

Abstract
Nitrile-type inhibitors are known to interact with cysteine proteases in a covalent-reversible manner. The chemotype of 3-cyano-3-aza-β-amino acid derivatives was designed in which the N-cyano group is centrally arranged in the molecule to allow for interactions with the nonprimed and primed binding regions of the target enzymes. These compounds were evaluated as inhibitors of the human cysteine cathepsins K, S, B, and L. They exhibited slow-binding behavior and were found to be exceptionally potent, in particular toward cathepsin K, with second-order rate constants up to 52 900 × 103 M–1 s–1.
Keywords: Cysteine proteases, human cathepsins, β-amino acids, nitrile inhibitors
Cysteine cathepsins, proteolytic enzymes located within the lysosomes, belong to the family of papain-like cysteine peptidases. The overexpression of theses enzymes has been implicated in a multitude of pathological conditions, such as tumor progression and metastasis, MHC class II antigen presentation, and bone remodeling.1,2 The cysteine protease cathepsin K degrades several components of the bone matrix and has emerged as a promising target for intervention by small molecules on the basis of the inhibition of osteoclastic resorption observed in human and knockout animal models. Several cathepsin K inhibitors have been developed to reduce the excessive bone matrix degradation associated with osteoporosis. Among them, the front-runner odanacatib (1, Figure 1), a nonbasic and nonlysosomotropic peptidomimetic nitrile, is currently being developed as a once weekly treatment for osteoporosis.3−6
Figure 1.

Peptidomimetic cathepsin inhibitors: structures of the cathepsin K inhibitors odanacatib (1) and balicatib (2) and of azadipeptide nitriles.
Odanacatib (1) exhibits characteristic features of low molecular weight inhibitors of cysteine cathepsins, which are mainly peptidomimetic structures containing an electrophilic warhead to allow for covalent interactions with the active-site cysteine. Among such inhibitors, dipeptide nitriles have attracted particular attention. In the course of the drug development, a dipeptide has typically been decorated with an N-terminal capping group and the carboxylic function has been replaced by a cyano group. The P2 and P1 amino acid side chains then allow for specific binding to the S2 and S1 pockets within the active site of the target enzymes, the capping group for an interaction with the S3 binding pocket. The reaction of dipeptide nitriles with cysteine proteases involves the nucleophilic attack of the thiolate moiety at the inhibitors’ nitrile carbon and thus the reversible formation of thioimidate adducts.7
For papain-like cysteine proteases, azadipeptide nitriles have been introduced as a new inhibitor class whose representatives exhibited Ki values toward human cysteine cathepsins in the nano and even picomolar range. Such inhibitors have been designed through an exchange of the α-CH moiety of the P1 amino nitrile by a nitrogen atom.8,9 Azadipeptide nitriles form covalent-reversible isothiosemicarbazide adducts upon being attacked by the active site cysteine nucleophile. Representative examples for azadipeptide nitriles are depicted in Figure 1. The nitrogen atoms of the aza-amino nitrile moiety of azadipeptide nitriles are essentially alkylated for reasons of synthetic access. Undesired cyclization reactions have been observed when the nitrogen of the P2–P1 peptide bond and/or the P1 α-nitrogen bear a hydrogen substituent (−CO–NH–NH–CN, −CO–NH–NR–CN, or −CO–NR–NH–CN).8 However, carbamate-protected aza-amino nitriles have been described as cathepsin K inhibitors, which contain the open-chain O–CO–NH–NR–CN motif.12
A systematic scan with respect to the P3 and P2 substituents was performed to elucidate the strong impact of the noncovalent interactions for cathepsin K inhibition by azadipeptide nitriles.13,14 The high electrophilic reactivity15 toward thiols is not a sufficient condition for cathepsin inhibition, as, for example, Gly in P2 position in place of a preferred amino acid reduced the potency by 3 orders of magnitude.13 The sulfone derivative 3 showed dual inhibition of human cathepsins S and K with picomolar Ki values.16 In further series of azadipeptide nitriles, leucine was maintained at P2 position, and several aryl moieties were introduced in the N-terminal amide portion;17 a direct linkage of these aryl groups to the α-carbon of the P2 building block caused a strong reduction of the affinity to cathepsin K.18 The azadipeptide nitrile chemotype was employed for the design of activity-based probes for in situ proteome profiling of Trypanosoma brucei(19) and was chemically converted into fluorescent derivatives to be used for cathepsin labeling.20 The fluorine-18 labeled inhibitor 4 was applied for positron emission tomography to image tumor-associated cathepsin activity.21 Moreover, a cathepsin inhibitor cargo was linked by click chemistry to the localization peptide Tat for organelle-specific targeting, and subcellular delivery of the inhibitor-Tat conjugate 5 to the lysosomes was reported.22
In the present study we aimed at synthesizing cysteine protease inhibitors with an N-cyano group, which is centrally arranged in the molecule. This group was expected to interact with the active site cysteine and the flanking parts with the nonprimed and primed subsites of the target protease. Such type of nitrile-based inhibitors has not been described so far and can be considered as a structural expansion of cathepsin inhibiting alkyl 2-cyano-2-methylhydrazinecarboxylates.12 It was intended to introduce the cyano group via electrophilic cyanation.23 Incorporation of an aza-α-amino acid would lead to a −CO–NH–NαH–CO– substructure, in which the α-nitrogen would not be susceptible to electrophilic cyanation. Therefore, we envisaged a 3-aza-β-amino acid as the core element of the desired compounds. The corresponding −CO–NH–NβH–CαH2–CO– substructure should then allow the preparation of the 3-cyano-3-aza-β-amino acid derivatives.
The synthesis of the final compounds is illustrated in Scheme 1. Condensation of the carbazates 6 and 7 with glyoxylic acid gave the hydrazones 8 and 9. Amide coupling of 8 and 9 with morpholine, N-benzylmethylamine, and N,N-dibenzylamine, respectively, furnished 10–15. These compounds were catalytically hydrogenated to hydrazinoacetic acid derivatives 16–21 in order to make the β-nitrogen accessible to cyanation. Reactions with cyanogen bromide produced the Boc-protected final compounds 22–27. At the C-terminus, this first series of 3-cyano-3-aza-β-amino acid derivatives contains a tertiary carboxamide moiety. Whether it would also be possible to exchange R2 and/or R3 by hydrogen without eliciting a concomitant cyclization was not examined in the course of this study.
Scheme 1. Synthesis of 3-Cyano-3-aza-β-amino Acid Derivatives 22–27.

Reagents and conditions: (a) glyoxylic acid monohydrate, EtOH, RT; (b) (i) ClCO2i-Bu, NMM, THF, −25 °C; (ii) HNR2R3, THF, −25 °C to RT; (c) H2, Pd/C (10%), MeOH, 3.3 bar, RT; (d) BrCN, NaOAc, MeOH, RT.
An alternative order of reactions was followed as outlined in Scheme 2. Starting with 9, the hydrogenation step was carried out prior to the amide coupling. Indeed, we obtained 29 and successively 19, the precursor to the final product 25. However, when applying the hydrogenation-coupling sequence to the demethylated analogue 8 and then obtaining 28, the subsequent coupling reaction with morpholine failed.
Scheme 2. Alternative Synthesis of the 3-Aza-β-amino Acid Derivative 19.

Reagents and conditions: (a) H2, Pd/C (10%), MeOH, 3.3 bar, RT; (b) (i) ClCO2i-Bu, NMM, THF, −25 °C; (ii) morpholine, THF, −25 °C to RT.
Concerning the structural elucidation we have initially paid attention to the intermediates 10–15. Signals for two isomers occurred in the 1H and 13C NMR spectra of the N-benzylmethylamino derivatives 11 and 14. This could result from two different functionalities. E/Z isomerism is possible (i) because of the C=N double bond or (ii) because of the partial double bond character of the CCO–N bond. A corresponding E/Z isomerism about the carbamate OCO–N bond was ruled out as this would only be valid in the case of 14. Thus, the amide bond in 11 and 14 was suspected to be responsible for the mixtures. This was also based on the findings that (i) hydrogenation to 17 and 20 did not lead to a disappearance of mixtures and (ii) such mixtures have neither been observed in the case of the morpholine-derived intermediates 10 and 13 and hydrazinoacetic acid derivatives 16 and 19 nor in case of the N-dibenzylamine-derived intermediates 12 and 15 and hydrazinoacetic acid derivatives 18 and 21. Hence, the appearance of the isomers resulted from the E/Z isomerism about the amide bond in the unequally substituted tertiary carboxamides 11, 14, 17, and 20; it was also observed in the corresponding final products 23 and 26.
In previous studies, it has been shown that the azadipeptide amide 30 forms atropisomers (Chart 1).32,33 The diastereotopic protons of the glycine fragment indicated a restricted rotation around the N–N single bond. This rotation was enforced with a variable temperature (VT) NMR experiment, and a value of 117 kJ/mol was determined for the rotational barrier. As atropisomers have rotational barriers higher than 95 kJ/mol, compound 30 exhibits chirality as a result of its atropisomerism.32 Moreover, the azadipeptide nitrile 31 showed diastereotopic glycine methylene protons13 and is structurally similar to the final compounds 25–27 of this study. Therefore, we addressed the question whether 25–27 would also form atropisomers. However, diastereotopic protons in the corresponding 1H NMR spectra were not observed. For example, in the 1H NMR spectrum of 27, three distinct singlets for the methylene protons appear. It could therefore be concluded that in 25–27 a free rotation around the N–N single bond is possible. With respect to such a bond rotation, the significant difference between the azadipeptides 30 and 31, on the one hand, and the 3-cyano-3-aza-β-amino acid derivatives 25–27, on the other hand, is the position next to the N–N–CO carbonyl group. In comparison to the methylene group (in 30 and 31), the oxygen atom (in 25–27) is less voluminous. Moreover, it is the methylation of the peptide bond in azadipeptides 30 and 31 that leads to the E configuration (Chart 2) and hence to the aforementioned atropisomerism.32 In 3-cyano-3-aza-β-amino acid derivatives 25–27, one more mesomeric structure is possible and the contribution of the third limiting structure to the resonance reduces the double bond character of the CO–N bond (Chart 2).34−36 Thus, the E configuration in 25–27 is supposed to be less defined, and consequently, the N–N single bond rotation is not hindered.
Chart 1. Atropisomeric Structures of Azadipeptides 30 and 31.

Chart 2. Mesomeric Structures of the E Configured Hydrazide and Carbazate Fragments of 30, 31, and 25–27.

The results of the biochemical evaluation of 22−25 and, for comparison purpose, balicatib at four human cysteine cathepsins are outlined in Table 1. Fluorogenic or chromogenic peptide substrates were employed in the activity assays. Our inhibitors showed time-dependent inhibition of the target cathepsins. This behavior was in agreement with the expected covalent and reversible enzyme–inhibitor interaction. In some cases, the progress curves were analyzed after achievement of steady-state by linear regression. Plots of the steady-state rates versus inhibitor concentration gave IC50 values. Assuming an active-site directed mode of action, those were corrected using the Cheng–Prusoff equation to obtain the true inhibition constants, Ki. In other cases, the progress curves were analyzed by means of the slow binding equation,37 and nonlinear regression analysis of the progress curves provided the first-order rate constants kobs, besides the steady-state rates. The kobs values were used to obtain second-order rate constants, kon, governing the enzyme–inhibitor association. As Ki values were also determined as noted above, first-order rate constants for the dissociation of the enzyme–inhibitor complex, koff, were also accessible (Ki = koff/kon). A representative kinetic analysis is illustrated in Figure 2.
Table 1. Kinetic Parameters of Inhibition of Cathepsins by 3-Cyano-3-aza-β-amino Acid Derivatives 22–25 and Balicatib.

IC50 values were obtained from duplicate measurements in the presence of five different inhibitor concentrations. IC50 values were determined by nonlinear regression using equation vs = v0/(1 + [I]/IC50), were vs is the steady-state rate, v0 is the rate in the absence of the inhibitor, and [I] is the inhibitor concentration. Ki values were calculated from IC50 values by applying the equation Ki = IC50/(1 + [S]/Km), where [S] is the substrate concentration.
The cathepsin K assay was performed with the fluorogenic substrate Z-Leu-Arg-AMC at a final concentration of 40 μM (13.3 × Km). The reaction was followed over 60 min.13
The cathepsin S assay was performed with the fluorogenic substrate Z-Phe-Arg-AMC at a final concentration of 40 μM (0.74 × Km). The reaction was followed over 60 min.38
The cathepsin B assay was performed with the chromogenic substrate Z-Arg-Arg-pNA at a final concentration of 500 μM (0.45 × Km). The reaction was followed over 30 min.13
The cathepsin L assay was performed with the chromogenic substrate Z-Phe-Arg-pNA at a final concentration of 100 μM (5.88 × Km). The reaction was followed over 30 min.13
Progress curves were analyzed by nonlinear regression using the slow-binding equation [P] = vst + (vi – vs)(1 – exp(−kobst))/(kobs + d) to give vs, vi, and kobs, where [P] is the product concentration, vi is the initial rate, kobs is the observed first-order rate constant, and d is the offset. To obtain IC50 values, vs values from reactions in the presence of the inhibitor and v0 values obtained by linear regression of the progress curves in the absence of the inhibitor were used. The second-order rate constants kon were obtained by linear regression according to equation kobs = kon[I]/(1 + [S]/Km) + koff, where koff is the first-order rate constant of dissociation.
Progress curves were analyzed by linear regression after steady-state was reached (5–30 min).
Figure 2.

Inhibition of human cathepsin K by 22. Top: Monitoring of the cathepsin K-catalyzed hydrolysis of Z-Leu-Arg-AMC in the presence of increasing concentrations of compound 22 (closed circles, 0 pM; open circles, 400 pM; closed squares, 800 pM; open squares, 1200 pM; closed triangles, 1600 pM; open triangles, 2000 pM). Fluorescence emission at 440 nm was measured after excitation at 360 nm. Bottom: Plot of the rates obtained in duplicate measurements versus concentrations of 22. Nonlinear regression gave an IC50 value of 158 (±14) pM. Inset: Plot of the first-order rate constants kobs versus concentrations of 22. Linear regression gave a value kon/(1 + [S]/Km) of 1.33 (±0.15) × 10–6 pM–1 s–1.
As a first result of the kinetic investigations, a major difference in the inhibitory potency of the nonmethylated and the methylated morpholine derivatives was ascertained (22 versus 25), which was particularly striking in the case of cathepsin K (Ki of 11 pM versus Ki of 31 nM). It has been described that a nonmethylated amide bond in nitrile-type cysteine protease inhibitors (−CO–NH–X–CN) resulted in a much stronger inhibition than the methylated analogues (−CO–NMe–X–CN).8,12
Thus, we focused on the three nonmethylated 3-cyano-3-aza-β-amino acid derivatives (22–24) to elucidate structure–activity relationships. Noteworthy, these three derivatives exhibited a gradual decrease in affinity toward cathepsins K, S, B, and L. The stepwise introduction of one or two bulky aromatic moieties resulted in an improved inhibitory potency (22 versus 23 versus 24), which was determined for the four enzymes. Obviously, the benzyl groups allow for hydrophobic or cation−π interactions with the cathepsins’ active sites.
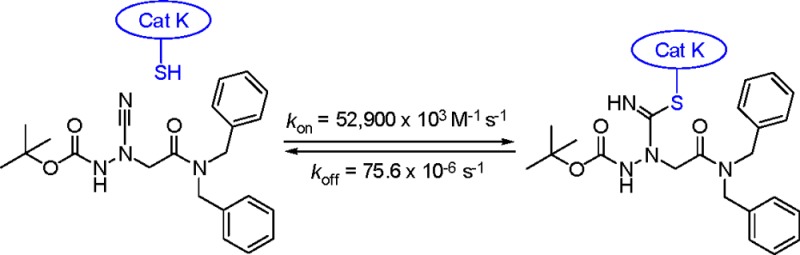
For human cathepsins K and S, inhibitors 22–24 exhibited very low Ki values down to 1.43 pM (24 at cathepsin K). This strong affinity is mainly due to an accelerated association to form the enzyme–inhibitor complex(es), as it can be inferred from the high second-order rate constants (e.g., kon = 52 900 × 103 M–1 s–1 for 24 at cathepsin K). For 22–24, half-lives of the cathepsin K-inhibitor and cathepsin S-inhibitor complexes could be calculated from the corresponding koff values and were in the range between 10 and 150 min (22 at cathepsin S and 24 at cathepsin K).
We propose that the Boc capping group of the 3-cyano-3-aza-β-amino acid derivatives is bound in the nonprimed binding region of the target enzymes and that the amide part is accommodated in the primed binding region. This assumption is based on the cocrystal structure of tert-butyl 2-cyano-2-methylhydrazinecarboxylate with cathepsin K.12 In this complex, the tert-butyl group occupies the S2 pocket formed by residues 67Tyr, 68 Met, 134Ala, 163Ala, and 209Leu. Hydrogen bonding interactions of the carbamate occur with the backbone NH of 66Gly and the backbone carbonyl oxygen of 161Asn.12 Although alkyl 2-cyano-2-methylhydrazinecarboxylates exhibit a pronounced cathepsin K inhibition, the cathepsin K selectivity of the inhibitors described herein is even stronger and is probably caused by hydrophobic or cation−π interactions with the nonprimed binding region of cathepsin K. In future studies, it is intended to expand the inhibitor structure to both directions. This will enable to better define the binding mode of this new type of cysteine protease inhibitors. On the one hand, prior to cyanation and hydrogenation, a Boc-deprotection would allow for the incorporation of moieties to interact with the S1 and S2 pockets. On the other hand, the introduction of amino acid derivatives, in place of the secondary amines used in this study, might direct the so-modified amide part to the S1′ and S2′ pockets.
Biography
Michael Gütschow studied Biochemistry and received his Ph.D. in Pharmaceutical Chemistry from the University of Leipzig, Germany. After postdoctoral training at the Georgia Institute of Technology, School of Chemistry and Biochemistry, he returned to the University of Leipzig and was appointed as a professor for Pharmaceutical Chemistry at the University of Bonn in 2001. His research interests include (i) synthesis of bioactive heterocycles, (ii) peptides and peptidomimetic drugs, (iii) development of inhibitors and activity-based probes of proteases and esterases, and (iv) biochemistry of enzyme−drug interactions. He published approximately 170 publications about the scientific topics mentioned.
Supporting Information Available
Synthetic procedures and characterization of compounds. 1H NMR and 13C NMR spectra. Enzyme inhibition assays. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Contributions
§ J.S. and A.M.B contributed equally to this work. The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
J.S. is supported by the Gender Equality Center of the Bonn-Rhein-Sieg University of Applied Sciences.
The authors declare no competing financial interest.
Supplementary Material
References
- Withana N. P.; Blum G.; Sameni M.; Slaney C.; Anbalagan A.; Olive M. B.; Bidwell B. N.; Edgington L.; Wang L.; Moin K.; Sloane B. F.; Anderson R. L.; Bogyo M. S.; Parker B. S. Cathepsin B inhibition limits bone metastasis in breast cancer. Cancer Res. 2012, 72, 1199–1209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costantino C. M.; Ploegh H. L.; Hafler D. A. Cathepsin S regulates class II MHC processing in human CD4+ HLA-DR+ T cells. J. Immunol. 2009, 183, 945–952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reginster J. Y.; Neuprez A.; Beaudart C.; Lecart M. P.; Sarlet N.; Bernard D.; Disteche S.; Bruyere O. Antiresorptive drugs beyond bisphosphonates and selective oestrogen receptor modulators for the management of postmenopausal osteoporosis. Drugs Aging 2014, 31, 413–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng K. W.; Martin T. J. New therapeutics for osteoporosis. Curr. Opin. Pharmacol. 2014, 16, 58–63. [DOI] [PubMed] [Google Scholar]
- Black W. C. Peptidomimetic inhibitors of cathepsin K. Curr. Top. Med. Chem. 2010, 10, 745–751. [DOI] [PubMed] [Google Scholar]
- Novinec M.; Lenarčič B. Cathepsin K: a unique collagenolytic cysteine peptidase. Biol. Chem. 2013, 394, 1163–1179. [DOI] [PubMed] [Google Scholar]
- Frizler M.; Stirnberg M.; Sisay M. T.; Gütschow M. Development of nitrile-based inhibitors of cysteine cathepsins. Curr. Top. Med. Chem. 2010, 10, 294–322. [DOI] [PubMed] [Google Scholar]
- Löser R.; Frizler M.; Schilling K.; Gütschow M. Azadipeptide nitriles: highly potent and proteolytically stable inhibitors of papain-like cysteine proteases. Angew. Chem., Int. Ed. 2008, 47, 4331–4334. [DOI] [PubMed] [Google Scholar]
- For structurally related thiosemicarbazone-type cysteine protease inhibitor, see refs (10) and (11).
- Song J.; Jones L. M.; Kishore Kumar G. D.; Conner E. S.; Bayeh L.; Chavarria G. E.; Charlton-Sevcik A. K.; Chen S. E.; Chaplin D. J.; Trawick M. L.; Pinney K. G. Synthesis and biochemical evaluation of thiochromanone thiosemicarbazone analogues as inhibitors of cathepsin L. ACS Med. Chem. Lett. 2012, 3, 450–453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song J.; Jones L. M.; Chavarria G. E.; Charlton-Sevcik A. K.; Jantz A.; Johansen A.; Bayeh L.; Soeung V.; Snyder L. K.; Lade S. D. Jr.; Chaplin D. J.; Trawick M. L.; Pinney K. G. Small-molecule inhibitors of cathepsin L incorporating functionalized ring-fused molecular frameworks. Bioorg. Med. Chem. Lett. 2013, 23, 2801–2807. [DOI] [PubMed] [Google Scholar]
- Barrett D. G.; Deaton D. N.; Hassell A. M.; McFadyen R. B.; Miller A. B.; Miller L. R.; Payne J. A.; Shewchuk L. M.; Willard D. H. Jr.; Wright L. L. Acyclic cyanamide-based inhibitors of cathepsin K. Bioorg. Med. Chem. Lett. 2005, 15, 3039–3043. [DOI] [PubMed] [Google Scholar]
- Frizler M.; Lohr F.; Furtmann N.; Kläs J.; Gütschow M. Structural optimization of azadipeptide nitriles strongly increases association rates and allows the development of selective cathepsin inhibitors. J. Med. Chem. 2011, 54, 396–400. [DOI] [PubMed] [Google Scholar]
- Frizler M.; Lohr F.; Lülsdorff M.; Gütschow M. Facing the gem-dialkyl effect in enzyme inhibitor design: preparation of homocycloleucine-based azadipeptide nitriles. Chem.—Eur. J. 2011, 17, 11419–11423. [DOI] [PubMed] [Google Scholar]
- Oballa R. M.; Truchon J. F.; Bayly C. I.; Chauret N.; Day S.; Crane S.; Berthelette C. A generally applicable method for assessing the electrophilicity and reactivity of diverse nitrile-containing compounds. Bioorg. Med. Chem. Lett. 2007, 17, 998–1002. [DOI] [PubMed] [Google Scholar]
- Frizler M.; Schmitz J.; Schulz-Fincke A. C.; Gütschow M. Selective nitrile inhibitors to modulate the proteolytic synergism of cathepsins S and F. J. Med. Chem. 2012, 55, 5982–5986. [DOI] [PubMed] [Google Scholar]
- Ren X. F.; Li H. W.; Fang X.; Wu Y.; Wang L.; Zou S. Highly selective azadipeptide nitrile inhibitors for cathepsin K: design, synthesis and activity assays. Org. Biomol. Chem. 2013, 11, 1143–1148. [DOI] [PubMed] [Google Scholar]
- Yuan X. Y.; Fu D. Y.; Ren X. F.; Fang X.; Wang L.; Zou S.; Wu Y. Highly selective aza-nitrile inhibitors for cathepsin K, structural optimization and molecular modeling. Org. Biomol. Chem. 2013, 11, 5847–5852. [DOI] [PubMed] [Google Scholar]
- Yang P. Y.; Wang M.; Li L.; Wu H.; He C. Y.; Yao S. Q. Design, synthesis and biological evaluation of potent azadipeptide nitrile inhibitors and activity-based probes as promising anti-Trypanosoma brucei agents. Chem.—Eur. J. 2012, 18, 6528–6541. [DOI] [PubMed] [Google Scholar]
- Frizler M.; Mertens M. D.; Gütschow M. Fluorescent nitrile-based inhibitors of cysteine cathepsins. Bioorg. Med. Chem. Lett. 2012, 22, 7715–7718. [DOI] [PubMed] [Google Scholar]
- Löser R.; Bergmann R.; Frizler M.; Mosch B.; Dombrowski L.; Kuchar M.; Steinbach J.; Gütschow M.; Pietzsch J. Synthesis and radiopharmacological characterisation of a fluorine-18-labelled azadipeptide nitrile as a potential PET tracer for in vivo imaging of cysteine cathepsins. ChemMedChem 2013, 8, 1330–1344. [DOI] [PubMed] [Google Scholar]
- Loh Y.; Shi H.; Hu M.; Yao S. Q. ″Click″ synthesis of small molecule-peptide conjugates for organelle-specific delivery and inhibition of lysosomal cysteine proteases. Chem. Commun. 2010, 46, 8407–8409. [DOI] [PubMed] [Google Scholar]
- For examples of electrophilic cyanations, see refs (24−28). For recent examples of nucleophilic metal-catalyzed or photoinduced direct cyanations, see refs (29−31).
- Hoshikawa T.; Yoshioka S.; Kamijo S.; Inoue M. Photoinduced direct cyanation of C(sp3)-H bonds. Synthesis 2013, 45, 874–887. [Google Scholar]
- Anbarasan P.; Neumann H.; Beller M. A novel and convenient synthesis of benzonitriles: electrophilic cyanation of aryl and heteroaryl bromides. Chem.—Eur. J. 2011, 17, 4217–4222. [DOI] [PubMed] [Google Scholar]
- Hughes T. V.; Cava M. P. Electrophilic cyanations using 1-cyanobenzotriazolo: sp2 and sp carbanions. J. Org. Chem. 1999, 64, 313–315. [DOI] [PubMed] [Google Scholar]
- Emmrich T.; Reinke H.; Langer P. Synthesis of 2-(arylsulfonyl)-4-hydroxypyridines by hetero-Diels–Alder reaction of 1,3-bis-silyl enol ethers with arylsulfonyl cyanides. Synthesis 2006, 2551–2555. [Google Scholar]
- Hussain I.; Yawer M. A.; Lalk M.; Lindequist U.; Villinger A.; Fischer C.; Langer P. Hetero-Diels–Alder reaction of 1,3-bis(trimethylsilyloxy)-1,3-butadienes with arylsulfonylcyanides. Synthesis and antimicrobial activity of 4-hydroxy-2-(arylsulfonyl)pyridines. Bioorg. Med. Chem. 2008, 16, 9898–9903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schareina T.; Zapf A.; Cotté A.; Müller N.; Beller M. A bio-inspired copper catalyst system for practical catalytic cyanation of aryl bromides. Synthesis 2008, 3351–3355. [Google Scholar]
- Seayad A. M.; Ramalinga B.; Yoshinaga K.; Nagata T.; Chai C. L. Highly enantioselective titanium-catalyzed cyanation of imines at room temperature. Org. Lett. 2010, 12, 264–267. [DOI] [PubMed] [Google Scholar]
- Hoshikawa T.; Yoshioka S.; Kamijo S.; Inoue M. Photoinduced direct cyanation of C(sp3)-H bonds. Synthesis 2013, 874–887. [Google Scholar]
- Ottersbach P. A.; Schnakenburg G.; Gütschow M. Induction of chirality: experimental evidence of atropisomerism in azapeptides. Chem. Commun. 2012, 48, 5772–5774. [DOI] [PubMed] [Google Scholar]
- Ottersbach P. A.; Schmitz J.; Schnakenburg G.; Gütschow M. An access to aza-Freidinger lactams and E-locked analogs. Org. Lett. 2013, 15, 448–451. [DOI] [PubMed] [Google Scholar]
- Kessler H. Detection of hindered rotation and inversion by NMR spectroscopy. Angew. Chem., Int. Ed. 1970, 9, 219–235. [Google Scholar]
- Smith B. D.; Goodenough-Lashua D. M.; D’Souza C. J.; Norton K. J.; Schmidt L. M.; Tung J. C. Substituent effects on the barrier to carbamate C-N rotation. Tetrahedron Lett. 2004, 45, 2747–2749. [Google Scholar]
- Pontes R. M.; Basso E. A.; dos Santos F. D. Medium effect on the rotational barrier of carbamates and its sulfur congeners. J. Org. Chem. 2007, 72, 1901–1911. [DOI] [PubMed] [Google Scholar]
- Morrison J. F.; Walsh C. T. The behavior and significance of slow binding enzyme inhibitors. Adv. Enzymol. Relat. Areas Mol. Biol. 1988, 61, 201–301. [DOI] [PubMed] [Google Scholar]
- Mertens M. D.; Schmitz J.; Horn M.; Furtmann N.; Bajorath J.; Mareš M.; Gütschow M. A coumarin-labeled vinyl sulfone as tripeptidomimetic activity-based probe for cysteine cathepsins. ChemBioChem 2014, 15, 955–959. [DOI] [PubMed] [Google Scholar]
- Falgueyret J. P.; Desmarais S.; Oballa R.; Black W. C.; Cromlish W.; Khougaz K.; Lamontagne S.; Massé F.; Riendeau D.; Toulmond S.; Percival M. D. Lysosomotropism of basic cathepsin K inhibitors contributes to increased cellular potencies against off-target cathepsins and reduced functional selectivity. J. Med. Chem. 2005, 48, 7535–7543. [DOI] [PubMed] [Google Scholar]
- Robichaud J.; Oballa R.; Prasit P.; Falgueyret J. P.; Percival M. D.; Wesolowski G.; Rodan S. B.; Kimmel D.; Johnson C.; Bryant C.; Venkatraman S.; Setti E.; Mendonca R.; Palmer J. T. A novel class of nonpeptidic biaryl inhibitors of human cathepsin K. J. Med. Chem. 2003, 46, 3709–3727. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.