

Abstract
In Alzheimer’s disease, cytochrome c-dependent apoptosis is a crucial pathway in neuronal cell death. Although beta-amyloid (Aβ) oligomers are known to be the neurotoxins responsible for neuronal cell death, the underlying mechanisms remain largely elusive. Here, we report that the oligomeric form of synthetic Aβ of 42 amino acids elicits death of HT-22 cells. But, when expression of a bcl-2 family protein BAK is suppressed by siRNA, Aβ oligomer-induced cell death was reduced. Furthermore, significant reduction of cytochrome c release was observed with mitochondria isolated from BAK siRNA-treated HT-22 cells. Our in vitro experiments demonstrate that Aβ oligomers bind to BAK on the membrane and induce apoptotic BAK pores and cytochrome c release. Thus, the results suggest that Aβ oligomers function as apoptotic ligands and hijack the intrinsic apoptotic pathway to cause unintended neuronal cell death.
Introduction
Alzheimer’s disease (AD), a progressive neurodegenerative disorder, may be identified by the accumulation of senile plaques, neurofibrillary tangles, and substantial loss of brain mass (1,2). Mounting evidence clearly indicates that intracellular beta-amyloid (Aβ) oligomers are the neurotoxins responsible for neuronal cell death in AD (3–7). Neuronal cell death, commonly found among AD patients and pathologically significant due to its irreversibility, may be a result of apoptosis, specifically the release of cytochrome c from the mitochondria (8,9). Cytochrome c release, which is a pivotal step in apoptosis, triggers a cascade of caspase activation that leads to cell death. Under normal apoptotic conditions, cytochrome c is thought to be released through apoptotic pores in the mitochondrial outer membrane (MOM), which are induced by the activation of BAK or BAX via caspase-8-cleaved BID (tBID, p15 BID) (10–12).
The notion that Aβ oligomers induce neuronal cell death via apoptosis in AD is based on apoptotic markers found in postmortem AD brain such as DNA fragmentations, increased bcl-2 family protein expression, increased caspase activities (13,14), and cell-level studies that show elevated levels of cytochrome c found in the cytosol (12,15). Moreover, some studies demonstrate that Aβ oligomers elicit cytochrome c release from isolated mitochondria (16,17). It has been reported that amyloid precursor protein and Aβ localize to the mitochondria and lead to mitochondrial dysfunction by interacting with mitochondrial proteins, by blocking mitochondrial import channels, by disrupting electron-transport chain, or by increasing reactive-oxygen species products (18,19). However, these mitochondrial dysfunctions do not satisfactorily explain the Aβ-induced cytochrome c release.
In this work, we investigate the direct involvement of a bcl-2 family protein BAK in Aβ-induced cytochrome c release and neuronal cell death. In cells, suppression of BAK expression with siRNA resulted in reduced cell death by Aβ oligomers. Isolated mitochondria from siRNA-treated cells showed decreased release of cytochrome c by Aβ oligomers, together linking BAK to Aβ oligomer-induced cytochrome c release and cell death. Further on, in a well-defined in vitro setting, we unambiguously show that Aβ oligomers interact with BAK and trigger the release of cytochrome c from MOM-like vesicles. The results raise the possibility that Aβ oligomers hijack the cytochrome c-dependent apoptotic pathway by interacting with BAK even in the absence of the death signal, leading to cell death.
Materials and Methods
Preparation of Aβ oligomers
The synthetic peptide wild-type Aβ (1–42) was purchased from American Peptide (Sunnyvale, CA). As described in Dahlgren et al. (20) and Ahmed et al. (21), the Aβ (1–42) peptide was initially dissolved to 1 mM in hexafluoroisopropanol (Sigma-Aldrich, St. Louis, MO) and separated into aliquots in sterile microcentrifuge tubes. Hexafluoroisopropanol was removed under vacuum in a SpeedVac (Thermo Fisher Scientific, Rockford, lL) and the peptide film was stored desiccated at −80°C. To make oligomers, cold phosphate-buffered saline was added to bring the peptide to a final concentration of 10 μM and incubated at 4°C for 18 h (see Fig. S1 in the Supporting Material).
Expression and purification of recombinant proteins
The mouse BAK protein containing amino acids 16–184 (BAKΔC) was prepared with a C-terminal histidine (6×) tag using pPosKJ-sBAK-cHis plasmid as described in Oh et al. (22). N-terminal His6-tagged Bid (p22 BID) was purified, and p7/p15 BID or cBID, an activated form of BID, was prepared from the p22BID protein after cleavage with caspase-8, as described in Oh et al. (23). The fragment p22BID forms a tight complex as p7/p15 BID (cBID). All protein preparations were stored in 18% glycerol, 20 mM Tris buffer (pH 7.4), and 150 mM KCl at −80°C. The protein concentration was determined using the bicinchoninic acid protein assay kit (Thermo Fisher Scientific), and purified proteins were examined with 15% sodium dodecyl-sulfate polyacrylamide gel electrophoresis (SDS-PAGE), and the purity was at least 95% for all proteins (see Fig. S3 B).
Cell culture and gene silencing
HT-22 cells were cultured in Dulbecco’s modified Eagle’s medium (Invitrogen, Carlsbad, CA) supplemented with 10% heat-inactivated fetal bovine serum, 1% penicillin/streptomycin at 37°C in a 5% CO2 incubator. After 24 h incubation, BAK siRNA transfection was performed with an siRNA transfection reagent (Santa Cruz Biotechnology, Dallas, TX) according to the manufacturer’s instructions. Briefly, transfection reagent and BAK siRNA or nonfunctional control siRNA were dissolved separately in a dilution buffer and the respective transfection mixtures were added to the antibiotic and serum-free cell culture medium to a final concentration of 20 nM siRNA. Six hours after transfections, the 2× medium (DMEM with 20% FBS and 2% antibiotics) was added to the siRNA-treated HT22 cells, and the cells were further incubated for 18 h.
Mitochondria isolation and cytochrome c release assay
Mitochondria were isolated using a Mitochondrial Isolation Kit for Cultured Cells (Thermo Fisher Scientific) according to the manufacturer’s instructions. Isolated mitochondria were incubated with the indicated concentrations of cBID and Aβ oligomers for 45 min at room temperature in mitochondria buffer (150 mM KCl, 5 mM MgCl2, 1 mM EGTA, 25 mM HEPES, 1 mM DTT pH 7.5) under constant stirring using a Thermomixer (Vfinal = 100 μL; Eppendorf, Hamburg, Germany). Reaction mixtures were centrifuged at 14,000 × g for 15 min, with cytochrome c quantitated using a colorimetric ELISA (Invitrogen) according to the manufacturer’s protocol. Briefly, supernatant fraction was added to the well of a microtiter plate precoated with monoclonal anti-cytochrome c antibody, followed by adding 100 μL of cytochrome c biotin conjugate solution for 1 h at room temperature (RT). Then, 100 μL of streptavidin-HRP working solution was added for 30 min at RT. After adding 100 μL of stabilized chromogen, equal volume of stop solution was also added and the product was measured on a spectrophotometer at 450 nm.
Immunoblotting
For Western blot analysis, HT-22 cells were lysed with RIPA buffer (Cell Signaling Technology, Danvers, MA) with Protease Inhibitor Cocktail (Calbiochem, Darmstadt, Germany). After centrifugation at 15,000 × g for 15 min at 4°C, total protein amounts were determined with the BCA Protein Assay kit (Bio-Rad, Hercules, CA). Equal amounts of protein lysates were separated by SDS-PAGE electrophoresis, and transferred to nitrocellulose membrane. The blot was probed with an anti-BAK rabbit polyclonal antibody (G-23) at 4°C overnight. Membranes were then exposed to the HRP-conjugated rabbit anti-goat secondary antibody (Sigma-Aldrich) followed by a chemiluminescence detection of antibody binding (Thermo Fisher Scientific). Equal protein loading was controlled by reprobing the membrane with a monoclonal anti-β-actin antibody (Sigma-Aldrich).
Cofloatation assay
To measure the binding properties of Aβ oligomers to proteoliposomes (liposomes reconstituted with BAKΔC only or BAKΔC/cBID or cBID only), each vesicle (100 μM) was incubated with Aβ oligomers for 30 min at room temperature, then air-fused at high speed (Airfuse; Beckman Instruments, Brea, CA). The amounts of bound Aβ oligomers were measured by Dot blot using anti-amyloid oligomer antibody (A11; Millipore, Billerica, MA). The proteins bound on the liposomes were detected by Western blot using the BAK (G-23) or BID (FL-195) antibodies purchased from Santa Cruz Biotechnology.
Cytochrome c labeling and MOM-like vesicle preparation
For dye labeling, cytochrome c (from equine heart, Sigma-Aldrich) in phosphate-buffered saline (137 mM NaCl, 10.13 mM Na2HPO4, 2.68 mM KCl, 1.76 mM KH2PO4, pH 7.4) was incubated with twofold excess of Cy3 succinimide ester (Lumiprobe, Hallandale Beach, FL) for 6 h at 4°C, such that ∼1–2 Cy3 dyes were labeled per cytochrome c. Free dyes were removed from the solution by using the PD-10 desalting column (GE Healthcare, Piscataway, NJ).
Lipids were purchased from Avanti Polar Lipids (Alabaster, AL). Lipids in organic solvent were mixed, in accordance with previous work (22,24), with the molar ratio 36:21.9:9:8:20:5 for POPC (1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine)/POPE (1-palmitoyl-2-oleoyl-sn-glycero-3-phosphoethanolamine)/POPS (1-palmitoyl-2-oleoyl-sn-glycero-3-phosphoserine)/cholesterol/cardiolipin (from beef heart)/DOGS-NTA-Ni (nickel salt; 1,2-dioleoyl-sn-glycero-3-([N-5-amino-1-carboxylpentyl)-iminodiacetic acid]succinyl).
For the surface-attachment, 0.1 mol % of biotin-DPPE (1,2-dipalmitoyl-sn-glycero-3-phosphoethanolamine-N-(cap biotinyl)) lipid was added to the lipid solution. The lipid mixture was completely dried using the nitrogen gas and further dried with a vacuum pump for at least 3 h. The dried lipid film was resuspended in ∼15 μM Cy3-labeled cytochrome c. After five freeze/thaw cycles, large unilamellar vesicles (∼200 nm diameter) were prepared by extrusion through polycarbonate filters (Avanti Polar Lipids) (see Fig. S3 C). Untrapped cytochrome c was removed by air-fused centrifugation (Airfuse; Beckman Instruments).
Single-vesicle cytochrome c release assay
The flow chambers were assembled on a quartz imaging surface functionalized with PEG (polyethylene glycol) and PEG-biotin molecules (molar ratio 40:1; Laysan Bio, Arab, AL) followed by streptavidin (0.2 mg/mL, Sigma-Aldrich) incubation for 10 min. After washing several times, the MOM-like vesicles are injected into the chamber and immobilized, followed by the washing of free vesicles with 500 μL injection of buffer (50 mM Tris-HCl, pH7.4, 150 mM KCl). Once immobilized, photobleaching safe laser intensity is secured, then BAKΔC, cBID, and other proteins are injected into the flow chamber. The vesicles were monitored with a prism-type total internal reflection fluorescence (TIRF) microscope attached to an electron-multiplying charge-coupled device camera for digital image processing (see Fig. S4 A).
Photobleaching test
Photobleaching is induced by the destruction of fluorophores via light exposure that results in a decrease in fluorescence emission. A laser intensity that does not alter the fluorescence intensity of the MOM-like vesicle for a 10- or 20-min observation period must be secured. We used the first flow chamber (out of a total of 10) in every slide to calibrate the maximum laser intensity that does not induce photobleaching (see Fig. S4 B). All experiments within the respective slide were carried out below the photobleaching safe intensity.
TIRF data processing
The image streaming of the sample chamber from the electron-multiplying charge-coupled device camera connected to the TIRF microscope was collected using single molecule software developed by Dr. Jong-Bong Lee at POSTECH (Pohang University of Science and Technology, Pohang, South Korea). The time traces of the fluorescence intensity of the MOM-like vesicles were then extracted using interactive data language. Once the time traces for the vesicles were obtained, preliminary categorizations of the fluorescence intensity, specifically detection of stepwise drops that correspond to cytochrome c release, were performed using a step-detection program (25). The categorized data were then manually examined one by one to exclude any false-positives.
Purification of α-synuclein and preparation of large α-synuclein oligomers
Recombinant GST-fused α-synuclein was expressed in Escherichia coli Rosetta (DE3) pLysS (Novagen, Darmstadt, Germany). The cells were grown at 37°C in LB medium with 100 μg/mL ampicillin until the absorbance at 600 nm reached 0.6–0.8. Isopropyl β-D-1-thiogalactopyranoside (0.5 mM final concentration) was then added to induce protein expression. Cells were grown for another 12 h at 16°C. GST fusion protein was purified with affinity chromatography using glutathione-agarose beads and then cleaved off from the resin by incubating with thrombin at room temperature for 2 h. To prepare large α-synuclein oligomers, 10 μM of α-synuclein was mixed with 100 μM dopamine in 20 mM sodium phosphate buffer (pH 7) at 37°C for 72 h. After incubation, the mixture was centrifuged to remove large aggregates at 13,000 × g for 5 min at 4°C. Then, the supernatant was concentrated using Ultracel 10 K membrane (Millipore). Large oligomers were purified by size exclusion chromatography using Superdex 200 10/300 GL (GE Healthcare) and concentrated again using Ultracel 10 K-membrane (26).
Dynamic light scattering (DLS)
The size distribution of MOM-like vesicles and Aβ oligomers were analyzed by DLS with Zetasizer nano Zs (Malvern Instruments, Malvern, Worcestershire, UK) at a fixed angle of 173° at 25°C. The MOM-like vesicles (10 μM lipid concentration) were subjected to the DLS measurement after 10-min centrifugation at 13,000 rpm. The data was analyzed by the cumulative method using software provided by the instrument.
Results
BAK is involved in cytochrome c release by Aβ oligomers
We prepared synthetic 42-amino-acid-long Aβ peptide and converted it to oligomers by incubation at 4°C for 16 h (21). The DLS analysis of Aβ oligomers shows a single peak at ∼17 nm (see Fig. S1 A), which is in coherence with previous studies that report 10∼20-nm-sized Aβ oligomers are pentamers/hexamers (21,27). We tested the toxicity of our synthetic Aβ oligomers on HT-22 cells with the MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) assay. We delivered Aβ oligomers directly into the cytoplasm of HT-22 cells using the protein transfection method and measured cell viability (Fig. 1 A). Although Aβ oligomers did not harm cell viability at concentrations up to 1000 nM (see Fig. S2) in the period of first 3 h, exogenous Aβ oligomers induce cell death at longer exposure times (Fig. 1 A) (28).
Figure 1.
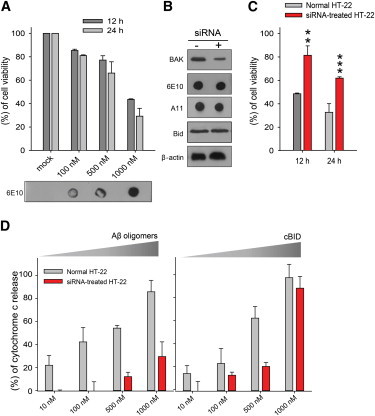
Aβ oligomer-induced cell death and cytochrome c release from siRNA-transfected and normal HT-22 cells. (A) Cell viability of HT-22 cells was evaluated with the MTT assay, 12 h and 24 h, after treatment of the indicated concentrations of Aβ oligomers. Transfected Aβ oligomers were confirmed with Dot blot using anti-β amyloid (6E10) antibody. The data represent the mean ± SD of three independent experiments. (B) Immunoblot analysis of BAK and Aβ oligomers protein in HT-22 cells using anti-BAK and anti-β amyloid (6E10) antibody. HT-22 cells were pretreated with/without the 20 nM BAK siRNA for18 h. A quantity of 1000 nM Aβ oligomers were treated with protein transfection regents. Anti-β-actin and anti-Bid antibodies used as control is shown. (C) Aβ oligomer-induced cell death was compared in normal and BAK KD HT-22 cells using the MTT assay. The data represent the mean ± SD of three independent experiments (∗∗∗P < 0.005; ∗∗P < 0.01 compared with Aβ oligomers-treated (1 μM) normal HT-22 cells pretreated with vehicle). (D) Measurement of cytochrome c release from isolated mitochondria of HT-22 cells using a colorimetric ELISA assay kit (Invitrogen). The data represent the mean ± SD of four independent experiments. Concentrations are given in moles of monomeric Aβ. To see this figure in color, go online.
To address the potential role of BAK in Aβ oligomers-induced cell death, we took a gene-silencing approach using BAK siRNA that reduced BAK mRNA and consequently, protein levels (Fig. 1 B). We observed that Aβ oligomer-induced cell death in BAK knockdown (KD) HT-22 cells was significantly reduced when compared to untreated HT-22 cells (Fig. 1 C), strongly suggesting that Aβ oligomers may play a role in the BAK-dependent apoptotic pathway.
To narrow down the potential targets of Aβ oligomers, we extracted the mitochondria from both normal and BAK KD HT-22 cells and compared the amount of cytochrome c release induced by Aβ oligomers with the ELISA assay (28,29). Remarkably, for mitochondria from BAK KD HT-22 cells, a significant reduction in cytochrome c release was observed when compared with that from normal cells (Fig. 1 D). Thus, our results show that Aβ oligomers most likely target BAK for cytochrome c release.
As controls, we induced the cytochrome c release from isolated mitochondria with cBID (p7/p15 BID), which is a potent apoptotic trigger for both BAK and BAX. As expected, we observed a cBID-concentration-dependent increase in cytochrome c release, suggesting that cytochrome c release machinery works normally in isolated mitochondria. Furthermore, significantly less cytochrome c release out of mitochondria extracted from BAK KD HT-22 cells than that from a normal cell at 500 nM cBID is consistent with reduced BAK protein levels in the siRNA-treated cells.
Single-vesicle cytochrome c release assay
To investigate the effects of Aβ oligomers on apoptotic BAK pores without involving other mitochondrial protein factors, we developed a well-defined in vitro single-vesicle assay that can monitor the real-time dynamics of cytochrome c release from single MOM-like vesicles (Fig. 2 A). MOM-like vesicles were encapsulated with Cy3-labeled cytochrome c and immobilized on the quartz imaging surface, which was monitored with a TIRF microscope. Next, a mixture of recombinant BAK without the transmembrane helix (BAKΔC) and cBID is injected into the flow chamber. BAKΔC is a recombinant form of BAK that has been frequently used for the structural and functional investigations of BAK (22,24,30,31). Hexa-histidine tag on the C-terminal end of BAKΔC tethers onto the Ni-NTA lipid, mimicking its membrane anchor (Fig. 2 A, inset below; and see Fig. S3 A). When BAKΔC is activated by cBID, we observed the release of Cy3-labeled cytochrome c, which resulted in a sudden drop in the fluorescence intensity (Fig. 2 B). Intensity drop due to release differs from slow intensity decline due to photobleaching (see Fig. S4 B).
Figure 2.
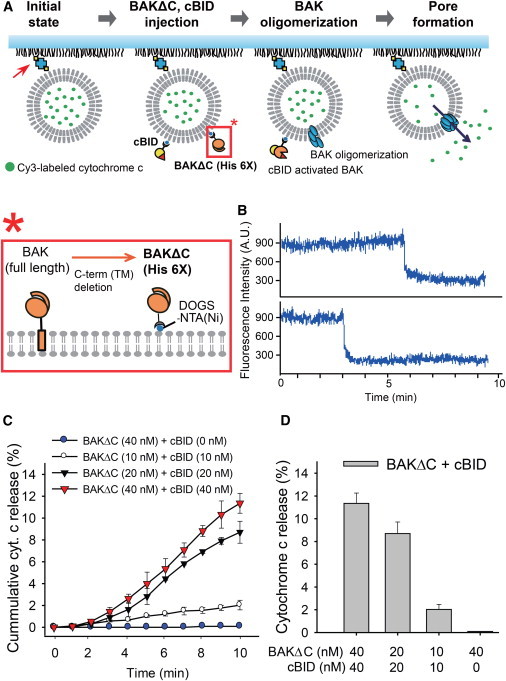
In vitro single-vesicle cytochrome c release assay. (A) Experimental design of the single-vesicle cytochrome c release assay system. The initial state starts with Cy3-labeled cytochrome c encapsulated MOM-like vesicles tethered to a PEG-coated quartz surface via biotin-streptavidin interactions (red arrow). Then BAKΔC and cBID are injected into the flow chamber, inducing apoptotic BAK pores followed by cytochrome c release. (Inset, below) Soluble form of recombinant mouse BAK protein (BAKΔC, residues 16–184) with a C-terminal hexa-histidine tag (see Fig. S3A in the Supporting Material) tethered onto an Ni-NTA lipid. (B) Representative time traces of Cy3-labeled cytochrome c release via apoptotic BAK pore formation. (C) The cumulative histogram of cytochrome c release when treated with the indicated concentrations of recombinant apoptotic proteins observed for 10 min (100-ms time binning). (D) Total cytochrome c release histogram of the indicated concentrations of recombinant apoptotic proteins after 10 min. Data in panels C and D represents the mean ± SD of five independent experiments. To see this figure in color, go online.
As expected, we observed an increase in cytochrome c release events as BAKΔC and cBID concentrations were increased. Specifically, after a 10-min observation period, ∼11% of the surface-immobilized vesicles exhibited cytochrome c release at 40 nM for both BAKΔC and cBID (Fig. 2, C and D). Cytochrome c release was not observed in the absence of either BAKΔC or cBID (see Table S1 in the Supporting Material), consistent with the results from previous in vitro experiments (22,30). The necessity of both BAKΔC and cBID was also confirmed by using sulforhodamine (SRB)-dextran fluorescent dyes in our in vitro single-vesicle assay (see Table S2). Thus, the results validate our assay for the study of apoptotic BAK pores and cytochrome c release (30,32–34).
Aβ oligomers induce formation of apoptotic BAK pores
Here, we take an in vitro approach to investigate whether Aβ oligomers and BAK alone can induce cytochrome c release. Because some studies relate Aβ oligomers with direct MOM permeabilization (15,35,36), we tested whether Aβ oligomers could independently induce cytochrome c release from protein-free MOM-like vesicles. High concentration (1 μM, in monomer concentration) injections of Aβ oligomers, into the flow, in the absence of BAKΔC did not exhibit any fluorescence intensity change (see Table S3), assuring that at concentrations lower than 1 μM, Aβ oligomers alone could not induce protein pores or membrane permeability that would enable cytochrome c release.
When Aβ oligomers and BAKΔC were injected into the flow chamber, cytochrome c release is clearly observed from the vesicles (Fig. 3 A and see Table S4). Specifically, ∼12% of the vesicles displayed cytochrome c release, after 10 min, when treated with 200 nM Aβ oligomers (in monomer concentration) and 40 nM BAKΔC.
Figure 3.
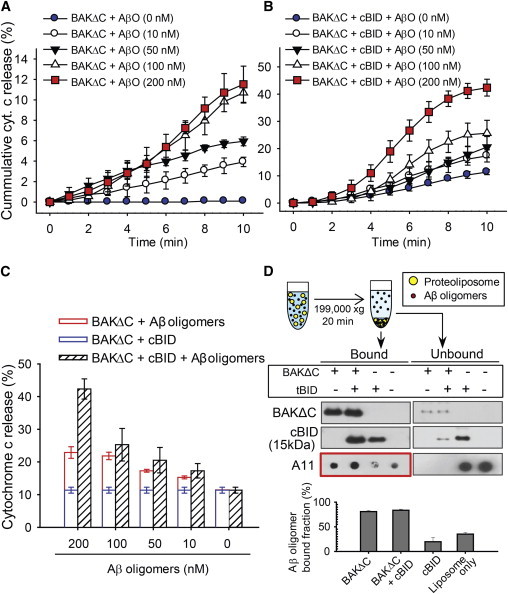
Cytochrome c release via Aβ oligomer-induced apoptotic BAK pores. (A) The cumulative histogram of cytochrome c release when treated with the indicated concentrations of Aβ oligomers and 40 nM BAKΔC observed for 10 min. The data represent the mean ± SD of five independent experiments. (B) Cofloatation assay of Aβ oligomers interaction with proteoliposomes. Liposomes treated with BAKΔC and/or cBID mixed with/without Aβ oligomers, and precipitated with high-speed centrifugation. The amount of vesicle-bound Aβ oligomer was quantified by Dot blot. The data represent the mean ± SD of three independent experiments. The quantification of the bound levels of Aβ oligomers (red box) was performed with the software QUANTITY ONE (Bio-Rad). (C) The cumulative histogram of cytochrome c release when treated with the indicated concentrations of Aβ oligomers 40 nM BAKΔC and 40 nM cBID, observed for 10 min. (D) Total cytochrome c release histogram of the indicated concentrations of Aβ oligomers 40 nM BAKΔC and 40 nM cBID, compared to the sum of 40 nM BAKΔC, 40 nM cBID, 40 nM BAKΔC Aβ oligomers (indicated concentration) after 10 min. Data in panels C and D represent the mean ± SD of five independent experiments. Concentrations are given in moles of monomeric Aβ. To see this figure in color, go online.
Our in vitro single-vesicle results suggest that Aβ oligomers directly induce apoptotic BAK pores and thus, it is highly likely that there is a physical interaction between Aβ oligomers and BAK. To test this hypothesis, we performed a modified cofloatation assay in a cell-free membrane environment. Aβ oligomers were incubated with BAKΔC, cBID, or BAKΔC/cBID proteoliposomes, respectively. Vesicles were collected from the pellets after centrifugation, then separated by SDS-PAGE and transferred onto Western blot.
The results show that Aβ oligomers bind strongly with BAKΔC and BAKΔC/cBID proteoliposomes. However, a weak interaction is also observed between Aβ oligomers and cBID proteoliposomes. The weak intensity of Aβ oligomers binding to cBID appears to represent nonspecific Aβ oligomers binding to the lipid bilayer as opposed to direct binding with cBID, because a comparable intensity was also observed in the mixture of Aβ oligomers and protein-free liposome. Consistently, the supernatant fractions contain a high concentration of unbound Aβ oligomers when incubated with cBID, whereas significantly fewer free Aβ oligomers were present when incubated with BAKΔC (Fig. 3 B). Therefore, our results confirm that Aβ oligomers indeed directly bind with BAKΔC tethered on the MOM-like vesicle.
Aβ oligomers accelerate BAK/cBID mediated apoptotic pores
We have identified that cBID and Aβ oligomers induce apoptotic BAK pores mutually independently. Now, we investigated whether the two can work synergistically. Experiments were performed with various concentrations of Aβ oligomers whereas BAKΔC and cBID concentrations were fixed at 40 nM. Although it is not obvious at low concentrations of Aβ oligomers, the results clearly show that cBID and Aβ oligomers are synergic in enhancing cytochrome c release at 200 nM (Fig. 3 C). There is a twofold increase compared to the linear sum of those when cBID and Aβ oligomers are individually present (Fig. 3 D). The results raise the possibility that Aβ oligomers have the capacity to accelerate cBID-induced apoptotic BAK pore formation, in addition to its ability to induce BAK pore formation on their own.
Do α-synuclein, tau induce apoptotic BAK pores?
Next, we investigated the specificity of Aβ oligomers by testing the effects of other well-known dementia-inducing proteins (DIPs) on apoptotic BAK pore formation (1,5,37). We first tested whether the various forms, either monomer or oligomers, of tau, α-synuclein, or Aβ (1 μM) could independently release cytochrome c from the vesicles without BAKΔC. After 10 min, cytochrome c release did not occur in all cases (see Table S3), providing grounds that the DIPs alone do not puncture the vesicles. Next, we injected BAKΔC with DIPs. Our results clearly show that only Aβ oligomers possess the ability to induce apoptotic BAK pores (Fig. 4 A). In addition, we also investigated the pro/anti-apoptotic effects of DIPs on cBID-induced apoptotic BAK pores. Except for Aβ oligomers, other DIPs did not cause any alterations in the cytochrome c release rates (Fig. 4 B). Thus, the results show that Aβ oligomers specifically induce apoptotic pores and accelerate the formation of cBID-induced apoptotic BAK pores.
Figure 4.
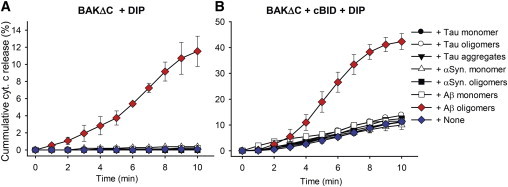
The effect of DIPs on apoptotic BAK pores. The cumulative histogram of cytochrome c release when treated with the indicated DIPs (200 nM) along with (A) 40 nM BAKΔC or (B) 40 nM BAKΔC and 40 nM cBID observed for 10 min. Data represent the mean ± SD of five independent experiments. Concentrations are given in moles of monomeric Aβ, tau, and α-synuclein. To see this figure in color, go online.
Discussion
Studies implicate that Aβ oligomers are the key modulators of cytochrome c release in relation to neuronal cell death in AD. Aβ is known to be produced by enzymatic cleavage of amyloid precursor protein, located on the plasma membrane, and normally excreted to the extracellular space (1,2,38). However, some studies show that Aβ is often cleaved prematurely from the ER and Golgi and is also trafficked back into the cytosol in the significant numbers sufficient to cause physiological difficulties (7,39,40). Cell-level studies link Aβ oligomers with elevation of reactive oxygen species, transition in the MOM permeability, mitochondrial morphological change, and alteration of the expression level of various bcl-2 family proteins (15,18,19,28). It is clear that intracellular Aβ oligomers target the mitochondria and exhibit neurotoxic effects. However, the underlying mechanisms by which Aβ oligomers induce cytochrome c release is yet to be elucidated. Here we show that Aβ oligomers physically target BAK and hijack the intrinsic apoptotic pathway (Fig. 5).
Figure 5.
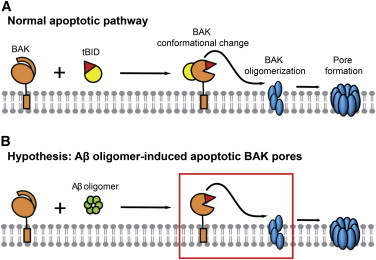
A proposed model of Aβ oligomer function on apoptotic BAK pore formation. (A) Normal apoptotic pathway; tBID induces conformational change of BAK, inducing membrane insertion followed by oligomerization, which results in apoptotic BAK pores. (B) Aβ oligomers hijack the apoptotic pathway by inducing BAK conformational change such that apoptotic BAK pores are formed in the absence of the death signal. To see this figure in color, go online.
We observed significantly reduced Aβ oligomer-induced cell death in BAK KD HT-22 cells. Moreover, Aβ oligomer-induced cytochrome c release out of isolated mitochondria from BAK KD HT-22 cells was reduced when compared with that from mitochondria from normal cells. Thus, these cell-level results establish the connection between Aβ oligomers and BAK-dependent apoptotic pathway. Our in vitro single-vesicle study goes much further by uncovering the underlying mechanisms by which Aβ oligomers induce cytochrome c release and identify that Aβ oligomers’ sole interaction with BAK is sufficient to induce apoptotic BAK pores. Using a cofloatation assay, we show that Aβ oligomers bind directly with BAK on the membrane, providing the molecular basis for its role in the cytochrome c-dependent apoptotic pathway.
Recently, however, Camilleri et al. (17) argued that the disruption of lipid membrane integrity may be the reason for the Aβ oligomer toxicity on isolated mitochondria. We note that much higher concentrations of Aβ oligomers were used in their in vitro study than in ours, which could have made the vesicles leaky for small contents in their long observation time. In sharp contrast, we did not observe the release of much larger cytochrome c in our single-vesicle assay. Furthermore, significantly reduced cytochrome c release levels was observed in isolated mitochondria from BAK KD HT-22 cells, which clearly substantiates the role of BAK in Aβ oligomers-induced cytochrome c release.
Interestingly, we also observed vesicle populations with multiple steps in our in vitro single-vesicle assay (see Fig. S5). Had abrupt membrane rupture been the cause of cytochrome c release, we would have observed only single-step time traces. Further, experiments employing SRB-dextran displayed content release for a fivefold-higher number of vesicles than when we used Cy3-labeled cytochrome c, indicating that some openings in the membrane are large enough to allow SRB-dextran but not sufficient for cytochrome c. Thus, our results suggest that Aβ oligomers indeed induce apoptotic BAK pores.
Our results were possible because of our in vitro single-vesicle cytochrome c release assay. Previous cell-based assays that measure the amount of cytochrome c released from the mitochondria (15,16,28) do not provide temporal information and the closeup examination of the relevant protein machinery is difficult. An ensemble, in vitro release assay using small fluorescence probes could provide real-time dynamics and identifications of relevant protein machinery, but are not able to discriminate physiological apoptotic pores (22,30,41). In contrast, the significance of our assay is that we can monitor the cytochrome c levels within individual MOM-like vesicles and detect pores sufficient for cytochrome c release. Our assay should be generally applicable to the studies of other pro/anti-apoptotic proteins and other protein translocation pores.
Although we show that Aβ oligomers are sufficient to induce apoptotic BAK pores in a minimalistic setting, we cannot completely exclude the possibility of Aβ oligomers interacting with other apoptotic bcl-2 family proteins such as BAX (10–12,33,42) or hindering BAK-inhibiting proteins such as VDAC2 (43,44). Also, our BAK KD experiments were conducted using HT-22 cells from the hippocampal neuronal cell line. Because neuronal cell death is common within the cerebral cortex (8,45), further experiments using primary cultured cortical neurons is warranted.
In conclusion, we show that even in the absence of normal apoptotic signals such as tBID, Aβ oligomers can directly interact with BAK and release cytochrome c. Thus, our results suggest that even without a death signal, the presence of Aβ oligomers alone can trigger neuronal cell death, which is in coherence with the progression of AD. Our results also suggest that Aβ oligomers have the capacity to amplify the death signal, which could transform a normally tolerable apoptotic signal to a faithful on-signal for cell death.
Acknowledgments
This work was, in whole or in part, supported by the National Institutes of Health (grant No. R01 GM051290 to Y.-K.S. and grant No. 5 R01 GM097508 to K.J.O.), the Korea Institute of Science and Technology (KIST Institutional Project No. 2E25000), the Electronic Paramagnetic Resonance Center at the Rosalind Franklin University of Medicine and Science (to K.J.O.), and the Startup Fund from the Rosalind Franklin University of Medicine and Science (to K.J.O.).
Footnotes
Jaewook Kim and Yoosoo Yang contributed equally to this work.
Supporting Material
References
- 1.Selkoe D.J. Alzheimer’s disease: genes, proteins, and therapy. Physiol. Rev. 2001;81:741–766. doi: 10.1152/physrev.2001.81.2.741. [DOI] [PubMed] [Google Scholar]
- 2.Hardy J.A., Higgins G.A. Alzheimer’s disease: the amyloid cascade hypothesis. Science. 1992;256:184–185. doi: 10.1126/science.1566067. [DOI] [PubMed] [Google Scholar]
- 3.Haass C., Selkoe D.J. Soluble protein oligomers in neurodegeneration: lessons from the Alzheimer’s amyloid β-peptide. Nat. Rev. Mol. Cell Biol. 2007;8:101–112. doi: 10.1038/nrm2101. [DOI] [PubMed] [Google Scholar]
- 4.McLean C.A., Cherny R.A., Masters C.L. Soluble pool of Aβ amyloid as a determinant of severity of neurodegeneration in Alzheimer’s disease. Ann. Neurol. 1999;46:860–866. doi: 10.1002/1531-8249(199912)46:6<860::aid-ana8>3.0.co;2-m. [DOI] [PubMed] [Google Scholar]
- 5.Huang Y., Mucke L. Alzheimer mechanisms and therapeutic strategies. Cell. 2012;148:1204–1222. doi: 10.1016/j.cell.2012.02.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Benilova I., Karran E., De Strooper B. The toxic Aβ oligomer and Alzheimer’s disease: an emperor in need of clothes. Nat. Neurosci. 2012;15:349–357. doi: 10.1038/nn.3028. [DOI] [PubMed] [Google Scholar]
- 7.LaFerla F.M., Green K.N., Oddo S. Intracellular amyloid-β in Alzheimer’s disease. Nat. Rev. Neurosci. 2007;8:499–509. doi: 10.1038/nrn2168. [DOI] [PubMed] [Google Scholar]
- 8.Mattson M.P. Apoptosis in neurodegenerative disorders. Nat. Rev. Mol. Cell Biol. 2000;1:120–129. doi: 10.1038/35040009. [DOI] [PubMed] [Google Scholar]
- 9.Castro R.E., Santos M.M., Rodrigues C.M. Cell death targets and potential modulators in Alzheimer’s disease. Curr. Pharm. Des. 2010;16:2851–2864. doi: 10.2174/138161210793176563. [DOI] [PubMed] [Google Scholar]
- 10.Bredesen D.E., Rao R.V., Mehlen P. Cell death in the nervous system. Nature. 2006;443:796–802. doi: 10.1038/nature05293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hail N., Jr., Carter B.Z., Andreeff M. Apoptosis effector mechanisms: a requiem performed in different keys. Apoptosis. 2006;11:889–904. doi: 10.1007/s10495-006-6712-8. [DOI] [PubMed] [Google Scholar]
- 12.Jiang X., Wang X. Cytochrome c-mediated apoptosis. Annu. Rev. Biochem. 2004;73:87–106. doi: 10.1146/annurev.biochem.73.011303.073706. [DOI] [PubMed] [Google Scholar]
- 13.Smale G., Nichols N.R., Horton W.E., Jr. Evidence for apoptotic cell death in Alzheimer’s disease. Exp. Neurol. 1995;133:225–230. doi: 10.1006/exnr.1995.1025. [DOI] [PubMed] [Google Scholar]
- 14.Ankarcrona M., Winblad B. Biomarkers for apoptosis in Alzheimer’s disease. Int. J. Geriatr. Psychiatry. 2005;20:101–105. doi: 10.1002/gps.1260. [DOI] [PubMed] [Google Scholar]
- 15.Sanz-Blasco S., Valero R.A., Núñez L. Mitochondrial Ca2+ overload underlies Aβ oligomers neurotoxicity providing an unexpected mechanism of neuroprotection by NSAIDs. PLoS ONE. 2008;3:e2718. doi: 10.1371/journal.pone.0002718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kim H.S., Lee J.H., Suh Y.H. Amyloid-β peptide induces cytochrome c release from isolated mitochondria. Neuroreport. 2002;13:1989–1993. doi: 10.1097/00001756-200210280-00032. [DOI] [PubMed] [Google Scholar]
- 17.Camilleri A., Zarb C., Vassallo N. Mitochondrial membrane permeabilization by amyloid aggregates and protection by polyphenols. Biochim. Biophys. Acta. 2013;1828:2532–2543. doi: 10.1016/j.bbamem.2013.06.026. [DOI] [PubMed] [Google Scholar]
- 18.Spuch C., Ortolano S., Navarro C. New insights in the amyloid-β interaction with mitochondria. J. Aging Res. 2012;2012:324968. doi: 10.1155/2012/324968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Liu T., Roh S.E., Kang D.E. Cooperative role of RanBP9 and P73 in mitochondria-mediated apoptosis. Cell Death Dis. 2013;4:e476. doi: 10.1038/cddis.2012.203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dahlgren K.N., Manelli A.M., LaDu M.J. Oligomeric and fibrillar species of amyloid-β peptides differentially affect neuronal viability. J. Biol. Chem. 2002;277:32046–32053. doi: 10.1074/jbc.M201750200. [DOI] [PubMed] [Google Scholar]
- 21.Ahmed M., Davis J., Smith S.O. Structural conversion of neurotoxic amyloid-β(1–42) oligomers to fibrils. Nat. Struct. Mol. Biol. 2010;17:561–567. doi: 10.1038/nsmb.1799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Oh K.J., Singh P., Walters D.E. Conformational changes in BAK, a pore-forming proapoptotic Bcl-2 family member, upon membrane insertion and direct evidence for the existence of BH3-BH3 contact interface in BAK homo-oligomers. J. Biol. Chem. 2010;285:28924–28937. doi: 10.1074/jbc.M110.135293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Oh K.J., Barbuto S., Korsmeyer S.J. Conformational changes in BID, a pro-apoptotic BCL-2 family member, upon membrane binding. A site-directed spin labeling study. J. Biol. Chem. 2005;280:753–767. doi: 10.1074/jbc.M405428200. [DOI] [PubMed] [Google Scholar]
- 24.Aluvila S., Mandal T., Oh K.J. Organization of the mitochondrial apoptotic BAK pore: oligomerization of the BAK homodimers. J. Biol. Chem. 2014;289:2537–2551. doi: 10.1074/jbc.M113.526806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kalafut B., Visscher K. An objective, model-independent method for detection of non-uniform steps in noisy signals. Comput. Phys. Commun. 2008;179:716–723. [Google Scholar]
- 26.Choi B.K., Choi M.G., Shin Y.K. Large α-synuclein oligomers inhibit neuronal SNARE-mediated vesicle docking. Proc. Natl. Acad. Sci. USA. 2013;110:4087–4092. doi: 10.1073/pnas.1218424110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bitan G., Kirkitadze M.D., Teplow D.B. Amyloid β-protein (Aβ) assembly: Aβ 40 and Aβ 42 oligomerize through distinct pathways. Proc. Natl. Acad. Sci. USA. 2003;100:330–335. doi: 10.1073/pnas.222681699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cha M.Y., Han S.H., Mook-Jung I. Mitochondria-specific accumulation of amyloid β induces mitochondrial dysfunction leading to apoptotic cell death. PLoS ONE. 2012;7:e34929. doi: 10.1371/journal.pone.0034929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Leshchiner E.S., Braun C.R., Walensky L.D. Direct activation of full-length proapoptotic BAK. Proc. Natl. Acad. Sci. USA. 2013;110:E986–E995. doi: 10.1073/pnas.1214313110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Landeta O., Landajuela A., Basañez G. Reconstitution of proapoptotic BAK function in liposomes reveals a dual role for mitochondrial lipids in the BAK-driven membrane permeabilization process. J. Biol. Chem. 2011;286:8213–8230. doi: 10.1074/jbc.M110.165852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dai H., Smith A., Kaufmann S.H. Transient binding of an activator BH3 domain to the Bak BH3-binding groove initiates Bak oligomerization. J. Cell Biol. 2011;194:39–48. doi: 10.1083/jcb.201102027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Karbowski M., Norris K.L., Youle R.J. Role of Bax and Bak in mitochondrial morphogenesis. Nature. 2006;443:658–662. doi: 10.1038/nature05111. [DOI] [PubMed] [Google Scholar]
- 33.Korsmeyer S.J., Wei M.C., Schlesinger P.H. Pro-apoptotic cascade activates BID, which oligomerizes BAK or BAX into pores that result in the release of cytochrome c. Cell Death Differ. 2000;7:1166–1173. doi: 10.1038/sj.cdd.4400783. [DOI] [PubMed] [Google Scholar]
- 34.Dewson G., Kluck R.M. Mechanisms by which Bak and Bax permeabilize mitochondria during apoptosis. J. Cell Sci. 2009;122:2801–2808. doi: 10.1242/jcs.038166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Parks J.K., Smith T.S., Parker W.D., Jr. Neurotoxic Aβ peptides increase oxidative stress in vivo through NMDA-receptor and nitric-oxide-synthase mechanisms, and inhibit complex IV activity and induce a mitochondrial permeability transition in vitro. J. Neurochem. 2001;76:1050–1056. doi: 10.1046/j.1471-4159.2001.00112.x. [DOI] [PubMed] [Google Scholar]
- 36.Lin H., Bhatia R., Lal R. Amyloid β protein forms ion channels: implications for Alzheimer’s disease pathophysiology. FASEB J. 2001;15:2433–2444. doi: 10.1096/fj.01-0377com. [DOI] [PubMed] [Google Scholar]
- 37.Tiraboschi P., Hansen L.A., Corey-Bloom J. The importance of neuritic plaques and tangles to the development and evolution of AD. Neurology. 2004;62:1984–1989. doi: 10.1212/01.wnl.0000129697.01779.0a. [DOI] [PubMed] [Google Scholar]
- 38.Hardy J. The amyloid hypothesis for Alzheimer’s disease: a critical reappraisal. J. Neurochem. 2009;110:1129–1134. doi: 10.1111/j.1471-4159.2009.06181.x. [DOI] [PubMed] [Google Scholar]
- 39.Sakono M., Zako T. Amyloid oligomers: formation and toxicity of Aβ oligomers. FEBS J. 2010;277:1348–1358. doi: 10.1111/j.1742-4658.2010.07568.x. [DOI] [PubMed] [Google Scholar]
- 40.Li M., Chen L., Zhang Y. The role of intracellular amyloid-β in Alzheimer’s disease. Prog. Neurobiol. 2007;83:131–139. doi: 10.1016/j.pneurobio.2007.08.002. [DOI] [PubMed] [Google Scholar]
- 41.Schafer B., Quispe J., Kuwana T. Mitochondrial outer membrane proteins assist Bid in Bax-mediated lipidic pore formation. Mol. Biol. Cell. 2009;20:2276–2285. doi: 10.1091/mbc.E08-10-1056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhang Y., McLaughlin R., LeBlanc A. Selective cytotoxicity of intracellular amyloid β peptide 1–42 through p53 and Bax in cultured primary human neurons. J. Cell Biol. 2002;156:519–529. doi: 10.1083/jcb.200110119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Plötz M., Gillissen B., Eberle J. Disruption of the VDAC2-Bak interaction by Bcl-x(S) mediates efficient induction of apoptosis in melanoma cells. Cell Death Differ. 2012;19:1928–1938. doi: 10.1038/cdd.2012.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cheng E.H., Sheiko T.V., Korsmeyer S.J. VDAC2 inhibits BAK activation and mitochondrial apoptosis. Science. 2003;301:513–517. doi: 10.1126/science.1083995. [DOI] [PubMed] [Google Scholar]
- 45.Niikura T., Tajima H., Kita Y. Neuronal cell death in Alzheimer’s disease and a neuroprotective factor, humanin. Curr. Neuropharmacol. 2006;4:139–147. doi: 10.2174/157015906776359577. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.