

Abstract
Mass-spectrometry (MS) based proteomics has become a key enabling technology for the systems approach to biology, providing insights into the protein complement of an organism. Bioinformatics analyses play a critical role in interpretation of large, and often replicated, MS datasets generated across laboratories and institutions. A significant amount of computational effort in the workflow is spent on the identification of protein and peptide components of complex biological samples, and consists of a series of steps relying on large database searches and intricate scoring algorithms. In this work, we share our efforts and experience in efficient handling of these large MS datasets through database indexing and parallelization based on multiprocessor architectures. We also identify important challenges and opportunities that are relevant specifically to the task of peptide and protein identification, and more generally to similar multi-step problems that are inherently parallelizable.
Keywords: Bioinformatics, High-throughput Proteomics, Indexing, Multiprocessing, Parallelization
I. Introduction
Advances in technology coupled with innovative experimental and computational workflows have transformed biology into a quantitative science, allowing elaborate inquiry into the molecular basis of health and disease. The field of Proteomics, which characterizes an organism’s proteome - the complete set of proteins expressed in various cells and tissues – comprises of several challenging workflows [1]. A key feature of such “omic” sciences is their high-throughput aspect, producing huge volumes of data in a short duration. For example, a moderate-sized proteomics lab can generate several GB of data each day, and this rate is continuously increasing with advancing technology [2]. Moreover, most experiments are replicated across laboratories and institutions, increasing the need for collaborative mining of such large-scale datasets.
Currently, significant effort in proteomics research is directed towards algorithms for identifying the many hundreds to thousands of proteins present in complex biological samples. This is done via the “shotgun” method using tandem mass spectrometry (MS/MS) technology, which generates large datasets of mass spectra with the goal being identification of the molecular entities that generated those spectra [3]. A single experiment from a modern mass-spectrometer can generate up to the order of 5-10K MS/MS spectra in less than an hour. Associating the spectra with their true peptide/protein identification involves searching large protein databases to retrieve, score and rank potential candidates. Depending upon the size of the database and constraints applied on the search, each spectrum may have to be evaluated against over 100K candidates to select the one that best explains the observed data.
Scoring and evaluation of candidates involves several steps, and requires significant computation time depending upon the algorithm applied. High noise content and variability in MS/MS datasets present difficult data analysis challenges that contribute to loss of identifications. Current state-of-the-art algorithms have a very low coverage and only < 30% of spectra in a large-scale experiment are statistically confidently assigned with a candidate [4]. Consequently, newer more complex scoring algorithms are constantly being researched and developed – these typically provide a significant boost in identification accuracies at the expense of greater computational cost. For example, a recent probabilistic scoring algorithm called CSPI (details to follow) confidently identifies more spectra at a controlled false discovery rate (FDR) as compared with popular state-of-the-art methods [5]. However, due to the complexity of the model, it takes several seconds to evaluate each spectrum under constrained searches, which is at least two orders of magnitude more than the closest competitor. Moreover, search time will rapidly increase due to greater number of candidates being evaluated, if constraints such as allowable post-translationally modified forms of proteins are removed or reduced, as will be necessary for a more thorough analysis of the data [6].
Fortunately this particular application is amenable to massive parallelization and can exploit large multiprocessor and/or distributed computing architectures to alleviate the computational bottleneck. This approach was followed for evaluating the recently developed CSPI framework in our lab [5]. In this paper, we identify some challenges that we encountered related to protein database indexing and multiprocessing-based parallelization, along with opportunities for further innovations, based on our experience with developing and implementing an efficient scoring framework for more confident assignment of peptides to MS/MS spectra. In the next section, we give a brief background on the shotgun proteomics approach and peptide identification problem. In section III, we present the methods that we used for efficiently handling MS/MS data. Section IV concludes with other potential avenues for future research.
II. Background
MS/MS workflow proceeds in the following three steps [2, 7] (Fig. 1A): a) break or digest large protein molecules, which are difficult to analyze using MS, into small manageable pieces called peptides; b) chemical-property-based separation of peptides using liquid chromatography, in order to reduce the mixture complexity [8]; c) isolation and fragmentation of these peptides using tandem mass-spectrometry (MS/MS) [8]. The fundamental unit of data in such experiments is a peptide MS/MS spectrum, which is generated by collision-induced fragmentation of the peptides inside the mass-spectrometer. The spectrum consists of a set of <mass-to-charge ratio or m/z vs. relative-abundance or Intensity> pairs (called peaks) that represent various detected fragments of the corresponding peptide as well as unexplainable noise peaks. The goal then is to confidently identify peptides responsible for large datasets of experimental MS/MS spectra followed by relating the identified peptides back to their parent proteins [7]. An example of how to evaluate an MS/MS spectrum against one (arbitrary) peptide is given in Figure 1B.
Fig. 1.

(A) Schematic of shotgun proteomics approach; (B) Peptide evaluation against MS/MS spectrum. Cleavage at any position can yield a left and/or right fragment, called b or y-ions respectively. The ions in the series have a different m/z, which can be located on the x-axis of the spectrum. Black peaks represent unexplained events or noise. Y-axis represents the relative abundance of respective entities.
A typical computational approach for assigning peptides to MS/MS spectra is called ‘Database Searching’, and consists of the following steps [9] (see Figure 2): a) From a protein sequence database, generate a list of candidate peptides for each MS/MS spectrum; b) Generate theoretical spectra for each candidate based on known rules of peptide fragmentation and compare with the experimental spectrum; c) Rank the candidates according to a scoring algorithm which gives higher scores to candidates that explain better a larger number of more intense peaks - this is done by comparing the expected (theoretical) peptide spectrum with the experimentally observed spectrum, and is the heart of peptide identification systems; d) Statistical evaluation for reducing false peptide identifications by controlling the FDR [10].
Fig. 2.
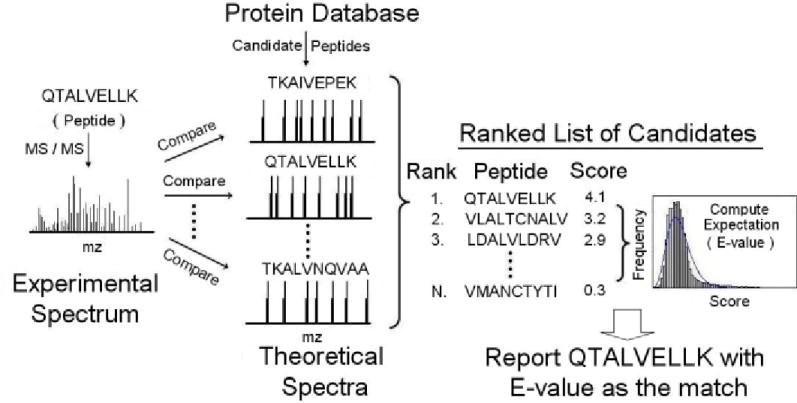
Schematic for Peptide identification by MS/MS via database searching (adapted from [3]). E-value, which stands for expectation value, is a statistical measure of significance and refers to the number of matches that are expected to obtain equal or better score by chance alone.
We have recently developed a novel probabilistic scoring algorithm called Context-Sensitive Peptide Identification (CSPI), which utilized Input-Output Hidden Markov Models (IO-HMM) to capture the effect of peptide physicochemical properties on their observed MS/MS spectra [5, 11]. IOHMMs are an extension of the classic hidden markov models (HMMs) and are used to stochastically model pairs of sequences, called input and output sequence. These models have been previously successfully applied to several sequential data-mining tasks, including financial data analysis [12], music processing [13] and gene regulation [14]. The graphical structure showing similarities and differences between HMM and IO-HMM is shown in Figure 3. As can be seen, IO-HMMs contain extra nodes (than HMMs) for the input sequence <x1, x2, …, xT>, which can probabilistically influence the output layer and/or the hidden states, represented as <y1, y2, …, yT> and <q1, q2, …, qT> respectively. They represent the joint conditional probability distribution P(y1y2…yT| x1x2…xT; Θ), where ‘Θ’ are the model parameters. The intermediate hidden layer <q1q2…qt…qT> facilitates modeling the sequential dependencies between the input-output sequence pair as complex probability distributions. Both xt and yt can be uni-variate or multi-variate, discrete or continuous, whereas the hidden states, qt, are typically discrete. In the case of CSPI framework, input layer is a representation of the peptide sequence while the output layer is a representation of their MS/MS spectrum intensities. Additionally, the input sequence can be constructed with arbitrary features (from the domain) that may or may not overlap in location, allowing rich contextual information at local (specific location) as well as global (sequence) level to be incorporated in the sequence mapping tasks. For notational convenience, we have used the same length ‘T’ for all sequences. This is easily adapted to more general cases).
Fig. 3.
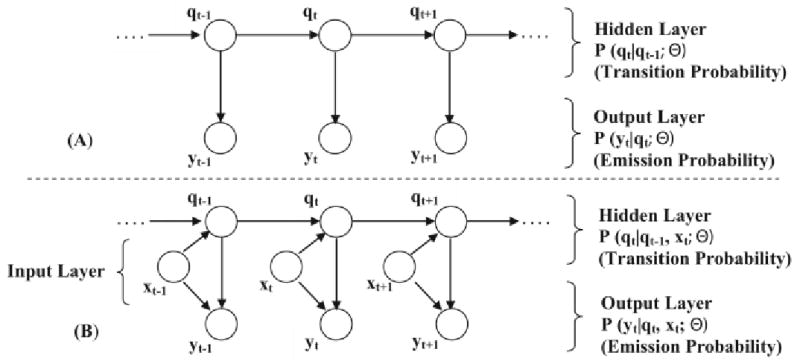
A) Classical Hidden Markov Model; B) Input-output Hidden Markov Model.
Due to conditioning on the input layer, the transition probability distributions are potentially non-stationary in location and must be computed afresh for each input sequence. In practice, there is one transition function for each hidden state, to compute the probability distribution of state at current location (qt) given the state at previous location (qt-1), i.e. P(qt | qt-1, xt). Within CSPI, these are modeled using logistic functions. In the current implementation, a constrained model structure is used such that the input layer influences only the transition probabilities and not emission probabilities. Accordingly, there is one emission function for every hidden state, to compute the probability distribution of the emission/observation at the current location, given the state at current location, i.e. P(yt | qt). These are modeled using simple distributions (Gaussian, Exponential or Beta) depending upon how the spectrum intensities are normalized. The parameters of the model are trained using Generalized Expectation Maximization algorithm (GEM). Trained CSPI models are used to score and rank candidate peptides obtained via Database Search for each spectrum. This is done using the Forward procedure, which follows similar mechanics as in HMMs with the exception that all computations must take into account the context presented in the input layer.
Empirical evaluation showed that scores based on CSPI significantly improve peptide identification performance, identifying up to ∼25% more peptides at 1% False Discovery Rate (FDR) as compared with popular state-of-the-art approaches. Superior performance of the CSPI framework has the potential to impact downstream proteomic investigations (like protein quantification and differential expression) that utilize results from peptide-level analyses. Being computationally intensive, the design and implementation of CSPI supports efficient handling of large MS/MS datasets, achieved through protein database indexing and parallelization of the computational workflow using multiprocessing architecture, as described in the next section.
III. Methods for efficient processing of MS/MS spectra
The first step in analysis involves extracting candidate peptides for each spectrum by querying a protein database. Protein databases are simple ASCII text files with a list of protein sequences or character strings (the protein alphabet is of size 20, with each character being of a different mass). Each sequence in the database is preceded with a single line header (identified with the “>” symbol in the beginning) uniquely identifying and describing the sequence, followed by the lines containing the actual sequence of amino-acids (characters) making the protein. An example of such a file is given in Figure 4. The sequences are of variable lengths, ranging from several tens to several thousands of characters.
Fig. 4.

Sample from Human protein database file (IPI stands for “International Protein Index” that provides a unique and stable identifier to track protein sequences and allows a mapping between variety of bioinformatics databases [15])
The extraction step amounts to a range query on the “expected mass” of the true peptide, where a peptide mass is computed by summing the masses of individual characters in the peptide. Hence, for a given mass query, the naïve approach would be to scan for sub-strings in these protein databases such that the mass is within some allowable tolerance of the query mass depending upon the accuracy of the data-generating instruments. For complex organisms like Humans, the protein database files are typically large with several thousands of protein sequences (the database used consisted of ∼89,575 protein sequences). Scanning them afresh for each spectrum for retrieving sub-strings of required mass is prohibitive in terms of time.
Database search as described above is performed with certain parameters that reflect the experimental protocol used for generating the data, and remain the same for the entire dataset of MS/MS spectra from the experiment. For example, all the sub-strings must end in characters K or R while allowing up to three internal Ks and/or Rs, or that certain characters are modified such that each occurrence of it will carry an extra mass than its native form. This kind of constrained search eliminates the need to enumerate all possible sub-strings from the protein database, and provides a way to speed up query and retrieval.
We process the protein database prior to analyzing the data and pre-compute an indexed version by first generating the list of all possible sub-strings satisfying the desired search constraints. For each peptide, we compute the mass up to one decimal point as well as note its location in the database (protein number as it appears in the text file and position number within the parent protein sequence) and its length in number of characters. This information is stored in a key-value store where the “key” is the string representation of the peptide’s mass while the “value” is the string concatenation of the auxiliary information using a separator (location and length). ‘Values’ of peptides with the same key are concatenated with a different separator. Additionally, in order to keep the size of index files small, the entire range of expected peptide masses is split into bins of size 25 mass units (arbitrarily chosen and may be optimized further), leading to multiple index files each storing a different mass region.
Now, for every new query, the index allows for fast retrieval of candidates, by first mapping the query mass (“key”) to the appropriate index file, followed by retrieval of peptides in the corresponding mass-region that meet the mass-tolerance search criterion, and reconstruction of the peptide sequences using the corresponding information stored in the “value” part of the key-value pair (in conjunction with the original protein database ASCII text file). Indexes were generated using the Berkeley DB key-value store [16] and was accessed using its Python language interface.
Challenge 1
This approach works well for constrained database searches (total of ∼10 million peptides, and ∼10-20K candidates per spectrum) that were employed in the current implementation and analysis. However, unconstrained searches can yield a total search space of several billion peptides, leading to larger index files and increased index generation as well as querying time. A potential scalable solution is a distributed index with capability for parallel generation and querying (using simple synchronization primitives) which is facilitated by split indexes (as described above) as well as the fact that each spectrum can be queried independently of others. Such schemes or variants thereof will be crucial for future large-scale proteomics and must be explored.
The next step in database searching evaluates all the candidates retrieved for each spectrum. This is computationally the most expensive step in the peptide identification workflow but comprises an embarrassingly parallel problem. Specifically, for each spectrum in the dataset, searching and scoring/ranking candidate peptides can be performed in parallel, independent of other spectra.
We used a simple multiprocessing application design using shared synchronized queues for inter-process communication. The flow diagram is shown in Figure 5. The main process reads in and preprocesses the spectra, queries the protein database stored as a pre-computed index on the hard disk (as described above) and places the retrieved candidates along with the preprocessed spectrum on a shared queue. From this queue, all the worker processes extract the objects, run the CSPI scoring algorithm, and store the results onto a shared output queue. Another child process extracts the results from this output queue and stores them in an output file. The algorithm was tested on a single machine with eight quad-core processors (total of 32 cores) as well as a large blade-based system (Blacklight server) with up to 64 requested cores at the Pittsburgh Supercomputing Center.
Fig. 5.

Workflow of the multiprocessing version of CSPI scoring algorithm [5]
Database search and candidate evaluation time depend upon the size of the MS/MS datasets as well as the number of candidates evaluated per spectrum (which in turn depends upon the search constraints applied). Figure 6 shows how our CSPI framework scales with addition of processor units, for the results presented in that work [5]. Specifically, the constrained searches performed resulted in between 10K and 20K candidates to be evaluated per spectrum.
Fig. 6.
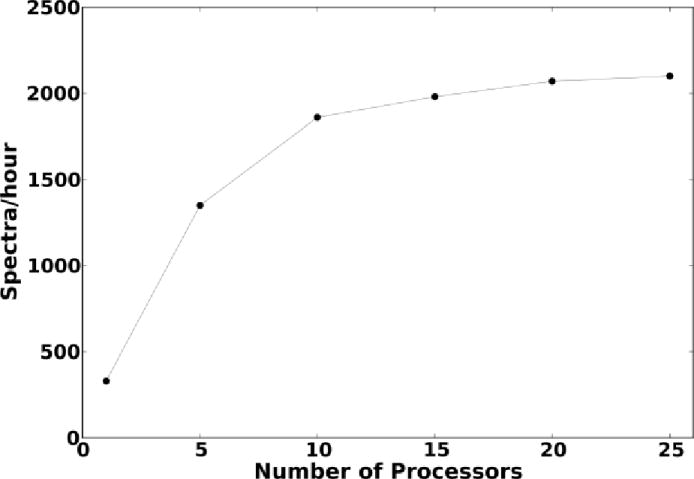
Scalability of the multiprocessing version of CSPI scoring algorithm [5]
We see that the throughput increases rapidly initially, although not linearly, but saturates at about 15 processors. Although simpler scoring systems can achieve much higher performance gains through parallelization [17], the gap can be possibly reduced with alternate schemes for task-distribution. These are worth investigating due to good performance characteristics of CSPI and other state-of-the-art complex algorithms for confident peptide identification.
Challenge 2
As described above, the current workflow breaks the tasks at the individual spectrum level, which means once a spectrum and its potential candidates are assigned to a child process, they are evaluated sequentially within the same process. However, since evaluation of each candidate against a spectrum itself requires several steps and can be performed independently of all other candidates for all other spectra, there is scope for much further optimization. It is important to note that although the entire process of peptide identification is inherently parallelizable, optimum task distribution and sharing between processes will need careful profiling of processing needs of individual steps and will also depend critically upon such factors as the size of the database searched as well as search constraints applied. Further, with greater granularity of tasks and number of processes, overhead due to inter-process communication will become an important factor to consider [17]. Automatically adjusting for all these dependencies within resource constraints is a non-trivial but interesting problem to investigate.
IV. Discussion and Concluding Remarks
Peptide, followed by protein identification is an important problem in current proteomics research and offers several unique computational challenges. Confident identification is critical to further downstream analyses like predictive biomarker discovery for disease classification and characterization, and relies on complex and computationally expensive scoring algorithms. With advancing technology and increasing trend towards integrated, personalized healthcare, timely and efficient handling of concomitant large datasets is an equally important aspect. Towards this end, we have presented insights based on our experience in addressing two key computational bottlenecks related to the peptide identification problem. Our methods have helped to significantly increase the efficiency of our previously developed probabilistic scoring algorithm, CSPI [5]. We have also identified important research challenges that will provide a further boost in that direction. All experiments and analyses were performed using the Python programming language. Parallelization was achieved using the multiprocessing package, while indexing was performed using Python’s ‘bsddb’ interface to the Berkeley DB library.
The CSPI framework demonstrates the feasibility of applying context-sensitive markov models to a complex real-world problem involving scoring and identification of peptides from high-throughput tandem mass-spectrometry experiments. More generally, it shows the applicability of IO-HMMs to handle big datasets that involve local and global sequential dependencies in the sequence pairs being modeled. Further, the ability to parallelize such problems demonstrated in this paper, allows for processing of collaborative big datasets involving experimental data from multiple laboratories.
One proposal for such analyses could involve the cloud-computing architecture, where the cloud would be the repository of centralized information on experimental outcomes/data, which can then be processed in a distributed manner using their easily accessible and integrated compute resources. The main issues relate to the complexity of the models themselves, in terms of the large number of parameters that must be estimated. For example, with four hidden states the number of parameters to be estimated for an IO-HMM model in CSPI is over 700. The ability to obtain data through collaborative architectures should greatly facilitate more accurate estimates of such parameters, and the efficient processing achieved through parallelization will be a necessary component in the overall analytical workflow.
Acknowledgments
The research reported in this publication was supported in part by the following two grants: National Institutes of Health Award Number P41RR006009 and National Library of Medicine Award Number R01LM010950. The content is solely the responsibility of the authors and does not necessarily represent the official views of the granting organizations.
Contributor Information
Himanshu Grover, Department of Biomedical Informatics, University of Pittsburgh, Pittsburgh, PA 15206-3701 USA (hig2@pitt.edu).
Vanathi Gopalakrishnan, Department of Biomedical Informatics, and has joint appointments with the Intelligent Systems Program and the Computational & Systems Biology Department, University of Pittsburgh, Pittsburgh, PA 15206-3701 USA. She is also the corresponding author (phone: 412-624-3290; fax: 412-624-5310; vanathi@pitt.edu).
References
- 1.Aebersold R, Goodlett DR. Mass spectrometry in proteomics. Chem Rev. 2001;101(2):269–295. doi: 10.1021/cr990076h. [DOI] [PubMed] [Google Scholar]
- 2.Noble WS, MacCoss MJ. Computational and statistical analysis of protein mass spectrometry data. PLoS Comput Biol. 8(1):e1002296. doi: 10.1371/journal.pcbi.1002296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nesvizhskii AI, Vitek O, Aebersold R. Analysis and validation of proteomic data generated by tandem mass spectrometry. Nat Methods. 2007;4(10):787–797. doi: 10.1038/nmeth1088. [DOI] [PubMed] [Google Scholar]
- 4.Marcotte E. How do shotgun proteomics algorithms identify proteins? Nature Biotechnology. 2007;25(7):755–757. doi: 10.1038/nbt0707-755. [DOI] [PubMed] [Google Scholar]
- 5.Grover H, Wallstrom G, Wu CC, Gopalakrishnan V. Context-sensitive Markov Models for Peptide Scoring and Identification from Tandem Mass Spectrometry. OMICS: A Journal of Integrative Biology. 2012 doi: 10.1089/omi.2012.0073. submitted for publication. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Li Y, Chi H, Wang LH, Wang HP, Fu Y, Yuan ZF, et al. Speeding up tandem mass spectrometry based database searching by peptide and spectrum indexing. Rapid Commun Mass Spectrom. 24(6):807–814. doi: 10.1002/rcm.4448. [DOI] [PubMed] [Google Scholar]
- 7.Steen H, Mann M. The ABC’s (and XYZ’s) of peptide sequencing. Nat Rev Mol Cell Biol. 2004;5(9):699–711. doi: 10.1038/nrm1468. [DOI] [PubMed] [Google Scholar]
- 8.Mann M, Hendrickson RC, Pandey A. Analysis of proteins and proteomes by mass spectrometry. Annu Rev Biochem. 2001;70:437–473. doi: 10.1146/annurev.biochem.70.1.437. [DOI] [PubMed] [Google Scholar]
- 9.Nesvizhskii AI. Protein identification by tandem mass spectrometry and sequence database searching. Methods Mol Biol. 2007;367:87–119. doi: 10.1385/1-59745-275-0:87. [DOI] [PubMed] [Google Scholar]
- 10.Kall L, Storey JD, MacCoss MJ, Noble WS. Assigning significance to peptides identified by tandem mass spectrometry using decoy databases. J Proteome Res. 2008;7(1):29–34. doi: 10.1021/pr700600n. [DOI] [PubMed] [Google Scholar]
- 11.Bengio Y, Frasconi P. Book An Input Output HMM Architecture. 1995. An Input Output HMM Architecture; pp. 427–434. in Editor (Ed.)∧(Eds.) [Google Scholar]
- 12.Bengio Y, Lauzon V-P, Ducharme Rj. Experiments on the Application of IOHMMs to Model Financial Returns Series. IEEE Trans Neural Networks. 2001;12(1):113–123. doi: 10.1109/72.896800. [DOI] [PubMed] [Google Scholar]
- 13.Paiement Jean-Fran\ o, Bengio S, Eck D. Probabilistic models for melodic prediction. Artif Intell. 2009;173(14):1266–1274. [Google Scholar]
- 14.Ernst J, Vainas O, Harbison CT, Simon I, Bar-Joseph Z. Reconstructing dynamic regulatory maps. Molecular Systems Biology. 2007;3:74. doi: 10.1038/msb4100115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kersey PJ, Duarte J, Williams A, Karavidopoulou Y, Birney E, Apweiler R. The International Protein Index: an integrated database for proteomics experiments. Proteomics. 2004;4(7):1985–1988. doi: 10.1002/pmic.200300721. [DOI] [PubMed] [Google Scholar]
- 16.Olsen MA, Bostic K, Seltzer M. Book Berkeley DB. 1999. Berkeley DB. in Editor (Ed.)∧(Eds.) pp. [Google Scholar]
- 17.Xu H, Freitas MA. MassMatrix: a database search program for rapid characterization of proteins and peptides from tandem mass spectrometry data. Proteomics. 2009;9(6):1548–1555. doi: 10.1002/pmic.200700322. [DOI] [PMC free article] [PubMed] [Google Scholar]