

Abstract

Development of peptide-based drugs has been severely limited by lack of oral bioavailability with less than a handful of peptides being truly orally bioavailable, mainly cyclic peptides with N-methyl amino acids and few hydrogen bond donors. Here we report that cyclic penta- and hexa-leucine peptides, with no N-methylation and five or six amide NH protons, exhibit some degree of oral bioavailability (4–17%) approaching that of the heavily N-methylated drug cyclosporine (22%) under the same conditions. These simple cyclic peptides demonstrate that oral bioavailability is achievable for peptides that fall outside of rule-of-five guidelines without the need for N-methylation or modified amino acids.
Keywords: Oral bioavailability, absorption, peptides, rule of five, permeability
Lack of oral bioavailability and plasma stability have been limiting factors in the development and use by patients of peptide-based drugs.1−3 Despite these deficiencies, peptides and proteins, especially antibodies, have become “blockbuster” injectable drugs in recent years.3 Protein and peptide-based drugs also suffer from high costs of manufacture and delivery, low patient compliance with injection schedules and nonresponsiveness in some patients. Oral delivery of drugs is still the preferred route for treating most chronic diseases where regular, cheaper, and longer term dosing is required.3 Here we describe a step toward understanding factors that govern peptide oral bioavailability, reporting on comparative oral bioavailability of a simple cyclic penta- and hexapeptide in rats.
In medicinal chemistry, compounds are more likely to be orally absorbed if they obey “rule-of-five” (Ro5) parameters (MW < 500, cLogP < 5, hydrogen bond donors (HBD) ≤ 5, and hydrogen bond acceptors (HBA) ≤ 10).4−6 A cyclic pentapeptide lies at the boundary of this range, with HBD = 5 and HBA = 10, while MW ≈ 500 and cLogP < 5 are dependent upon amino acid side chain composition. The chances of oral bioavailability are further increased if peptides are not rapidly cleaved before or after membrane penetration. Cyclic peptides are more resistant than linear peptides to degradation by proteases because there are no N- or C- termini available for recognition and cleavage by aminopeptidases and carboxypeptidases, respectively.7−9 Nevertheless, most peptides, including cyclic peptides, have < 1% oral bioavailability. There are a handful of exceptions, mainly those containing N-methyl amino acids and thus fewer HBD atoms.10−12 The best known exception is the widely used 11-residue immunosuppressive cyclic peptide drug cyclosporin A (CSA), with seven N-methylated amino acids, four hydrogen-bonded amide NH protons, and oral bioavailability (F = 15%–50%) that is dependent on the animal model, excipient/solvent formulation, and the concentration administered.13−19
Here we compare structures, membrane permeabilities, and oral
bioavailabilities
of simple cyclic peptides with properties that lie just outside rule-of-five
parameters. Cyclic pentaleucine, cyclo-[Leu]5 (1) (MW = 566, HBD = 5, HBA = 10, and cLogP = 7.6) is compared with
cyclo-hexaleucine isomers cyclo-[(l-Leu)5(d-Leu)] (2) (MW = 679, HBD = 6, HBA = 12, and cLogP
= 9.1) and cyclo-[Leu]6 (3). Leucines were
chosen to ensure cLogP > 5. A d-leucine was inserted only
in the hexpeptide, as it is known to be less constraining than in
a pentapeptide where it induces both a hydrogen bond and a beta turn.20,21 The compounds violate rule-of-five guidelines4−6 yet have some
membrane permeability and oral bioavailability in rats. These findings
provide a valuable platform for future studies aimed at understanding
factors that determine peptide oral bioavailability.
Compounds 1–3 were synthesized using standard solid phase peptide synthesis protocols and Fmoc protected amino acids in DMF to produce linear precursors. These were cyclized under dilute conditions (10–3 M) and the cyclic peptides purified by rpHPLC (Supporting Information). Epimerization was observed duration formation of 3, but diastereomers 2 and 3 were separable by rpHPLC and could be purified.
The membrane permeability of compounds 1–3 vs CSA was measured in RRCK cell monolayers (Figure 1), which lack active transporters, and in CACO-2 cell monolayers, which have been more widely used to measure membrane permeability but also involve active transport.10,11 Membrane permeability is often used to predict whether a compound will be orally absorbed since the first barrier after surviving the GI tract is membrane permeation and resistance to enterocyte metabolism.22−25 In the RRCK assay, compound 1 had lower permeability (Papp = 1.7 × 10–6 cm·s–1) than CSA (Papp = 5 × 10–6 cm·s–1),12 while cyclic hexapeptide isomers 2 and 3 had 6–7-fold greater permeability than 1 and 2–3-fold higher RRCK permeability than CSA (Figure 1A). By contrast, all three compounds were similarly permeable in CACO-2 cells (Figure 1B). When CACO-2 permeability was measured at 50 μM, compound 1 was not detected, while compounds 2 and 3 retained permeability (Figure S1, Supporting Information).
Figure 1.
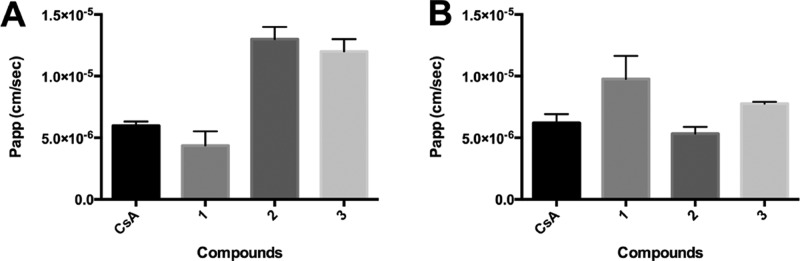
Apparent permeability (cm·s–1) of cyclic peptides 1–3 (A) at 8 μM vs CSA (8 μM) in RRCK cells or (B) at 10 μM vs CSA (30 μM) in CACO-2 cells. Cell medium = 0.05% DMSO in HEPES buffer, pH 7.4, 37 °C.
Given the differences in membrane permeability of 1 versus 2 and 3 and the reduced CACO-2 cell permeability of 1 at higher concentration, their structures were examined for potential influences on membrane permeability. The crystal structure of 1, isolated from aqueous methanol, showed intermolecular (but no intramolecular) hydrogen bonds (Figure 2A) indicating some propensity for 1 to aggregate. Concentration-dependent changes in the line-shapes of circular dichroism spectra for 1 (30–500 μM) in TFE were consistent with a tendency to aggregate at concentrations > 60 μM in solution (Figure 2B), although this may not occur at physiologically relevant concentrations in aqueous conditions and so may not affect cell permeation in vivo.
Figure 2.
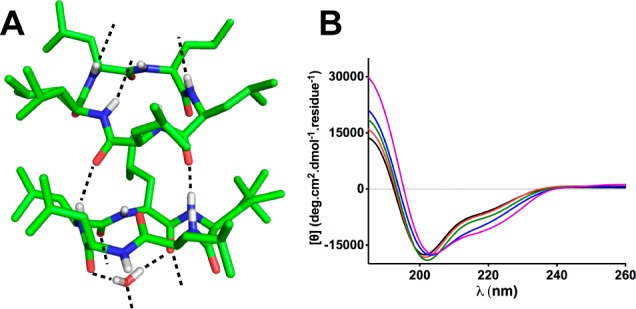
Aggregation of compound 1. (A) Crystal structure of 1 isolated from MeOH/H2O (80:20), showing intermolecular hydrogen bonds (dashes) (CCDC 1002286). (B) Concentration-dependent CD spectra of 1 (30 μM, purple; 60 μM, blue; 150 μM, green; 250 μM, red; 500 μM, black) in TFE at 298 K.
By contrast the CD spectral line-shapes for compound 3 were concentration independent over the same range (Figure 3A), consistent with no aggregation for the cyclic hexapeptide. To understand differences in aggregation properties between cyclic penta- and hexaleucine, NMR structures of the symmetrical 1 and 3 were also determined (in DMSO-d6) using dihedral angle constraints (Supporting Information). Interestingly, whereas compound 1 had a pseudoplanar circular structure, compound 3 formed a more rectangular structure with two identical turns at each end. The leucine side chains in 1 were projected outward (Figure 3B), exposing the polar groups to solvent and allowing formation of intermolecular hydrogen bonds between exposed NH and CO groups, thereby encouraging aggregation. In contrast, for compound 3, two of the leucine side chains were projected inward (Figure 3B) enabling shielding of the amide backbone from solvent. These leucine side chains were also on the same face as the amide NHs (Figure 3B), preventing formation of intermolecular hydrogen bonds, thereby hindering aggregation. These results are consistent with higher membrane permeability for 3 (no aggregation) than 1 (aggregation).
Figure 3.
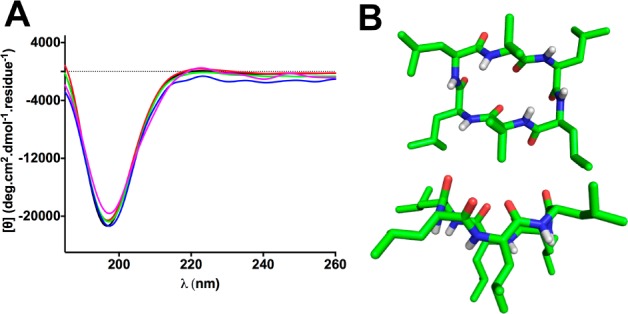
(A) Concentration-independent CD spectra for 3 in TFE (30 μM, purple; 60 μM, blue; 150 μM, green; 250 μM, red; 500 μM, black; 298 K). (B) NMR-derived structure of 3 in DMSO-d6. Top and side views show two Leu side chains approaching closely to prevent intermolecular amide H-bonds.
Since compounds 1–3 showed detectable permeability (Papp >1 × 10–6 cm·s–1) in RRCK or CACO-2 cells, oral bioavailability was measured in male Wistar rats (Figure 4) for 1–3 at 10 mg/kg/p.o. in olive oil vs 1 mg/kg/i.v. in DMSO (Supporting Information). The systemic fraction of peptide (oral bioavailability, F%) was determined by comparing areas under the plasma concentration–time traces (AUC) over 8 h after i.v. and oral doses (D), namely, by F% = (AUCp.o. × Di.v.)/(AUCi.v. × Dp.o.). The rank order of oral bioavailability (F%) was 1 (4%) < 2 (9%) < 3 (17%) < CsA (23%) and did not correlate with cell permeability (Figure 1). However, oral bioavailability is dependent on other factors in addition to membrane permeability, including solubility, metabolic stability (GI tract, enterocytes, plasma, and liver), clearance, tissue distribution, and protein binding.
Figure 4.
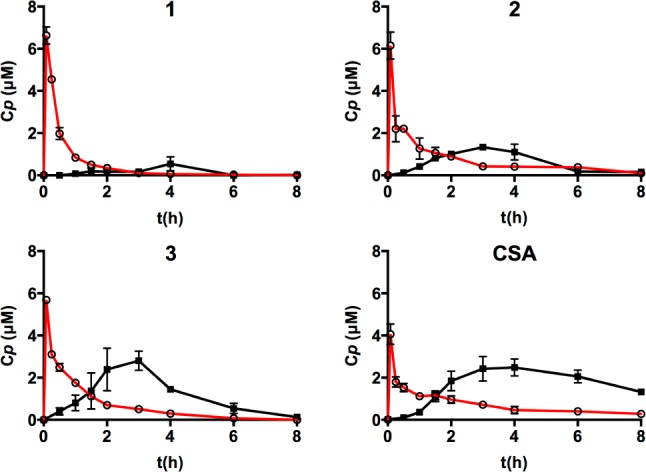
Relative concentrations in plasma (Cp) assessed with time (t) by LCMS up to 8 h after administration of peptides 1–3 or cyclosporin-A at 10 mg/kg/p.o. in olive oil (black) vs 1 mg/kg/i.v. in DMSO (red) to male Wistar rats. Oral bioavailability (F%) = 4.0 ± 1.7%, n = 3 (1); 8.5 ± 1.1%, n = 2 (2); 17.3 ± 5.7%, n = 3 (3); 22.5 ± 7.8%, n = 4 (CSA).
The lower bioavailability of 2 vs 3 is consistent with the higher plasma clearance of 2 (24.1 mL/min/kg) vs 3 (4.7 mL/min/kg) after intravenous dosing. Different clearances of the two diastereomers might be due in part to differences in solvent exposure of the peptide backbone. For example, compound 3 had only one amide NH resonance in its 1H NMR spectrum in DMSO-d6, and it exchanged rather slowly (t1/2 = 480 min, DMSO-d6/D2O 9:1, 298 K; Supporting Information). By contrast, 2 displayed well-dispersed resonances with different 3JNHHα coupling constants for all six amide NHs, consistent with a well-defined asymmetric structure, with three more rapidly exchanging (t1/2 = 60, 120, and 150 min) and three slowly exchanging (t1/2 > 150 min) amide NH protons. This suggests that conformational influences of amino acid substitutions might reduce plasma clearance rates by controlling solvent exposure, and this should be investigated in detail in the future.
Conclusions
Very few peptides are orally bioavailable. They typically have backbone N-methylated amides and intramolecular hydrogen bonds. Here we find, surprisingly, that even simple cyclic peptides without N-methyl groups or intramolecular hydrogen bonds are able to permeate cell membranes and have a degree of oral bioavailability. Hydrophobic side chains of amino acids can sufficiently shield polar amides from solvation to permit membrane permeation and oral absorption. The peptide conformation may also impact oral bioavailability by influencing exposure to solvent and to proteins that dictate plasma clearance and metabolism. Peptide aggregation may hinder oral absorption despite reduced solvent exposure; this is likely due to reduced solubility and formation of higher molecular weight aggregates. This study adds to our understanding of oral bioavailability of compounds with different physicochemical properties than traditional drugs.
Supporting Information Available
Synthetic methods, compound characterization, RRCK and CACO-2 experiments, rat pharmacokinetic experiments and data, CD methods, NMR calculations, NMR spectra, and crystal structure coordinates. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Contributions
⊥ These authors (T.A.H. and R.-J.L) contributed equally to this work. All authors contributed to experiments or writing or reviewing of results or manuscript. All authors have given approval to the final version of the manuscript.
We acknowledge funding from Australian Research Council (FF0668733, DP1096290, LP110200213, and CE140100011), the Queensland Government (CIF), and the Australian National Health and Medical Research Council (for Senior Principal Research Fellowships to D.C. (1026501) and D.F. (1027369)).
Oral bioavailabilities were disclosed at the 241st American Chemical Society National Meeting, “Towards orally bioavailable peptides and peptidomimetics” (Beyond the Rule of Five session), Anaheim, CA, March, 2011.
The authors declare no competing financial interest.
Supplementary Material
References
- Danho W.; Swistok J.; Khan W.; Chu X.-J.; Cheung A.; Fry D.; Sun H.; Kurylko G.; Rumennik L.; Cefalu J.; Cefalu G.; Nunn P. Opportunities and challenges of developing peptide drugs in the pharmaceutical industry. Peptides for Youth 2009, 611, 467–469. [DOI] [PubMed] [Google Scholar]
- Vlieghe P.; Lisowski V.; Martinez J.; Khrestchatisky M. Synthetic therapeutic peptides: science and market. Drug Discovery Today 2010, 15, 40–56. [DOI] [PubMed] [Google Scholar]
- Craik D. J.; Fairlie D. P.; Liras S.; Price D. The Future of Peptide-based Drugs. Chem. Biol. Drug Design 2013, 81, 136–147. [DOI] [PubMed] [Google Scholar]
- Lipinski C. A. Drug-like properties and the causes of poor solubility and poor permeability. J. Pharmacol. Toxicol. Methods 2001, 44, 235–249. [DOI] [PubMed] [Google Scholar]
- Lipinski C. A.; Lombardo F.; Dominy B. W.; Feeney P. J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Delivery Rev. 1997, 23, 3–25. [DOI] [PubMed] [Google Scholar]
- Lipinski C. A.; Lombardo F.; Dominy B. W.; Feeney P. J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Delivery Rev. 2001, 46, 3–26. [DOI] [PubMed] [Google Scholar]
- Bockus A. T.; McEwen C. M.; Lokey R. S. Form and function in cyclic peptide natural products: A pharmacokinetic perspective. Curr. Top. Med. Chem. 2013, 13, 821–836. [DOI] [PubMed] [Google Scholar]
- Giordanetto F.; Kihlberg J. Macrocyclic drugs and clinical candidates: What can medicinal chemists learn from their properties?. J. Med. Chem. 2014, 57, 278–295. [DOI] [PubMed] [Google Scholar]
- Gracia S. R.; Gaus K.; Sewald N. Synthesis of chemically modified bioactive peptides: recent advances, challenges and developments for medicinal chemistry. Future Med. Chem. 2009, 1, 1289–1310. [DOI] [PubMed] [Google Scholar]
- White T. R.; Renzelman C. M.; Rand A. C.; Rezai T.; McEwen C. M.; Gelev V. M.; Turner R. A.; Linington R. G.; Leung S. S. F.; Kalgutkar A. S.; Bauman J. N.; Zhang Y. Z.; Liras S.; Price D. A.; Mathiowetz A. M.; Jacobson M. P.; Lokey R. S. On-resin N-methylation of cyclic peptides for discovery of orally bioavailable scaffolds. Nat. Chem. Biol. 2011, 7, 810–817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rand A. C.; Leung S. S. F.; Eng H.; Rotter C. J.; Sharma R.; Kalgutkar A. S.; Zhang Y. Z.; Varma M. V.; Farley K. A.; Khunte B.; Limberakis C.; Price D. A.; Liras S.; Mathiowetz A. M.; Jacobson M. P.; Lokey R. S. Optimizing PK properties of cyclic peptides: the effect of side chain substitutions on permeability and clearance. MedChemComm 2012, 3, 1282–1289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guimaraes C. R. W.; Mathiowetz A. M.; Shalaeva M.; Goetz G.; Liras S. Use of 3D properties to characterize beyond rule-of-5 property space for passive permeation. J. Chem. Inf. Model. 2012, 52, 882–890. [DOI] [PubMed] [Google Scholar]
- Borel J. F. History of the discovery of cyclosporin and of its early pharmacological development. Wien. Klin. Wochenschr. 2002, 114, 433–437. [PubMed] [Google Scholar]
- Noble S.; Markham A. Cyclosporine: A review of the pharmacokinetic properties, clinical efficacy and tolerability of a microemulsion-based formulation (Neoral). Drugs 1995, 50, 924–941. [DOI] [PubMed] [Google Scholar]
- Lindbergfreijs A.; Karlsson M. O. Dose-dependent absorption and linear disposition of cyclosporine-a in rat. Biopharm. Drug Dispersion 1994, 15, 75–86. [DOI] [PubMed] [Google Scholar]
- Kim S. J.; Choi H. K.; Lee Y. B. Pharmacokinetic and pharmacodynamic evaluation of cyclosporin A O/W-emulsion in rats. Int. J. Pharm. 2002, 249, 149–156. [DOI] [PubMed] [Google Scholar]
- Kim C. K.; Shin H. J.; Yang S. G.; Kim J. H.; Oh Y. K. Once-a-day oral dosing regimen of cyclosporin A: Combined therapy of cyclosporin A premicroemulsion concentrates and enteric coated solid-state premicroemulsion concentrates. Pharm. Res. 2001, 18, 454–459. [DOI] [PubMed] [Google Scholar]
- Gonzalez R. C. B.; Huwyler J.; Walter I.; Mountfield R.; Bittner B. Improved oral bioavailability of cyclosporin A in male Wistar rats: Comparison of a Solutol HS 15 containing self-dispersing formulation and a microsuspension. Int. J. Pharm. 2002, 245, 143–151. [DOI] [PubMed] [Google Scholar]
- Hedayati S.; Bernareggi A.; Rowland M. Absorption kinetics of cyclosporine in the rat. J. Pharm. Pharmacol. 1992, 44, 122–124. [DOI] [PubMed] [Google Scholar]
- Beck J. G.; Chatterjee J.; Laufer B.; Kiran M. U.; Frank A. O.; Neubauer S.; Ovadia O.; Greenberg S.; Gilon C.; Hoffman A.; Kessler H. Intestinal permeability of cyclic peptides: Common key backbone motifs identified. J. Am. Chem. Soc. 2012, 134, 12125–12133. [DOI] [PubMed] [Google Scholar]
- Chatterjee J.; Mierke D.; Kessler H. N-methylated cyclic pentaalanine peptides as template structures. J. Am. Chem. Soc. 2006, 128, 15164–15172. [DOI] [PubMed] [Google Scholar]
- Wilson G.; Hassan I. F.; Dix C. J.; Williamson I.; Shah R.; Mackay M.; Artursson P. Transport and permeability properties of human Caco-2 cells: an in vitro model of the intestinal epithelial cell barrier. J. Controlled Release 1990, 11, 25–40. [Google Scholar]
- Artursson P.; Palm K.; Luthman K. Caco-2 monolayers in experimental and theoretical predictions of drug transport. Adv. Drug Delivery Rev. 2012, 64, 280–289. [DOI] [PubMed] [Google Scholar]
- Ovadia O.; Greenberg S.; Chatterjee J.; Laufer B.; Opperer F.; Kessler H.; Gilon C.; Hoffman A. The effect of multiple N-methylation on intestinal permeability of cyclic hexapeptides. Mol. Pharmaceutics 2011, 8, 479–487. [DOI] [PubMed] [Google Scholar]
- Di L.; Whitney-Pickett C.; Umland J. P.; Zhang H.; Zhang X.; Gebhard D. F.; Lai Y. R.; Federico J. J.; Davidson R. E.; Smith R.; Reyner E. L.; Lee C.; Feng B.; Rotter C.; Varma M. V.; Kempshall S.; Fenner K.; El-Kattan A. F.; Liston T. E.; Troutman M. D. Development of a new permeability assay using low-efflux MDCKII cells. J. Pharm. Sci. 2011, 100, 4974–4985. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.