

Abstract
During β-adrenergic stimulation, cardiac troponin I (cTnI) is phosphorylated by protein kinase A (PKA) at sites S23/S24, located at the N-terminus of cTnI. This phosphorylation has been shown to decrease KCa and pCa50, and weaken the cTnC-cTnI (C-I) interaction. We recently reported that phosphorylation results in an increase in the rate of early, slow phase of relaxation (kREL,slow) and a decrease in its duration (tREL,slow), which speeds up the overall relaxation. However, as the N-terminus of cTnI (residues 1–40) has not been resolved in the whole cardiac troponin (cTn) structure, little is known about the molecular-level behavior within the whole cTn complex upon phosphorylation of the S23/S24 residues of cTnI that results in these changes in function. In this study, we built up the cTn complex structure (including residues cTnC 1–161, cTnI 1–172, and cTnT 236–285) with the N-terminus of cTnI. We performed molecular-dynamics (MD) simulations to elucidate the structural basis of PKA phosphorylation-induced changes in cTn structure and Ca2+ binding. We found that introducing two phosphomimic mutations into sites S23/S24 had no significant effect on the coordinating residues of Ca2+ binding site II. However, the overall fluctuation of cTn was increased and the C-I interaction was altered relative to the wild-type model. The most significant changes involved interactions with the N-terminus of cTnI. Interestingly, the phosphomimic mutations led to the formation of intrasubunit interactions between the N-terminus and the inhibitory peptide of cTnI. This may result in altered interactions with cTnC and could explain the increased rate and decreased duration of slow-phase relaxation seen in myofibrils.
Introduction
Troponin (Tn) and tropomyosin (Tm) regulate thin-filament interactions with the thick filament in a Ca2+-dependent manner (1,2). Cardiac Tn (cTn) serves as a critical regulator of contraction in cardiac muscle and consists of three distinct subunits (cTnC, cTnI, and cTnT), each named according to their function (3). Contractile activation of cardiac muscle is initiated by Ca2+ binding to the regulatory domain of cTnC (site II). The resulting conformational change triggers the movement of the switch peptide of the inhibitory subunit cTnI (residues 147–163) toward hydrophobic residues exposed within the N-terminal portion of cTnC (NcTnC). The movement of the cTnI switch peptide pulls the adjacent inhibitory peptide (residues 137–146) away from the actin-Tm complex. Consequently, the mobility of Tm on the thin filament is increased, resulting in increased exposure of myosin-binding sites on actin.
β-Adrenergic stimulation is a primary physiological mechanism for meeting increased circulatory demands via positive inotropic and lusitropic effects (4). During β-adrenergic stimulation, cTnI is phosphorylated by protein kinase A (PKA) at sites S23/S24, which are located within the cardiac-specific N-terminus of cTnI (NcTnI) (5). The cTnC-cTnI (C-I) interaction is critical for contractile modulation of beat-to-beat systolic needs and during adrenergic stimulation (6). When cTnI is phosphorylated, the C-I interaction is weakened (4,7), leading to a reduction in the Ca2+ affinity of the regulatory domain of cTnC and the Ca2+ sensitivity of force generation, as well as an increase in the rates of muscle relaxation (4,5,8).
Little is known about the molecular-level structural changes that occur in the whole Tn complex upon phosphorylation of the S23/S24 residues of cTnI and result in these changes in function. NMR and x-ray crystallography have shed light on the atomic-level structures of the cTnC regulatory domain and the cTn complex (9–11). Spyracopoulos et al. (9) and Li et al. (10) solved NMR structures of the regulatory domain of cTnC in both Ca2+-saturated and Ca2+-free states, as well as the regulatory domain of cTnC in complex with the switch peptide of cTnI. Based on these NMR structures, Lindert et al. (12) and Kekenes-Huskey et al. (13) studied the dynamics of NcTnC, as well as Ca2+ association with NcTnC, via both conventional and accelerated molecular dynamics (MD) simulations, and examined the exposure dynamics and kinetics of cTnC hydrophobic residues via microsecond MD simulations (14). Wang et al. (15,16) applied experimental and computational approaches to study the interactions of cTnC variants with altered Ca2+-binding affinities with the switch peptide of cTnI. In 2003, Takeda et al. (11) published the first crystal structure of the human cTn complex in the Ca2+-saturated form. Varughese et al. (17) used this structure to study the binding of the drug Bepridil to cTnC, design a set of potential cTnC-binding ligands (18), and study the interactions and correlated motions among the three components of cTn (19). Using fluorescence resonance energy transfer techniques in combination with MD simulations, Jayasundar et al. (20) studied the molecular details of how a Ca2+ signal received at cTnC is transmitted to cTnI. All of these studies used either partial models of cTnC with the cTnI switch peptide or the Tn model without the N-terminus of cTnI. Thus, the basis of how PKA phosphorylation influences cTn structure and Ca2+ binding remains elusive. Recently, Howarth et al. (21) used solution NMR to determine the structure of the N-terminal extension of cTnI for both nonphosphorylated and bis-phosphorylated species, thereby making computational models to study PKA-mediated phosphorylation feasible. They then combined the N-terminal NMR structure with the crystal structure of core cTn to create an atomic model of cTn consistent with the spectroscopic data.
A computational investigation of how cTnI phosphorylation influences C-I interactions and Ca2+ binding to cTnC is an important step toward understanding the molecular basis of myofilament function during β-adrenergic stimulation. In this study, we built up the structure of the cTn complex (including residues cTnC 1–161, cTnI 1–172, and cTnT 236–285) containing the cardiac-specific N-terminus of cTnI. This structure also includes the inhibitory peptide of cTnI (residues 138–148) and the C-terminal residues (residues 282–285) of cTnT that were not present in Takeda et al.’s (11) crystal structure (PDB ID 1J1E) and Howarth et al.’s (21) NMR structure. In previous reports, other investigators (22–28) and our group (8,29) demonstrated that mutation of Ser-23 and Ser-24 to aspartic acid (S23D/S24D) changes the contractile properties of myofibrils (and trabeculae) and Tn function (in solution) in a manner consistent with PKA treatment of cTn. Additionally, the use of these bis-phosphomimics allows investigation of the specific role of cTnI S23/24 phosphorylation in cardiac muscle contraction, as titin and myosin-binding protein C (cMyBP-C) are also phosphorylated by PKA during β-adrenergic stimulation (30,31). Thus, to mimic phosphorylation and correlate the structure with our functional results, we performed all-atom/explicit-solvent MD simulations on both wild-type (WT) and cTnI-S23D/S24D cTn models in triplicate for 150 ns each. Simulation with the cTnI-S23D/S24D model showed minimal changes in the site-II Ca2+-binding loop. In contrast, there were significant changes in C-I interactions, particularly in the inhibitory-switch peptide regions. These changes were accompanied by an N-terminal cTnI interaction with the inhibitory peptide of cTnI that was not present in the WT cTn simulations, and are consistent with previous predictions based on the NMR data (7). These data suggest that the phosphomimics mutant may alter the C-I interaction by directly contacting the cTnI inhibitory peptide, precipitating an alteration in switch-peptide interaction with NcTnC.
Materials and Methods
Initial cTn complex building
The Tn model was built up from the core crystal structure of Takeda et al. (11) with addition of the N-terminal domain of cTnI, based on the NMR structure provided by Howarth et al. (21). In this NMR model, the NcTnI is in close proximity to the NcTnC and interacts with the C-terminus of cTnI. The crystal structure of the core domain of human cTn in the Ca2+-saturated form (Protein Data Bank (PDB) ID 1J1E) (11) was retrieved from the RCSB PDB. To provide as complete a starting model as possible, we used the following computational strategy to rebuild crucial residues missing in the crystal and NMR structures: Only some residues in the cTnC domain were mutated. All of the mutated residues of cTnC (residues 35, 84, and 115) were modeled back to the WT state. Both the cTnI and cTnT domains had larger fragments of missing residues. The inhibitory peptide (residues 137–146) and C-terminus of cTnI (residues 192–210), and the N-terminus (residues 1–211) and C-terminus (residues 280–288) of cTnT were missing in the crystal structure. The missing residues of the cTnI inhibitory peptide and the C-terminus were modeled using Modeler and Rosetta (32,33). To avoid changing the structure of the N-terminus of cTnI, one redundant residue (Ala-25) in the NMR structure was also retained in the built-up model. Residues 4, 10, 13, and 19, which were mutated in the NMR structure, were modeled back to their WT state. Additionally, the C-terminal residues of cTnT were modeled using Rosetta; however, because they are associated with Tm, the residues at the N-terminal region were not modeled (34). The final model for the MD simulation was the compacted core structure, including residues cTnC 1–161, cTnI 1–172, and cTnT 236–285 (see Fig. 1 A), and hereafter is referred to as the WT model.
Figure 1.
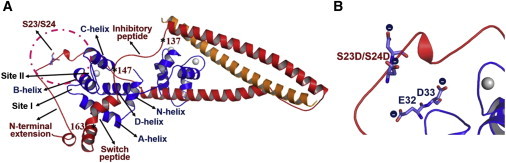
(A) The initial structures of the WT cTn complex were built up based on Takeda et al.’s (11) crystal structure and Howarth et al.’s (21) NMR structure. The inhibitory-peptide region (residues 138–147) of cTnI and the C-terminus (residues 280–285) of cTnT were modeled using Rosetta. cTnC (1–161) is shown in blue, cTnI (residues 1–172) is in red, and cTnT (residues 236–285) is in gold. Asterisks indicate the key positions in cTnI. (B) Close-up of the region around sites 23/24 of cTnI in the cTnI-S23D/S24D cTn model. The substitutions result in close apposition of negative charges on D23/D24 of cTnI with E32 and D33 between the A- and B-helices of cTnC. To see this figure in color, go online.
System preparation
To mimic phosphorylation, we constructed a bis-phosphomimics model by mutating S23/S24 of cTnI to aspartic acid (D). We prepared two systems of human cTn for simulations: 1), WT Ca2+-bound cTnC1-161-cTnI1-172-cTnT236-285 (WT model); and 2), cTnI-S23D/S24D Ca2+-bound cTnC1-161-cTnI1-172-cTnT236-285 (cTnI-S23D/S24D cTn model). cTnI mutations were performed using the Mutate Residue module in VMD (35). The constructed models were immersed with TIP3P water molecules in a truncated rectangular box that extended at least 14 Å away from any solute atoms (36). Then, all systems were neutralized and brought to 150 mM ionic strength by addition of K+ and Cl− ions. The fully solvated systems contained 112,742 (WT model) or 112,743 (cTnI-S23D/S24D cTn model) atoms. In subsequent molecular-mechanics minimizations and MD simulations, the CHARMM27 force field was applied to establish the potentials of the proteins (37).
MD simulations
Before conducting the MD simulations, we performed three steps of minimization using NAMD 2.9 (38): 1), a 10,000-step minimization of the solvent and ions (with the protein restrained using a force constant of 500 kcal/mol/Å2); 2), a 10,000-step minimization of the side chain of the protein (with the backbone of the protein restrained using a force constant of 500 kcal/mol/Å2); and 3), a 10,000-step minimization of the entire system without any restraints. Then, we used a short (380 ps) initial MD simulation with consecutively weaker restraints (10–0.1 kcal/mol/Å2) on the protein atoms to gradually heat up the entire system to a temperature of 300 K. The equilibration was finished by a short (20 ps) NPT simulation without any restraints.
Next, we performed 150 ns MD simulations in the NPT ensemble at 1 atm pressure and 300 K using NAMD 2.9 (38) and the CHARMM27 force field (37). For constant pressure control, a default setting was applied. For constant temperature control, the langevinDamping was set to 1, the langevinTemp was set to 300 K, and the langevinhydrogen was set to Off. Particle mesh Ewald (PME) was employed to calculate the long-range electrostatic interactions, along with a nonbonded interaction cutoff of 12 Å (39). The PMETolerance was set to the default value (10−6), the PMEInterpOrder was set to 4, and the PMEGridSpacing was set to 1. Center-of-mass drift due to PME was not removed. Periodic boundary conditions (PBCs) were applied in the simulations, and the wrapWater and wrapAll were both set to On. The cell origin was defined as (in Å) 8.6, 45.9, and 25.2; the cellBasisVector1 was defined as (in Å) 82.4, 0, and 0; the cellBasisVector2 was defined as (in Å) 0, 131.3, and 0; and the cellBasisVector3 was defined as (in Å) 0, 0, and 110.3. The SHAKE procedure was applied on the bonds involving hydrogen atoms and the time step was set to 2.0 fs (40). The geometric tolerance used by SHAKE was 10−8 Å. During the sampling process, the coordinates were saved every 10 ps.
Calcium coordination
To monitor the stability of coordination between site II Ca2+ and its binding residues of cTnC, we calculated the time evolution of the following distances for each 150 ns simulation: Ca2+-Asp-65 OD2, Ca2+-Asp-67 OD2, Ca2+-Ser-69 OG, Ca2+-Thr-71 OG1, Ca2+-Asp-73 OD2, and Ca2+-Glu-76 OE2. All of these atoms correspond to the atoms that coordinate with the site II Ca2+ ion in the crystal structure (11). Considering that the coordinating atoms switched occasionally during the course of the simulations, we also recorded additional distances between the following alternate possible coordinating atoms: Ca2+-Asp-65 OD1, Ca2+-Asp-67 OD1, Ca2+-Asp-73 OD1, and Ca2+-Glu-76 OE1. The shorter distance between Ca2+ and the oxygen atom of residues Asp-65, Asp-67, and Asp-73 was recorded. Considering that both OE1 and OE2 of Glu-76 coordinate with Ca2+, we recorded the distances for both of them. Distances were measured every 10 ps in the two models.
Contact analysis
The residue-residue contacts between cTnC and key regions of cTnI (i.e., the N-terminus and inhibitory-peptide and switch-peptide regions) were monitored over the course of the entire simulations. Contacts between two residues were defined as described previously (15), with a carbon-carbon distance of ≤5.4 Å and a distance between any other noncarbon atoms of ≤4.6 Å being considered a contact. Contacts between NcTnC-NcTnI, between NcTnC and the switch-peptide of cTnI, as well as between cTnC and the inhibitory-peptide of cTnI were monitored. Intrasubunit interactions between the N-terminus and the inhibitory-peptide region of cTnI were also recorded. Additionally, contacts between the B- and C-helices of cTnC, a region that is important for Ca2+ handling (site II Ca2+ binding) were monitored. For each residue contact pair, the fraction of simulation time during which these residues were in contact was calculated for both simulation systems. Residue contact pairs for which that fraction in contact changed most dramatically upon introduction of the bis-phosphomimic mutations were identified as hotspot residues.
Interhelical angle analysis and solvent-accessible surface area calculation
The interhelical angles and distances were calculated using interhlx (K. Yap, University of Toronto). To quantify the exposure of the hydrophobic surface in cTnC, which has been shown to be correlated with binding to the switch peptide of cTnI, 14 hydrophobic residues (Phe-20, Ala-23, Phe-24, Ile-26, Phe-27, Ile-36, Leu-41, Val-44, Leu-48, Leu-57, Met-60, Phe-77, Met-80, and Met-81) were selected to calculate the solvent-accessible surface area values via VMD (41) as described previously (15).
Results
Global structural behavior
The starting structure for cTn is shown in Fig. 1 A and is referred to as the WT model. Simulations for cTnI-S23D/S24D cTn were initiated from the same structure following the two-residue substitution. Fig. 1 B shows that these substitutions result in close apposition of negative charges on position 23/24 of cTnI with E32 and D33 between the A- and B-helices of cTnC, which may cause repulsive electrostatic interactions.
We compared the dynamics of the two systems from triplicate 150 ns simulations. The results are demonstrated by the values of the root mean-square displacement (RMSD) of the protein backbone atoms (C, CA, and N), calculated based on the starting structure (see Fig. S1 in the Supporting Material). The RMSD plot indicates that the backbone atoms in both complexes were stable after 80 ns. We then calculated the root mean-square fluctuations (RMSFs) versus the protein residue numbers of each subunit. Fig. 2 A shows the average (± SD) RMSF of the cTnC and cTnI subunits for both the WT and cTnI-S23D/S24D cTn systems in triplicate rounds of MD simulations. Fluctuations increased for the cTnI-S23D/S24D cTn model with respect to the WT model. Interestingly, changes were not distributed equally over the individual subunits. A large change was observed in NcTnC (cTnC residues 1–89) and an even more dramatic change was observed for NcTnI (cTnI residues 1–41). Fig. 2 A highlights site I (blue) and site II (the Ca2+-binding loop; pink) of cTnC, and the inhibitory-peptide (green) and switch-peptide regions (orange) of cTnI. These regions were influenced differently upon introduction of the phosphomimic mutations. Site I of cTnC was more flexible in the cTnI-S23D/S24D cTn model than in the WT model (the average RMSF for site I increased from 2.4 Å to 4.4 Å upon phosphomimic mutations). The average RMSF of cTnC site II also increased slightly upon introduction of the phosphomimic mutations (2.2 Å for WT vs. 3.2 Å for the cTnI-S23D/S24D cTn model). Similarly, the cTnI switch peptide became more flexible upon introduction of the phosphomimic mutations (the average RMSF for the switch peptide increased from 2.8 Å to 5.1 Å). Additionally, the RMSFs of the cTnI inhibitory region and the I-T arm (cTnI residues 42–137) were virtually unchanged for the phosphomimic mutants.
Figure 2.
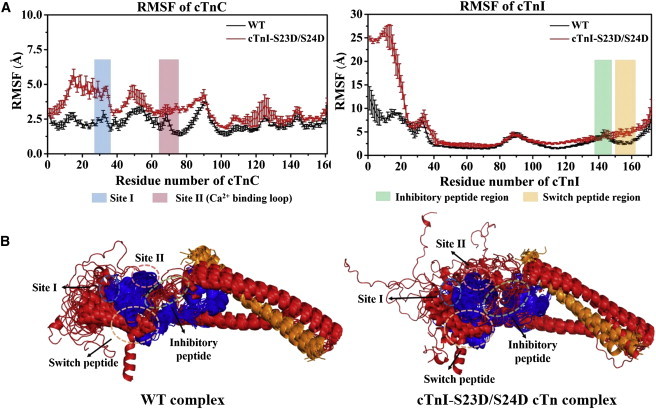
(A) Comparison of average (± SD) RMSF values of cTnC and cTnI for both the WT and cTnI-S23D/S24D cTn systems in triplicate rounds of MD simulations. Site I and site II (the Ca2+-binding loop) of cTnC are highlighted in blue and pink, respectively, and the inhibitory-peptide and switch-peptide regions of cTnI are highlighted in green and orange, respectively. (B) Superposition of snapshots (in cartoon representation) extracted every 10 ns during 150 ns MD simulations for both complexes. cTnC is shown in blue, cTnI is in red, cTnT is in gold, and all key regions are highlighted with dashed circles. To see this figure in color, go online.
To better visualize how phosphorylation influences subunit interactions in the cTn complex, we overlaid snapshots taken every 10 ns during the 150 ns MD simulations, as shown in Fig. 2 B. For clarity, cTnC is shown in blue, cTnI is in red, and cTnT is in gold. It can be seen in Fig. 2 B that the N-terminus of cTnI in the cTnI-S23D/S24D cTn model exhibited greater flexibility with respect to the WT model. This led to increased fluctuations of the NcTnC domain. In addition, NcTnI lost interactions with the C-terminus of cTnI in the cTnI-S23D/S24D cTn model, whereas some interactions were maintained in the WT model. As discussed further below, we speculate that this difference allows NcTnI to become available for interactions with other regions of the cTn complex.
Calcium-binding loop
It is difficult to obtain a detailed computational description of all Ca2+-related processes, but we measured two parameters that are associated with Ca2+ binding in site II: 1), the time evolution of distances between the bound Ca2+ ion and its coordinating residues; and 2), the contacts between the B-helix (cTnC residues 38–48) and C-helix (cTnC residues 54–64) of cTnC. Fig. 3 A shows the site II Ca2+-binding loop, as well as the six coordinating residues at the beginning of the simulations. We monitored the distances between Ca2+ and its six coordinating residues over the course of each 150 ns simulation. For four of the residues (Asp-65, Asp-67, Asp-73, and Glu-76), there was no significant difference in fluctuation between the cTnI-S23D/S24D and WT models in any of the simulations (Fig. S2), and these residues were always coordinated with Ca2+. Fig. 3 B shows the distances for the other two coordinating residues, Ser-69 and Thr-71. Distances fluctuated much more for these two residues and varied for each run in both models. The coordinating time for Ser-69 was reduced equally for both models (Fig. 3 C), which is in agreement with our previous observation (16).
Figure 3.
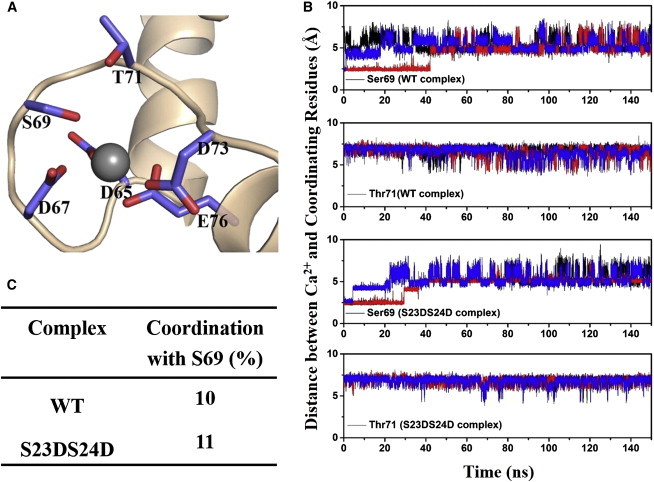
(A) Structure of the site II Ca2+-binding loop at the beginning of the simulation. Six coordinating residues are in stick representation. (B) Distances between Ca2+ and its coordinating cTnC site II residues (Ser-69 and Thr-71) over the course of each MD simulation for both complexes. The first run result is shown in black, the second run result is in red, and the third run result is in blue. (C) Coordinating time (percentage) of Ser-69 with Ca2+ during totally 450 ns MD simulations for the WT and cTnI-S23D/S24D cTn complexes. To see this figure in color, go online.
The contact maps of residue-residue pairs between the B- and C-helices of cTnC were calculated and are plotted in Fig. S3 (see legend for color coding). There was no significant change of contacts between the B- and C-helices upon introduction of the phosphomimic mutations. Two residues of the C-helix (Leu-57 and Ile-61) formed strong interactions with the B-helix, indicating that these residues may be crucial for structural stability and Ca2+ binding. This result is consistent with our previous observation that mutation of either residue (L57Q or I61Q) reduced Ca2+ binding (16). In summary, our analyses suggest that phosphomimic mutations do not impact the behavior of coordinating residues or the structure of the Ca2+-binding loop.
cTnI-S23D/S24D affects interactions of NcTnI with NcTnC
To identify the C-I contacts that are most affected by phosphomimic mutations, we calculated the fraction of contact time for every residue-residue pair. We first focused on the binding region between NcTnC (cTnC residues 1–89) and NcTnI (cTnI residues 1–41). In the WT model (Fig. 4 A), NcTnI mainly contacts the loop between the A- and B-helices (LoopA-B) of NcTnC (residues 29–37), as well as the D-helix (residues 74–83). In contrast, in the cTnI-S23D/S24D cTn model (Fig. 4 A), NcTnI binds with NcTnC D-helix residues and the C-helix (residues 54–64), but not with residues in LoopA-B. This large reduction in contact time with LoopA-B of cTnC, as well as the greater contact time with the C-helix of NcTnC, is more clearly shown in Fig. 4 C, which maps the contact difference between Fig. 4, A and B.
Figure 4.
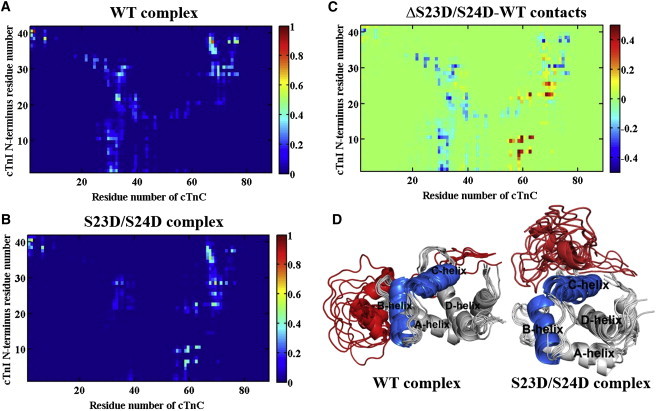
(A and B) Average contact maps of residue-residue pairs between NcTnC and NcTnI during 450 ns MD simulations for the (A) WT and (B) cTnI-S23D/S24D complexes. The blue end of the spectrum (value = 0) reflects no contact between the residue-residue pair, and the red end of the spectrum (value = 1) represents 100% contact between the residue-residue pair. (C) Difference contact map of residue-residue pairs between NcTnC and NcTnI that were most affected upon introduction of the phosphomimic mutations. The color green (value = 0) reflects no difference between the two systems, the red end of the spectrum (values > 0) reflects more contacts in the cTnI-S23D/S24D cTn system, and the blue end of the spectrum (values < 0) indicates more contacts in the WT model. (D) Comparison of interactions between NcTnC and NcTnI during the last 70 ns (from 80 ns to 150 ns) of MD simulations for WT and cTnI-S23D/S24D complexes. The B-helix (residues 38–48) and C-helix (residues 54–64) of NcTnC (residues 1–89) are highlighted in blue, the other region of NcTnC is shown in gray, and NcTnI (residues 1–41) is in red. To see this figure in color, go online.
To further examine the binding interface between NcTnC and NcTnI, we aligned and compared the structures of these regions for every 10 ns during the last 70 ns of simulations (Fig. 4 D). In the WT model, NcTnI was always in close proximity to the NcTnC region, whereas in the cTnI-S23D/S24D cTn model, NcTnI flipped over and was in close proximity to the C-helix of cTnC. As mentioned above, in the initial WT structure, cTnI sites S23/S24 were close to the B-helix of cTnC, and residues E32 and D33 were located at LoopA-B of NcTnC. For the cTnI-S23D/S24D cTn model, the close apposition of negative charges on D23/D24 of cTnI with E32/D33 of cTnC may result in repulsive electrostatic interactions that disfavor the WT configuration (see Fig. 1 B) and thus repel NcTnI. We also calculated the angles and distances between the B- and C-helices of NcTnC (Fig. S5), and found no significant change between the two models.
cTnI-S23D/S24D affects cTnI switch peptide-NcTnC and cTnI inhibitory peptide-cTnC interactions
We subsequently studied how interactions between the cTnI switch peptide (cTnI residues 148–164) and NcTnC, as well as interactions between the cTnI inhibitory peptide (cTnI residues 138–147) and cTnC, were affected upon introduction of the S23/S24 phosphomimic mutations into cTnI. Average contact maps for interactions between the cTnI switch peptide and NcTnC are shown in Fig. 5, A (WT model) and B (cTnI-S23D/S24D model). Most of the residue-residue pairs displayed in the figure have an altered percentage of contact time with the cTnI-S23D/S24D mutation model (Fig. 5 C). We also calculated the angles and distances between the A- and B-helices of cTnC, as well as the exposure of the hydrophobic patch in cTnC. No significant differences were found between the two complexes (the results are summarized in Figs. S7 and S8). On the other hand, the average contact maps for interactions between the cTnI inhibitory peptide and cTnC (Fig. 5, D (WT model) and E (cTnI-S23D/S24D model)) show that the most pronounced cluster of residues that were affected consists of residues 55–60, 85–95, and 150–160 of cTnC, as demonstrated in the difference contact map (Fig. 5 F).
Figure 5.
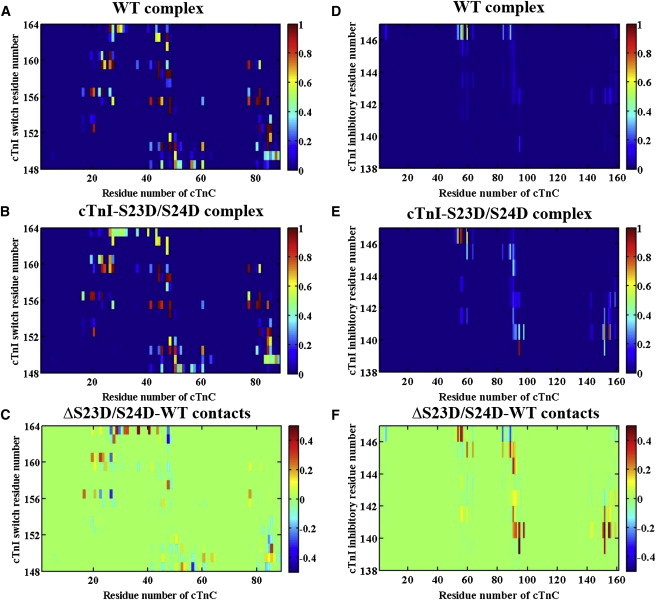
(A and B) Average contact maps of residue-residue pairs between NcTnC and cTnI switch peptides for (A) WT and (B) cTnI-S23D/S24D complexes. (C) Difference contact map of residue-residue pairs between NcTnC and cTnI switch peptides that were most affected upon introduction of the phosphomimic mutations. (D and E) Average contact maps of residue-residue pairs between cTnC and cTnI inhibitory peptides for the (D) WT and (E) cTnI-S23D/S24D complexes. (F) Difference contact map of residue-residue pairs between cTnC and cTnI inhibitory peptides that were most affected upon introduction of the phosphomimic mutations. To see this figure in color, go online.
cTnI-S23D/S24D leads to the formation of intrasubunit binding between the N-terminus and the inhibitory-peptide region of cTnI
In a previous study, Howarth et al. (21) used solution NMR to determine the structures of the N-terminal extension of cTnI for both nonphosphorylated and bis-phosphorylated species. They applied comparative docking, which was designed to maximize the cardiac N-terminus to interact with opposite polarity, to determine the low-energy docking structure. In the low-energy structure, cTnI1-32 resulted in a weakening interaction with the N-lobe of cTnC and a repositioning of the cTnI1-32 for favorable interactions with basic regions of cTnI (most likely the inhibitory region of cTnI). Specifically, the acidic residues of the N-terminus (Asp-3, Glu-4, Asp-7, and Glu-11) interacted with residues Arg-142 and Arg-146 of the inhibitory region (21). To test the hypothesis that the acidic N′ region of cTnI (residues 2–11) helps regulate myocardial function, Sadayappan et al. (42) generated cardiac-specific transgenic mice in which residues 2–11 of cTnI (cTnI (Δ2–11)) were deleted. The mouse hearts displayed significantly decreased contraction and relaxation under basal and β-adrenergic stress compared with nontransgenic hearts, with a reduction in maximal Ca2+-dependent force and maximal Ca2+-activated Mg2+-ATPase activity. However, the Ca2+ sensitivity of force development and the cTnI-S23/S24 phosphorylation were not affected. These observations suggest that residues 2–11 of cTnI, comprising the acidic N′ region, do not play a direct role in the Ca2+-induced transition in the N-lobe of cTnC. Deletion of the conserved acidic N′ region in cTnI (Δ2–11) in mice also resulted in a decrease in myocardial contractility, demonstrating the importance of the acidic N′ region for regulating myocardial contractility and mediating the response of the heart to β-adrenergic stimulation. These results provided further support for the notion that the acidic N-terminus plays a role in decreasing cardiac contractility and mediating the response of the heart to β-adrenergic stimulation by interacting directly with the inhibitory region of cTnI. Based on this finding (42) and the NMR structural data (21), Solaro et al. (7) predicted that the phosphorylated N-terminal extension of cTnI can interact with the inhibitory region of cTnI.
To quantitatively assess whether this interaction can form upon introduction of the two phosphomimic mutations into S23/S24 in our MD simulations, we monitored the intrasubunit contacts between the N-terminus and the inhibitory-peptide region of cTnI for both systems. In the WT model (Fig. 6 A), no interaction was detected between these two regions (same finding for all three runs), and this lack of interaction can be clearly seen in the WT model structure (Fig. 6 E). Interestingly, in the cTnI-S23D/S24D model, we found that phosphomimic mutations led to the formation of an intrasubunit interaction between the N-terminus and the inhibitory-peptide region of cTnI in all three independent runs of the MD simulations (Figs. 6, B–D). In our cTnI-S23D/S24D cTn model, residues 9–14 of the N-terminus could interact with residues 140–142 of the inhibitory peptide of cTnI (for the detailed binding pattern, see Fig. 6 F).
Figure 6.
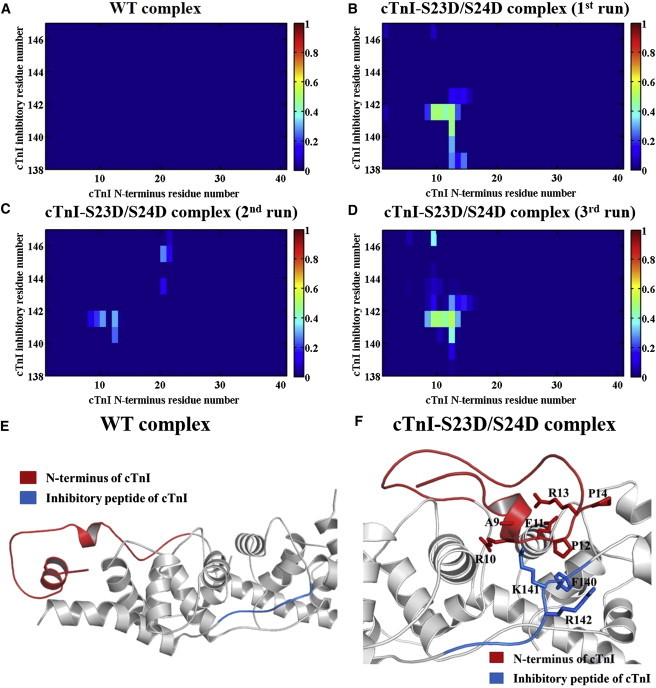
(A) Average contact map of residue-residue pairs between the N-terminus and the inhibitory-peptide region of cTnI for the WT model. (B–D) Contact maps of residue-residue pairs between the N-terminus and inhibitory-peptide region of cTnI in cTnI-S23D/S24D complexes for three runs of simulations. (E and F) Representative binding pattern between the N-terminus (red) and the inhibitory-peptide region (blue) of cTnI in the (E) WT and (F) cTnI-S23D/S24D cTn models. To see this figure in color, go online.
Discussion
Our goal in this study was to elucidate how the phosphomimic mutant of cTnI (cTnI-S23D/S24D) induces changes in the cTn structure (via parallel MD simulations) that underlie changes in function reported in the literature (4,5,8,29). To mimic phosphorylation, we constructed a bis-phosphomimic model (cTnI-S23D/S24D cTn model) by mutating S23/S24 of cTnI to aspartic acid. The cTnI-S23D/S24D mutant was previously demonstrated to mimic the PKA phosphorylation effects on S23/S24 of cTnI (cTnI-pS23/pS24) both structurally and functionally (22–25). Finley et al. (22) reported that cTnI (1–80)DD, which has phosphorylated serine residues mutated to aspartic acid, served as a good structural mimetic of the phosphorylated state and would facilitate future biophysical studies. Subsequently, several groups used the cTnI-S23D/S24D mutation instead of cTnI-pS23/pS24 to study the impact of PKA phosphorylation on cardiac function because it is a useful tool for studying the specific effect of PKA phosphorylation of cTnI (8,25–29) in the absence of cMyBP-C and titin phosphorylation, which also occurs during β-adrenergic stimulation (30,31). The major findings of the study presented here are that the phosphomimic mutations 1), resulted in minimal changes in the site-II Ca2+ binding loop; 2), significantly altered the C-I interactions, particularly in the inhibitory-switch peptide region; and 3), importantly, led to the formation of an intrasubunit interaction between the N-terminus and inhibitory-peptide regions of cTnI. Taken together, these data suggest that the introduction of phosphomimic mutations into the N-terminus of cTnI may alter the C-I interaction by forming a direct contact with the cTnI inhibitory peptide, resulting in an altered switch-peptide interaction with NcTnC.
The effects of bis-phosphorylation of cTnI on the overall cTn structure have been studied experimentally. In fluorescence studies, Dong et al. (43) found that bis-phosphorylation resulted in a reduction of the axial ratio of cTnI and the formation of a more compact structure upon phosphorylation. Heller et al. (44) reported that bis-phosphorylation induced a dramatic bending of the rod-like cTnI at the N-terminal extension that binds with cTnC, resulting in a significant decrease in the axial ratio of cTnI and the cTn complex overall. Using surface plasmon resonance, Reiffert et al. (45) demonstrated that the shape of cTnI changed from an asymmetrical structure to a more symmetrical one upon phosphorylation, which is consistent with the bending that results in a shorter and broader structure. All of these experimental findings suggest that there is an alternative binding for the bis-phosphorylated cardiac-specific N-terminus. Our simulations also suggest a bending at the N-terminal extension of cTnI and a more compact cTn structure upon introduction of the phosphomimic mutations, consistent with previous biochemical studies (43–45).
Recently, our group studied how PKA phosphorylation of cTnI modulates the activation and relaxation kinetics of ventricular myofibrils (8). A major finding of that study was that PKA phosphorylation of cTnI is responsible for the reduced duration and increased rate of slow-phase relaxation, which speeds overall relaxation, and that this was correlated with decreased C-I interaction and binding affinity of Ca2+ to cTnC. We also found that the rate of myofibril contraction was slowed by PKA phosphorylation at submaximal levels of Ca2+ activation, which is more representative of the normal heart twitch in vivo. We hypothesized that changes in the C-I interaction, rather than Ca2+ sensitivity, are responsible for the PKA-modulated thin-filament activation and relaxation. In support of the idea that cTn properties can affect relaxation, Narolska et al. (46) reported that the C-terminus of cTnI increased the duration of slow-phase relaxation, and our previous study (47) indicated that a cTnC mutation increased the Ca2+-binding affinity and C-I interaction, thereby prolonging the duration of slow-phase relaxation. In the study presented here, we found that introducing the two phosphomimic mutations into S23/S24 had little effect on the site-II Ca2+-binding loop, whereas it altered the C-I interactions, especially near the inhibitory-switch region of cTnI. The most interesting finding is that the phosphomimic mutants led to the formation of an intrasubunit interaction between the N-terminus and the inhibitory-peptide regions of cTnI. Howarth et al. (21) applied molecular docking designed for maximum interactions of the N-terminal extension with the opposite polarity region, and provided evidence that the phosphorylated N-terminal extension of cTnI can interact with basic regions of cTnI (most likely the inhibitory region). Sadayappan et al. (42) provided further support for the notion that the acidic N-terminus plays a role in decreasing cardiac contractility and mediating the response of the heart to β-adrenergic stimulation by directly interacting with the inhibitory region of cTnI. Also, Solaro et al. (7) predicted the potential interaction between the phosphorylated N-terminal extension of cTnI and the inhibitory-peptide region. Together, these findings support our finding that the phosphomimic mutations led to the intrasubunit interaction between the N-terminus and inhibitory regions of cTnI. Burkart et al. (48) reported that phosphorylation of Thr-144, which is located at the inhibitory region of cTnI, differentially depresses myofilament tension and shortens the velocity, providing indirect evidence that the intrasubunit interaction between the N-terminal extension and the inhibitory-peptide region may alter cross-bridge kinetics. We speculate that this intrasubunit interaction may subsequently weaken interactions between the switch peptide of cTnI and NcTnC, allowing a stronger interaction between the inhibitory peptide of cTnI and actin (Scheme 1), and thus modulate the cross-bridge activity (i.e., increase the slow-phase relaxation). In the future, it would be interesting to compare the dynamics of simulated phosphorylated serines with our current findings regarding phosphomimic mutations.
Scheme 1.
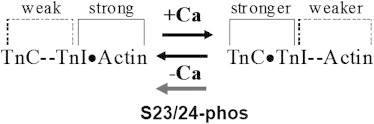
Flow chart of interactions among cTnC, cTnI, and actin in the presence and absence of Ca2+, as well as phosphorylation of the S23/S24 residues of cTnI.
Conclusions
β-Adrenergic stimulation serves as a primary physiological mechanism for meeting increased circulatory demands via positive inotropic and lusitropic effects (4). During β- adrenergic stimulation, cTnI is phosphorylated by PKA at sites S23/S24, which are located at the N-terminus of cTnI. This PKA-mediated phosphorylation on sites S23/S24 of cTnI has been shown to decrease KCa and pCa50, and weaken the C-I interaction (4). A previous study demonstrated that PKA phosphorylation of a cardiac skinned muscle resulted in a decrease in the Ca2+ sensitivity of muscle contraction, as well as an increased rate of cardiac muscle relaxation (4,5). In another study (8), we demonstrated that this phosphorylation resulted in an accelerated slow-phase relaxation in cardiac myofibril, which sped up the overall relaxation. However, since the N-terminus of cTnI has not been resolved in the whole cTn structure, the molecular-level structural changes that occur in the whole cTn complex upon cTnI phosphorylation at sites S23/S24 and result in these functional changes have remained elusive. Our current findings from MD simulations of cTn containing the N-terminal extension of cTnI provide structural evidence that supports previous NMR work on partial protein complexes, as well as previously proposed structure-function relationships (7,8,29,42). Taken together, these results show that the combination of solution protein biochemistry, myofibril mechanics/kinetics measurements, and computational modeling offers a powerful approach for determining the molecular mechanisms underlying how PKA-mediated phosphorylation of myofilament proteins during β-adrenergic stimulation affects the contraction and relaxation of both normal and diseased cardiac muscle.
Acknowledgments
We thank J.W. Howarth and colleagues for providing their NMR structure, which enabled us to build up the whole cTn complex model. We appreciate the support of Prof. Peter Arzberger and helpful discussions with other members of M.R.’s lab.
This research was supported by NIH grants R01 HL-65497 and HL-11197 (M.R.), PO1 HL-062426 (R.J.S.), AHA 11POST7400069 (V.S.R.), and AHA 12POST11570005 (S.L.). Funding and support from the National Biomedical Computational Resource (NBCR) is provided through NIH grant P41 GM103426 to Y.C., S.L., and P.K.-H. Work by J.A.M.’s group is supported in part by the NSF, NIH, HHMI, and NSF Supercomputer Centers.
Supporting Material
References
- 1.Gordon A.M., Homsher E., Regnier M. Regulation of contraction in striated muscle. Physiol. Rev. 2000;80:853–924. doi: 10.1152/physrev.2000.80.2.853. [DOI] [PubMed] [Google Scholar]
- 2.Davis J.P., Norman C., Tikunova S.B. Effects of thin and thick filament proteins on calcium binding and exchange with cardiac troponin C. Biophys. J. 2007;92:3195–3206. doi: 10.1529/biophysj.106.095406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Farah C.S., Reinach F.C. The troponin complex and regulation of muscle contraction. FASEB J. 1995;9:755–767. doi: 10.1096/fasebj.9.9.7601340. [DOI] [PubMed] [Google Scholar]
- 4.Zhang R., Zhao J., Potter J.D. Cardiac troponin I phosphorylation increases the rate of cardiac muscle relaxation. Circ. Res. 1995;76:1028–1035. doi: 10.1161/01.res.76.6.1028. [DOI] [PubMed] [Google Scholar]
- 5.Kentish J.C., McCloskey D.T., Solaro R.J. Phosphorylation of troponin I by protein kinase A accelerates relaxation and crossbridge cycle kinetics in mouse ventricular muscle. Circ. Res. 2001;88:1059–1065. doi: 10.1161/hh1001.091640. [DOI] [PubMed] [Google Scholar]
- 6.Abbott M.B., Dvoretsky A., Rosevear P.R. Cardiac troponin I inhibitory peptide: location of interaction sites on troponin C. FEBS Lett. 2000;469:168–172. doi: 10.1016/s0014-5793(00)01271-0. [DOI] [PubMed] [Google Scholar]
- 7.Solaro R.J., Rosevear P., Kobayashi T. The unique functions of cardiac troponin I in the control of cardiac muscle contraction and relaxation. Biochem. Biophys. Res. Commun. 2008;369:82–87. doi: 10.1016/j.bbrc.2007.12.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rao, V. S., Y. Cheng, …, M. Regnier. PKA phosphorylation of cardiac troponin I modulates activation and relaxation kinetics of ventricular myofibrils. In press. (http://dx.doi.org/10.1016/j.bpj.2014.07.027). [DOI] [PMC free article] [PubMed]
- 9.Spyracopoulos L., Li M.X., Sykes B.D. Calcium-induced structural transition in the regulatory domain of human cardiac troponin C. Biochemistry. 1997;36:12138–12146. doi: 10.1021/bi971223d. [DOI] [PubMed] [Google Scholar]
- 10.Li M.X., Spyracopoulos L., Sykes B.D. Binding of cardiac troponin-I147-163 induces a structural opening in human cardiac troponin-C. Biochemistry. 1999;38:8289–8298. doi: 10.1021/bi9901679. [DOI] [PubMed] [Google Scholar]
- 11.Takeda S., Yamashita A., Maéda Y. Structure of the core domain of human cardiac troponin in the Ca(2+)-saturated form. Nature. 2003;424:35–41. doi: 10.1038/nature01780. [DOI] [PubMed] [Google Scholar]
- 12.Lindert S., Kekenes-Huskey P.M., McCammon J.A. Long-timescale molecular dynamics simulations elucidate the dynamics and kinetics of exposure of the hydrophobic patch in troponin C. Biophys. J. 2012;103:1784–1789. doi: 10.1016/j.bpj.2012.08.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kekenes-Huskey P.M., Lindert S., McCammon J.A. Molecular basis of calcium-sensitizing and desensitizing mutations of the human cardiac troponin C regulatory domain: a multi-scale simulation study. PLOS Comput. Biol. 2012;8:e1002777. doi: 10.1371/journal.pcbi.1002777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lindert S., Kekenes-Huskey P., McCammon J.A. Analysis of free energy of opening the troponin C binding pocket for troponin I using microsecond molecular dynamics simulations. Biophys. J. 2013;104:331A–332A. [Google Scholar]
- 15.Wang D., Robertson I.M., Regnier M. Structural and functional consequences of the cardiac troponin C L48Q Ca(2+)-sensitizing mutation. Biochemistry. 2012;51:4473–4487. doi: 10.1021/bi3003007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang D., McCully M.E., Regnier M. Structural and functional consequences of cardiac troponin C L57Q and I61Q Ca(2+)-desensitizing variants. Arch. Biochem. Biophys. 2013;535:68–75. doi: 10.1016/j.abb.2013.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Varughese J.F., Baxley T., Li Y. A computational and experimental approach to investigate bepridil binding with cardiac troponin. J. Phys. Chem. B. 2011;115:2392–2400. doi: 10.1021/jp1094504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Varguhese J.F., Li Y. Molecular dynamics and docking studies on cardiac troponin C. J. Biomol. Struct. Dyn. 2011;29:123–135. doi: 10.1080/07391102.2011.10507378. [DOI] [PubMed] [Google Scholar]
- 19.Varughese J.F., Chalovich J.M., Li Y. Molecular dynamics studies on troponin (TnI-TnT-TnC) complexes: insight into the regulation of muscle contraction. J. Biomol. Struct. Dyn. 2010;28:159–174. doi: 10.1080/07391102.2010.10507350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jayasundar J.J., Xing J., Dong W.-J. Molecular dynamics simulations of the cardiac troponin complex performed with FRET distances as restraints. PLoS ONE. 2014;9:e87135. doi: 10.1371/journal.pone.0087135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Howarth J.W., Meller J., Rosevear P.R. Phosphorylation-dependent conformational transition of the cardiac specific N-extension of troponin I in cardiac troponin. J. Mol. Biol. 2007;373:706–722. doi: 10.1016/j.jmb.2007.08.035. [DOI] [PubMed] [Google Scholar]
- 22.Finley N., Abbott M.B., Rosevear P.R. NMR analysis of cardiac troponin C-troponin I complexes: effects of phosphorylation. FEBS Lett. 1999;453:107–112. doi: 10.1016/s0014-5793(99)00693-6. [DOI] [PubMed] [Google Scholar]
- 23.Sakthivel S., Finley N.L., Robbins J. In vivo and in vitro analysis of cardiac troponin I phosphorylation. J. Biol. Chem. 2005;280:703–714. doi: 10.1074/jbc.M409513200. [DOI] [PubMed] [Google Scholar]
- 24.Dohet C., al-Hillawi E., Rüegg J.C. Reconstitution of skinned cardiac fibres with human recombinant cardiac troponin-I mutants and troponin-C. FEBS Lett. 1995;377:131–134. doi: 10.1016/0014-5793(95)01319-9. [DOI] [PubMed] [Google Scholar]
- 25.Takimoto E., Soergel D.G., Murphy A.M. Frequency- and afterload-dependent cardiac modulation in vivo by troponin I with constitutively active protein kinase A phosphorylation sites. Circ. Res. 2004;94:496–504. doi: 10.1161/01.RES.0000117307.57798.F5. [DOI] [PubMed] [Google Scholar]
- 26.Biesiadecki B.J., Kobayashi T., de Tombe P.P. The troponin C G159D mutation blunts myofilament desensitization induced by troponin I Ser23/24 phosphorylation. Circ. Res. 2007;100:1486–1493. doi: 10.1161/01.RES.0000267744.92677.7f. [DOI] [PubMed] [Google Scholar]
- 27.Hanft L.M., Biesiadecki B.J., McDonald K.S. Length dependence of striated muscle force generation is controlled by phosphorylation of cTnI at serines 23/24. J. Physiol. 2013;591:4535–4547. doi: 10.1113/jphysiol.2013.258400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wijnker P.J.M., Foster D.B., van der Velden J. Impact of site-specific phosphorylation of protein kinase A sites Ser23 and Ser24 of cardiac troponin I in human cardiomyocytes. Am. J. Physiol. Heart Circ. Physiol. 2013;304:H260–H268. doi: 10.1152/ajpheart.00498.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rao V.S., Korte F.S., Martyn D.A. N-terminal phosphorylation of cardiac troponin-I reduces length-dependent calcium sensitivity of contraction in cardiac muscle. J. Physiol. 2013;591:475–490. doi: 10.1113/jphysiol.2012.241604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yamasaki R., Wu Y., Granzier H. Protein kinase A phosphorylates titin’s cardiac-specific N2B domain and reduces passive tension in rat cardiac myocytes. Circ. Res. 2002;90:1181–1188. doi: 10.1161/01.res.0000021115.24712.99. [DOI] [PubMed] [Google Scholar]
- 31.Colson B.A., Bekyarova T., Moss R.L. Protein kinase A-mediated phosphorylation of cMyBP-C increases proximity of myosin heads to actin in resting myocardium. Circ. Res. 2008;103:244–251. doi: 10.1161/CIRCRESAHA.108.178996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rohl C.A., Strauss C.E.M., Baker D. Modeling structurally variable regions in homologous proteins with rosetta. Proteins. 2004;55:656–677. doi: 10.1002/prot.10629. [DOI] [PubMed] [Google Scholar]
- 33.Sood V.D., Baker D. Recapitulation and design of protein binding peptide structures and sequences. J. Mol. Biol. 2006;357:917–927. doi: 10.1016/j.jmb.2006.01.045. [DOI] [PubMed] [Google Scholar]
- 34.Tobacman L.S. Thin filament-mediated regulation of cardiac contraction. Annu. Rev. Physiol. 1996;58:447–481. doi: 10.1146/annurev.ph.58.030196.002311. [DOI] [PubMed] [Google Scholar]
- 35.Humphrey W., Dalke A., Schulten K. VMD: visual molecular dynamics. J. Mol. Graph. 1996;14:33–38. doi: 10.1016/0263-7855(96)00018-5. 27–28. [DOI] [PubMed] [Google Scholar]
- 36.Jorgensen W.L., Chandrasekhar J., Madura J.D., Impey R.W., Klein M.L. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 1983;79:926. [Google Scholar]
- 37.MacKerell A.D., Jr., Banavali N., Foloppe N. Development and current status of the CHARMM force field for nucleic acids. Biopolymers. 2000-2001;56:257–265. doi: 10.1002/1097-0282(2000)56:4<257::AID-BIP10029>3.0.CO;2-W. [DOI] [PubMed] [Google Scholar]
- 38.Phillips J.C., Braun R., Schulten K. Scalable molecular dynamics with NAMD. J. Comput. Chem. 2005;26:1781–1802. doi: 10.1002/jcc.20289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Darden T., York D., Pedersen L. Particle mesh Ewald—an N⋅log(N) method for Ewald sums in large systems. J. Chem. Phys. 1993;98:10089–10092. [Google Scholar]
- 40.Ryckaert J.-P., Ciccotti G., Berendsen H.J.C. Numerical integration of the cartesian equations of motion of a system with constraints: molecular dynamics of n-alkanes. J. Comput. Phys. 1977;23:327–341. [Google Scholar]
- 41.Lee B., Richards F.M. The interpretation of protein structures: estimation of static accessibility. J. Mol. Biol. 1971;55:379–400. doi: 10.1016/0022-2836(71)90324-x. [DOI] [PubMed] [Google Scholar]
- 42.Sadayappan S., Finley N., Robbins J. Role of the acidic N′ region of cardiac troponin I in regulating myocardial function. FASEB J. 2008;22:1246–1257. doi: 10.1096/fj.07-9458com. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Dong W.J., Chandra M., Cheung H.C. Phosphorylation-induced distance change in a cardiac muscle troponin I mutant. Biochemistry. 1997;36:6754–6761. doi: 10.1021/bi9622276. [DOI] [PubMed] [Google Scholar]
- 44.Heller W.T., Finley N.L., Trewhella J. Small-angle neutron scattering with contrast variation reveals spatial relationships between the three subunits in the ternary cardiac troponin complex and the effects of troponin I phosphorylation. Biochemistry. 2003;42:7790–7800. doi: 10.1021/bi0341509. [DOI] [PubMed] [Google Scholar]
- 45.Reiffert S.U., Jaquet K., Herberg F.W. Stepwise subunit interaction changes by mono- and bisphosphorylation of cardiac troponin I. Biochemistry. 1998;37:13516–13525. doi: 10.1021/bi980280j. [DOI] [PubMed] [Google Scholar]
- 46.Narolska N.A., Piroddi N., Stienen G.J.M. Impaired diastolic function after exchange of endogenous troponin I with C-terminal truncated troponin I in human cardiac muscle. Circ. Res. 2006;99:1012–1020. doi: 10.1161/01.RES.0000248753.30340.af. [DOI] [PubMed] [Google Scholar]
- 47.Kreutziger K.L., Piroddi N., Regnier M. Calcium binding kinetics of troponin C strongly modulate cooperative activation and tension kinetics in cardiac muscle. J. Mol. Cell. Cardiol. 2011;50:165–174. doi: 10.1016/j.yjmcc.2010.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Burkart E.M., Sumandea M.P., Solaro R.J. Phosphorylation or glutamic acid substitution at protein kinase C sites on cardiac troponin I differentially depress myofilament tension and shortening velocity. J. Biol. Chem. 2003;278:11265–11272. doi: 10.1074/jbc.M210712200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.