

Abstract
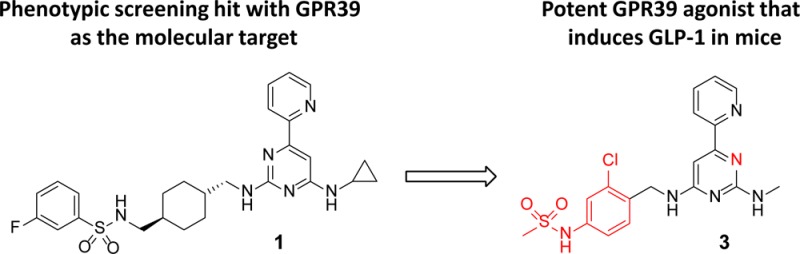
The identification of highly potent and orally bioavailable GPR39 agonists is reported. Compound 1, found in a phenotypic screening campaign, was transformed into compound 2 with good activity on both the rat and human GPR39 receptor. This compound was further optimized to improve ligand efficiency and pharmacokinetic properties to yield GPR39 agonists for the potential oral treatment of type 2 diabetes. Thus, compound 3 is the first potent GPR39 agonist (EC50s ≤ 1 nM for human and rat receptor) that is orally bioavailable in mice and robustly induced acute GLP-1 levels.
Keywords: GPR39 agonists, GPR39 receptor, Zn2+-sensing receptor, antidiabetic treatment, β-cell protection, GLP-1 secretion, insulin secretion
GPR39 is a putative zinc-sensing G protein-coupled receptor (GPCR) related to the ghrelin/neurotensin peptide receptor subfamily.1,2 The functional full-length receptor is widely expressed in peripheral, endocrine, and metabolic organs such as the stomach, gastrointestinal (GI) tract, pancreas, liver, kidney and the white adipose tissue.3 Zn2+ is the only known stimulator of GPR39 activity4,5 via the Gαq, Gα12/13, and Gαs pathways with an EC50 value in the micromolar range. GPR39 signaling is implicated in cellular processes such as insulin secretion, protection from cell death,6 gastric emptying,7 and epithelial repair.8 Initial reports about obestatin as the natural ligand for this receptor have not been verified.4 Information relating to the pancreatic function of GPR39 has come from multiple mouse knockout studies.9−11 Deletion of the GPR39 gene is associated with impaired insulin secretion and glucose homeostasis. These knockout studies suggested that GPR39 may play a role in the development of type 2 diabetes, and therefore, activating the receptor could exert a β-cell protective effect. This hypothesis was tested with a transgenic mouse strain with inducible overexpression of human GPR39 selectively in pancreatic β-cells.12 Because of the high degree of constitutive activity, increased expression leads to increased receptor signaling mimicking the action of an endogenous agonistic ligand. This study by Egerod et al. showed that GPR39 overexpression in the β-cells by itself impaired glucose tolerance slightly and decreased β-cell mass, but it completely protected against gradual hyperglycemia in streptozotocin-treated animals. This suggests that GPR39 functions in a β-cell protective manner and could be a target for antidiabetic drug treatment with long-term benefits. GPR39 agonists would help to test this hypothesis pharmacologically. The first GPR39 agonists have been recently identified by screening of GPCR-focused compound libraries.13 However, the reported chemical probes are moderately potent, and their properties are not suitable for in vivo studies.
Herein, we report the first highly potent and orally bioavailable GPR39 agonists. Compound 1 was discovered through a reporter gene assay aimed at identifying inhibitors of Hedgehog signaling. The compound was employed in a chemogenomics approach to identify GPR39 as its molecular target.14 Compound 1 had good activity via the Gαq pathway in an assay that measured inositol-1-phosphate (IP1) in HEK293 cells overexpressing rat or mouse GPR39; however, it showed moderate cross-reactivity with the human receptor. Whereas the sequence identity between rat and mouse receptor is very high (93.6%), the overall homology for human GPR39 with mouse (80.5%) and rat (79.6%) is lower.15 Additionally, 1 suffered from high metabolic clearance (Cl(h) = 51.2 mL min–1 kg–1) in rat liver microsomes and low aqueous solubility (<5 μM at pH 6.8). Our objective was to optimize the potency of these compounds against human and rodent receptors and improve their pharmacokinetic properties in order to validate the target in vivo and ultimately to utilize them as therapeutic agents.
Compound 2, a pyrimidine with a regioisomeric presentation of the central pyrimidine ring compared to 1, resulted in improved activity on all three receptors and a much smaller shift in potency between the human and rodent receptors. This finding prompted us to investigate more thoroughly the role of the biaryl moiety (boxed area of compound 2, Figure 1) for potency on human and rodent receptors. None of the investigated biaryl compounds including regioisomeric pyridylpyrimidines matched the potency of compound 2 and further work focused on the 4-(pyridin-2-yl)pyrimidines described below.
Figure 1.

Discovery of 2-pyridylpyrimidines as GPR39 agonists.
Although compound 2 had very good potency, the pharmacokinetic properties (clearance of 42 mL min–1 kg–1 in Sprague–Dawley rats after a single iv dose of 2 mg/kg in a formulation of 10% NMP, 30% PEG200, and 5% Solutol in water and a bioavailability of 25% after a oral dose of 5 mg/kg of the above formulation) were not favorable for in vivo studies. As this molecule has a molecular weight of 497 and a clogP of 5.6 we focused on “deconstructing” this lead with the goal of identifying the minimal pharmacophore and increasing the apparent ligand efficiency (LE).16 In a second step we planned to build upon this and further optimize relevant drug properties. Compound 4 with a low molecular weight of 255 was found to be the most ligand efficient molecule with a LE = 0.49 for the human GPR39 receptor compared to compound 2 with a LE = 0.3. “Reconstruction” with the goal to maintain the high apparent ligand efficiency involved two small compound libraries, one with modifications to the cyclopropylmethylamino substituent R1 and one with variations of the methylamino substituent R2 (Table 1). As demonstrated by representative compounds 5–9, changes to the methylamino group (R2) resulted in partial or significant loss of activity. By contrast, modifications to substituent R1 leaving the methylamino substituent R2 unchanged resulted mostly in improvements in EC50 values: Whereas a neopentyl group (compound 10) increased potency slightly (EC50 = 73 nM), this improvement was more distinct for the compounds containing the cyclohexylmethyl (11) and para-chlorobenzyl (12) substituent at this position (EC50s = 11 and 15 nM, respectively). The 4-pyridyl group in compound 13, instead of a phenyl moiety, resulted in reduced activity (EC50 = 87 nM). Direct attachment of an aryl substituent, exemplified by compound 14, led to a significant drop in activity into the micromolar range. Exploration of the regioisomeric positions on the phenyl ring of R1 resulted in a >10-fold increase in potency for compound 15, which exhibits an additional 2-chloro substituent in comparison to compound 12. Remarkably, compound 15 combines the high LE of our starting compound 4 (LE = 0.49 and 0.50 for compounds 4 and 15, respectively) with subnanomolar potency. However, as observed for compounds 1 and 2, the rapid clearance of compound 15 in rat liver microsomes (RLM) did not predict sufficient exposure upon oral dosing in rats. Prediction of metabolic sites for compound 15 indicated the methylamino substituent and the benzylic position as preferred sites for oxidative metabolism.17 None of the attempts to block these metabolic weak spots by replacing the methylamino group with a trifluoroethylamino group (compound 16), methyl group (compound 17), or blocking the benzylic position with a cyclobutyl group (compound 18), provided the desired outcome as these compounds continued to have high in vitro clearance in RLMs. Comparison of various molecular descriptors such as clogP, clogD, and polar surface area (PSA) against in vitro clearance revealed an interesting correlation useful for further optimization of metabolic stability: Compounds 15 to 20 with lower polar surface area (PSA ≤ 72 Å2) had high clearance, but compound 20, with a PSA of 96.9 Å2, showed a reduced clearance in RLM (Cl(h) = 34.1 mL min–1 kg–1). A pharmacokinetic study in Sprague–Dawley rats confirmed the improved clearance in vivo (18 mL min–1 kg–1 after a single iv dose of 1 mg/kg, F = 45% after a 3 mg/kg oral dose) and supporting further use of rat liver microsome stability for optimization. We utilized this hypothesis to synthesize a number of compounds (21 to 23) with PSAs of ≥91.8 Å2. Gratifyingly, we observed improved in vitro clearance in all three compounds without compromising their ability to cross cell membranes. For example, compound 21 demonstrated high permeability in Caco-2 cells (Papp A–B = 6.9 × 10–6 cm s–1) and the parallel artificial membrane permeability assay (calculated fraction absorbed = 89%). To further improve potency, we made use of an earlier observation that substituents in 2-position on the phenyl ring in R1 increased potency as seen by the comparison between compounds 12 and 15. Compounds 24 and 3 show increased potency while retaining their in vitro metabolic stability.
Table 1. SAR of 2-Pyridylpyrimidine Analogues.
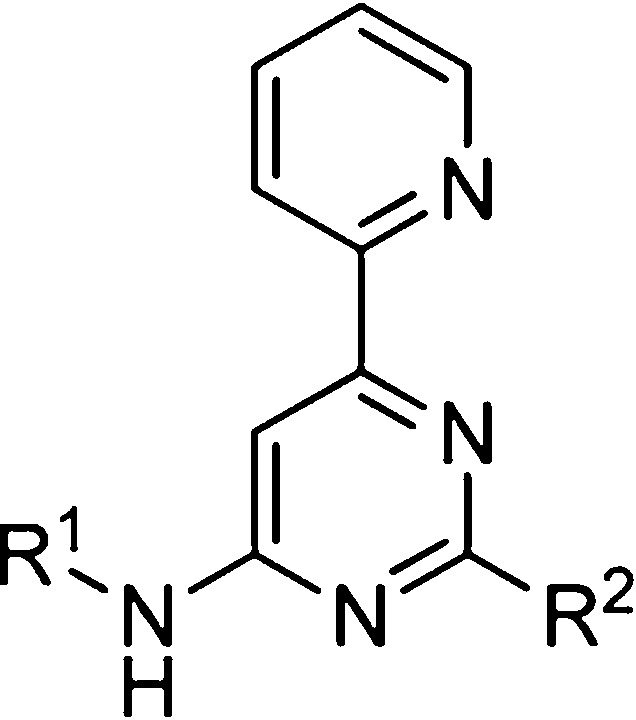
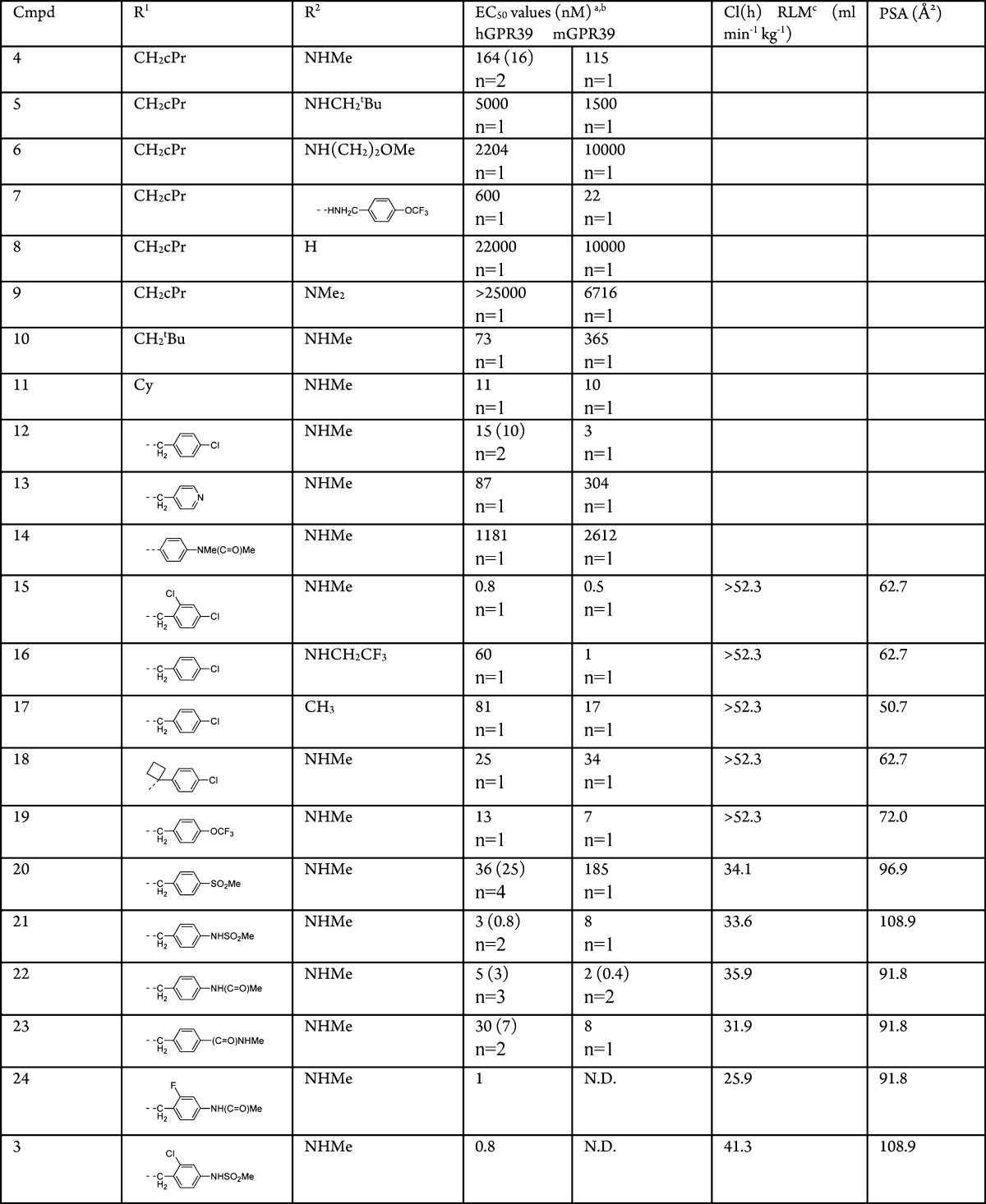
Values derived from a 11-point dose–response curve performed in triplicate. The number of replicates range from 1 to 4 as indicated, with standard deviation reported in parentheses where available.
HEK293 cells expressing human or mouse GPR39.
Predicted hepatic clearance based on intrinsic clearance in rat liver microsomes. Scaling factors for Cl(int) → Cl(h): 45 mg of microsomal protein/g of liver; 40 g of liver/kg of body weight; and 55 mL of portal blood flow/min/kg. N.D. = not determined.
Some of the most potent compounds, with Zn2+ for comparison, were evaluated in additional cell assays to further assess their potency. GPR39 is known to signal through Gαs in addition to Gαq.4 Thus, we tested compounds in a HEK293 cell line overexpressing the human GPR39 receptor and used forskolin as a reference compound to measure the maximal cAMP release. All compounds were active in Gαs signaling, with compound 3 as the most active both in terms of EC50 value and maximal efficacy. Both the IP1 assay and the cAMP assay relied on recombinant cell lines. In order to more accurately assess the functional activity we employed a mouse GLP-1 secreting enteroendocrine cell line (STC-1) that expresses endogenous GPR39.18 Additionally, we used a rat insulinoma cell line (INS-1E)19 that has a beta cell phenotype and tested for protection against cytokine-induced cell death. All four compounds showed activity in both assays with a rank-order as seen in the cAMP assay (Table 2). Compound 22 had an unexpectedly high EC50 value in the STC-1 assay, yet achieved the highest maximal GLP-1 secretion of all four compounds, suggesting that it might be working through an additional target on these cells. Compound 3 was the most potent compound across the assays. Rat and mouse plasma protein binding for 3 was measured as 99.3% and 99.1%, respectively. Considering off-targets, 2,4-diamino-6-(pyridin-2-yl) pyrimidines have been described as kinase inhibitors,20 but 3 and other compounds described in this letter did not show inhibitory effects (IC50s > 10 μM) on a panel of kinases nor did they exhibit relevant binding affinity for the related ghrelin and neurotensin-1 receptors (IC50s > 30 μM) and other enzymes, transporters, and GPCRs (data given in Supporting Information). On the basis of mutagenesis studies, Zn2+ has been proposed to act as a GPR39 agonist by binding to His-17 and His-19 located in the extracellular domain.21 Mutagenesis studies and pharmacological characterization of the mutants with the 2-pyridylpyrimidines described herein are not yet available. However, studies with sub-EC50 concentrations of Zn2+ in the presence of a potent 2-pyridylpyrimidine GPR39 agonist resulted in generation of IP1 greater than the amount produced by either substance alone (data given in Supporting Information). This suggests that our compounds and Zn2+ do not compete for binding to the receptor, which is most likely due to binding to separate sites resulting in a receptor response. More detailed data to this point will be reported in due course.
Table 2. In Vitro Potencies of Selected Compounds in Various Cell Assaysa.
| compd | EC50, nM rGPR39b | EC50, μM hcAMPc [% efficacy] | EC50, μM GLP1/STCd [fold max] | EC50, μM protection/INS-1Ee |
|---|---|---|---|---|
| ZnCl2 | 4038 | 3.6 [48%] | N.D. | N.D. |
| 21 | 3 (1.5) n = 3 | 0.18 (0.05) n = 3 [55%] | 1.3 (0.37) n = 16 [3×] | 0.4 (0.13) n = 3 |
| 22 | 2 (0.5) n = 2 | 0.55 (0.08) n = 3 [48%] | 4.4 (0.32) n = 2 [5.5×] |
1.2 n = 1 |
| 24 | 0.6 n = 1 |
0.35 (0.07) n = 2 [55%] | 0.2 n = 1 [3.5×] |
0.04 n = 1 |
| 3 | 0.4 n = 1 |
0.06 (0.03) n = 3 [64%] |
0.06 (0.03) n = 2 [3.5×] |
0.02 n = 1 |
Values derive from a dose–response curve performed in triplicate. The number of replicates range from 1 to 16 as indicated, with standard deviation reported in parentheses where available.
HEK293 cells expressing rat GPR39.
HEK293 cells expressing human GPR39. The percent efficacy is reported relative to the cAMP response with 25 μM forskolin.
Mouse enteroendocrine STC-1 cells secreting GLP-1. The maximal activity of the test sample was expressed as fold increase of GLP-1 compared with the DMSO control.
Caspase 3/7 activity was measured in cytokine-treated rat insulinoma INS-1E cells. N.D. = not determined.
Compound 3 was prepared as described in Scheme 1. Mesylation of 4-amino-2-chlorobenzonitrile (25) in pyridine provided 26, which was reduced to the benzylamine 27(22) by reaction with lithium aluminum hydride in 54% yield over the 2 steps. Reaction of amine 27 with 2,4-dichloro-6-(pyridin-2-yl)pyrimidine23 yielded compound 28 as the major regioisomer, which after displacement of the second chlorine with methylamine furnished compound 3 in 61% yield for the two displacement reactions.
Scheme 1. Synthesis of 3.

Reagents and conditions: (a) MsCl, pyridine, 0 °C, 80% yield; (b) LiAlH4, THF, 0 °C, 68% yield; (c) 2,4-dichloro-6-(pyridin-2-yl)pyrimidine, MeCN, triethylamine 60 °C, chromatographic separation on silica gel, 70% yield; (d) 15% MeNH2 in EtOH, 130 °C, microwave heating, 87% yield.
Since compound 3 exhibited promising in vitro properties, the pharmacokinetic properties were evaluated in male C57BL/6 mice. Upon single oral doses of 10, 30, and 100 mg/kg of aqueous suspensions in 0.5% methylcellulose/0.1% Tween 80, compound 3 achieved, between 1 and 1.5 h, maximal exposures of 1.4, 6.1, and 25.3 μM, respectively, indicating slightly overproportional exposures. A half-life of 7 h for compound 3 in mice was calculated from a separate study with 10 mg/kg. The potency of compound 3 and the less active analogue 21 were assessed in mice by measuring a pharmacodynamic marker, GLP-1 levels (Figure 2). Consistent with the EC50s observed in vitro, compound 3 induced very high levels of active GLP-1 (6-fold), whereas the less potent compound 21 modestly induced (<2-fold) active GLP-1 levels at comparable plasma exposure for both compounds (1.6 and 2.0 μM, respectively).
Figure 2.

Induction of a PD Marker, active GLP-1, for compounds 21 and 3.
Male C57BL/6 mice were concurrently dosed with DPP-IV inhibitor (S)-1-(2-(adamantan-2-ylamino)acetyl)pyrrolidine-2-carbonitrile24 and the indicated compound. Animals were challenged after 1 h with a glucose bolus (3 g/kg, po), and active GLP-1 levels were measured 30 min later. Data were statistically evaluated with one-way ANOVA followed by Dunnett’s post-test. P < 0.001 versus the vehicle group.
The current study demonstrates that a low molecular weight agonist with drug-like properties can be found for the GPR39 receptor. We identified the first GPR39 agonists with high potency for the human and rodent receptor and excellent functional activity in physiologically relevant rodent cells and in vivo. An acute study in normal mice with orally administrated GPR39 agonists confirmed in vitro findings by demonstrating an increase of the relevant pharmacodynamic marker GLP-1.
Acknowledgments
We thank the whole GPR39 team for stimulating scientific discussions and in particular Heather Burks, Robert Damon, MooJe Sung, and Scott Tria for valuable contributions to the medicinal chemistry strategy as well as Lawrence Hamann for the strong support of the program. We thank Fred Bassilana for providing HEK293 cells expressing human GPR39 and Martin Marro for discussions in regards to in vitro assays.
Glossary
ABBREVIATIONS
- HEK
human embryonic kidney
- EC50
half maximal effective concentration
- IP1
inositol-1-phosphate
- NMP
N-methyl-2-pyrrolidone
- PEG
polyethylene glycol
- DMSO
dimethyl sulfoxide
- LE
ligand efficiency, calculated from −RT ln(EC50)/number of heavy atoms in the molecule
- RLM
rat liver microsome
- Cl(h)
hepatic clearance
- F
bioavailability
- PSA
polar surface area
- cAMP
cyclic adenosine monophosphate
- GLP-1
glucagon-like peptide-1
- PD
pharmacodynamic
- DPP-IV
dipeptidyl peptidase IV
Supporting Information Available
Experimental procedures and characterization data for compounds 1–24, additional biological data, PK and PD study information, and experimental details for the in vitro assays. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Supplementary Material
References
- McKee K. K.; Tan C. P.; Palyha O. C.; Liu J.; Feighner S. D.; Hreniuk D. L.; Smith R. G.; Howard A. D.; Van der Ploeg L. H. T. Cloning and characterization of two human G protein coupled receptor genes (GPR38 and GPR39) related to the growth hormone secretagogue and neurotensin receptors. Genomics 1997, 46, 426–434. [DOI] [PubMed] [Google Scholar]
- Review:Popovics P.; Stewart A. J. GPR39: a Zn2+-activated G protein-coupled receptor that regulates pancreatic, gastrointestinal and neuronal functions. Cell. Mol. Life Sci. 2011, 68, 85–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egerod K. L.; Holst B.; Petersen P. S.; Hansen J. B.; Mulder J.; Hökfelt T.; Schwartz T. W. GPR39 splice variants versus antisense gene LYPD1: expression and regulation of gastrointestinal tract, endocrine pancreas, liver and white adipose tissue. Mol. Endocrinol. 2007, 21, 1685–1698. [DOI] [PubMed] [Google Scholar]
- Holst B.; Egerod K. L.; Schild E.; Vickers S. P.; Cheetham S.; Gerlach L.-O.; Storjohann L.; Stidsen C. E.; Jones R.; Beck-Sickinger A. G.; Schwartz T. W. GPR39 signaling is stimulated by zinc ions but not by obestatin. Endocrinology 2007, 148, 13–20. [DOI] [PubMed] [Google Scholar]
- Yasuda S.-I.; Miyazaki T.; Munechika K.; Yamashita M.; Ikeda Y.; Kamizono A. Isolation of Zn2+ as an endogenous agonist of GPR39 from fetal bovine serum. J. Recept. Signal Transduction Res. 2007, 27, 235–246. [DOI] [PubMed] [Google Scholar]
- Dittmer S.; Sahin M.; Pantlen A.; et al. The constitutively active orphan G-protein-coupled receptor GPR39 protects from cell death by increasing secretion of pigment epithelium-derived growth factor. J. Biol. Chem. 2008, 283, 7074–7081. [DOI] [PubMed] [Google Scholar]
- Moechars D.; Depoortere I.; Moreaux B.; de Smet B.; Goris I.; Hoskens L.; Daneels G.; Kass S.; Ver Donck L.; Peeters T.; Coulie B. Altered gastroinstestinal and metabolic function in the GPR39-obestatin receptor-knockout mouse. Gastroenterology 2006, 131, 1131–1141. [DOI] [PubMed] [Google Scholar]
- Sharir H.; Zinger A.; Nevo A.; Sekler I.; Hershfinkel M. Zn2+ released from injured cells via the Zn2+-sensing receptor, ZnR, to trigger signaling leading to epithelial repair. J. Biol. Chem. 2010, 285, 26097–26106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holst B.; Egerod K. L.; Jin C.; Petersen P. S.; Østergaard V.; Hald J.; Sprinkel A. M. E.; Størling J.; Mandrup-Poulsen T.; Holst J. J.; Thams P.; Ørskov C.; Wierup N.; Sundler F.; Madsen O. D.; Schwartz T. W. G protein-coupled receptor 39 deficiency is associated with pancreatic islet dysfunction. Endocrinology 2009, 150, 2577–2585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tremblay F.; Richard A. M.; Will S.; Syed J.; Stedman N.; Perreault M.; Gimeno R. E. Disruption of G protein-coupled receptor 39 impairs insulin secretion in vivo. Endocrinology 2009, 150, 2586–2595. [DOI] [PubMed] [Google Scholar]
- Verhulst P. J.; Lintermans A.; Janssen S.; Loeckx D.; Himmelreich U.; Buyse J.; Tack J.; Depoortere I. GPR39, a receptor of the ghrelin receptor family, plays a role in the regulation of glucose homeostasis in a mouse model of early onset diet-induced obesity. J. Neuroendocrinol. 2011, 23, 490–500. [DOI] [PubMed] [Google Scholar]
- Egerod K. L.; Jin C.; Petersen P. S.; Wierup N.; Sundler F.; Holst B.; Schwartz T. W. β-cell specific overexpression of GPR39 protects against streptozotocin-induced hyperglycemia. Int. J. Endocrinol. 2011, 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boehm M.; Hepworth D.; Loria P. L.; Norquay L. D.; Filipski K. J.; Chin J. E.; Cameron K. O.; Brenner M.; Bonnette P.; Cabral S.; Conn E.; Ebner D. C.; Gautreau D.; Hadcock J.; Lee E. C. Y.; Mathiowetz A. M.; Morin M.; Rogers L.; Smith A.; VanVolkenburg M.; Carpino P. A. Chemical probe identification platform for orphan GPCRs using focused compound screening: GPR39 as a case example. ACS Med. Chem. Lett. 2013, 4, 1079–1084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bassilana F.; Carlson A.; DaSilva J. A.; Grosshans B.; Vidal S.; Beck V.; Wilmeringwetter B.; Llamas L. A.; Showalter T. B.; Rigollier P.; Bourret A.; Ramamurthy A.; Wu X.; Harbinski F.; Plonsky S.; Lee L.; Ruffner H.; Grandi P.; Schirle M.; Jenkins J.; Sailer A.; Bouwmeester T.; Porter J. A.; Meyer V.; Finan P. M.; Tallarico J. A.; Kelleher J. F. III; Seuwen K.; Jain R. K.; Luchansky S. J. Target identification for a hedgehog pathway inhibitor reveals a new role for the orphan receptor GPR39. Nature Chem. Biol. 2014, 105343–349. [DOI] [PubMed] [Google Scholar]
- Sequence homology was calculated in UniProt (www.uniprot.org) using the Clustal W algorithm and entries O43194, Q5U431, and E9PTT1 for human, mouse, and rat GPR39 protein, respectively.
- Hopkins A. L.; Groom C. R.; Alex A. Ligand-efficiency: a useful metric for lead selection. Drug Discovery Today 2004, 9, 430–431. [DOI] [PubMed] [Google Scholar]
- Cytochrom P450 mediated metabolism has been predicted using Metasite from Molecular Discovery. See alsoCruciani G.; Carosati E.; De Boeck B.; Ethirajulu K.; Mackie C.; Howe T.; Vianello R. MetaSite: Understanding metabolism in human cytochromes from the perspective of the chemist. J. Med. Chem. 2005, 48, 6970–6979. [DOI] [PubMed] [Google Scholar]
- Synthesis and secretion of intestinal proglucagon-derived peptides by the STC-1 enteroendocrine cell line.Brubaker P. L.; Izzo A.; Rocca A. S. Can. J. Diabetes 2003, 27, 141–148. [Google Scholar]
- Merglen A.; Theander S.; Rubi B.; Chaffard G.; Wollheim C. B.; Maechler P. Glucose sensitivity and metabolism-secretion coupling studied during two-year continuous culture in INS-1E insulinoma cells. Endocrinology 2004, 145, 667–678. [DOI] [PubMed] [Google Scholar]
- Nagarathnam D.; Dumas J.; Hatoum-Mokdad H.; Boyer S.; Pluempe H.. Preparation of pyrimidine derivatives for use in pharmaceutical compositions as Rho-kinase inhibitors. PCT patent application WO2003062227, 2003.
- Storjohann L.; Holst B.; Schwartz T. W. Molecular mechanism of Zn2+ agonism in the extracellular domain of GPR39. FEBS Lett. 2008, 582, 2583–2588. [DOI] [PubMed] [Google Scholar]
- Ding Y.; Longdregan A. T.; Marino J. P. Jr.. Preparation of 2-amino-1,3,5-triazine derivatives as soluble epoxide hydrolase (sEH) inhibitors and their use. PCT patent application WO2009049154, 2009.
- Harden D. B.; Mokrosz M. J.; Strekowski L. Addition and substitution reactions of chloropyrimidines with lithium reagents. J. Org. Chem. 1988, 53, 4137–4140. [Google Scholar]
- Akarte A. S.; Srinivasan B. P.; Gandhi S. A novel long acting DPP-IV inhibitor PKF-275-055 stimulates β-cell proliferation resulting in improved glucose homeostasis in diabetic rats. Biochem. Pharmacol. 2012, 83, 241–252. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.