

Abstract
Cathepsin S (Cat S) plays an important role in many pathological conditions, including abdominal aortic aneurysm (AAA). Inhibition of Cat S may provide a new treatment for AAA. To date, several classes of Cat S inhibitors have been reported, many of which form covalent interactions with the active site Cys25. Herein, we report the discovery of a novel series of noncovalent inhibitors of Cat S through a medium-throughput focused cassette screen and the optimization of the resulting hits. Structure-based optimization efforts led to Cat S inhibitors such as 5 and 9 with greatly improved potency and drug disposition properties. This series of compounds binds to the S2 and S3 subsites without interacting with the active site Cys25. On the basis of in vitro potency, selectivity, and efficacy in a CaCl2-induced AAA in vivo model, 5 (LY3000328) was selected for clinical development.
Keywords: Cathepsin, abdominal aortic aneurysm, development candidate, noncovalent
Abdominal aortic aneurysm (AAA) is a frequently fatal age-related disease in which the abdominal aorta, often between the renal and iliac arteries, enlarges and eventually ruptures. The risk of rupture increases with increase in aortic diameter, particularly with a diameter larger than 5.5 cm. The prevalence of AAA in patients over the age of 60 years is 4–8% and 0.5–1.5%, in men and women, respectively.1−7 The most frequently recognized risk factors for AAA are advanced age, history of cigarette smoking, male gender, and family history. AAA is typically asymptomatic, so early detection is a challenge.
Between rupture and postsurgical complications, AAA is responsible for at least 15,000 deaths annually in the USA. There are no approved drugs for the treatment of AAA; the only options for AAA patients are open surgery and endovascular stents. Patients are eligible for surgery when the abdominal aorta reaches a diameter of 5.5 cm because the risk–benefit balance is not in favor of intervention below this threshold. Thus, AAA patients with an aortic diameter of less than 5.5 cm have no viable treatment options.
Cat S is a 225 amino acid, 24.8 kDa lysosomal cysteine protease that belongs to the papain superfamily of proteases.8 Cat S controls MHC class II-mediated antigen presentation by epithelial cells.9 Immune cells such as macrophages and microglia secrete Cat S in response to inflammatory mediators such as lipopolysaccharides, proinflammatory cytokines, and neutrophils. Cat S is expressed by antigen-presenting cells including macrophages, B-lymphocytes, dendritic, microglia, and some epithelial cells. Although many lysosomal proteases function only under acidic conditions, Cat S is an exception; its pH optimum is between 6.0 and 7.5, and the enzyme is active outside the lysosome.
In a 28 day chronic angiotensin II infusion model of AAA with apolipoprotein E (ApoE) deficient (ApoE–/–) mice and ApoE/Cat S double-knockout (ApoE–/–Cat S–/–) mice, AAA lesions were analyzed.10 It was found that Cat S expression increased significantly in mouse AAA lesions. The AAA incidence in ApoE–/–Cat S–/– mice was much lower than that in ApoE–/–Cat S+/+ mice (10% vs 80%). There was significant reduction in external and luminal abdominal aortic diameters, medial elastin fragmentation, and adventitial collagen in the double knockout mice. Cat S deficiency reduced AAA lesion media smooth muscle cell (SMC) apoptosis, adventitial microvessel content, and inflammatory cell accumulation. Cat S also has been shown to play important roles in autoimmune disorders and inhibition of Cat S enzyme activity in such pathological conditions might have therapeutic value.11
In Cat S, it is known that Cys25 is capable of forming a reversible or irreversible covalent bond with electrophilic centers, so several classes of inhibitors reported in the literature contain electrophiles.12,13 Representative examples of Cat S inhibitors from the literature (Chart 1) contain different electrophilic groups, which are capable of forming this bond. For example, 1(14) is a Michael acceptor, whereas 2(15) contains a cyclic ketone. Similarly, 3(13) contains an α-ketoamide as an electrophilic center, and 4(16) contains a nitrile, which is capable of forming a thioimidate with Cys25. Compound 3 is a very potent inhibitor of Cat S, but it is also a very potent inhibitor of cathepsins L, V, and B. In some cases, the reactive nature of the inhibitors creates challenges in optimization of selectivity during lead optimization. Nonspecific reactivity with off-target biomolecules can potentially contribute to toxicity for molecules containing electrophilic centers. There are reports on noncovalent Cat S inhibitors that do not interact with the active site Cys25.17 We set out likewise to identify Cat S inhibitors that lack an electrophilic center and do not form covalent bond with active site Cys25. Herein, we report the structure of noncovalent Cat S inhibitor 5 (Chart 2), which does not form covalent bond with Cys25 and occupies the S2 and S3 pockets of the Cat S enzyme. Inhibitor 5 has potency, selectivity, drug disposition, toxicology, physicochemical, and in vivo properties suitable for clinical development.
Chart 1. Selected Cathepsin S Inhibitors.

Chart 2. Structures of Nonpeptidic, Noncovalent, Selective Cat S Inhibitors.

Test compounds were evaluated for their activity in Cat S and mCat S enzyme inhibition assays (Table 1).18,19 Similarly, selectivity against other cysteine proteases such as Cat L, Cat K, Cat B, and Cat V was determined in enzyme inhibition assays where compounds 5–9 exhibited very high selectivity.20
Table 1. hCat S and mCat S Inhibition Data.
| compd | hCat S IC50 (nM) | mCat S IC50 (nM) |
|---|---|---|
| 6 | 1170 ± 227 (n = 7) | 1570 ± 108 (n = 3) |
| 7 | 1290 ± 453 (n = 6) | 2780 ± 230 (n = 3) |
| 8 | 4000 ± 1630 (n = 4) | 3930 ± 553 (n = 3) |
| 9 | 12.4 ± 6.48 (n = 10) | 3.91 ± 0.71 (n = 8) |
| 5 | 7.70 ± 5.85 (n = 11) | 1.67 ± 1.17 (n = 9) |
The series of compounds disclosed in this publication are inactive against rCat S due to a G137C mutation.21 This mutation reduces the size of the S2 pocket, thereby rejecting the binding of Cat S inhibitors with larger P2 groups.
After a medium-throughput screen of a structurally diverse cassette of approximately 120,000 compounds, several actives were identified. We focused our attention on nonpeptidic compound 6(22) (Chart 2) with a Cat S IC50 of 1.3 μM (Table 1) and selectivity against human cathepsin L (Cat L IC50 of >100 μM). The amidine in 6 was judged undesirable for a drug-like molecule, so it was readily replaced with a urea linker to provide 7, with no loss of potency. From what was previously known about Cat S inhibitors, we were unable to predict a reasonable binding mode for 7. Consequently, there was impetus to understand how 7 interacted with the enzyme. The 3-dimensional (3D) structure of the Cat S-7 complex was resolved by protein X-ray crystallography at a 1.8 Å resolution and R factors of 0.185 (working) and 0.210 (free). The 3D structure of the complex (Figure 1A) revealed a novel binding mode in which the phenyl ring of the benzamide occupied the S3 binding pocket and formed a face-to-edge interaction with Phe70. In addition, the phenyl moiety of the benzamide formed a π-stacking interaction with the peptide bond between Gly68 and Gly69. The aryl ring of the tetrahydronaphthalene substructure occupied the S2 pocket and formed a face-to-edge interaction with Phe70. The N-methylpiperazine was partly in proximity with Phe211 and partly outside the S2 subsite. There was only one apparent electrostatic interaction observed between 7 and Cat S: the NH residue of the benzamide formed a strong hydrogen bond (3 Å) with the carbonyl oxygen of the Gly69. It was noteworthy that the active site Cys25 residue did not make any contact with the inhibitor; the closest non-hydrogen atom of 7 was at a distance of 4.3 Å from the sulfur of Cys25.
Figure 1.
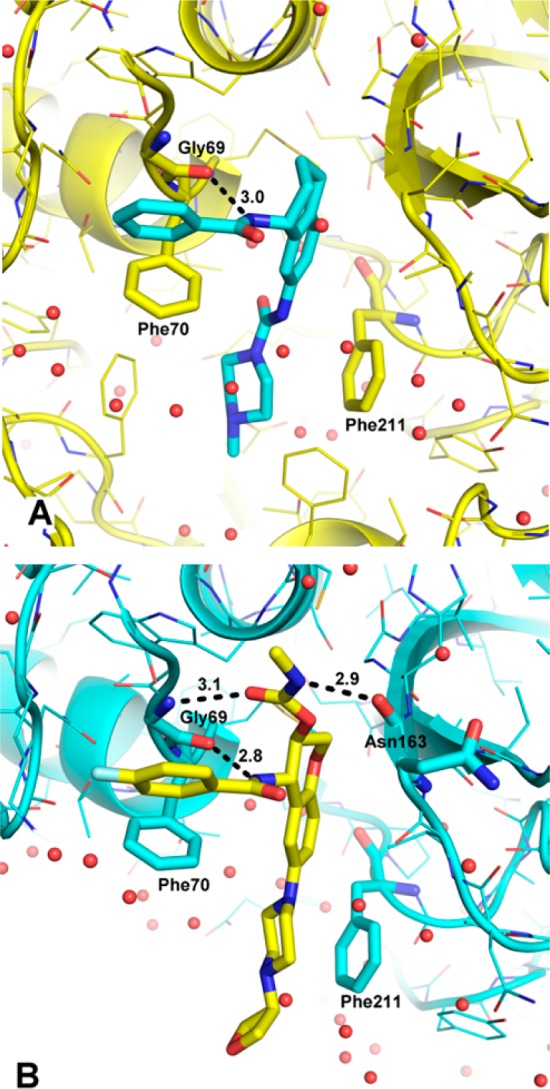
Ribbon figure of (A) Cat S (yellow) in complex with 7 (cyan) and (B) Cat S (cyan) in complex with 5 (yellow).
The availability of the 3D protein crystal structure of the Cat S-7 complex opened the door for structure-based drug design. With this structure in hand, we turned our focus toward the optimization of the potency, selectivity, drug disposition, biopharmaceutical, and toxicological properties. The structure suggested that the urea linker between the tetralin and the N-methylpiperazine served little purpose in contributing to electrostatic interactions. Indeed, compound 8 has comparable potency to 7. The purpose of linking N-methylpiperazine directly to phenyl ring in 8 was to improve solubility properties of 8 over 7. Further examination of the 3D structure of the Cat S-7 complex revealed that the NH residue of Gly69, usually forming a H-bond with the amide carbonyl of a P1 group, had no complementary hydrogen bond acceptors from 7. We hypothesized that placing an O-linked carbamate group on the homobenzylic carbon might provide a hydrogen bond acceptor complementary to the NH of Gly69. In addition, we posited that replacing the N-methyl group of the piperazine in 8 with a 3-oxetanyl group would reduce the pKa of the distal piperazine nitrogen while lowering the overall lipophilicity of the target compound 9. Indeed, we found 9 to be about 300-fold more potent than 8. The benzylic carbon in 9 was replaced with oxygen to provide 5 with reduced potential for metabolic oxidation. Compound 5 maintained excellent in vitro potency and selectivity. Compared to 7, the protein–crystal structure of the Cat S-5 complex (Figure 1B) revealed that 5 not only maintains the hydrogen bond between the NH of the benzamide and the carbonyl oxygen of the Gly69 residue (2.8 Å), but also makes two additional hydrogen bonds with Cat S. The carbonyl oxygen of the carbamate group of 5 formed a hydrogen bond (3.1 Å) with the NH of Gly69, and the NH of the carbamate group formed a hydrogen bond (2.9 Å) with the backbone carbonyl group of Asn163. While the structures of 5 and 7 were nearly identical, there were some subtle differences in relative positioning between them. For example, the oxygen atom in the chromane moiety of 5 was closer to the sulfur atom of Met71 at 3.3 Å than the corresponding distance of 3.9 Å for the benzylic carbon of 7. This S–O distance was near the sum of their van der Waals radii, and its geometry reflects a possible orbital overlap. Also, 5 had a basic nitrogen atom located 4.9 Å from the center of phenyl ring of Phe211 indicating a possible cation−π interaction between the piperazine moiety and Phe211 side chain.
Compounds 5 and 9 were found to have desirable physicochemical properties (Table 2). Preclinical pharmacokinetic evaluation of 5 and 9 demonstrated that both compounds had a similar profile in dog.20 However, compared to 9, the clearance of 5 was 3-fold lower, and the exposure was 3-fold higher in rat. In dog, both compounds were highly bioavailable (F > 75%), possessed low clearance (CL < 4 mL/min/kg), low volume of distribution (Vd < 1 L/kg), and relatively short half-lives. Additionally, 5 and 9 showed low in vitro CYP450 inhibition (<15% at 10 μM for CYP3A4, CYP2D6, and CYP2C9); low in vitro metabolism in mouse, rat, dog, and human liver microsomes (<20% after 30 min incubation at 4 μM); and good permeability (MDCK A-B > 4%). At a 100 μM concentration of 5 there was only 6% displacement of [3H]-astemizole in an assay with HEK293 membrane preparation, indicating low potential of hERG blockade. Also, when incubated in human or rat plasma for up to 30 min, there was no statistically significant loss of parent compound.
Table 2. Physicochemical Properties of 5 and 9.
| compd | logP | logD (pH 4) | most basic pKa | SGF solubility (mg/mL) |
|---|---|---|---|---|
| 5 | 2.24 | 1.0 | 5.11 | 1.44 |
| 9 | 2.24 | 1.2 | 5.0 | 1.13 |
The in vivo efficacies of 5 and 9 were studied in a mouse model of AAA (Figure 2). In this model, inflammation was induced using CaCl2 applied to the ablumenal surface. It was shown that features of the disease state in this model resemble those of human AAA.23 Both compounds exhibited a dose-responsive aortic diameter reduction at 1, 3, 10, and 30 mg/kg. However, 9 had a less profound effect than did 5. At the lowest dose of 1 mg/kg of 5, the aortic diameter was reduced by 58%, then 83% at 3 mg/kg, and 87% at 10 mg/kg. The exposure (AUC) for both compounds increased in a dose-dependent manner, suggesting that the drug disposition properties of both 5 and 9 were favorable. On a per dose basis, the percent reduction in aortic diameter for 5 was superior to 9, which may be driven by improved pharmacokinetics properties of 5. Based in part on these data, 5 was selected for development as a clinical candidate, and it has completed phase I single ascending dose studies.24−26
Figure 2.
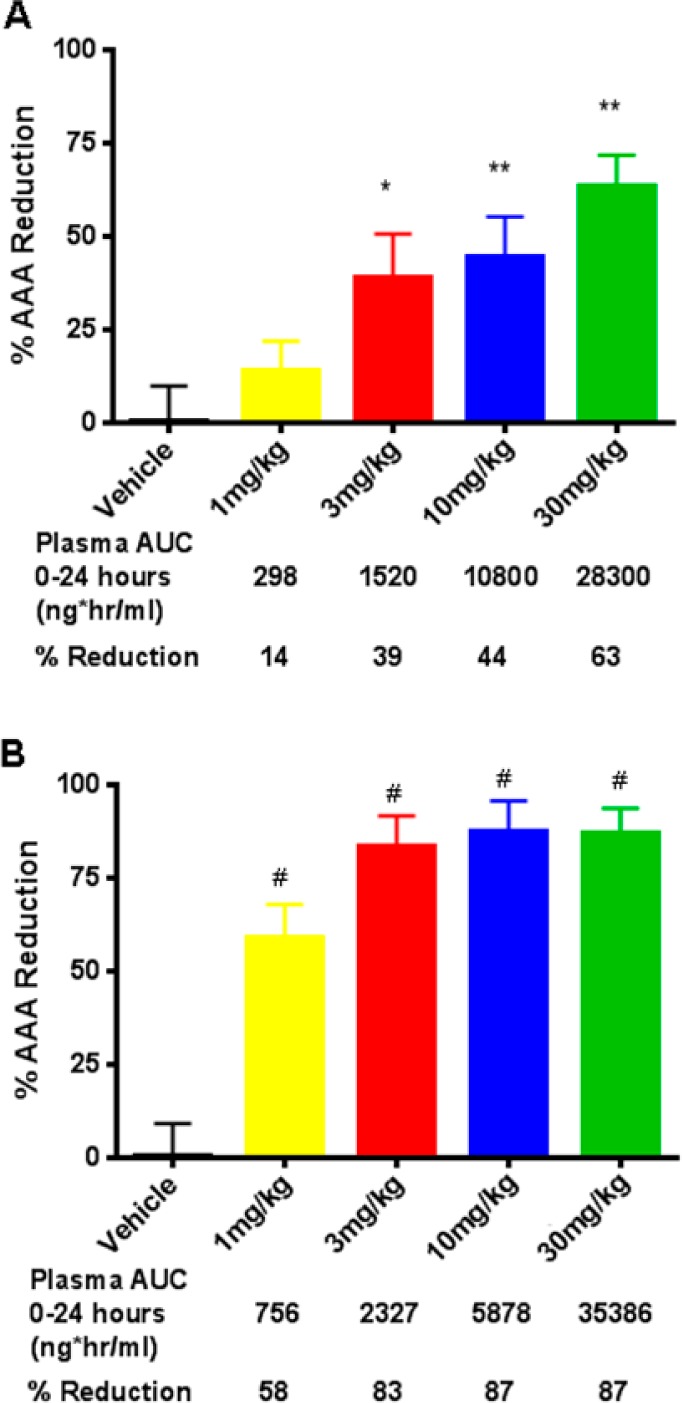
Effect of Cat S inhibitors 9 (A) and 5 (B) following 28 days of oral, BID dosing in mouse CaCl2 AAA model.
In conclusion, a medium-throughput screen of a focused compound cassette yielded 6. X-ray protein crystallography of 7 bound to Cat S revealed no interactions with Cys25. Compound 7 was further optimized primarily through structure-based design to yield compounds 5 and 9, Cat S inhibitors with excellent in vitro potency and selectivity against other cysteine proteases. After analysis of in vivo drug disposition and efficacy in the CaCl2 model, 5 emerged as the leading molecule, which was selected for clinical development.
Glossary
ABBREVIATIONS
- AAA
abdominal aortic aneurysm
- BID
twice daily
- Cat B
human cathepsin B
- Cat K
human cathepsin K
- Cat L
human cathepsin L
- Cat S
human cathepsin S
- Cat V
human cathepsin V
- CD4
cluster of differentiation 4
- compd
compound
- CYP450
cytochrome P450
- mCat S
mouse cathepsin S
- PO
by mouth
- rCat S
rat cathepsin S
- SGF
simulated gastric fluid
Supporting Information Available
Experimental procedures and analytical data for the preparation of compounds 5–9, in vitro ADME data for 5 and 9, and preclinical in vivo ADME data for 5 and 9. This material is available free of charge via the Internet at http://pubs.acs.org.
Accession Codes
Coordinates and structure factors are available from the Protein Data Bank with accession codes 4P6G and 4P6E for Cat S/5 and Cat S/7 binary complexes, respectively.
Use of the Advanced Photon Source, an Office of Science User Facility operated for the U.S. Department of Energy (DOE) Office of Science by Argonne National Laboratory, was supported by the U.S. DOE under Contract No. DE-AC02-06CH11357. Use of the Lilly Research Laboratories Collaborative Access Team (LRL-CAT) beamline at Sector 31 of the Advanced Photon Source was provided by Eli Lilly Company, which operates the facility.
The authors declare no competing financial interest.
Supplementary Material
References
- Wilmink T. B.; Quick C. R.; Hubbard C. S.; Day N. E. The influence of screening on the incidence of ruptured abdominal aortic aneurysms. J. Vasc. Surg. 1999, 30, 203–208. [DOI] [PubMed] [Google Scholar]
- Lederle F. A.; Johnson G. R.; Wilson S. E.; Chute E. P.; Hye R. J.; Makaroun M. S.; Barone G. W.; Bandyk D.; Moneta G. L.; Makhoul R. G. Aneurysm detection and management veterans affairs cooperative study investigators. The aneurysm detection and management study screening program: validation cohort and final results. Arch. Int. Med. 2000, 160, 1425–1430. [DOI] [PubMed] [Google Scholar]
- Ashton H. A.; Buxton M. J.; Day N. E.; Kim L. G.; Marteau T. M.; Scott R. A.; Thompson S. G.; Walker N. M. The Multicentre Aneurysm Screening Study (MASS) into the effect of abdominal aortic aneurysm screening on mortality in men: a randomised controlled trial. Lancet 2002, 360, 1531–1539. [DOI] [PubMed] [Google Scholar]
- Lawrence-Brown M. M.; Norman P. E.; Jamrozik K.; Semmens J. B.; Donnelly N. J.; Spencer C.; Tuohy R. Initial results of ultrasound screening for aneurysm of the abdominal aorta in Western Australia: relevance for endoluminal treatment of aneurysm disease. Cardiovasc. Surg. 2001, 9, 234–240. [DOI] [PubMed] [Google Scholar]
- Wilmink A. B.; Quick C. R. Epidemiology and potential for prevention of abdominal aortic aneurysm. Br. J. Surg. 1998, 85, 155–162. [DOI] [PubMed] [Google Scholar]
- Lindholt J. S.; Juul S.; Fasting H.; Henneberg E. W. Hospital costs and benefits of screening for abdominal aortic aneurysms: results from a randomised population screening trial. Eur. J. Vasc. Endovasc. Surg. 2002, 23, 55–60. [DOI] [PubMed] [Google Scholar]
- Kent K. C.; Zwolak R. M.; Jaff M. R.; Hollenbeck S. T.; Thompson R. W.; Schermerhorn M. L.; Sicard G. A.; Riles T. S.; Cronenwett J. L. Screening for abdominal aortic aneurysm: a consensus statement. J. Vasc. Surg. 2004, 39, 267–269. [DOI] [PubMed] [Google Scholar]
- Shi G.-P.; Munger J. S.; Meara J. P.; Rich D. H.; Chapman H. A. Molecular cloning and expression of human alveolar macrophage cathepsin S, an elastinolytic cysteine protease. J. Biol. Chem. 1992, 267, 7258–7262. [PubMed] [Google Scholar]
- Beers C.; Burich A.; Kleijmeer M. J.; Griffith J. M.; Wong P.; Rudensky A. Y. Cathepsin S controls MHC class II-mediated antigen presentation by epithelial cells in vivo. J. Immunol. 2005, 174, 1205–1212. [DOI] [PubMed] [Google Scholar]
- Qin Y.; Cao X.; Guo J.; Zhang Y.; Pan L.; Zhang H.; Li H.; Tang C.; Du J.; Shi G.-P. Deficiency of cathepsin S attenuates angiotensin II-induced abdominal aortic aneurysm formation in apolipoprotein E-deficient mice. Cardiovasc. Res. 2012, 96, 401–410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saegusa K.; Ishimaru N.; Yanagi K.; Arakaki R.; Ogawa K.; Saito I.; Katunuma N.; Hayashi Y. Cathepsin S inhibitor prevents autoantigen presentation and autoimmunity. J. Clin. Invest. 2002, 110, 361–369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiener J. J.; Sun S.; Thurmond R. L. Recent advances in the design of cathepsin S inhibitors. Curr. Top. Med. Chem. 2010, 10, 717–732. [DOI] [PubMed] [Google Scholar]
- Lee-Dutra A.; Wiener D. K.; Sun S. Cathepsin S inhibitors: 2004–2010. Expert Opin. Ther. Pat. 2011, 21, 311–337. [DOI] [PubMed] [Google Scholar]
- Palmer J. T.; Rasnick D.; Klaus J. L.; Brömme D. Vinyl sulfones as mechanism-based cysteine protease inhibitors. J. Med. Chem. 1995, 38, 3193–3196. [DOI] [PubMed] [Google Scholar]
- Ayesa S.; Lindquist C.; Agback T.; Benkestock K.; Classon B.; Henderson I.; Hewitt E.; Jansson K.; Kallin A.; Sheppard D.; Samuelsson B. Solid-phase parallel synthesis and SAR of 4-amidofuran-3-one inhibitors of cathepsin S: Effect of sulfonamides P3 substituents on potency and selectivity. Bioorg. Med. Chem. 2009, 17, 1307–1324. [DOI] [PubMed] [Google Scholar]
- Irie O.; Kosaka T.; Ehara T.; Yokokawa F.; Kanazawa T.; Hirao H.; Iwasaki A.; Sakaki J.; Teno N.; Hitomi Y.; Iwasaki G.; Fukaya H.; Nonomura K.; Tanabe K.; Koizumi S.; Uchiyama N.; Bevan S. J.; Malcangio M.; Gentry C.; Fox A. J.; Yaqoob M.; Culshaw A. J.; Hallett A. Discovery of orally bioavailable cathepsin S inhibitors for the reversal of neuropathic pain. J. Med. Chem. 2008, 51, 5502–5505. [DOI] [PubMed] [Google Scholar]
- Ameriks M. K.; Bembenek S. D.; Burdett M. T.; Choong I. C.; Edwards J. P.; Gebauer D.; Gu Y.; Karlsson L.; Purkey H. E.; Staker B. L.; Sun S.; Thurmond R. L.; Zhu J. Diazinones as P2 replacements for pyrazole-based cathepsin S inhibitors. Bioorg. Med. Chem. Lett. 2010, 20, 4060–4064and references cited therein. [DOI] [PubMed] [Google Scholar]
- Brömme D.; Okamoto K.; Wang B. B.; Biroc S. Human cathepsin O2, a matrix protein-degrading cysteine protease expressed in osteoclasts. Functional expression of human cathepsin O2 in Spodoptera frugiperda and characterization of the enzyme. J. Biol. Chem. 1996, 271, 2126–2132. [DOI] [PubMed] [Google Scholar]
- Brömme D.; Li Z.; Barnes M.; Mehler E. Human cathepsin V functional expression, tissue distribution, electrostatic surface potential, enzymatic characterization, and chromosomal localization. Biochemistry 1999, 38, 2377–2385. [DOI] [PubMed] [Google Scholar]
- See Supporting Information for more detailed data.
- Mason C. S.; Lamers M. B. A. C.; Henderson I. M. J.; Monk T.; Williams D. H. Baculoviral expression and characterization of rodent cathepsin S. Protein Expression Purif. 2001, 23, 45–54. [DOI] [PubMed] [Google Scholar]
- Experimental procedures for the preparation of 5–9 appear in Supporting Information.
- Longo G. M.; Xiong W.; Greiner T. C.; Zhao Y.; Fiotti N.; Baxter T. B. Matrix metalloproteinases 2 and 9 work in concert to produce aortic aneurysms. J. Clin. Invest. 2002, 110, 625–632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Payne C. D.; Deeg M. A.; Chan M.; Tan L. H.; LaBell E. S.; Shen T.; DeBrota D. J.. A single-dose, dose escalation study to evaluate the pharmacokinetics, pharmacodynamics, safety and tolerability of cathepsin S inhibitor, LY3000328, in healthy subjects. Br. J. Clin. Pharmacol. 2014, submitted [DOI] [PMC free article] [PubMed]
- A Study to Evaluate the Safety and Tolerability of LY3000328 in Healthy Participants. http://clinicaltrials.gov/show/NCT01515358.
- Deng G.; Gavardinas K.; Kim E.; Jadhav P. K.; Schiffler M. A.. Cathepsin S Inhibitor Compounds. U.S. Patent 8,227,486, July 24, 2012.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.