

Abstract
A-1155463, a highly potent and selective BCL-XL inhibitor, was discovered through nuclear magnetic resonance (NMR) fragment screening and structure-based design. This compound is substantially more potent against BCL-XL-dependent cell lines relative to our recently reported inhibitor, WEHI-539, while possessing none of its inherent pharmaceutical liabilities. A-1155463 caused a mechanism-based and reversible thrombocytopenia in mice and inhibited H146 small cell lung cancer xenograft tumor growth in vivo following multiple doses. A-1155463 thus represents an excellent tool molecule for studying BCL-XL biology as well as a productive lead structure for further optimization.
Keywords: BCL-XL, BCL-2, apoptosis, cancer
Apoptosis is a highly conserved process that is critical for cellular homeostasis and development. Dysregulation in apoptotic signaling is a common requirement for oncogenesis, tumor maintenance, and chemoresistance.1 Dynamic binding interactions between the pro-apoptotic (e.g., BAX, BAK, BAD, BIM, NOXA) and antiapoptotic (e.g., BCL-2, BCL-XL, MCL-1) BCL-2 family proteins control commitment to programmed cell death.2,3 BCL-2 plays a dominant role in the survival of lymphoid malignancies where it is frequently overexpressed,4 while overexpression of BCL-XL has been correlated with drug resistance and disease progression of multiple solid tumors and hematological malignancies.5 Targeting these proteins via small molecule inhibitors has thus been a longstanding goal in the oncology community.
Despite the inherent difficulties in targeting protein–protein interactions, a combination of nuclear magnetic resonance (NMR)-based screening and structure-based design afforded ABT-263 (navitoclax).6−8 Navitoclax is a first-in-class, orally bioavailable inhibitor of both BCL-2 and BCL-XL, which has demonstrated clinical antitumor activity in lymphoid malignancies that are believed to be dependent upon BCL-2 for survival.9,10 Consistent with preclinical studies showing the role of BCL-XL in platelet survival,11,12 thrombocytopenia was the predominant clinical dose-limiting toxicity of navitoclax when dosed as a single agent.9 This observation prompted the generation of the BCL-2 selective inhibitor ABT-199,13 which has shown antileukemic activity in refractory chronic lymphocytic leukemia patients without the dose limiting thrombocytopenia elicited by navitoclax treatment. These clinical data demonstrate that selective inhibitors of a single BCL-2 family protein can have particular promise in targeting specific cancer types.
We previously reported that the synergy observed between navitoclax and chemotherapies is driven primarily by the inhibition of BCL-XL in solid tumors.14,15 Additionally, a BCL-XL selective inhibitor offered the potential for reduced immunosuppressive effects relative to dual inhibition of BCL-2 and BCL-XL.9,10 A program to develop selective inhibitors was thus initiated with the immediate goal of identifying a compound suitable for key in vivo studies.
Previously, a series of selective BCL-XL inhibitors containing hydrazinylbenzothiazole cores 1 or 2 (Figure 1) was reported.16,17 Optimization led to the identification of WEHI-539 (3), which demonstrated selective and mechanism-based cell killing in mouse embryo fibroblast (MEF) cells engineered to be dependent on BCL-XL via loss of MCL-1 (mcl-1–/–).16 WEHI-539 also demonstrated cell killing in the BCL-XL dependent human small cell lung cancer (SCLC) cell line H146 (Figure 1). However, the utility of WEHI-539 as a tool molecule was limited by the presence of a labile and potentially toxic hydrazone moiety. Additionally, the poor physicochemical properties of this compound made in vivo dosing untenable. Initial efforts to redesign the hydrazone-based cores 1 and 2 afforded an amide-based pharmacophore 4.18 Although 4 showed some loss in BCL-XL affinity relative to the parent hydrazone it demonstrated acceptable pharmacokinetic (PK) properties in rats and had no inherent pharmaceutical liabilities.18 The generation of compound 5 via extension into the critical “P4” binding pocket demonstrated that analogues with on-target cellular activity could be obtained;18 however, the lack of potency in the higher bar human tumor H146 cell line indicated that further optimization was necessary.
Figure 1.

First generation hydrazone-based BCL-XL inhibitors and second generation amide-based analogues.
The P4 binding pocket in BCL-XL has previously been demonstrated to be critical for tight binding of BH3-peptides and inhibitors.6,7,16 Thus, we targeted this region for our optimization efforts. In order to facilitate the identification of productive P4-binding moieties beyond the methylamine represented in WEHI-539,16 we employed a structure–activity relationship (SAR) by NMR fragment-based approach.19 The crystal structures of the hydrazone (1 and 2) and amide (4) cores with BCL-XL demonstrated that these molecules bound the P2 pocket of BCL-XL in very similar fashion and could thus be used interchangeably as first site ligands.16−18 Consequently, the slightly higher solubility of core 1 in the aqueous conditions of the NMR experiment prompted its nomination as the first site ligand. To identify second site ligands, NMR screening was undertaken (see Supporting Information) with a library of 875 highly soluble fragments in the presence of 1.
Of the various second site ligands that bound in the presence of 1, bicyclic heterocycle 6 (Figure 2) demonstrated good affinity (Kd ≈ 4 mM) along with multiple vectors for attachment and modification. Six protein–ligand NOEs observed in a 13C-edited, 12C-filtered 3D NOESY spectrum were then used to dock 6 into the crystal structure of BCL-XL bound to 1. This model demonstrated the phenyl group of 6 to be less than 4 Å away from the picolinic acid (Figure 2), indicating that a small tether could be used for linking the two molecules. Furthermore, the proximity of the meta- and para-positions of the phenyl moiety of 6 suggested these vectors to be optimal for linkage.
Figure 2.

Left: Highly soluble fragment 6 identified as a 2nd site binder by NMR in the presence of core 1. Right: NMR-derived ternary structure of 1 (green) and 6 (magenta) bound to BCL-XL.
A small set of compounds (7–11, Table 1) with varying linker lengths and attachment points were thus synthesized to explore these hypotheses. The combination of para-substitution and a 4-atom tether clearly emerged as the local optimum with respect to BCL-XL affinity, as the resultant compound (10) exhibited a subnanomolar Ki value while preserving excellent binding selectivity (>4000-fold) over BCL-2. However, as predicted by the large and hydrophobic nature of the pharmacophore (MW = 688.8, ClogP = 6.2) and the presence of a carboxylic acid, the addition of 1% human serum (HS) to the TR-FRET assay conditions right-shifted the apparent binding affinities of all analogues by approximately 10- to 30-fold. Despite this attenuated binding, 10 showed potent cell killing in mcl-1–/– MEF cells in the presence of 10% fetal bovine serum (FBS).
Table 1. SAR of Fragment-Linked Analoguesa.

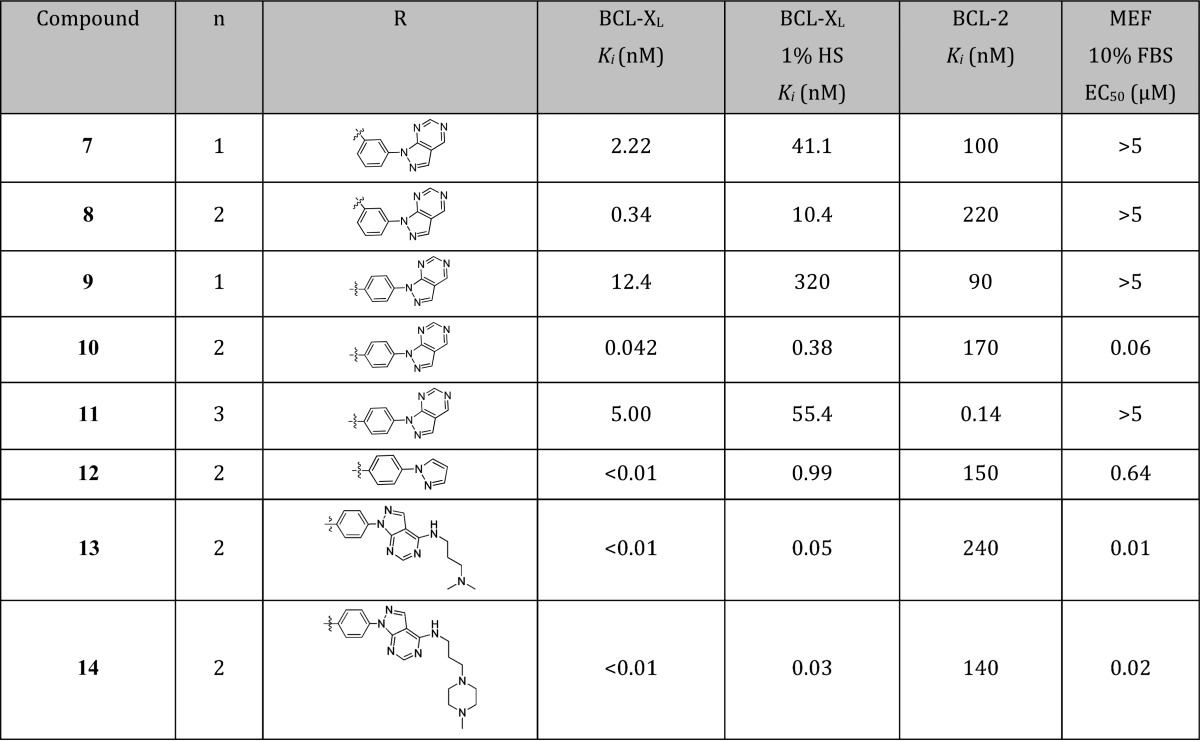
All assays were run in triplicate.
To further understand the binding mode, an X-ray cocrystal structure of 10 bound to BCL-XL was obtained (1.8 Å resolution, Figure 3). The core of compound 10 resided within the P2 pocket as expected.16−18 The benzothiazole ring nitrogen and amide proton formed the same key hydrogen bonds with Leu108 and Ser106, respectively, of the BCL-XL protein as observed with the hydrazone-based inhibitors.16 Similarly, the carboxylic acid of 10 formed a hydrogen bond with the side chain of Arg139 within the protein. As predicted from the NMR-derived model, the tethered phenyl-1H-pyrazolo[3,4-d]pyrimidine bound within the hydrophobic P4 pocket of BCL-XL, with the phenyl and pyrazole rings lying in closest proximity to hydrophobic residues Tyr101 and Tyr195, respectively.
Figure 3.

X-ray crystal structure of compound 10 (green) bound to BCL-XL. Protein is shown as a ribbon diagram where key amino acids are colored orange, oxygen atoms are red, and nitrogen atoms are blue. Key hydrogen bonds are shown in red dotted lines. PDB code: 4TUH.
Because the pyrimidine ring of 10 was solvent exposed, the contribution of this moiety to BCL-XL affinity was unclear. This prompted the synthesis and evaluation of analogue 12 (Table 1), which retained the pyrazole ring but lacked the pyrimidine ring. While 12 showed slightly improved binding affinity relative to 10, it suffered from a substantial diminution in BCL-XL binding in the presence of 1% HS and a 10-fold decrease in activity in mcl-1–/– MEF cells compared to pyrazolopyrimidine 10. These data suggested that the pyrimidine ring of the latter compound was conferring a reduction in protein binding via increased polarity7 while adding no additional benefit in target affinity.
To extend this hypothesis, the pyrimidine ring of 10 was next utilized as a scaffold for the incorporation of polar groups. In previous optimization efforts toward dual BCL-2/BCL-XL inhibitors, human serum albumin (HSA) binding could be alleviated via the incorporation of amine groups at a specific position.6,7 Furthermore, the basicity of the amine group could be optimized to offset the acidic portion of the pharmacophore to afford substantial benefits in cellular potency and oral absorption. Inspired by this example, we synthesized a pair of analogues with tethered amines projecting from the solvent-exposed pyrimidine moiety (13 and 14, Table 1).
Interestingly, the addition of tethered amines increased the target affinity of compounds 13 and 14 relative to 10. This was exemplified by the Ki values to BCL-XL, which were below the limits of detection (10 picomolar) in the TR-FRET assay and dramatically improved in the presence of 1% HS. The enhanced target affinity of 13 and 14 translated into significant gains in cellular activity, with both analogues showing highly potent killing of mcl-1–/– MEF cells (Table 1).
The presence of the pyrazole moiety within analogues 10–14 appeared to drive target affinity via favorable π-stacking interactions within the P4 pocket of BCL-XL. The tethered amines within analogues 13 and 14, in turn, were putatively beneficial in reducing HSA binding while simultaneously increasing target affinity, although the reason for the latter is unclear given the solvent-exposed nature of these moieties. These observations lead to the design and preparation of aminoalkyne-linked 15 (Table 2), a simplified analogue that could both achieve π-stacking interactions as well as reduce HSA-binding. The affinity of this analogue was encouraging, albeit slightly lower than that of 13 and 14. However, the excellent potency of 15 against mcl-1–/– MEF cells (EC50 = 8 nM) suggested a possible increase in permeability, a result that was ascribed to the lower basicity of the terminal alkynyl amine. We then tested the ability of 15 to kill H146 cells. Gratifyingly, in the presence of 10% HS, 15 demonstrated an EC50 of 600 nM (Table 2).
Table 2. Biological Activity of Alkyne 15 and Halogenated Analoguesa.
| compound | halogen position | BCL-XLKi (nM) | BCL-XL 1% HS Ki (nM) | BCL-2 Ki (nM) | H146 10% HS EC50 (μM) |
|---|---|---|---|---|---|
| 15 | none | 0.02 | 0.28 | 60 | 0.60 |
| 16 | 2-Cl | 0.04 | 0.41 | 280 | 0.18 |
| 17 (A-1155463) | 2-F | <0.01 | 0.10 | 80 | 0.065b |
| 18 | 3-F | 0.01 | 1.12 | 88 | 0.051 |
| 19 | 2-F, 5-F | 0.03 | 0.33 | 90 | 0.126 |
All assays were run in triplicate.
Geometric mean of over 35 assays run in triplicate.
Alkyne analogue 15 was an attractive lead structure given its straightforward synthesis and decreased molecular weight (MW = 652) relative to pyrazolopyrimidine analogues 13 (MW = 789) and 14 (MW = 844). The electron-rich 4-alkynyl phenoxy moiety was thus targeted for additional modification, with the hypothesis that halogens could create additional interactions in the hydrophobic P4 pocket while conferring beneficial effects on metabolic stability.20 To this end, a set of analogues with a single halogen on either the 2 or 3 position (16–18) were prepared along with the 2,5-difluorinated analogue 19 (Table 2). In all cases, halogenation of the phenyl ring enhanced the cytotoxic activity of the corresponding analogues in H146 cells as compared to 15. The single fluorine analogues 17 (A-1155463) and 18 in particular showed an approximately 10-fold boost in cell killing activity.
An X-ray cocrystal structure of BCL-XL-bound to A-1155463 (2.1 Å resolution, Figure 4) revealed the 2-fluoro substituent to be proximal to the side chains of Val141 and Phe97 within the hydrophobic P4 pocket. These close C–F distances (<4.0 Å) suggested the presence of significant van der Waals contacts.20
Figure 4.
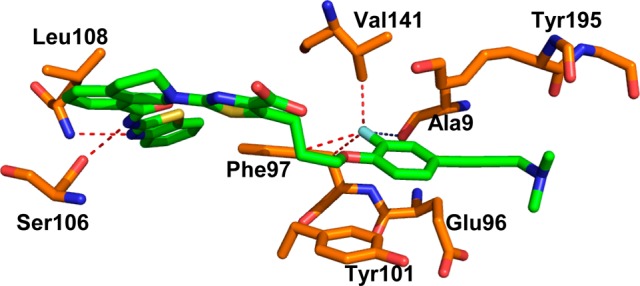
X-ray crystal structure of A-1155463 (green) bound to BCL-XL. Key amino acids are colored orange, oxygen atoms are red, and nitrogen atoms are blue. Dotted contacts are <4 Å. PDB code: 4QVX.
A-1155463 showed picomolar binding affinity to BCL-XL, and >1000-fold weaker binding to BCL-2 (Table 2) and related proteins BCL-W (Ki = 19 nM) and MCL-1 (Ki > 440 nM). Moreover, the cellular activity in the BCL-XL-dependent H146 cell line was improved by nearly an order of magnitude relative to the starting hydrazone WEHI-539. Because the aqueous solubility of this and related compounds compromised the generation of quality in vitro ADME data, we elected to directly investigate the pharmacokinetic properties of A-1155463 in nontumor bearing SCID-Beige mice following a single intraperitoneal (IP) dose. As shown in Table 3, a 5 mg/kg dose afforded target coverage of 12 h relative to the H146 EC50, which was sufficient for an expectation of in vivo activity.6−8
Table 3. Pharmacokinetic Properties of A-1155463 in SCID-Beige Micea.
| Cmax (μM) | Tmax (hr) | AUC (μM·h) | C12h (μM) |
|---|---|---|---|
| 1.8 | 2 | 9.19 | 0.18 |
A-1155463 was dosed at 5 mg/kg IP in a vehicle containing 5% DMSO, 10% EtOH, 20% Cremaphor ELP, and 65% D5W.
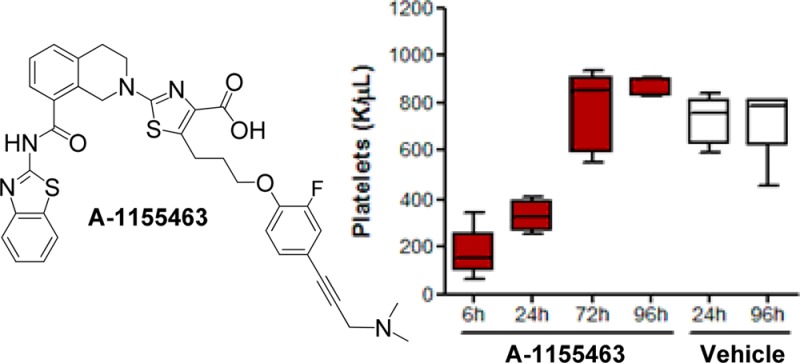
We next measured the effects of A-1155463 on platelets, which serve as a convenient biomarker for BCL-XL inhibition in vivo.8,9,11,12 Following a single 5 mg/kg IP dose of A-1155463 in nontumor bearing SCID-Beige mice, platelet counts fell dramatically as measured at 6 h postdose and then rebounded to normal levels within 72 h (Figure 5a). The kinetics of platelet depletion and recovery were similar to those previously reported with the dual inhibitor navitoclax.8 To provide additional evidence that A-1155463 was conferring on-target in vivo activity, we then administered this compound to SCID-Beige mice that had been inoculated with BCL-XL-dependent H146 tumor cells. Daily dosing at 5 mg/kg IP for 14 days caused a statistically significant inhibition of tumor growth (maximum tumor growth inhibition = 44%), which was alleviated upon cessation of dosing (Figure 5b).
Figure 5.
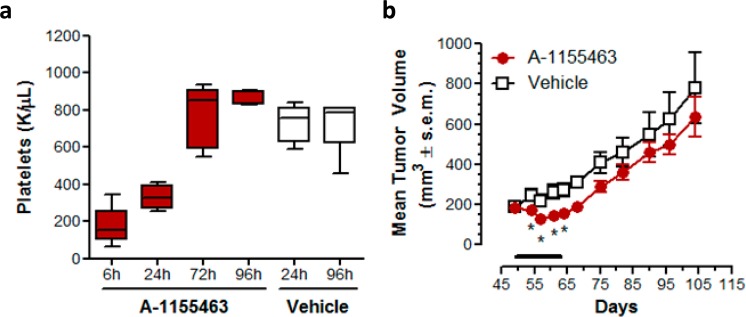
(a) Kinetics of platelet reduction and rebound following a single IP dose of A-1155463 in SCID-Beige mice. Nontumor bearing female SCID-Beige mice were treated with a single dose of A-1155463 (5 mg/kg, IP). Blood samples were collected by means of retro-orbital bleeds at regular time intervals. The number of platelets was plotted as a function of time. Each point reflects the average of 5 biological replicates. Each box indicates the median ± SEM. The error bars reflect minimum and maximum values (n = 5 per time point). The colored boxes represent upper and lower quartiles. The horizontal lines within each colored box represent the mean, and the whiskers are the upper and lower range. (b) Inhibition of H146 SCLC xenograft growth by A-1155463. Female SCID-Beige mice bearing NCI-H146 subcutaneous xenografts were treated with A-1155463 (5 mg/kg IP, QDx14) or an equal volume of the vehicle (5% DMSO, 10% EtOH, 20% Cremophor ELP, and 65% D5W; n = 5 per time point) and changes in tumor volume determined as a function of time. Each point reflects the average volume of 5 tumors. Points with significant (Student’s t test, p < 0.05) difference between treated and untreated groups are indicated by means of asterisks. Tumor volume was calculated by W2·L·0.5 where W and L are two perpendicular diameters indicating width and length, respectively.
In summary, we used iterative structure-based design to generate a highly potent and selective BCL-XL inhibitor that is devoid of the labile hydrazone linkage of the earlier tool compound WEHI-539. The ability of A-1155463 to exert in vivo on-target activity was demonstrated through a rapid and reversible reduction in platelets in SCID-Beige mice following a single IP dose. Additionally, administration of A-1155463 to tumor bearing SCID-Beige mice afforded modest but statistically significant tumor growth inhibition. Detailed mechanistic studies of A-1155463 and combination activity with relevant chemotherapy across multiple tumor types will be reported in an accompanying manuscript.
Acknowledgments
We thank Drs. Mike Michaelides, Andrew Judd, Kent Stewart, Michael Wendt, and Gerard Sullivan at AbbVie for critical reading of the manuscript and Dr. David Huang for helpful discussion during this work. We thank the beamline staff at the Australian Synchrotron where diffraction data for the BCL-XL-compound 10 complex were collected.
Glossary
ABBREVIATIONS
- BCL-2
B-cell lymphoma 2
- BCL-XL
B-cell lymphoma-extra large
- MCL-1
Myeloid cell leukemia sequence 1
Supporting Information Available
Fragment screening procedures, biological assay protocols, synthetic description and procedures, and analytic data including NMR, MS, and crystallographic methods. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Present Address
# (B.J.S.) La Trobe Institute for Molecular Science, La Trobe University, VIC 3086, Australia.
This work was sponsored by AbbVie, Inc., and Genentech, a member of the Roche group. Part of this work was supported by fellowships and grants from the Australian Research Council (fellowship to P.E.C.), the National Health and Medical Research Council (NHMRC, fellowships to P.M.C.; development grant 305536 and program grants 257502, 461221, and 1016701), the Leukemia and Lymphoma Society (specialized center of research grant nos. 7015 and 7413), the Cancer Council of Victoria (fellowship to P.M.C.; grant-in-aid 461239), and the Australian Cancer Research Foundation. Infrastructure support from the NHMRC Independent Research Institutes Infrastructure Support Scheme grant no. 361646 and a Victorian State Government OIS grant are gratefully acknowledged.
The authors declare the following competing financial interest(s): Z.-F.T., L.H., L.W., X.W., A.M.P., C.H.P., E.R.B., N.D.C., J.C., S.J., R.A.J., P.J.K., M.J.M., P.N., A.O., D.C.P., M.L.S., S.K.T., Y.X., J.X., H.Z., S.H.R., C.T., J.D.L., S.W.E., and A.J.S. are employees of AbbVie Inc.; K.D., W.J.F., J.A.F., S.G.H., M.F.T.K., C.O.N., H.E.P., and K.Z. are employees of Genentech Inc.; P.M.C., P.E.C., G.L., B.E.S., B.J.S., and K.G.W. are employees of The Walter and Eliza Hall Institute of Medical Research. AbbVie, Genentech, and WEHI participated in the study conduct, design, interpretation of data, review, and approval of the publication.
Supplementary Material
References
- Hanahan D.; Weinberg R. A. The hallmarks of cancer. Cell 2000, 100, 57–70. [DOI] [PubMed] [Google Scholar]
- Adams J. M.; Cory S. The BCL-2 apoptotic switch in cancer development and therapy. Oncogene 2007, 26, 1324–1337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Czabotar P. E.; Lessene G.; Strasser A.; Adams J. M. Control of apoptosis by the BCL-2 protein family: implications for physiology and therapy. Nat. Rev. Mol. Cell Biol. 2014, 15, 49–63. [DOI] [PubMed] [Google Scholar]
- Vaux D. L.; Cory S.; Adams J. M. BCL-2 gene promotes haemopoietic cell survival and cooperates with c-myc to immortalize pre-B cells. Nature 1988, 335, 440–442. [DOI] [PubMed] [Google Scholar]
- Amundson S. A.; Myers T. G.; Scudiero D.; Kitada S.; Reed J. C.; Fornace A. J. Jr. An informatics approach identifying markers of chemosensitivity in human cancer cell lines. Cancer Res. 2000, 60, 6101–6110. [PubMed] [Google Scholar]
- Oltersdorf T.; Elmore S. W.; Shoemaker A. R.; Armstrong R. C.; Augeri D. J.; Belli B. A.; Bruncko M.; Deckwerth T. L.; Dinges J.; Hayduk P. J.; Joseph M. K.; Kitada S.; Korsmeyer S. J.; Kunzer A. R.; Letai A.; Li C.; Mitten J. M.; Nettesheim D. G.; Ng S.; Nimmer P. M.; O’Connor J. M.; Oleksijew A.; Petros A. M.; Reed J. C.; Shen W.; Tahir S. K.; Thompson C. B.; Tomaselli K. J.; Wang B.; Wendt M. D.; Zhang H.; Fesik S. W.; Rosenberg S. H. An inhibitor of Bcl-2 family proteins induces regression of solid tumours. Nature 2005, 435, 677–681. [DOI] [PubMed] [Google Scholar]
- Wendt M. D.; Shen W.; Kunzer A.; McClellan W. J.; Bruncko M.; Oost T. K.; Ding H.; Joseph M. K.; Zhang H.; Nimmer P. M.; Ng S.-C.; Shoemaker A. R.; Petros A. M.; Oleksijew A.; Marsh K.; Bauch J.; Oltersdorf T.; Belli B. A.; Martineau D.; Fesik S. W.; Rosenberg S. H.; Elmore S. W. Discovery and structure-activity relationship of antagonists of B-cell lymphoma 2 family proteins with chemopotentiation activity in vitro and in vivo. J. Med. Chem. 2006, 49, 1165–1181. [DOI] [PubMed] [Google Scholar]
- Tse C.; Shoemaker A. R.; Adickes J.; Anderson M. G.; Chen J.; Jin S.; Johnson E. F.; Marsh K. C.; Mitten M. J.; Nimmer P.; Roberts L.; Tahir S. K.; Xiao Y.; Yang X.; Zhang H.; Fesik S.; Rosenberg S. H.; Elmore S. W. ABT-263: a potent and orally bioavailable BCL-2 family inhibitor. Cancer Res. 2008, 68, 3421–3428. [DOI] [PubMed] [Google Scholar]
- Wilson W. H.; O’Connor O. A.; Czuczman M. S.; LaCasce A. S.; Gerecitano J. F.; Leonard J. P.; Tulpule A.; Dunleavy K.; Xiong H.; Chiu Y. L.; Cui Y.; Busman T.; Elmore S. W.; Rosenberg S. H.; Krivoshik A. P.; Enschede S. H.; Humerickhouse R. Navitoclax, a targeted high-affinity inhibitor of BCL-2, in lymphoid malignancies: a phase 1 dose-escalation study of safety, pharmacokinetics, pharmacodynamics, and antitumour activity. Lancet Oncol. 2010, 11, 1149–1159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts A. W.; Seymour J. F.; Brown J. R.; Wierda W. G.; Kipps T. J.; Khaw S. L.; Carney D. A.; He S. Z.; Huang D. C. S.; Xiong H.; Cui Y.; Busman T. A.; McKeegan E. M.; Krivoshik A. P.; Enschede S. H.; Humerickhouse R. Substantial susceptibility of chronic lymphocytic leukemia to BCL2 inhibition: results of a phase I study of navitoclax in patients with relapsed or refractory disease. J. Clin. Oncol. 2012, 30, 488–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H.; Nimmer P. M.; Tahir S. K.; Chen J.; Fryer R. M.; Hahn K. R.; Iciek L. A.; Morgan S. J.; Nasarre M. C.; Nelson R.; Preusser L. C.; Reinhart G. A.; Smith M. L.; Rosenberg S. H.; Elmore S. W.; Tse C. BCL-2 family proteins are essential for platelet survival. Cell Death Differ. 2007, 14, 943–951. [DOI] [PubMed] [Google Scholar]
- Mason K. D.; Carpinelli M. R.; Fletcher J. I.; Collinge J. E.; Hilton A. A.; Ellis S.; Kelly P. N.; Ekert P. G.; Metcalf D.; Roberts A. W.; Huang D. C. S.; Kile B. T. Programmed anuclear cell death delimits platelet life span. Cell 2007, 128, 1173–1186. [DOI] [PubMed] [Google Scholar]
- Souers A. J.; Leverson J. D.; Boghaert E. R.; Ackler S. L.; Catron N. D.; Chen J.; Dayton B. D.; Ding H.; Enschede S. H.; Fairbrother W. J.; Huang D. C.; Hymowitz S. G.; Jin S.; Khaw S. L.; Kovar P. J.; Lam L. T.; Lee J.; Maecker H. L.; Marsh K. C.; Mason K. D.; Mitten M. J.; Nimmer P. M.; Oleksijew A.; Park C. H.; Park C. M.; Phillips D. C.; Roberts A. W.; Sampath D.; Seymour J. F.; Smith M. L.; Sullivan G. M.; Tahir S. K.; Tse C.; Wendt M. D.; Xiao Y.; Xue J. C.; Zhang H.; Humerickhouse R. A.; Rosenberg S. H.; Elmore S. W. ABT-199, a potent and selective Bcl-2 inhibitor, achieves antitumor activity while sparing platelets. Nat. Med. 2013, 19, 202–208. [DOI] [PubMed] [Google Scholar]
- Lin X.; Morgan-Lappe S.; Huang X.; Li L.; Zakula D. M.; Vernetti L. A.; Fesik S. W.; Shen Y. ‘Seed’ analysis of off-target siRNAs reveals an essential role of MCL-1 in resistance to the small-molecule BCL-2/BCL-XL inhibitor ABT-737. Oncogene 2007, 26, 3972–3979. [DOI] [PubMed] [Google Scholar]
- Wong M.; Tan N.; Zha J.; Peale F. V.; Yue P.; Fairbrother W. J.; Belmont L. D. Navitoclax (ABT-263) reduces Bcl-XL-mediated chemoresistance in ovarian cancer models. Mol. Cancer Ther. 2012, 11, 1026–1035. [DOI] [PubMed] [Google Scholar]
- Lessene G.; Czabotar P. E.; Sleebs B. E.; Zobel K.; Lowes K. N.; Adams J. M.; Baell J. B.; Colman P. E.; Deshayes K.; Fairbrother W. J.; Flygare J. A.; Gibbons P.; Kersten W. J. A; Kulasegaram S.; Moss R. M.; Parisot J. P.; Smith B. J.; Street I. S.; Yang H.; Huang D. C. S.; Watson K. G. Structure-guided design of a selective BCL-XL inhibitor. Nat. Chem. Biol. 2013, 9, 390–397. [DOI] [PubMed] [Google Scholar]
- Sleebs B. E.; Kersten W. J. A.; Kulasegaram S.; Nikolakopoulos G.; Hatzis E.; Moss R. M.; Parisot J. P.; Yang H.; Czabotar P. E.; Fairlie W. D.; Lee E. F.; Adams J. M.; Chen L.; van Delft M. F.; Lowes K. N.; Wei A.; Huang D. C. S.; Colman P. M.; Street I. P.; Baell J. B.; Watson K.; Lessene G. Discovery of potent and selective benzothiazole hydrazone inhibitors of Bcl-XL. J. Med. Chem. 2013, 56, 5514–5540. [DOI] [PubMed] [Google Scholar]
- Koehler M. F. T.; Bergeron P.; Choo E. F.; Lau K.; Ndubaku C.; Dudley D.; Gibbons P.; Sleebs B. E.; Rye C. S.; Nikolakopoulos G.; Bui C.; Kulasegaram S.; Kersten W. J. A.; Smith B. J.; Czabotar P. E.; Colman P. M.; Huang D. C. S.; Baell J. B.; Watson K. G.; Hasvold L.; Tao Z.-F.; Wang L.; Souers A. J.; Elmore S. W.; Flygare J. A.; Fairbrother W. J.; Lessene G. Structure-guided rescaffolding of selective antagonists of BCL-XL. ACS Med. Chem. Lett. 2014, 5, 662–667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shuker S. B.; Hajduk P. J.; Meadows R. P.; Fesik S. W. Discovering high-affinity ligands for proteins: SAR by NMR. Science 1996, 274, 1531–1534. [DOI] [PubMed] [Google Scholar]
- Müller K.; Faeh C.; Diederich F. Fluorine in pharmaceuticals: looking beyond intuition. Science 2007, 317, 1881–1886. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.