after axotomy [55–57], several signaling pathways regulate axon regeneration in both RGCs and corticospinal neurons. These and other prominent intrinsic regulators recently identified by genetic studies in mice are discussed below. Understanding the effects of each intrinsic regulator on post-injury cell survival and/or axon growth (Figure 1) will help determine how the corresponding signaling pathway can be best manipulated for therapeutic interventions targeting specific CNS neuron types.
Figure 1. Representative neuron-intrinsic regulators of post-axotomy cell survival and/or axon regeneration.
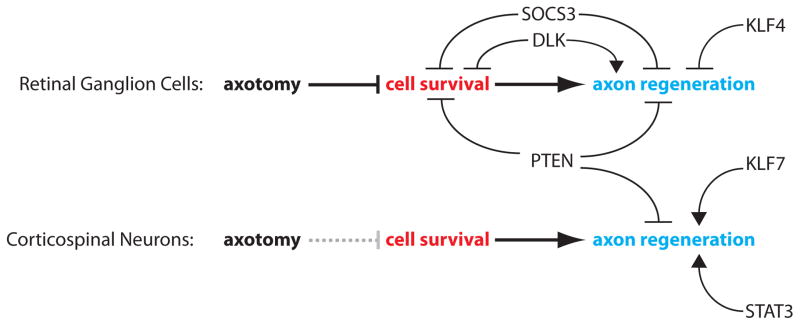
Response to axotomy and its regulators are compared between retinal ganglion cells (RGCs) and corticospinal neurons. RGCs undergo significant cell death following axotomy, rendering cell survival a prerequisite for axon regeneration. It is debatable whether axon injury compromises survival of corticospinal neurons, as indicated by dotted grey line. Neuron-intrinsic regulators of post-axotomy cell survival and axon regeneration validated in vivo are shown.
PTEN
Deletion of phosphatase and tensin homologue (PTEN) stimulates optic nerve and CST regeneration in adult mice [53, 58]. PTEN is a well studied tumor suppressor that dephosphorylates phosphatidylinositol-3,4,5-triphosphate to antagonize the pro-proliferative PI3K-AKT pathway [59]. Its deletion in RGCs increases cell survival from 20% to 45%; and 8–10% of the surviving RGCs re-grew axons [53]. This suggests PTEN deletion largely supports survival but also primes neurons for axon regeneration requiring additional unknown signals. Two studies have examined whether the robust optic nerve regeneration in PTEN-deficient mice restores visual function. While one reported success of regenerated axons to reach visual targets and recovery of visual behaviors [60], the other reported failure of reinnervation [61]. Whether the use of behavior testing in the former, but not in the latter, study elicited a training effect that promoted proper axon reinnervation remains to be tested. Regardless, proper target innervation following axon regeneration is an important topic to explore in future studies.
Increased protein synthesis downstream of mammalian target of rapamycin (mTOR) is hypothesized to mediate the effect of PTEN deletion on axon regeneration. However, it is difficult to distinguish the general effect of protein synthesis on cell survival from its proposed specific effect on axon extension. GSK3, another downstream effector of the PI3K-Akt pathway, also regulates axon regeneration of sensory neurons [62]. This suggests PTEN deletion recruits multiple mechanisms to enhance axon regeneration.
DLK
Dual leucine zipper kinase (DLK) is an upstream activator of the mitogen activated protein (MAP) kinase pathway that signals through p38 in lower organisms and JNK-cJun in mammals [63–65]. Intriguingly, its function straddles regeneration, cell death, and Wallerian degeneration. In the mouse optic nerve, it is required for apoptosis and the regeneration-enhancing effect of PTEN deletion [66, 67]. A unifying explanation for the dichotomous effects of DLK manipulation emerging from these studies is that DLK activates both regenerative and apoptotic programs in response to injury, but the final outcome depends on the regenerative competence of the system. In mammalian PNS neurons capable of re-growing injured axons, DLK is required for axon regeneration [68]; whereas in mammalian CNS neurons that naturally cannot regenerate axons, DLK is required for cell death [66]. Consistent with this hypothesis, when the CNS is primed to regenerate by PTEN deletion, DLK is required for this regeneration [67]. In addition, DLK is required for Wallerian degeneration in the PNS [69]. Whether activating DLK promotes axon regeneration in the CNS remains to be tested.
KLFs
Kruppel-like factors (KLFs) are a family of 17 transcription factors that play critical roles in cell proliferation and differentiation [70], a subset of which regulate axon growth in a development-dependent manner [71]. KLF4 deletion increases optic nerve regeneration [71], whereas overexpressing an engineered enhanced version of KLF7 increases CST regeneration [72]. In vitro, KLF6 is the only member that when overexpressed significantly enhances neurite growth in cortical neurons, whereas KLFs 1, 2, 4, 5, 9, 13, 14, 15, and 16 inhibit neurite extension. Interestingly, it has been suggested that KLFs regulate intrinsic regenerative capacity independently of cell survival, although the role of several KLF members in cell growth is well documented in other systems [70, 73]. Key transcriptional targets that mediate the effect of KLFs on axon growth remain to be identified.
JAK-STAT-SOCS
The Janus kinase–signal transducer and activator of transcription (JAK-STAT) pathway regulates cytokine signaling in the immune system [74] and was recently found to promote axon regeneration in the mammalian CNS. In the conditioning lesion model, whereby peripheral axotomy of sensory neurons enhances the regenerative potential of their central axons in the spinal cord, pharmacological inhibition of JAK abolishes the conditioning effects on central axon regeneration [75]. Consistent with this, deletion of suppressor of cytokine signaling 3 (SOCS3), an inhibitor of the JAK-STAT axis, significantly increases neuronal survival and axon regeneration after optic nerve injury [54]. Enhanced axon growth in the absence of SOCS3 is dependent on receptor gp130 upstream of JAK-STAT, whose activation after injury is suggested to involve injury-induced upregulation of its ligands such as CNTF [54]. Furthermore, overexpression of STAT3 in the CST increased the growth of collaterals at the injury site as well as distal CST sprouting (discussed below) [76]. The JAK-STAT pathway acts independently of but synergizes with the mTOR pathway to stimulate axon growth [77]. In addition to acting as a neuron-intrinsic regulator of axonal growth, STAT3 also positively contributes to motor recovery after spinal cord injury by modulating astrogliosis [78].
Circuit rewiring away from the injury site
A roundabout alternative for the motor cortex to reconnect with its severed targets following SCI is through the establishment of detour circuits that bypass the injury site. In contrast to the regeneration of transected CNS axons that generally fails, sprouting of CNS axons – canonically defined as growth of an intact axon but also used to describe growth of transected axons, especially those proximal to the injury site – spontaneously occurs following injury and can form compensatory connections (Box 1) [79–81]. Based on findings from animal models, compensatory structural and functional reorganizations are thought to occur through: 1) sprouting that re-establishes contact with the original target by relays with spared tracts; or 2) injury-induced activation of pre-existing latent tracts (Figure 2). These mechanisms likely contribute to spontaneous neurological recovery that occurs in 40% of human patients with partial sensory sparing, and at least 60% of those with partial motor sparing [34]. Although spontaneous recovery is limited, these encouraging findings raise the possibility that such innate adaptability of the CNS can be harnessed to augment recovery. Understanding patterns of structural changes and the underlying molecular mechanisms will be the first steps to evaluating the therapeutic potential of intraspinal rewiring and/or functional substitution by latent systems.
Box 1. Shaping and mapping functional relay circuits.
Axonal sprouting as a spontaneous response to CNS injury in adult mammals renders it a form of endogenous plasticity that can be harnessed to promote functional recovery. When left to occur spontaneously after spinal cord injury, axonal sprouts may form ectopic connections that alter the motor representation map in the cortex [81] and lead to neuronal dysfunction in the chronic stage [116]. However, when electrochemical stimulation is provided together with treadmill training to increase motor output, the resultant intraspinal relay restores voluntary locomotion [116]. These findings underscore activity-dependent formation of adaptive relay circuits.
Promoting circuit rewiring of functional consequence by stimulating neuromuscular activity, followed by mapping of these relays and identification of molecular mechanisms regulating their formation, will facilitate the development of therapies that direct endogenous plasticity towards functional recovery (Figure 3). However, hurdles to achieving this goal are 1) traditional neuroanatomical techniques limit the identification of compensatory relays, and 2) lack of method(s) to distinguish relays that contribute to functional recovery from those that do not. Recent technological advances now provide solutions to these problems. In principle, the use of retrograde trans-synaptic tracer coupled with tissue clearing methods such as 3DISCO [117] and CLARITY [118] will enable unbiased mapping of relay networks in the CNS (Figure 3), although viral tracer-induced neuronal cell death will need to be overcome. In addition, selective and reversible genetic inactivation of specific neuronal populations [119] will allow identification of relays of functional significance. With optimization of these available tools, it is feasible to map adaptive circuitry and subsequently determine the molecular regulators of its formation, which may lead to therapeutic interventions that potentiate activity-dependent recovery.
Figure 2. Circuit rewiring in the corticospinal tract (CST) and phrenic system after injury.

(A) Left, In the intact CST (blue lines), axons of corticospinal neurons whose cell bodies reside in the motor cortex (blue circles) decussate at the medullary pyramids, descend contralaterally mainly in the dorsal and dorsolateral columns of the spinal cord, and synapse directly or indirectly on motor neurons (green) to control voluntary movements. For illustration purpose, only direct synapses are shown, which are more prevalent in primates than in rodents. Right, In the injured CST with unilateral lesion rostral to decussation, pyramidotomy (bold red line) results in contralateral denervation. Spontaneous compensatory sprouting from intact axons (orange) has been shown to re-establish detour connections with motor neurons through spinal interneurons (purple). (B) Left, In the intact phrenic system that controls respiration, medullary neurons in the rostral ventral respiratory group RVRG (blue circles) descend bilaterally through the bulbospinal tract (blue lines) to the phrenic nuclei (light green ovals) that project axons (dark green) to control the diaphragm. Bulbospinal axons also form “silent” connections (dotted blue lines) with the contralateral phrenic nuclei. Right, Spinal cord hemisection (long bold red line) paralyzes the ipsilateral hemidiaphragm. In the crossed phrenic phenomenon (CPP), subsequent lesion of the contralateral phrenic nerve (phrenicotomy – short bold red line) induces activation of the latent phrenic pathway (orange) to restore respiratory function of the hemidiaphram paralyzed by spinal cord injury. In addition, sprouting of the crossed phrenic pathway (dotted orange lines) may occur [115].
Circuit rewiring: detour formation by axon sprouting
The CST has served as a model to study routes of axonal detour, given the defined anatomical locations and physiological role of the CST in the control of voluntary movements. The CST originates from the sensorimotor cortex and, in different species, projects through different regions of the spinal white matter. In rodents, the main part of the CST (with >95%) decussates at the medullary pyramids and descend contralaterally in the dorsal column (dCST), with minor components in the dorsolateral, lateral, and/or ipsilateral ventral white matter [79, 82]. In primates, about 90% of the CST descends contralaterally in the dorsolateral columns, with the remaining axons traveling ipsilaterally in the same region or in the ventral column [83, 84]. Primate CST axons extensively cross the spinal cord midline in the grey matter [85], whereas few do in rodents, which may lead to observed inter-species differences in the formation of different injury-induced axonal detours.
Early evidence for sprouting-associated functional recovery in rodents after spinal cord injury demonstrated a compensatory role of spared axons. Cervical lesion of dCST in rats induces local sprouting of vCST axons making contacts with medial motor neurons in lamina IX of the grey matter. Subsequent abolishment of post-sprouting functional improvements by vCST lesion suggests this effect contributes to post-injury recovery. [86]. Surprisingly, lesion of vCST alone, consisting of only 5% of the total CST axons, also results in a functional deficit comparable to dCST lesion. Whether and how dCST undergoes structural reorganization to compensate for vCST is unknown.
Although sprouting is most frequently used to refer to the growth from spared CST axons, the term is also used to refer to growth from injured axons occurring proximal to the injury site as sprouting (or collateral sprouting). In practice, it is not always straightforward to distinguish between proximal collateral sprouting from injured axons and sprouting from spared axons that normally terminate proximal to the injury site. Mid-thoracic lesion of dCST leads to proximal sprouting of transected hindlimb CST axons into the intermediate laminae at high cervical level [80]. The extent and timing of such anatomical rearrangement correlates with a shift in cortical motor representation, in which stimulation of the hindlimb motor cortex aberrantly activated responses in the forelimbs, whiskers, and trunk at 3 weeks post-injury. One neuronal target of these CST axonal sprouts is a type of spinal interneurons called propriospinal neurons (PSNs), which project ventrally from the cervical enlargement to the lower cervical level (short PSNs) or lumbosacral enlargement (long PSNs) to terminate on spinal motor neurons [81]. Interestingly, whereas initial contacts were made between dCST axonal sprouts and both the short and long PSNs, only the latter bridging the lesion site were maintained, and their terminal arborization with motor neurons increased. This suggests both need-based and activity-dependent strengthening of contacts. Retrograde trans-synaptic tracing together with electrophysiological and behavioral assessment of hindlimb response demonstrated anatomical and functional integrity of the hindlimb CST neuron-PSN-lumbar motor neuron relay circuit at 12 weeks after injury [81]. However, in this study, not all hindlimb motor neurons were correctly rewired to the corresponding motor cortex, and a substantial fraction was relayed to the forelimb motor cortex. It is unclear if and how these ectopic connections contribute to regaining hindlimb function. Using single or staggered lateral hemisections at the thoracic level, it was later shown that propriospinal relays can bypass more than one injury site to re-establish supraspinal control of stepping [87]. It remains to be seen if there are other neural systems, beyond PSNs, that also contribute to spontaneous functional recovery in these models.
Directing the formation of functionally meaningful axonal detours as a therapeutic strategy for CNS injury requires an understanding of spontaneous injury-induced circuit remodeling as well as identification of molecular regulators of sprouting. The long history of studies on Nogo has firmly established its role as a classic modulator of mammalian axon sprouting [15]. Inhibition of Nogo-A by neutralizing antibodies stimulated neuronal reorganization via sprouting observed in several descending tracts after SCI including the raphespinal, corticospinal, and rubrospinal systems, concomitant with improved motor and/or sensory recovery in rats and monkeys [22, 23, 88–94]. The translational potential of Nogo-regulated circuit remodeling was perhaps best shown when anti-Nogo treatment was combined with rehabilitative training in a stroke model [95] (Box 2). Nogo-A suppresses CNS plasticity in the presence as well as in the absence of injury. It is suggested that Nogo-A limits CNS plasticity under basal conditions through regulation of the cytoskeleton machinery and the transcriptional program of neuronal growth [96]. Understanding whether and how the injury signal modifies the basal growth-inhibitory mechanisms of Nogo-A could reveal new molecular targets that may complement anti-Nogo therapy to enhance post-injury plasticity.
Box 2. Combining rehabilitation and axon growth-enhancing strategies to augment recovery after CNS injury.
Currently, the most widely applied strategy to promote functional recovery in SCI patients is rehabilitative training. Through repetition, rehabilitation aims to stimulate and reinforce engagement of residual neural circuitries in performing functions disrupted by injury. Major paradigms for spinal cord rehabilitation are: motor training, pharmacological stimulation, and electrical stimulation. Locomotor (step) training was developed based on the principle that the spinal cord can autonomously process sensory information to generate appropriate motor responses without conscious control [120]. It has proved efficacious to various extents in SCI patients [120]. The second approach, pharmacologic stimulation, targets neurotransmitter systems to modulate the excitability of locomotor circuits, thereby potentiating the effects of locomotor training. The third rehabilitation strategy of electrical stimulation can be applied to 1) restore muscle tone, 2) increase excitatory drive of neural circuits, 3) enhance axon outgrowth, and 4) directly activate muscle movement. Spinal epidural stimulation lowers the activation threshold of locomotor neurons, thereby facilitating the generation of movements when combined with motor training. Excitingly, this paradigm was shown to enable voluntary movements in rats and human patient with complete paralysis [121, 122]. In light of studies reporting enhanced compensatory sprouting of the CST that connect with denervated motor circuits to restore skilled motor function following electrical stimulation of the motor cortex [123], it would be interesting to see if epidural stimulation similarly elicits intraspinal axon outgrowth (Box 1: mapping circuits). Another area in the brain whose stimulation has the potential to improve locomotor function after SCI is the mesencephalic locomotor region (MLR) in the brainstem. In intact animals, MLR stimulation regulates stepping [120]. In spinally injured rats, MLR stimulation improved walking and swimming through spared reticulospinal fibers [124]. Finally, high-level electrical stimulation can directly activate muscle movements [120]. Its reliance on external control for muscle activation however may render the coordination of multiple muscle groups challenging.
Given that most rehabilitation paradigms strengthen spared circuits relevant to trained tasks and/or increase excitability of motor circuits, pre-conditioning with axon growth-promoting strategies may provide more substrates for behavioral training to act on, thereby augmenting functional recovery. Indeed, anti-Nogo treatment followed by motor training enhanced compensatory sprouting that underlies the nearly complete restoration of skilled forelimb use after stroke in rats [95]. Notably, concurrent application of anti-Nogo antibody and training worsened motor performance following both SCI and stroke [95, 125], highlighting the importance of treatment sequence in determining the functional outcome of combination therapy. Another key parameter is the type of training involved. ChABC treatment improved forelimb function after CST lesion, but only when combined with forelimb use-specific training [126]. This illustrates that rehabilitation may only enhance the type of behavior reinforced by the training and may interfere with behaviors that are not trained. ChABC increased CST sprouting after injury regardless of training paradigm, with no additional sprouting from task-specific training. This supports a role in strengthening functional connections, not in increasing axon growth, for the training applied in this study. Transfer of specifically trained skills to novel tasks has also been shown [98]. The degree of similarity between trained and novel activities, rigor of training, difficulty of the tasks, and motivation likely factor into the transferability of acquired skills. Combination of anti-Nogo antibody, ChABC, and training was shown to more effectively enhance axon sprouting, regeneration, and motor function after SCI than applying either biologic with training alone [127].
Combination treatment can be efficacious in achieving functional recovery when applied appropriately. The interactive effects of the constituent interventions depend on the type of treatments involved, as well as the sequence and timing of application, which must be carefully evaluated to maximize desirable outcome. Combining interventions that each targets a different mechanism of repair will likely generate synergistic effect on recovery. In this regard, inclusion of neuroprotection in combination therapy with growth-promoting strategies and rehabilitation may be beneficial. Finally, recent genetic strategies that reprogram the injured CNS neurons’ intrinsic capacity of axon growth [6] offer exciting therapeutic avenues for exploration, alone or with rehabilitation training. Our ever-growing battery of repair strategies lends an optimistic outlook on treating SCI.
Degradation of CSPG by ChABC also enhances sprouting that is now considered as the primary factor contributing to functional improvement observed with ChABC treatment alone. ChABC promotes collateral sprouting of the lesioned CST rostral to the injury site, as well as compensatory sprouting of the intact CST, serotonergic, and sensory fibers [97, 98]. In contrast to the ability of anti-Nogo treatment to induce sprouting of CST and sensory fibers in the intact spinal cord, ChABC seems to only promote sprouting in the injured spinal cord. While this led to the suggestion that denervation is required of ChABC-dependent sprouting to occur, the alternative explanation is simply that CSPG is an injury-induced barrier to plasticity, which is consistent with its production by reactive astrocytes following injury. Given that CSPG acts as an injury-specific repressor of axon growth whereas Nogo acts a general repressor, blocking both may synergistically boost CNS plasticity after injury, as shown when combined with rehabilitation (Box 2).
Intrinsic regulators of axon regeneration also modulate sprouting. Sustained activation of the transcription factor STAT3 increases sprouting of thoracically lesioned CST axons both locally at the injury site and remotely at the cervical level in mice [76]. STAT3 activation also induces cervical sprouting of spared CST fibers after pyramidotomy [76]. These sprouts cross over to the injured side to form functional synapses with both short PSNs and forelimb motor neurons to improve forelimb function on the injured side, as verified by electrophysiological stimulation. PTEN increases CST sprouting after unilateral pyramidotomy in mice [58]. However, whether it regulates cervical or thoracic lesion-induced sprouting in a more clinically relevant model remains to be tested.
Spontaneous CST remodeling after injury occurs not only in rodents, but also in primates. In monkeys, 24 weeks after unilateral lesion of the spinal cord at the low cervical level, as much as 60% of the axon density, compared with the intact control, was restored on the lesioned side [84]. Such extensive reconstitution of axonal density by injury-induced sprouting of CST fibers correlated with substantial recovery in hindlimb locomotion and forelimb use during locomotion and in object retrieval. The positive contribution of spontaneous sprouting to functional recovery in both rodents and primates underlines the potential of promoting such endogenous plasticity as a therapeutic approach, which will require elucidation of injury-induced adaptive detours and the neuronal populations involved (Box 1).
Circuit rewiring: latent pathway activation
Another form of neural modulation and plasticity following injury is the activation of a latent pathway, as exemplified by the crossed phrenic phenomenon following disruption of the bulbospinal tract (BST) that controls breathing (Figure 2) [99, 100]. Axons of the BST descend bilaterally from the respiratory regions in the brainstem to the phrenic nuclei at cervical levels C3–C6, which innervate the diaphragm by the phrenic nerves. Disruption of this respiratory pathway, as occurs in spinal cord injuries at the cervical level, is life threatening because it compromises breathing. Experimentally, unilateral lesion of the BST above the phrenic nucleus paralyzes the ipsilateral half of the diaphragm (hemidiaphram), whose function can be restored by subsequent transection of the contralateral phrenic nerve (phrenicotomy). Such functional restoration of the previously paralyzed hemidiaphram, attributed to the activation of a spared, latent crossed pathway of the BST, is termed the crossed phrenic phenomenon (CPP). CPP induction depends on increased respiratory drive, which likely strengthens the crossed phrenic synapses [99]. Furthermore, CPP can be reversible or persistent, depending on the stimulus and its regimen [99].
Alternative methods to increase respiratory drive without phrenicotomy are of therapeutic value in restoring breathing in the spinally injured and other conditions that impair respiration. These include pharmacological intervention and intermittent hypoxia. Theophylline, a drug already in use to treat asthma and chronic obstructive pulmonary disease, activates CPP by blocking adenosine receptors that inhibit cAMP synthesis and phosphodiesterases that break down cAMP. This dual inhibition thereby upregulates cAMP, whose administration alone improves respiratory function. Notably, chronic oral administration of theophylline elicits respiratory recovery that persists after treatment [101], rendering it an appealing candidate drug.
Exposure to acute intermittent hypoxia (AIH) is another approach being explored to induce respiratory plasticity. AIH enhances respiratory drive for hours after exposure, a phenomenon called phrenic long-term facilitation (pLTF). Central to the activation of pLTF is hypoxia-induced release of serotonin from brainstem raphe neurons, which binds 5-HT2 receptors of phrenic motor neurons to trigger protein synthesis of BDNF and subsequent activation of TrkB signaling that involves the downstream effectors extracellular signal-regulated protein kinase (ERK) and Akt [102–104]. ERK and Akt are also common effectors downstream of vascular endothelial growth factor (VEGF) and erythropoietin (EPO), two transcriptional targets of hypoxia inducible factor 1 (HIF-1) recently shown to induce phrenic motor facilitation in response to AIH [105, 106]. Whereas serotonin induces pLTF, activation of NMDA receptors maintains it [107, 108].
Surprisingly, in rats with chronic spinal injury at the cervical level, daily AIH not only improves respiratory function, but also forelimb motor performance [109]. It is unclear whether the latter is a secondary effect of the former or AIH directly induces plasticity in both systems, although hypoxia-dependent upregulation of growth factors is suggested to play a role. Such a general effect of AIH on plasticity in motor neurons was also observed in humans. Specifically, intermittent hypoxia safely increases plantar flexion torque, improves walking speed and endurance, and elicits brief pLTF in humans sustaining incomplete spinal cord injuries [110–112]. Future studies refining AIH dosing to optimize pLTF in humans and elucidating the mechanisms underlying the effect of AIH on locomotor recovery will be of scientific and clinical importance.
Concluding remarks and future directions
Human spinal cord injury is characterized by a highly variable and complex pathology that demands a complex treatment, likely requiring a combinatorial approach involving multiple mechanisms impinging on circuit plasticity. Achieving functional recovery largely depends on bridging circuits above and below the lesion site through regeneration of severed axons and/or sprouting-mediated formation of axonal relays. However, indiscriminately promoting regeneration and/or sprouting may cause adverse effects: axon regeneration can lead to spasticity and even worsen motor function [113], while maladaptive sprouting may increase sensitivity to pain [114]. These side effects of exuberant plasticity highlight the need for 1) inclusion of pain assessment in neural regeneration studies, especially when the treatment of interest is globally applied to the whole spinal cord; 2) identification of factors that guide the formation of adaptive plasticity, such as stimulation of neuromuscular activity by rehabilitative training, that can be coupled to manipulations that promote plasticity; and 3) better understanding of axonal tract-dependent response to injury and pro-regenerative treatments so as to enable selective enhancement of plasticity in specific axonal pathways. Current challenges that shape future directions in neural regeneration research are discussed in “Outstanding questions and future directions” (Box 3).
Box 3. Outstanding questions and future directions.
Dual roles of axon growth inhibitors and cell type-specific functions
Emerging dichotomous functions of classic extrinsic inhibitors –such as MAG and RPTPσ– in regulating injury-induced axonal growth raises the questions of what other inhibitors also play such dual roles and how future analysis can be better designed to capture their divergent functions. Furthermore, involvement of the same signaling pathway such as STAT3 in regulating both extrinsic (astrogliosis) and intrinsic (axon regeneration) processes highlight the need to study cell type specific effects of pathways. It is possible that a pathway may need to be differentially modulated in neurons versus glia to maximize beneficial outcomes. The evolving and diverse functions of extrinsic and intrinsic regulators need to be elucidated for effective therapeutic design.
Identification of novel regulators of CNS axon regeneration
To date, the search for neuron-intrinsic regulators of regeneration has mainly employed a hypothesis-driven approach, focusing on select candidates with established roles in cell growth or developmental axon elongation. This strategy leaves many potentially important regulators in the dark. While a completely unbiased and systematic in vivo genetic screen for proteins that modulate CNS axon regeneration may presently be too ambitious of a goal, the use of genomic and proteomic approaches to profile regeneration-associated genes is being pursued [128–130]. One of these system-wide analyses led to an in vitro screening effort that ultimately resulted in the in vivo validation of KLFs as regulators of mammalian axon growth [71, 72, 130]. The effectiveness of these “-omics” efforts in identifying novel regulators of axon growth remains to be seen with future validation.
Integration of regenerated axons into functional circuitry and translational application
Currently, a major challenge in the study of intrinsic regulators is functional demonstration of their effects on axon repair. However, it is difficult to meet this challenge because 1) the number of regenerating fibers is so few that their effects may escape detection by current behavior assays and electrophysiology, and 2) dual regulation of both axon regeneration and sprouting by the same intrinsic factor, as was shown with PTEN and STAT3, confounds evaluation of the individual contribution of these processes to functional recovery. So far, some of the best evidence suggesting integration of regenerated axons into a circuitry remains anatomical, such as the demonstration of co-localization of boutons of regenerated axons with synaptic markers caudal to the injury site [58]. Furthermore, the clinical value of promising intrinsic regulators like PTEN and others identified by genetic studies need to be determined. Future efforts towards developing clinically relevant strategies to manipulate these proteins in the spinally injured and optimizing their therapeutic efficacy based on timing, dosing, and interactive effects with rehabilitation strategies will be necessary to realize the therapeutic application of basic scientific findings.
Future of personalized therapy for SCI
Human spinal cord injuries are highly heterogeneous as a result of variability in the mechanisms, levels, and severity of injuries. Axon regenerative potential varies depending on the identity of the axonal tract; and formation of compensatory detour circuits is likely determined by the identity and amount of spared axons. Therefore, promoting axon repair may require different strategies depending on the injury. Systematic evaluation on the contribution of axon regeneration, sprouting, and signaling pathways to axonal repair following different types of injuries using defined animal models will begin to address whether this is indeed the case. It is possible that different axonal systems require different signaling molecules to stimulate their regenerative response, which would support the need for treatments customized to specific injury types. Based on the heterogeneity of human spinal cord injuries and axonal tract-specific regenerative capacity, it is reasonable to envisage development of personalized therapy in the future for enhanced efficacy as the field works towards finding a treatment for spinal cord injury.
Figure 3. Shaping and mapping functional relay circuits.

Illustrated is a proposed approach to guide the formation of adaptive sprouting and map the resultant relay circuit by combining post-injury rehabilitation, retrograde trans-synaptic labeling, tissue clearing, and three-dimensional imaging of cleared brain and spinal cord. (A) In an uninjured animal, retrograde trans-synaptic tracing with fluorescent label from spinal motor neurons (green) –as can be achieved by injection of viral tracer into the muscle of interest (brown)– followed by tissue clearing to reveal proper connection of spinal motor neurons to corticospinal neurons (blue) in the corresponding motor cortex. Rodent spinal interneurons are not shown. (B) In a spinally injured animal (red “X”), spontaneous sprouting occurs but is undirected, leading to nonfunctional or maladaptive connections (indicated by blue arrow) that may change cortical motor representation and worsen motor function among other adverse effects. (C) In a spinally injured animal subjected to post-injury rehabilitation (exercise and/or electrochemical stimulation), spontaneous sprouting occurs and training strengthens adaptive circuits (indicated by blue arrow) exemplified by the establishment of relay connections to motor neurons through spinal interneurons (purple) to enhance functional recovery. Retrograde transneuronal tracing and tissue clearing allow subsequent visualization of such functional relay network. Furthermore, molecular interventions may enhance the sprouting response that in combination with rehabilitation could increase formation of new functional connections (not shown).
HIGHLIGHTS.
Neuron extrinsic and intrinsic factors regulate axon plasticity.
Circuit rewiring involves formation of detour circuits or activation of latent tracts.
Rehabilitation may strengthen adaptive connections after injury.
Transneuronal tracing and 3D imaging may reveal post-injury network alterations.
Acknowledgments
Research in the authors’ laboratory is supported by grants from NIH/NINDS (R01NS054734), CIRM (RB3-02143), the Craig H. Neilsen Foundation and Wings for Life Spinal Cord Research Foundation. M.C. is supported by a Ruth L. Kirschstein NRSA Individual Postdoctoral Fellowship from NIH/NINDS (F32NS083186).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.David S, Aguayo AJ. Axonal elongation into peripheral nervous system “bridges” after central nervous system injury in adult rats. Science. 1981;214:931–933. doi: 10.1126/science.6171034. [DOI] [PubMed] [Google Scholar]
- 2.Lee JK, Zheng B. Role of myelin-associated inhibitors in axonal repair after spinal cord injury. Experimental neurology. 2012;235:33–42. doi: 10.1016/j.expneurol.2011.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Oudega M, et al. Regeneration of adult rat sensory axons into intraspinal nerve grafts: promoting effects of conditioning lesion and graft predegeneration. Experimental neurology. 1994;129:194–206. doi: 10.1006/exnr.1994.1161. [DOI] [PubMed] [Google Scholar]
- 4.Richardson PM, Issa VM. Peripheral injury enhances central regeneration of primary sensory neurones. Nature. 1984;309:791–793. doi: 10.1038/309791a0. [DOI] [PubMed] [Google Scholar]
- 5.Goldberg JL, et al. Amacrine-signaled loss of intrinsic axon growth ability by retinal ganglion cells. Science. 2002;296:1860–1864. doi: 10.1126/science.1068428. [DOI] [PubMed] [Google Scholar]
- 6.Liu K, et al. Neuronal intrinsic mechanisms of axon regeneration. Annual review of neuroscience. 2011;34:131–152. doi: 10.1146/annurev-neuro-061010-113723. [DOI] [PubMed] [Google Scholar]
- 7.Yiu G, He Z. Glial inhibition of CNS axon regeneration. Nature reviews Neuroscience. 2006;7:617–627. doi: 10.1038/nrn1956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fernandez-Valle C, et al. Schwann cells degrade myelin and proliferate in the absence of macrophages: evidence from in vitro studies of Wallerian degeneration. Journal of neurocytology. 1995;24:667–679. doi: 10.1007/BF01179817. [DOI] [PubMed] [Google Scholar]
- 9.George R, Griffin JW. Delayed macrophage responses and myelin clearance during Wallerian degeneration in the central nervous system: the dorsal radiculotomy model. Experimental neurology. 1994;129:225–236. doi: 10.1006/exnr.1994.1164. [DOI] [PubMed] [Google Scholar]
- 10.Vargas ME, et al. Endogenous antibodies promote rapid myelin clearance and effective axon regeneration after nerve injury. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:11993–11998. doi: 10.1073/pnas.1001948107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fitch MT, Silver J. CNS injury, glial scars, and inflammation: Inhibitory extracellular matrices and regeneration failure. Experimental neurology. 2008;209:294–301. doi: 10.1016/j.expneurol.2007.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Giger RJ, et al. Guidance molecules in axon regeneration. Cold Spring Harbor perspectives in biology. 2010;2:a001867. doi: 10.1101/cshperspect.a001867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Benowitz LI, Popovich PG. Inflammation and axon regeneration. Current opinion in neurology. 2011;24:577–583. doi: 10.1097/WCO.0b013e32834c208d. [DOI] [PubMed] [Google Scholar]
- 14.Geoffroy CG, Zheng B. Myelin-associated inhibitors in axonal growth after CNS injury. Current opinion in neurobiology. 2014;27C:31–38. doi: 10.1016/j.conb.2014.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Schwab ME, Strittmatter SM. Nogo limits neural plasticity and recovery from injury. Current opinion in neurobiology. 2014;27C:53–60. doi: 10.1016/j.conb.2014.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lehmann M, et al. Inactivation of Rho signaling pathway promotes CNS axon regeneration. The Journal of neuroscience: the official journal of the Society for Neuroscience. 1999;19:7537–7547. doi: 10.1523/JNEUROSCI.19-17-07537.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Niederost B, et al. Nogo-A and myelin-associated glycoprotein mediate neurite growth inhibition by antagonistic regulation of RhoA and Rac1. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2002;22:10368–10376. doi: 10.1523/JNEUROSCI.22-23-10368.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yamashita T, et al. The p75 receptor transduces the signal from myelin-associated glycoprotein to Rho. The Journal of cell biology. 2002;157:565–570. doi: 10.1083/jcb.200202010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fournier AE, et al. Rho kinase inhibition enhances axonal regeneration in the injured CNS. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2003;23:1416–1423. doi: 10.1523/JNEUROSCI.23-04-01416.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Caroni P, Schwab ME. Antibody against myelin-associated inhibitor of neurite growth neutralizes nonpermissive substrate properties of CNS white matter. Neuron. 1988;1:85–96. doi: 10.1016/0896-6273(88)90212-7. [DOI] [PubMed] [Google Scholar]
- 21.Schnell L, Schwab ME. Axonal regeneration in the rat spinal cord produced by an antibody against myelin-associated neurite growth inhibitors. Nature. 1990;343:269–272. doi: 10.1038/343269a0. [DOI] [PubMed] [Google Scholar]
- 22.Bregman BS, et al. Recovery from spinal cord injury mediated by antibodies to neurite growth inhibitors. Nature. 1995;378:498–501. doi: 10.1038/378498a0. [DOI] [PubMed] [Google Scholar]
- 23.GrandPre T, et al. Nogo-66 receptor antagonist peptide promotes axonal regeneration. Nature. 2002;417:547–551. doi: 10.1038/417547a. [DOI] [PubMed] [Google Scholar]
- 24.Liebscher T, et al. Nogo-A antibody improves regeneration and locomotion of spinal cord-injured rats. Annals of neurology. 2005;58:706–719. doi: 10.1002/ana.20627. [DOI] [PubMed] [Google Scholar]
- 25.Pan B, et al. Myelin-associated glycoprotein and complementary axonal ligands, gangliosides, mediate axon stability in the CNS and PNS: neuropathology and behavioral deficits in single- and double-null mice. Experimental neurology. 2005;195:208–217. doi: 10.1016/j.expneurol.2005.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nguyen T, et al. Axonal protective effects of the myelin-associated glycoprotein. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2009;29:630–637. doi: 10.1523/JNEUROSCI.5204-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jones MV, et al. Accelerated axon loss in MOG35-55 experimental autoimmune encephalomyelitis (EAE) in myelin-associated glycoprotein-deficient (MAGKO) mice. Journal of neuroimmunology. 2013;262:53–61. doi: 10.1016/j.jneuroim.2013.06.008. [DOI] [PubMed] [Google Scholar]
- 28.Lee JK, et al. Assessing spinal axon regeneration and sprouting in Nogo-, MAG-, and OMgp-deficient mice. Neuron. 2010;66:663–670. doi: 10.1016/j.neuron.2010.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dergham P, et al. Rho signaling pathway targeted to promote spinal cord repair. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2002;22:6570–6577. doi: 10.1523/JNEUROSCI.22-15-06570.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lingor P, et al. Inhibition of Rho kinase (ROCK) increases neurite outgrowth on chondroitin sulphate proteoglycan in vitro and axonal regeneration in the adult optic nerve in vivo. Journal of neurochemistry. 2007;103:181–189. doi: 10.1111/j.1471-4159.2007.04756.x. [DOI] [PubMed] [Google Scholar]
- 31.Dubreuil CI, et al. Rho activation patterns after spinal cord injury and the role of activated Rho in apoptosis in the central nervous system. The Journal of cell biology. 2003;162:233–243. doi: 10.1083/jcb.200301080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bertrand J, et al. Application of Rho antagonist to neuronal cell bodies promotes neurite growth in compartmented cultures and regeneration of retinal ganglion cell axons in the optic nerve of adult rats. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2005;25:1113–1121. doi: 10.1523/JNEUROSCI.3931-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fehlings MG, et al. A phase I/IIa clinical trial of a recombinant Rho protein antagonist in acute spinal cord injury. Journal of neurotrauma. 2011;28:787–796. doi: 10.1089/neu.2011.1765. [DOI] [PubMed] [Google Scholar]
- 34.Fawcett JW, et al. Guidelines for the conduct of clinical trials for spinal cord injury as developed by the ICCP panel: spontaneous recovery after spinal cord injury and statistical power needed for therapeutic clinical trials. Spinal cord. 2007;45:190–205. doi: 10.1038/sj.sc.3102007. [DOI] [PubMed] [Google Scholar]
- 35.Silver J, Miller JH. Regeneration beyond the glial scar. Nature reviews Neuroscience. 2004;5:146–156. doi: 10.1038/nrn1326. [DOI] [PubMed] [Google Scholar]
- 36.Sofroniew MV. Molecular dissection of reactive astrogliosis and glial scar formation. Trends in neurosciences. 2009;32:638–647. doi: 10.1016/j.tins.2009.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Condic ML, et al. Embryonic neurons adapt to the inhibitory proteoglycan aggrecan by increasing integrin expression. The Journal of neuroscience: the official journal of the Society for Neuroscience. 1999;19:10036–10043. doi: 10.1523/JNEUROSCI.19-22-10036.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Afshari FT, et al. Schwann cell migration is integrin-dependent and inhibited by astrocyte-produced aggrecan. Glia. 2010;58:857–869. doi: 10.1002/glia.20970. [DOI] [PubMed] [Google Scholar]
- 39.Shen Y, et al. PTPsigma is a receptor for chondroitin sulfate proteoglycan, an inhibitor of neural regeneration. Science. 2009;326:592–596. doi: 10.1126/science.1178310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Fisher D, et al. Leukocyte common antigen-related phosphatase is a functional receptor for chondroitin sulfate proteoglycan axon growth inhibitors. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2011;31:14051–14066. doi: 10.1523/JNEUROSCI.1737-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Dickendesher TL, et al. NgR1 and NgR3 are receptors for chondroitin sulfate proteoglycans. Nature neuroscience. 2012;15:703–712. doi: 10.1038/nn.3070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sharma K, et al. Scar-mediated inhibition and CSPG receptors in the CNS. Experimental neurology. 2012;237:370–378. doi: 10.1016/j.expneurol.2012.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Coles CH, et al. Proteoglycan-specific molecular switch for RPTPsigma clustering and neuronal extension. Science. 2011;332:484–488. doi: 10.1126/science.1200840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bradbury EJ, et al. Chondroitinase ABC promotes functional recovery after spinal cord injury. Nature. 2002;416:636–640. doi: 10.1038/416636a. [DOI] [PubMed] [Google Scholar]
- 45.Yick LW, et al. Axonal regeneration of Clarke’s neurons beyond the spinal cord injury scar after treatment with chondroitinase ABC. Experimental neurology. 2003;182:160–168. doi: 10.1016/s0014-4886(02)00052-3. [DOI] [PubMed] [Google Scholar]
- 46.Houle JD, et al. Combining an autologous peripheral nervous system “bridge” and matrix modification by chondroitinase allows robust, functional regeneration beyond a hemisection lesion of the adult rat spinal cord. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2006;26:7405–7415. doi: 10.1523/JNEUROSCI.1166-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Cafferty WB, et al. Functional axonal regeneration through astrocytic scar genetically modified to digest chondroitin sulfate proteoglycans. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2007;27:2176–2185. doi: 10.1523/JNEUROSCI.5176-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bartus K, et al. Large-scale chondroitin sulfate proteoglycan digestion with chondroitinase gene therapy leads to reduced pathology and modulates macrophage phenotype following spinal cord contusion injury. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2014;34:4822–4836. doi: 10.1523/JNEUROSCI.4369-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Takeuchi K, et al. Chondroitin sulphate N-acetylgalactosaminyl-transferase-1 inhibits recovery from neural injury. Nature communications. 2013;4:2740. doi: 10.1038/ncomms3740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bush TG, et al. Leukocyte infiltration, neuronal degeneration, and neurite outgrowth after ablation of scar-forming, reactive astrocytes in adult transgenic mice. Neuron. 1999;23:297–308. doi: 10.1016/s0896-6273(00)80781-3. [DOI] [PubMed] [Google Scholar]
- 51.Faulkner JR, et al. Reactive astrocytes protect tissue and preserve function after spinal cord injury. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2004;24:2143–2155. doi: 10.1523/JNEUROSCI.3547-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Berkelaar M, et al. Axotomy results in delayed death and apoptosis of retinal ganglion cells in adult rats. The Journal of neuroscience: the official journal of the Society for Neuroscience. 1994;14:4368–4374. doi: 10.1523/JNEUROSCI.14-07-04368.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Park KK, et al. Promoting axon regeneration in the adult CNS by modulation of the PTEN/mTOR pathway. Science. 2008;322:963–966. doi: 10.1126/science.1161566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Smith PD, et al. SOCS3 deletion promotes optic nerve regeneration in vivo. Neuron. 2009;64:617–623. doi: 10.1016/j.neuron.2009.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hains BC, et al. Primary cortical motor neurons undergo apoptosis after axotomizing spinal cord injury. The Journal of comparative neurology. 2003;462:328–341. doi: 10.1002/cne.10733. [DOI] [PubMed] [Google Scholar]
- 56.Nielson JL, et al. Unexpected survival of neurons of origin of the pyramidal tract after spinal cord injury. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2010;30:11516–11528. doi: 10.1523/JNEUROSCI.1433-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Nielson JL, et al. A reassessment of whether cortical motor neurons die following spinal cord injury. The Journal of comparative neurology. 2011;519:2852–2869. doi: 10.1002/cne.22661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Liu K, et al. PTEN deletion enhances the regenerative ability of adult corticospinal neurons. Nature neuroscience. 2010;13:1075–1081. doi: 10.1038/nn.2603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Song MS, et al. The functions and regulation of the PTEN tumour suppressor. Nature reviews Molecular cell biology. 2012;13:283–296. doi: 10.1038/nrm3330. [DOI] [PubMed] [Google Scholar]
- 60.de Lima S, et al. Full-length axon regeneration in the adult mouse optic nerve and partial recovery of simple visual behaviors. Proceedings of the National Academy of Sciences of the United States of America. 2012;109:9149–9154. doi: 10.1073/pnas.1119449109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Luo X, et al. Three-dimensional evaluation of retinal ganglion cell axon regeneration and pathfinding in whole mouse tissue after injury. Experimental neurology. 2013;247:653–662. doi: 10.1016/j.expneurol.2013.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Saijilafu, et al. PI3K-GSK3 signalling regulates mammalian axon regeneration by inducing the expression of Smad1. Nature communications. 2013;4:2690. doi: 10.1038/ncomms3690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Hammarlund M, et al. Axon regeneration requires a conserved MAP kinase pathway. Science. 2009;323:802–806. doi: 10.1126/science.1165527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Yan D, et al. The DLK-1 kinase promotes mRNA stability and local translation in C. elegans synapses and axon regeneration. Cell. 2009;138:1005–1018. doi: 10.1016/j.cell.2009.06.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Xiong X, et al. Protein turnover of the Wallenda/DLK kinase regulates a retrograde response to axonal injury. The Journal of cell biology. 2010;191:211–223. doi: 10.1083/jcb.201006039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Welsbie DS, et al. Functional genomic screening identifies dual leucine zipper kinase as a key mediator of retinal ganglion cell death. Proceedings of the National Academy of Sciences of the United States of America. 2013;110:4045–4050. doi: 10.1073/pnas.1211284110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Watkins TA, et al. DLK initiates a transcriptional program that couples apoptotic and regenerative responses to axonal injury. Proceedings of the National Academy of Sciences of the United States of America. 2013;110:4039–4044. doi: 10.1073/pnas.1211074110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Shin JE, et al. Dual leucine zipper kinase is required for retrograde injury signaling and axonal regeneration. Neuron. 2012;74:1015–1022. doi: 10.1016/j.neuron.2012.04.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Miller BR, et al. A dual leucine kinase-dependent axon self-destruction program promotes Wallerian degeneration. Nature neuroscience. 2009;12:387–389. doi: 10.1038/nn.2290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.McConnell BB, Yang VW. Mammalian Kruppel-like factors in health and diseases. Physiological reviews. 2010;90:1337–1381. doi: 10.1152/physrev.00058.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Moore DL, et al. KLF family members regulate intrinsic axon regeneration ability. Science. 2009;326:298–301. doi: 10.1126/science.1175737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Blackmore MG, et al. Kruppel-like Factor 7 engineered for transcriptional activation promotes axon regeneration in the adult corticospinal tract. Proceedings of the National Academy of Sciences of the United States of America. 2012;109:7517–7522. doi: 10.1073/pnas.1120684109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Tetreault MP, et al. Kruppel-like factors in cancer. Nature reviews Cancer. 2013;13:701–713. doi: 10.1038/nrc3582. [DOI] [PubMed] [Google Scholar]
- 74.Ghoreschi K, et al. Janus kinases in immune cell signaling. Immunological reviews. 2009;228:273–287. doi: 10.1111/j.1600-065X.2008.00754.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Qiu J, et al. Conditioning injury-induced spinal axon regeneration requires signal transducer and activator of transcription 3 activation. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2005;25:1645–1653. doi: 10.1523/JNEUROSCI.3269-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Lang C, et al. STAT3 promotes corticospinal remodelling and functional recovery after spinal cord injury. EMBO reports. 2013;14:931–937. doi: 10.1038/embor.2013.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Sun F, et al. Sustained axon regeneration induced by co-deletion of PTEN and SOCS3. Nature. 2011;480:372–375. doi: 10.1038/nature10594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Herrmann JE, et al. STAT3 is a critical regulator of astrogliosis and scar formation after spinal cord injury. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2008;28:7231–7243. doi: 10.1523/JNEUROSCI.1709-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Tuszynski MH, Steward O. Concepts and methods for the study of axonal regeneration in the CNS. Neuron. 2012;74:777–791. doi: 10.1016/j.neuron.2012.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Fouad K, et al. Cervical sprouting of corticospinal fibers after thoracic spinal cord injury accompanies shifts in evoked motor responses. Current biology: CB. 2001;11:1766–1770. doi: 10.1016/s0960-9822(01)00535-8. [DOI] [PubMed] [Google Scholar]
- 81.Bareyre FM, et al. The injured spinal cord spontaneously forms a new intraspinal circuit in adult rats. Nature neuroscience. 2004;7:269–277. doi: 10.1038/nn1195. [DOI] [PubMed] [Google Scholar]
- 82.Steward O, et al. The dorsolateral corticospinal tract in mice: an alternative route for corticospinal input to caudal segments following dorsal column lesions. The Journal of comparative neurology. 2004;472:463–477. doi: 10.1002/cne.20090. [DOI] [PubMed] [Google Scholar]
- 83.Lemon RN. Descending pathways in motor control. Annual review of neuroscience. 2008;31:195–218. doi: 10.1146/annurev.neuro.31.060407.125547. [DOI] [PubMed] [Google Scholar]
- 84.Rosenzweig ES, et al. Extensive spontaneous plasticity of corticospinal projections after primate spinal cord injury. Nature neuroscience. 2010;13:1505–1510. doi: 10.1038/nn.2691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Rosenzweig ES, et al. Extensive spinal decussation and bilateral termination of cervical corticospinal projections in rhesus monkeys. The Journal of comparative neurology. 2009;513:151–163. doi: 10.1002/cne.21940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Weidner N, et al. Spontaneous corticospinal axonal plasticity and functional recovery after adult central nervous system injury. Proceedings of the National Academy of Sciences of the United States of America. 2001;98:3513–3518. doi: 10.1073/pnas.051626798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Courtine G, et al. Recovery of supraspinal control of stepping via indirect propriospinal relay connections after spinal cord injury. Nature medicine. 2008;14:69–74. doi: 10.1038/nm1682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Thallmair M, et al. Neurite growth inhibitors restrict plasticity and functional recovery following corticospinal tract lesions. Nature neuroscience. 1998;1:124–131. doi: 10.1038/373. [DOI] [PubMed] [Google Scholar]
- 89.Mullner A, et al. Lamina-specific restoration of serotonergic projections after Nogo-A antibody treatment of spinal cord injury in rats. The European journal of neuroscience. 2008;27:326–333. doi: 10.1111/j.1460-9568.2007.06006.x. [DOI] [PubMed] [Google Scholar]
- 90.Li S, Strittmatter SM. Delayed systemic Nogo-66 receptor antagonist promotes recovery from spinal cord injury. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2003;23:4219–4227. doi: 10.1523/JNEUROSCI.23-10-04219.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Fouad K, et al. Regenerating corticospinal fibers in the Marmoset (Callitrix jacchus) after spinal cord lesion and treatment with the anti-Nogo-A antibody IN-1. The European journal of neuroscience. 2004;20:2479–2482. doi: 10.1111/j.1460-9568.2004.03716.x. [DOI] [PubMed] [Google Scholar]
- 92.Freund P, et al. Anti-Nogo-A antibody treatment enhances sprouting of corticospinal axons rostral to a unilateral cervical spinal cord lesion in adult macaque monkey. The Journal of comparative neurology. 2007;502:644–659. doi: 10.1002/cne.21321. [DOI] [PubMed] [Google Scholar]
- 93.Raineteau O, et al. Reorganization of descending motor tracts in the rat spinal cord. The European journal of neuroscience. 2002;16:1761–1771. doi: 10.1046/j.1460-9568.2002.02243.x. [DOI] [PubMed] [Google Scholar]
- 94.Raineteau O, et al. Functional switch between motor tracts in the presence of the mAb IN-1 in the adult rat. Proceedings of the National Academy of Sciences of the United States of America. 2001;98:6929–6934. doi: 10.1073/pnas.111165498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Wahl AS, et al. Neuronal repair. Asynchronous therapy restores motor control by rewiring of the rat corticospinal tract after stroke. Science. 2014;344:1250–1255. doi: 10.1126/science.1253050. [DOI] [PubMed] [Google Scholar]
- 96.Kempf A, Schwab ME. Nogo-A represses anatomical and synaptic plasticity in the central nervous system. Physiology (Bethesda) 2013;28:151–163. doi: 10.1152/physiol.00052.2012. [DOI] [PubMed] [Google Scholar]
- 97.Barritt AW, et al. Chondroitinase ABC promotes sprouting of intact and injured spinal systems after spinal cord injury. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2006;26:10856–10867. doi: 10.1523/JNEUROSCI.2980-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Starkey ML, et al. Chondroitinase ABC promotes compensatory sprouting of the intact corticospinal tract and recovery of forelimb function following unilateral pyramidotomy in adult mice. The European journal of neuroscience. 2012;36:3665–3678. doi: 10.1111/ejn.12017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Goshgarian HG. The crossed phrenic phenomenon: a model for plasticity in the respiratory pathways following spinal cord injury. J Appl Physiol (1985) 2003;94:795–810. doi: 10.1152/japplphysiol.00847.2002. [DOI] [PubMed] [Google Scholar]
- 100.Sharma H, et al. Treatments to restore respiratory function after spinal cord injury and their implications for regeneration, plasticity and adaptation. Experimental neurology. 2012;235:18–25. doi: 10.1016/j.expneurol.2011.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Nantwi KD, et al. Effects of long-term theophylline exposure on recovery of respiratory function and expression of adenosine A1 mRNA in cervical spinal cord hemisected adult rats. Experimental neurology. 2003;182:232–239. doi: 10.1016/s0014-4886(03)00109-2. [DOI] [PubMed] [Google Scholar]
- 102.Hetman M, et al. Neuroprotection by brain-derived neurotrophic factor is mediated by extracellular signal-regulated kinase and phosphatidylinositol 3-kinase. The Journal of biological chemistry. 1999;274:22569–22580. doi: 10.1074/jbc.274.32.22569. [DOI] [PubMed] [Google Scholar]
- 103.Atwal JK, et al. The TrkB-Shc site signals neuronal survival and local axon growth via MEK and P13-kinase. Neuron. 2000;27:265–277. doi: 10.1016/s0896-6273(00)00035-0. [DOI] [PubMed] [Google Scholar]
- 104.Baker-Herman TL, et al. BDNF is necessary and sufficient for spinal respiratory plasticity following intermittent hypoxia. Nature neuroscience. 2004;7:48–55. doi: 10.1038/nn1166. [DOI] [PubMed] [Google Scholar]
- 105.Dale-Nagle EA, et al. Spinal vascular endothelial growth factor induces phrenic motor facilitation via extracellular signal-regulated kinase and Akt signaling. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2011;31:7682–7690. doi: 10.1523/JNEUROSCI.0239-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Dale EA, et al. Cervical spinal erythropoietin induces phrenic motor facilitation via extracellular signal-regulated protein kinase and Akt signaling. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2012;32:5973–5983. doi: 10.1523/JNEUROSCI.3873-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.McGuire M, et al. Phrenic long-term facilitation requires NMDA receptors in the phrenic motonucleus in rats. The Journal of physiology. 2005;567:599–611. doi: 10.1113/jphysiol.2005.087650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.McGuire M, et al. Formation and maintenance of ventilatory long-term facilitation require NMDA but not non-NMDA receptors in awake rats. J Appl Physiol (1985) 2008;105:942–950. doi: 10.1152/japplphysiol.01274.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Lovett-Barr MR, et al. Repetitive intermittent hypoxia induces respiratory and somatic motor recovery after chronic cervical spinal injury. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2012;32:3591–3600. doi: 10.1523/JNEUROSCI.2908-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Trumbower RD, et al. Exposure to acute intermittent hypoxia augments somatic motor function in humans with incomplete spinal cord injury. Neurorehabilitation and neural repair. 2012;26:163–172. doi: 10.1177/1545968311412055. [DOI] [PubMed] [Google Scholar]
- 111.Hayes HB, et al. Daily intermittent hypoxia enhances walking after chronic spinal cord injury: a randomized trial. Neurology. 2014;82:104–113. doi: 10.1212/01.WNL.0000437416.34298.43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Tester NJ, et al. Long-term facilitation of ventilation in humans with chronic spinal cord injury. American journal of respiratory and critical care medicine. 2014;189:57–65. doi: 10.1164/rccm.201305-0848OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Lu P, et al. Motor axonal regeneration after partial and complete spinal cord transection. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2012;32:8208–8218. doi: 10.1523/JNEUROSCI.0308-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Romero MI, et al. Extensive sprouting of sensory afferents and hyperalgesia induced by conditional expression of nerve growth factor in the adult spinal cord. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2000;20:4435–4445. doi: 10.1523/JNEUROSCI.20-12-04435.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Alilain WJ, et al. Functional regeneration of respiratory pathways after spinal cord injury. Nature. 2011;475:196–200. doi: 10.1038/nature10199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Beauparlant J, et al. Undirected compensatory plasticity contributes to neuronal dysfunction after severe spinal cord injury. Brain: a journal of neurology. 2013;136:3347–3361. doi: 10.1093/brain/awt204. [DOI] [PubMed] [Google Scholar]
- 117.Erturk A, et al. Three-dimensional imaging of solvent-cleared organs using 3DISCO. Nature protocols. 2012;7:1983–1995. doi: 10.1038/nprot.2012.119. [DOI] [PubMed] [Google Scholar]
- 118.Chung K, et al. Structural and molecular interrogation of intact biological systems. Nature. 2013;497:332–337. doi: 10.1038/nature12107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Kinoshita M, et al. Genetic dissection of the circuit for hand dexterity in primates. Nature. 2012;487:235–238. doi: 10.1038/nature11206. [DOI] [PubMed] [Google Scholar]
- 120.Fong AJ, et al. Recovery of control of posture and locomotion after a spinal cord injury: solutions staring us in the face. Progress in brain research. 2009;175:393–418. doi: 10.1016/S0079-6123(09)17526-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Angeli CA, et al. Altering spinal cord excitability enables voluntary movements after chronic complete paralysis in humans. Brain: a journal of neurology. 2014;137:1394–1409. doi: 10.1093/brain/awu038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.van den Brand R, et al. Restoring voluntary control of locomotion after paralyzing spinal cord injury. Science. 2012;336:1182–1185. doi: 10.1126/science.1217416. [DOI] [PubMed] [Google Scholar]
- 123.Carmel JB, Martin JH. Motor cortex electrical stimulation augments sprouting of the corticospinal tract and promotes recovery of motor function. Frontiers in integrative neuroscience. 2014;8:51. doi: 10.3389/fnint.2014.00051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Bachmann LC, et al. Deep brain stimulation of the midbrain locomotor region improves paretic hindlimb function after spinal cord injury in rats. Science translational medicine. 2013;5:208ra146. doi: 10.1126/scitranslmed.3005972. [DOI] [PubMed] [Google Scholar]
- 125.Maier IC, et al. Differential effects of anti-Nogo-A antibody treatment and treadmill training in rats with incomplete spinal cord injury. Brain: a journal of neurology. 2009;132:1426–1440. doi: 10.1093/brain/awp085. [DOI] [PubMed] [Google Scholar]
- 126.Garcia-Alias G, et al. Chondroitinase ABC treatment opens a window of opportunity for task-specific rehabilitation. Nature neuroscience. 2009;12:1145–1151. doi: 10.1038/nn.2377. [DOI] [PubMed] [Google Scholar]
- 127.Zhao RR, et al. Combination treatment with anti-Nogo-A and chondroitinase ABC is more effective than single treatments at enhancing functional recovery after spinal cord injury. The European journal of neuroscience. 2013;38:2946–2961. doi: 10.1111/ejn.12276. [DOI] [PubMed] [Google Scholar]
- 128.Roet KC, et al. A meta-analysis of microarray-based gene expression studies of olfactory bulb-derived olfactory ensheathing cells. Experimental neurology. 2011;229:10–45. doi: 10.1016/j.expneurol.2011.03.001. [DOI] [PubMed] [Google Scholar]
- 129.Michaelevski I, et al. Signaling to transcription networks in the neuronal retrograde injury response. Science signaling. 2010;3:ra53. doi: 10.1126/scisignal.2000952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Arlotta P, et al. Neuronal subtype-specific genes that control corticospinal motor neuron development in vivo. Neuron. 2005;45:207–221. doi: 10.1016/j.neuron.2004.12.036. [DOI] [PubMed] [Google Scholar]