

Abstract
Aurora A kinase regulates early mitotic events through phosphorylation and activation of a variety of proteins. Specifically, Aur-A is involved in centrosomal separation and formation of mitotic spindles in early prophase. The effect of Aur-A on mitotic spindles is mediated by the modulation of microtubule dynamics and association with microtubule binding proteins. In this study we show that Aur-A exerts its effects on spindle organization through the regulation of the actin cytoskeleton. Aurora A phosphorylates Cofilin at multiple sites including S3 resulting in the inactivation of its actin depolymerizing function. Aur-A interacts with Cofilin in early mitotic phases and regulates its phosphorylation status. Cofilin phosphorylation follows a dynamic pattern during the progression of prophase to metaphase. Inhibition of Aur-A activity induced a delay in the progression of prophase to metaphase. Aur-A inhibitor also disturbed the pattern of Cofilin phosphorylation, which correlated with the mitotic delay. Our results establish a novel function of Aur-A in the regulation of actin cytoskeleton reorganization, through Cofilin phosphorylation during early mitotic stages.
Keywords: Aurora A kinase, Cofilin, mitosis, phosphorylation, actin cytoskeleton
1. Introduction
Aurora A (Aur-A) is a member of the family of Aurora serine/threonine kinases, which play important roles in the mitotic process. Expression of Aur-A is significantly increased during late G2 when it is targeted to the centrosomes. Aur-A is responsible for centrosomal maturation and separation by recruiting α-tubulin, centrosomin, NDEL1, TACC, and LATS2 to the centrosomes [1–4]. Aur-A also regulates mitotic spindle assembly through interactions with LIMK1, TPX2, Eg5, Hurp, and XMAP215 [5–8]. Although the function of Aur-A is essential during early prophase, spindle pole localization of Aur-A is sustained through the mitotic phases, suggesting its involvement in later mitotic events. Recent studies showed a cooperative function of Aur-A and Aur-B on anaphase microtubule dynamics [9]. Aur-A expression is tightly regulated and altered expression of Aur-A results in mitotic spindle defects. Inhibition of Aur-A expression resulted in chromosome misalignment and multinucleated cells [10], whereas overexpression of Aur-A induced generation of supernumerary centrosomes, multipolar spindles, and aneuploidy. Importantly, overexpression of Aur-A is seen a variety of cancers including, breast, ovarian and prostate [11, 12], which may lead the development of aneuploidy in the cancerous cells.
In addition to its regulation of microtubule dynamics and chromosome segregation during mitosis, Aur-A has been implicated in the regulation of actin cytoskeleton. Activation of Drosophila Aur-A has been suggested to play a role in actin dependent asymmetric protein localization during mitosis [13]. Overexpression of Aur-A was shown to induce the up-regulation of SSH-1 leading to dephosphorylation and activation of the actin depolymerizing protein, Cofilin [14]. Aur-A also interacts with LIMK1 and Ajuba, proteins that are involved in the reorganization of the actin cytoskeleton [8, 15]. Recent studies showed an indirect relationship between Aur-A and regulation of actin-dependent processes through phosphorylation of Rho kinases in Drosophila [16]. Nonetheless, the role of Aur-A regulation of the actin cytoskeleton has not been clearly defined.
Although not widely studied, actin has an important function throughout mitosis. During G2 phase, the actin cytoskeleton is involved in centrosome separation [17, 18]. Cortical actin plays a role in the anchoring and orientation of the mitotic spindle [19, 20]. Additionally the regulation of actin dynamics is essential for the completion of cytokinesis through formation of the contractile ring [21, 22]. The dynamics of the actin cytoskeleton is regulated by the actin depolymerizing protein, Cofilin. Kinases, such as LIMK1/2 and TESK1/2, regulate Cofilin activity through phosphorylation, which prevents its binding to actin [23–27]. However, functionally active Cofilin is essential for the completion of cytokinesis. Also, LIMK1 mediated inactivating phosphorylation of Cofilin during mitosis is necessary for proper mitotic spindle orientation [28], however, the exact function of Cofilin during mitosis has yet to be determined. In this study, we identified Cofilin as a novel substrate of Aur-A. Aur-A regulates Cofilin activity through phosphorylation, thereby regulating actin polymerization. Additionally, we found that Aur-A is involved in the regulation of Cofilin phosphorylation during mitosis.
2. Materials and Methods
2.1 Cell Culture, G2/M Phase Enrichment, and Transfection
PC-3 prostate cancer cells (ATCC) were cultured in F12 HAM containing 10% fetal bovine serum and 1% antibiotic/antimycotic. M12 prostate cancer cells (a gift from Jay Ware) were cultured in RPMI containing 5% fetal bovine serum, 10ng/mL EGF, ITS mix (5μg/mL insulin, 5μg/mL transferrin, 5ng/ml selenium), 50μg/mL gentamycin, and 0.1μM dexamethasone. NIH-3T3 cells (ATCC) were cultured in DMEM containing 10% fetal bovine serum and 1% antibiotic/antimycotic. MCF7 cells obtained as a gift from James Turkson Univ. of Hawaii were cultured in DMEM containing 10% fetal bovine serum and 1% antibiotic/antimycotic. Cells were enriched at G2/M phase as follows: Cells were synchronized at the G2/M boundary by treatment with nocodazole (3T3 600ng/ml, 16hrs; M12 80ng/ml, 24 hrs). Synchronized cells were isolated by mitotic shake off and released into mitotic phases using fresh complete media. Cells were harvested at 0, 30, and 60 mins after release. For experiments with Aur-A inhibitor, M12 cells were treated with MLN8237 (100nM), BMS-5 (5μM), both BMS-5 (5μM) and MLN8237 (100nM) or DMSO for 24 hrs, then nocodazole was added for an additional 24 hrs. Cells were collected by mitotic shake off and were released into mitosis with fresh media containing MLN8237 (100nM), BMS-5 (5μM), both BMS-5 (5μM) and MLN8237 (100nM), or DMSO and harvested at specific time points. For ectopic expression of Cofilin, M12 cells were transfected with Cofilin-RFP using X-tremeGENE HP (Roche) and used between 48–72 hrs.
2.2 Inhibitors and Antibodies
Specific inhibitors and reagents used were BMS-5 (Synkinase), DMSO (Sigma), Nocodazole (Sigma), MLN8237 (a gift from Selleck), and VX-680 (a gift from Selleck). The primary and secondary antibodies used were mouse anti-human-Aur-A (Sigma), rabbit anti-human-Cofilin (Novus), rabbit anti-human-Cofilin (Pierce), rabbit anti-human-pS3-Cofilin (Cell Signaling), mouse anti-human-GAPDH (Sigma), AlexaFluor-488 Phalloidin (Molecular Probes), rabbit anti-human-pT505/T508-LIMK1/2 (Cell Signaling), mouse anti-human-α-tubulin (Sigma), rabbit anti-human-SSH1 (Cell Signaling), horseradish peroxidase (HRP) conjugated goat anti-rabbit (Jackson Laboratories), HRP conjugated goat anti-mouse (Jackson, Laboratories), anti-mouse AlexaFluor-488 (Invitrogen) and anti-rabbit Cy3 (Jackson Laboratories).
2.3 Immunoblotting and Immunoprecipitation
Cells were harvested by trypsinization and lysed with RIPA lysis buffer. For immunoblots, 50μg whole cell extracts were separated by SDS-PAGE and proteins were transferred to a PVDF membrane. Specific polypeptides were detected by reacting with the appropriate primary and secondary antibodies and protein bands were visualized using a substrate from an Immun-Star WesternC Kit (Biorad). For immunoprecipitates, 300μg–500μg whole cell extract was pre-cleared with Sepharose A/G beads (Santa Cruz) overnight at 4°C. Aur-A or Cofilin was precipitated from the extract with 1.5μg–2μg Aur-A or Cofilin antibody at 4°C for 6 hrs. Mouse IgG or rabbit IgG was used as a control. Protein-antibody complexes were pulled down with Sepharose A/G beads at 4°C overnight. Unbound protein was removed by washing with RIPA buffer. Bound proteins were separated by SDS-PAGE and Cofilin was detected by immunoblotting using anti-Cofilin antibodies.
2.4 Recombinant Protein Production and Purification
The coding sequence of Cofilin was cloned into the pET-30 vector as previously described [29], and in pCMV6-AC-RFP vector (Origene) to generate His- and RFP-tagged Cofilin. Cofilin mutants (CofilinS3A, CofilinS3A/S8A/T25A and CofilinS3EE) were produced using the QuickChange XL site-directed mutagenesis kit (Agilent). The truncated Cofilin construct Cofilin90-166 was produced by PCR amplification of the DNA fragment containing bases 268–501 of the Cofilin ORF and cloning into the pET-30 vector. The wild-type Aur-A and inactive Aur-AK162M mutant were cloned into the pET-30 vector as previously described [8]. Recombinant Cofilin and Aur-A were expressed and purified as previously described [8, 29].
2.5 Kinase Assays
For in vitro kinase assays, purified recombinant His-tagged proteins were incubated in kinase assay buffer (50mM MOPS pH 7.2, 25mM β-glycerophosphate, 10mM EGTA, 4mM EDTA, 50mM MgCl2, 0.5mM DTT, 250μM ATP) and 5nM γ-32P-ATP for 30 mins at room temperature. The reaction was stopped by the addition of Lamelli sample buffer and proteins were separated by SDS-PAGE and stained with Coomassie Brilliant Blue dye. Phosphorylation was detected by autoradiography. For immunocomplex kinase assays, Aur-A was immunoprecipitated as described above and sepharose bead-bound protein-antibody complexes were washed with kinase assay buffer. Beads were resuspended in kinase assay buffer and the assay was performed as described above, using recombinant His-Cofilin as the substrate.
2.6 Phosphopeptide Analysis
Recombinant His-Aur-A or His-Aur-AK162M was incubated with recombinant His-Cofilin in kinase assay buffer and cold ATP for 30 mins at room temperature. Proteins were separated by SDS-PAGE and the polypeptide bands were visualized by staining with Coomassie Blue. Cofilin bands from both samples were excised from the gel and used for LC MS/MS with titanium oxide enrichment at the W.M Keck Foundation Biotechnology Resource Laboratory (Yale Cancer Center Mass Spectroscopy Resources) as previously described [8].
2.7 Dual Label Immunofluorescence
M12 cells (4×104) were seeded onto poly-L-lysine coated glass coverslips and transfected with Cofilin-RFP constructs 24 hrs later as described above. At 48 hrs post-transfection, cells were washed with PBS, fixed using 4% paraformaldehyde for 10 mins at room temperature and permeabilized with 4% paraformaldehyde containing 0.2% Tween-20 for 10 mins at room temperature. Permeabilized cells were blocked in blocking solution (10% goat serum, 0.2% tween, 2% BSA in PBS) for 90 mins at room temperature. Cells were stained with Phalloidin for F-actin, washed in sodium phosphate buffer and coverslips were mounted with DAPI Fluoromount G (Southern Biotech). For localization of Cofilin in mitotic cells with or without treatment with Aur-A inhibitor, M12 cells (3×104) were seeded onto poly-L-lysine coated coverslips and after 24 hrs treated with either 100nM MLN8237 or DMSO for an additional 24 hrs. Next, cells were synchronized at G2/M phase and released into mitosis as described above. Coverslips were washed with PBS and fixed/permeabilized with 100% methanol for 10 mins at −20°C. Coverslips were blocked and treated with anti-Cofilin and anti-α-tubulin primary antibodies and anti-mouse AlexaFluor-488 and anti-rabbit Cy3 secondary antibodies. Cells were counterstained with DAPI and mounted as described above. For F-actin staining of MCF7 cells, 3×104 cells were seeded onto poly-L-lysine coated glass coverslips. Coverslips were washed with PBS and fixed with 4% paraformaldehyde and permeabilized with 4% paraformaldehyde containing 0.2% Tween-20 as described above. Coverslips were stained with Phalloidin and DAPI and mounted as described above. Positive signals were visualized in a Leica TCS SP5II confocal microscope.
2.8 Actin Polymerization Assay
Pyrene labeled actin (Cytoskeleton) was polymerized in the presence of polymerization buffer (2mM MgCl2, 0.5mM ATP, 0.2M KCl, pH 7.0) for 2 hrs at RT. The polymerized actin was then incubated with His-Cofilin, His-CofilinS3Aand His-CofilinS3A/S8A/T25A that had previously been phosphorylated by Aur-A as described above for 10 mins at room temperature. F-actin was stained with Phalloidin in a microfuge tube as described above and the actin/protein mixture was mounted on coverslips. Actin filaments were visualized by confocal microscopy.
3. Results
3.1. Cofilin acts as a substrate of Aurora A
LIMK1/2 act as the bona-fide kinases for inactivating phosphorylation of Cofilin [23] but treatment with BMS-5, a specific inhibitor of LIMK1/2 catalytic activity did not completely inhibit Cofilin phosphorylation. Although a significantly decreased phosphorylation of Cofilin was noted after treatment with BMS-5 compared to the vehicle control, DMSO (Fig. 1A) a small amount of phosphorylated Cofilin was still detectable. This suggests that either the kinase activity of LIMK1 is not completely blocked by BMS-5 or a different kinase, may be responsible for Cofilin phosphorylation. In our previous studies we identified a novel interaction between LIMK1 and Aur-A at the centrosomes [8], which prompted us to investigate if Aur-A is responsible for the remaining Cofilin phosphorylation. To determine if Cofilin is a substrate of Aur-A, we performed in vitro kinase assays with recombinant His-tagged Cofilin and Aur-A (Fig. 1B&C). A radioactive polypeptide band corresponding to the size of Cofilin was detected after incubation with Aur-A (Fig. 1C, lane 3). To further confirm that Cofilin is a substrate of Aur-A, we performed an immunocomplex kinase assay (Fig. 1D&E). Endogenous Aur-A was immunoprecipitated from asynchronous PC-3 cell lysate with anti-Aur-A antibodies and incubated with recombinant His-tagged Cofilin and γ-32P-ATP (Fig. 1D&E). Results showed phosphorylation of recombinant Cofilin by the immunoprecipitated Aur-A (Fig. 1E, lane 2). However, our previous studies showed that LIMK1 co-precipitates with Aur-A so it is possible that the phosphorylation seen may be due to a combination of both LIMK1 and Aur-A activities on Cofilin [8]. Together, this data confirms that Cofilin acts as a substrate of Aur-A.
Figure 1. Phosphorylation of Cofilin by Aurora A.

A: Western blot analysis of PC3 cells treated with either DMSO or BMS-5 (5μM) (LIMK1/2 inhibitor) for 24hr. Immunoblotting with anti-pS3-Cofilin and anti-GAPDH (loading control) antibodies show reduced cofilin phosphorylation after treatment with BMS-5 compared to the DMSO control. B & C: In vitro kinase assays with recombinant His-Cofilin (1μg) and His-Aurora A (0.22μg). B: SDS-PAGE showing the location of polypeptide bands. C: Autoradiogram showing phosphorylation of His-Cofilin. D & E: Immunocomplex kinase assays of immunoprecipitated Aur-A and His-Cofilin (1μg). Aurora A was immunoprecipitated from PC3 whole cell lysates (500μg) with anti-Aur-A antibodies and used in a kinase assay with recombinant His-Cofilin. D: SDS-PAGE showing the location and loading of Cofilin polypeptides. E: Autoradiogram of phosphorylated Cofilin.
3.2. Aurora A Phosphorylated Cofilin at S3, S8, and T25
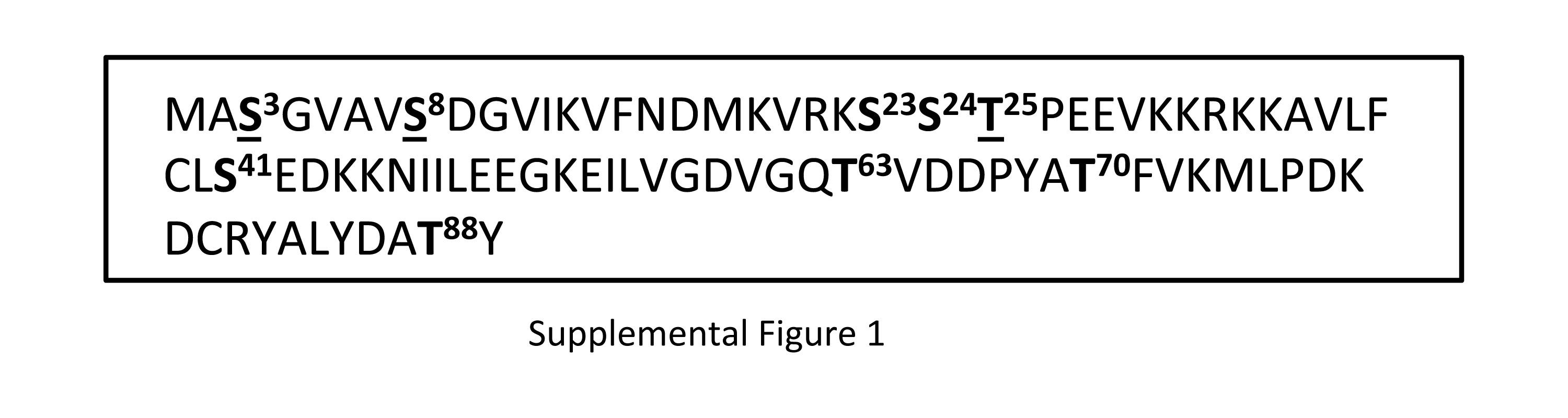
Cofilin activity is regulated by phosphorylation/dephosphorylation of its main phosphorylation site, S3. To determine if Aur-A phosphorylates Cofilin at S3, we performed in vitro kinase assays with a nonphosphorylatable S3A mutant Cofilin (CofilinS3A) (Fig. 2A&B). Phosphorylation of recombinant His-CofilinS3A by His-Aur-A (lane 2) was reduced compared to phosphorylation of wild-type His-Cofilin, suggesting that S3 is a site of phosphorylation by Aur-A. Because phosphorylation of CofilinS3A was reduced compared to wild-type Cofilin but not eliminated, it can be speculated that Aur-A phosphorylates Cofilin at additional residue(s). To identify the additional sites of phosphorylation, we performed phosphopeptide analysis of recombinant wild type full-length Cofilin subjected to in vitro non-radioactive kinase assays with recombinant wild type His-tagged Aur-A or catalytically inactive His-Aur-AK162M. Mass spectrometric analysis detected two phosphopeptides containing the phosphorylated residues S3, S8, and T25 in the sample incubated with active Aur-A (Fig. 2C). Phosphorylation at these sites was not detected in the sample incubated with inactive Aur-AK162M (data not shown). To confirm these results, we expressed recombinant His-tagged triple mutant Cofilin (CofilinS3A/S8A/T25A) in which these three residues were mutated to Alanine and used for in vitro kinase assays. Results showed CofilinS3A/S8A/T25A was still phosphorylated by Aur-A (Fig. 2E, lane 3), which was not detected when incubated with inactive Aur-AK162M (lane 7). Phosphorylation of CofilinS3A/S8A/T25A by Aur-A was reduced compared to phosphorylation of CofilinS3A, suggesting that these sites are phosphorylated by Aur-A but additional site(s) may also be phosphorylated by Aur-A. To broadly identify Cofilin fragments containing other possible phosphorylation sites, we expressed recombinant His-tagged C-terminal fragment of Cofilin containing amino acids 90-166 (Cofilin90-166) and used for in vitro kinase assays. Our results showed that Cofilin90-166 was not phosphorylated by Aur-A (Fig 2D, lane 4), suggesting that putative additional phosphorylation sites in Cofilin are between amino acids 1–89. Other than S3, S8, and T25, possible additional phosphorylation sites within this region are S23, S24, S41, T63, T70, and T88 (Supplemental Fig. 1).
Figure 2. Aurora A Phosphorylated Cofilin at Specific Sites.

A & B: In vitro kinase assays with recombinant His-Cofilin, His-CofilinS3A mutant, and His-Aur-A. A: Coomassie stained SDS-PAGE showing protein location and loading. B: Autoradiogram showing reduced phosphorylation of His-CofilinS3A compared to His-Cofilin. C: Phosphopeptide analysis of phosphorylated cofilin by mass spectroscopy. Two phosphopeptides were detected containing a total of three sites phosphorylated by Aur-A. D & E: In vitro kinase assays of recombinant wild type His-Cofilin and His-CofilinS3A, His-CofilinS3A/S8A/T25A, and His-Cofilin90-166 mutants using His-Aurora A or His-Aurora AK162M mutant. D: Coomassie stained SDS-PAGE showing protein location and loading. E: Autoradiogram showing phosphorylation of His-Cofilin, His-CofilinS3A, and His-CofilinS3A/S8A/T25A. No phosphorylation of His-Cofilin90-166 could be detected.
3.3. Phosphorylation by Aurora A Reduced the Actin Depolymerizing Activity of Cofilin
To examine the effect of phosphorylation of Cofilin by Aur-A on its actin modulatory function, we performed actin polymerization assays to assess the functional status of Cofilin. Wild-type recombinant His-Cofilin depolymerized F-actin as reduced Phalloidin staining and reduced lengths of F-actin were noted compared to the actin only control (Fig. 3A). Next, we examined the depolymerizing activity of His-Cofilin, His-CofilinS3A, and His-CofilinS3A/S8A/T25A after phosphorylation by His-Aur-A (Fig. 3B&C). His-Cofilin incubated with inactive His-Aur-AK162M was more active than His-Cofilin incubated with His-Aur-A as noted by the reduced length of F-actin and the reduced intensity of Phalloidin staining. Phosphorylation of His-CofilinS3A by His-Aur-A reduced its activity compared to His-CofilinS3A incubated with His-Aur-AK162M. Additionally, His-CofilinS3A/S8A/T25A incubated with His-Aur-A was significantly more active than His-CofilinS3A incubated with Aur-A, suggesting that phosphorylation at S8 or T25 may regulate Cofilin activity. Together, this data suggests that phosphorylation by Aur-A negatively regulates Cofilin activity via phosphorylation.
Figure 3. Phosphorylation by Aurora A Reduced Actin Depolymerizing Activity of Cofilin.

A: Images showing the depolymerization of actin by Cofilin. Decreased Phalloidin staining of F-actin could be noted in the presence of His-Cofilin. B: Recombinant His-Cofilin, His-CofilinS3A, or His-CofilinS3A/S8A/T25A mutants were in vitro phosphorylated by His-Aur-AK162M (top panels) or His-Aur-A (bottom panels) and incubated with polymerized actin and stained with Phalloidin. C: Quantification of actin filament length from B. Incubation with phosphorylated His-Cofilin or His-CofilinS3A mutant by inactive Aur-A reduced Phalloidin staining compared to His-cofilin or His-CofilinS3A phosphorylated with active Aur-A. Incubation with phosphorylated His-CofilinS3A/S8A/T25A by active Aur-A partially retained Cofilin activity as noted by shorter fragments of Phalloidin stained F actin compared to His-Cofilin or His-CofilinS3A. Data is representative of ten longest actin filaments each in 15 fields of two independent experiments. Scale bar: 25μm, *p<0.05
3.4. Inhibition of Aurora Kinases Decreased the Distribution of F-Actin
Next, we wanted to examine the effect of Aurora A activity on actin polymerization in vivo. MCF7 cells were treated with the pan-Aurora inhibitor, VX-680, or the vehicle and F-actin status was monitored by staining with Phalloidin (Fig. 4A–C). The mean intensity of F-actin was reduced to ~50% in cells treated with VX-680 compared to vehicle treated cells. This data suggests that actin depolymerizing activity of Cofilin was higher in cells treated with VX-680.
Figure 4. Inhibition of Aurora Kinases Reduced the Levels of F-Actin.

A: Immunofluorescence analysis of MCF7 cells treated with either VX-680 (100nM) or DMSO for 24 hrs. F-actin (green) was visualized by staining with Phalloidin. DNA was stained with DAPI (blue). B: Phalloidin staining from cells in A was imaged after increasing exposure time to show actin staining in detail within the cell. C: Quantitation of the mean intensity of Phalloidin staining. Data is representative of 150 cells from two independent experiments. Scale bar: 10μm.
3.5. Mutation of Aurora A Phosphorylation Sites on Cofilin Caused Mislocalization of Cofilin
To examine the effect of phosphorylation at S3, S8, and T25 by Aur-A we prepared a mammalian expression construct of non-phosphorylatable RFP-tagged Cofilin in which all three phosphorylation sites were mutated to alanines (CofilinS3A/S8A/T25A-RFP). M12 cells were transfected with either wild type RFP-tagged Cofilin (Cofilin-RFP) (Fig. 5A) or RFP-tagged CofilinS3A/S8A/T25A (Fig. 5B) for 48 hours. In cells expressing lower amounts of Cofilin-RFP (top panel), Cofilin-RFP localized primarily to the perinuclear region (white arrows). In cells expressing higher amounts of Cofilin-RFP (bottom panel), the expressed protein was also localized throughout the cell although in some areas accumulation of Cofilin-RFP could be seen. CofilinS3A/S8A/T25A-RFP, however, did not show specific localization to the perinuclear region (bottom panels). Cells expressing lower amounts of CofilinS3A/S8A/T25A-RFP (top panel) showed punctate localization of the expressed Cofilin throughout the cytoplasm while in cells with higher amounts of expressed protein (bottom panel), diffuse localization of CofilinS3A/S8A/T25A-RFP throughout the cytoplasm could be noted. Both proteins colocalized with F-actin (yellow arrows), but to a lesser extent for CofilinS3A/S8A/T25A-RFP. This data suggests that phosphorylation by Aur-A regulates subcellular localization of Cofilin.
Figure 5. Mutation of Aurora A Phosphorylation Sites Resulted in the Mislocalization of Cofilin.

Fluorescence microscopy of M12 cells transfected with Cofilin-RFP (A) or CofilinS3A/S8A/T25A-RFP (B). F-actin was stained with Phalloidin-488 (green) and DNA was stained with DAPI (blue). Cofilin-RFP localized to the perinuclear region (white arrows) while CofilinS3A/S8A/T25A-RFP showed diffuse staining throughout the cell. Colocalization of the wild type Cofilin and the mutant Cofilin with F-actin could be noted (yellow arrows). Scale bar: 10μm. Top panel: cells expressing lower amounts of Cofilin RFP or CofilinS3A/S8A/T25A-RFP; bottom panel: cells expressing higher amounts of Cofilin-RFP or CofilinS3A/S8A/T25A-RFP.
3.6. Aurora A Physically Associates with Cofilin During Mitosis
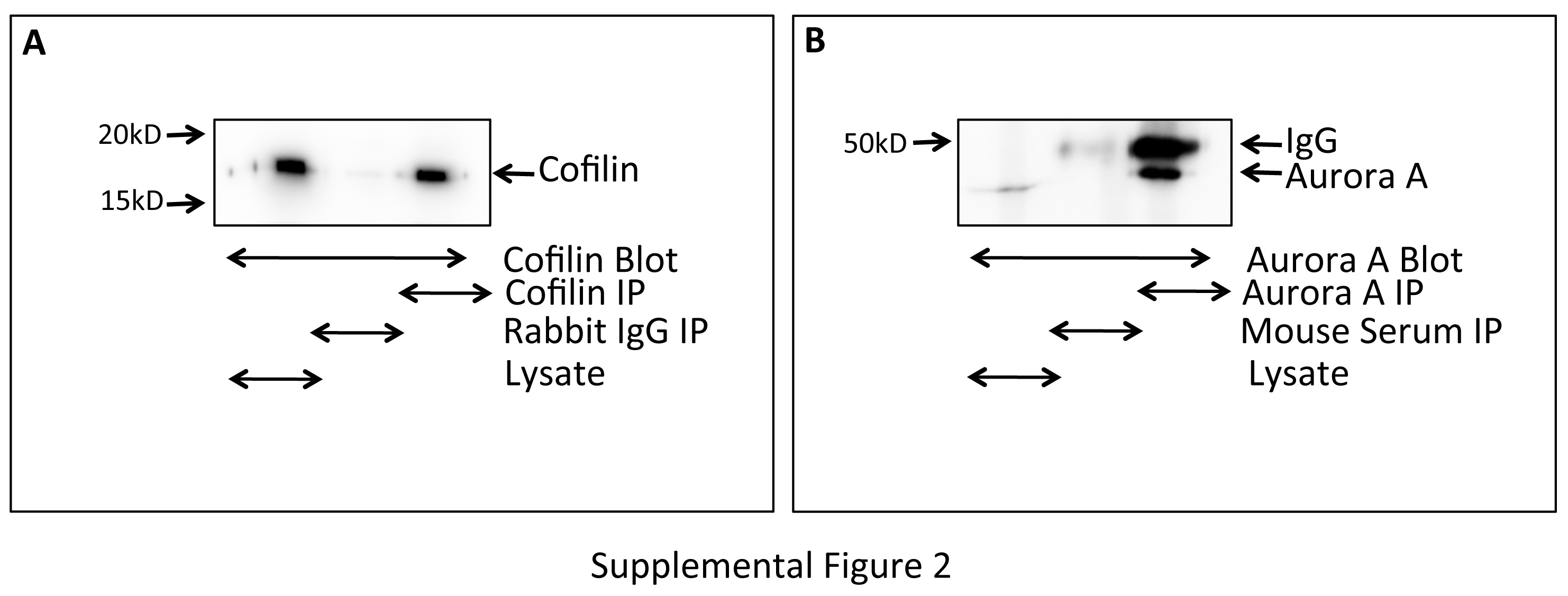
Aur-A is primarily expressed from late G2 throughout mitosis. In our next experiment, we wanted to examine if Aur-A and Cofilin interact during mitosis. M12 cells synchronized at the G2/M boundary were isolated by shake off and released into mitosis for 0, 30, and 60 mins. Aur-A was immunoprecipitated from mitotic cell extracts using anti-Aur-A antibodies and co-precipitated Cofilin was detected by immunoblotting. Cofilin was precipitated equally in all time points, which suggests that Cofilin and Aur-A interact throughout the early mitotic phases (Fig. 6A). The interaction was confirmed using NIH-3T3 cell extracts in which Cofilin was precipitated with Aur-A in all time points (Fig. 6B). Specificity of the antibodies was detected by immunoprecipitating Cofilin and Aurora from Nocodazole treated extracts. Immunoprecipitated antigens were detected by immunoblotting with anti-Cofilin or anti-Aurora A antibodies (Supplemental Fig. 2). This result suggests that Aur-A may play a role in the regulation of Cofilin activity during mitosis.
Figure 6. Interaction of Aurora A with Cofilin During Mitosis.

Coimmunoprecipitation of Cofilin with Aur-A in Nocodazole treated M12 or NIH-3T3 cell extracts harvested at different times after release. Aur-A was immunoprecipitated using anti-Aurora A antibodies, and Cofilin was detected by immunoblotting using anti-Cofilin antibodies. Mouse IgG was used as a control. Data represents the results of three independent experiments.
3.7. Inhibition of Aurora A Activity Altered Cofilin Phosphorylation During Mitosis
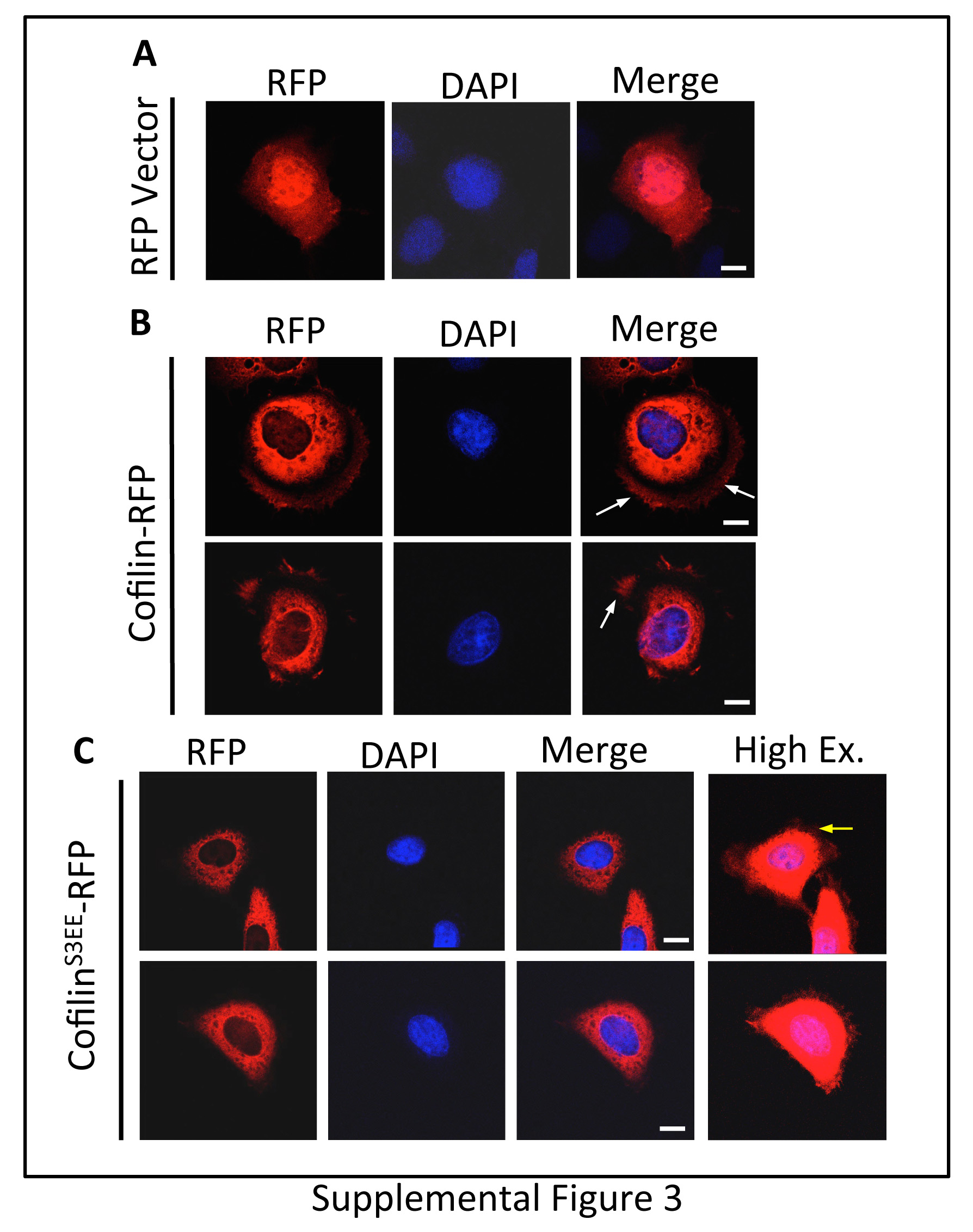
Next, we examined the association of Aur-A catalytic activity with Cofilin phosphorylation during mitosis. M12 cells were treated with the Aur-A specific inhibitor, MLN8237, or DMSO and synchronized at the G2/M boundary with nocodazole. Mitotic cells were collected and released for 0, 30, and 60 mins. Phosphorylated-Cofilin (pS3) and total Cofilin were detected in mitotic cell extracts by immunoblotting (Fig. 7A&B). Total Cofilin levels in DMSO and MLN8237 treated cells remained relatively constant in all time points but phospho-Cofilin levels fluctuated. In DMSO treated cells, Cofilin phosphorylation was highest at 30 mins (~1.5-fold increase compared to 0 hr) and barely detectable at 60 mins (~0.5-fold decrease compared to 0 hr). This is in support of an earlier study showing Cofilin phosphorylation during mitosis [30]. Interestingly, MLN8237 treated cells had low levels of phospho-Cofilin at 0 hr, but a > 4-fold increased level at 30 and 60 mins. Total Cofilin decreased slightly at 30 and 60 mins in MLN8237 treated cells compared to DMSO treated cells (Fig. 7C). MLN8237 treated cells contained ~70% less phospho-Cofilin compared to DMSO treated cells at 0 hr (Fig. 7C). From 0 to 30 mins, Cofilin phosphorylation increased ~4-fold in MLN8237 treated cells to a level about equal to that in DMSO treated cells. However, between 30 and 60 mins Cofilin phosphorylation in DMSO treated cells decreased while phosphorylation in MLN8237 treated cells did not change, causing ~2.5-fold difference in phosphorylation between the two treatments. This data suggests that Aur-A plays a role in the regulation of Cofilin activity during mitosis. To assess any change in the localization of phospho-Cofilin in the early mitotic stage, we expressed RFP-tagged phosphomimic mutant of Cofilin (CofilinS3EE) in M12 cells. Cells were transfected in nocodazole containing medium, incubated for 24 hrs and released for 30 mins. A distinct difference in sub-cellular localization of CofilinS3EE-RFP (inactive) compared to the wild type Cofilin-RFP was noted in the early mitotic stage. The cells expressing wild type Cofilin- RFP showed a punctate staining at the periphery, while the distribution of CofilinS3EE-RFP is more homogeneous (Fig. 7D, Supplemental Fig. 3). Differential targeting of phospho-Cofilin may have implication in altered actin dynamics in the early mitotic stages.
Figure 7. Inhibition of Aurora A Activity Altered Cofilin Phosphorylation and Phosphor-Cofilin Showed Differential Targeting in the Mitotic Cells.

Western blot analysis of endogenous Cofilin (A) and phospho-Cofilin (pS3) (B) in nocodazole treated M12 cell extracts released at different times with treatment with MLN8237 (100nM) or the vehicle. Anti-Cofilin and anti-phospho-Cofilin antibodies were used for the immunoblots. GAPDH expression was used as the loading control. Values below each figure indicates relative protein levels normalized to 0 minute expression (not released from G2/M boundary). C: Densitometric analysis of Cofilin and phospho-Cofilin in MLN8237 treated cells compared to DMSO treated cells. Data shows mean±SD of three independent experiments. D: Localization of CofilinS3EE-RFP and Cofilin-RFP in nocodazole synchronized M12 cells released for 30 mins. Spindle α-tubulin (green) was visualized by staining with anti-α-tubulin antibodies. DNA was stained with DAPI (blue). Cofilin-RFP showed a distinct punctate localization at the cell periphery whereas CofilinS3EE-RFP distribution was homogeneous surrounding the DNA at the early mitotic stages. Scale bar: 10 μm E: Western blot analysis of pS3-Cofilin in extracts of nocodazole synchronized M12 cells treated with BMS-5 (5μM) singly or in combination with MLN8237 (100nM) using anti-pS3-Cofilin antibodies. Cells were released into mitosis and harvested at different times. GAPDH expression was used as the loading control Lys: untreated whole cell lysate. Data is representative of at least three independent experiments.
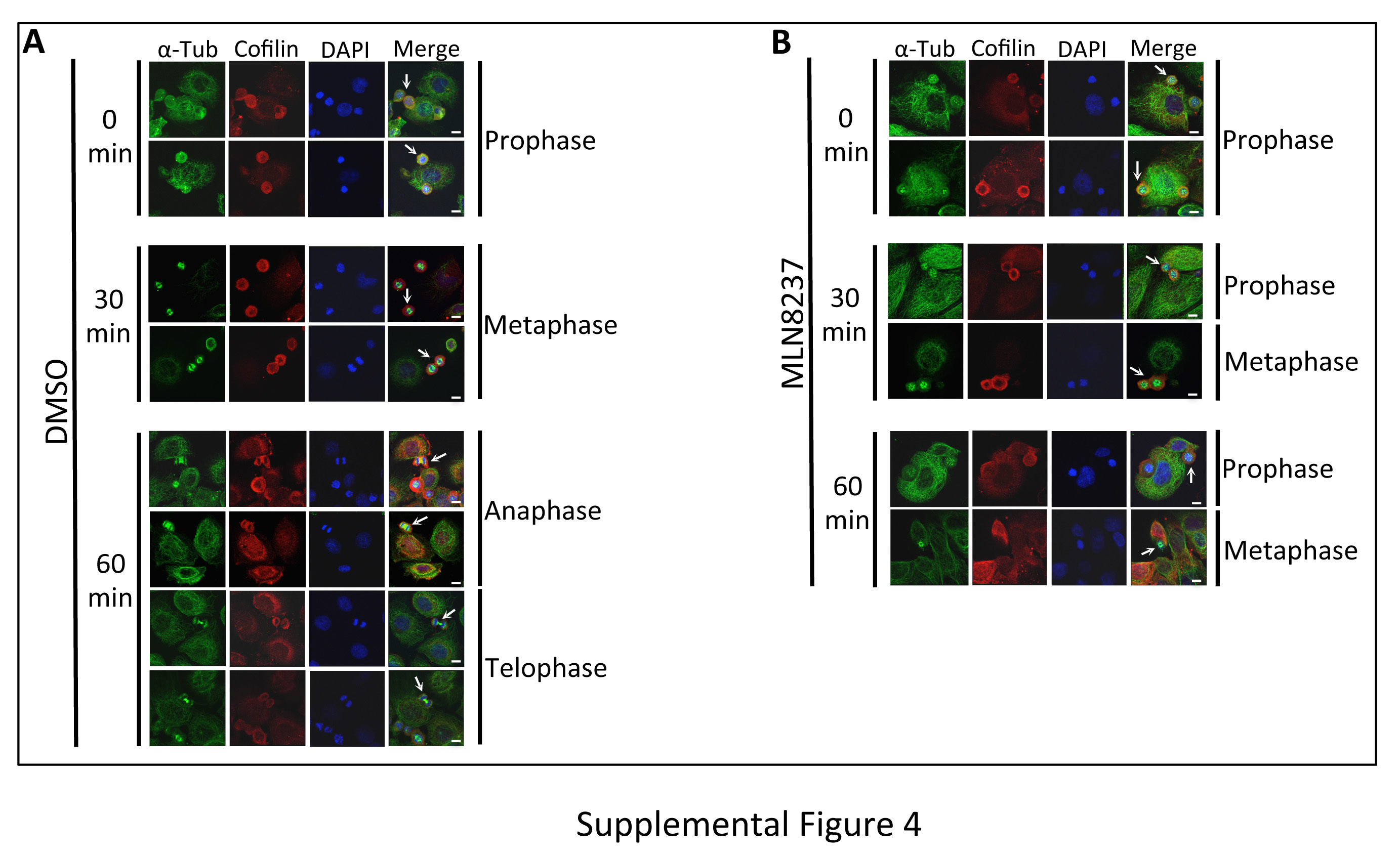
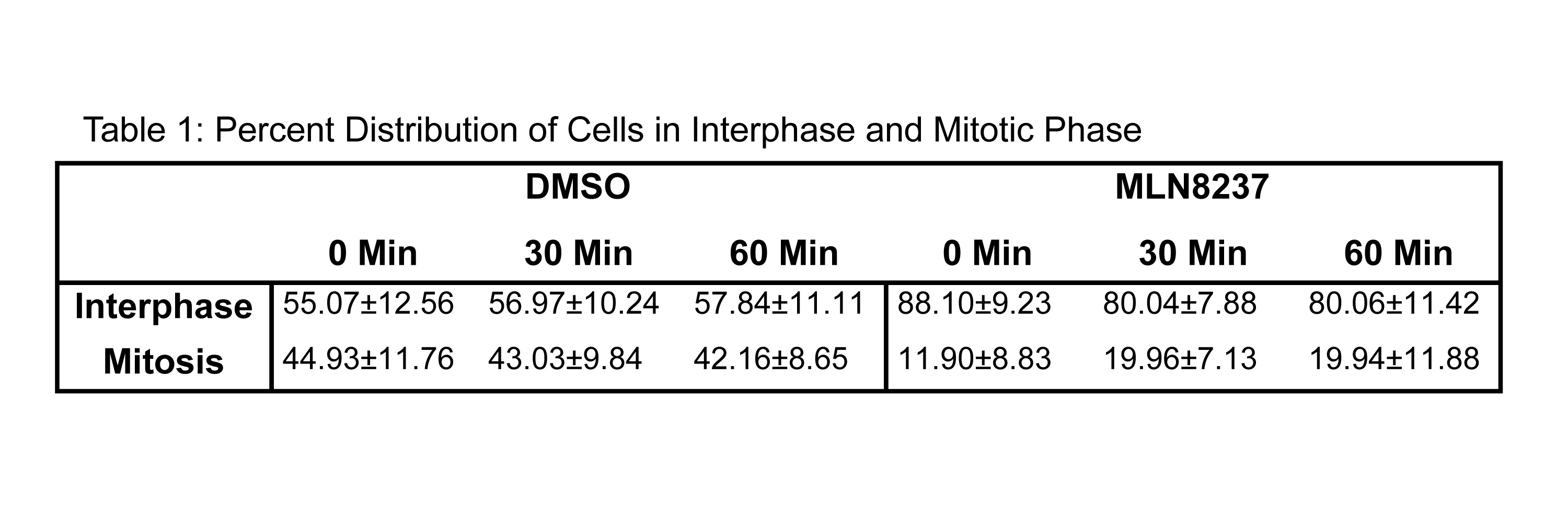
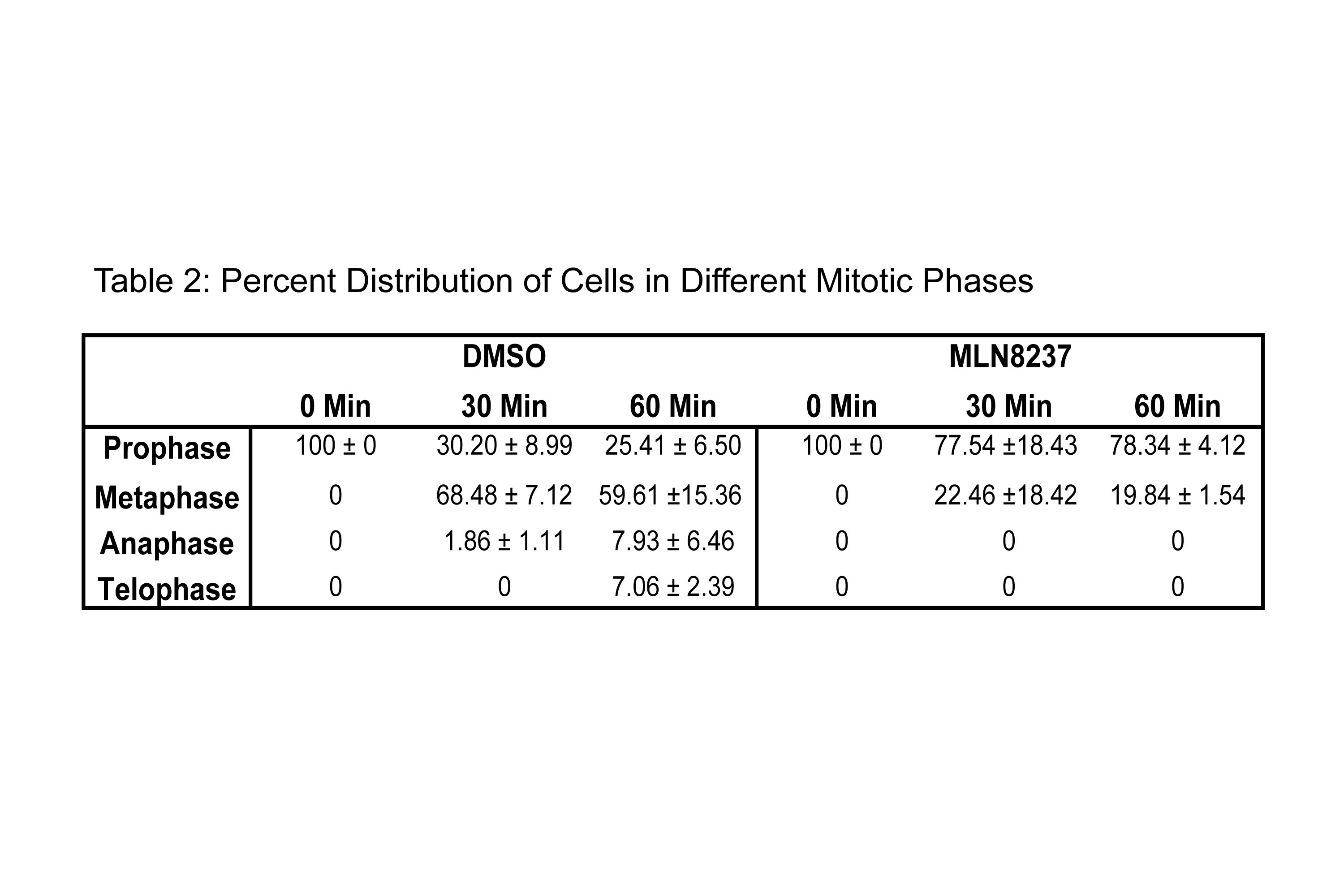
To coordinate the mitotic phases with Cofilin phosphorylation, we evaluated the stages of mitosis in MLN8237 treated cells as Aur-A inhibition has been shown to cause a mitotic delay [10]. We used immunofluorescence analysis to quantify the distribution of cells released in fresh medium in each mitotic phase at each time point in MLN8237 treated cells (Fig. 8A–D, Supplemental Fig. 4 and Table S1&2). DMSO treated cells had a higher percentage of cells in mitosis (~40% at each time point) compared to MLN8237 treated cells (~20% of cells at each time point) (Fig. 8C and Table S1). In DMSO treated cells, quantitative analysis of mitotic phases in DMSO treated cells showed that ~30.2%, ~68.48%, ~1.86%, and 0% of cells were in prophase, metaphase, anaphase, and telophase respectively, at 30 mins. At 60 mins, cells progressed to anaphase and telophase as evident from ~25.41%, ~59.61%, ~7.93%, and ~7.06% of cells in prophase, metaphase, anaphase, and telophase, respectively (Fig. 8D & Table S2). Treatment with Aur-A inhibitor caused a delay in mitotic progression as evident from ~77.54% and ~22.46% of cells at 30 mins and ~78.34% and ~19.84% of cells at 60 mins in prophase and metaphase, respectively. No cells in anaphase or telophase were noted at 60 mins. This data suggests that the alteration of Cofilin phosphorylation and phospho-Cofilin localization may be associated with the mitotic delay induced by the inhibition of Aur-A activity.
Figure 8. Treatment with Aur-A Inhibitor Delayed Progression of Cells Through Prophase.

Immunofluorescence analysis of DMSO (A) or MLN8237 (100nM) (B) treated and nocodazole synchronized M12 cells released into mitosis for 0, 30, or 60 mins. α-tubulin (green) and Cofilin (red) were visualized by staining with anti-α-tubulin and anti-Cofilin antibodies. DNA was stained with DAPI (blue). Mitotic cells presented are enlarged images from Supplemental Figure 4. C: Quantitation of the percent of cells in interphase or mitosis. Data shows average numbers of cells counted in 20 fields each from two separate experiments. D: Quantitation of the percent of cells in each mitotic phase. Data shows average numbers of cells counted in 20 random fields each from two separate experiments. Scale bar: 5μm.
3.8 Inhibition of Aurora A Activity Altered Slingshot-1 Expression During Mitosis
It has been shown that overexpression of Aur-A can increase the expression of Slingshot-1 phosphatase (SSH-1) [31]. Hence, we wanted to examine if inhibition of Aur-A altered expression of SSH-1. M12 cells were treated with either MLN8237 or DMSO and synchronized to the G2/M boundary with nocodazole. Mitotic cells were isolated by mitotic shake off and released into mitosis with fresh media containing either MLN8237 or DMSO. SSH-1 expression was detected in mitotic extracts by immunoblotting (Fig. 9A&B). In DMSO treated cells, SSH-1 expression increased through 60 mins. SSH-1 expression in MLN8237 treated cells followed a similar trend but expression was significantly lower in all time points compared to DMSO treated cells. Together, this data confirms that Aur-A modulates SSH-1 expression during early mitotic phases.
Figure 9. Inhibition of Aurora A Activity Altered Slingshot-1 Expression.

A: Western blot analysis of SSH-1 expression in nocodazole treated M12 extracts released at different times with treatment of MLN8237 (100nM) or vehicle. Anti-SSH-1 antibodies were used for the immunoblots and α-tubulin expression was used as the loading control. B: Densitometric analysis of SSH-1 expression in MLN8237 and DMSO treated cells. Data is representative of at least three independent experiments. *p=<0.05. **p=<0.005.
3.9. Both Aurora A and LIMK1 contribute to Cofilin Phosphorylation in the Early Mitotic Phase
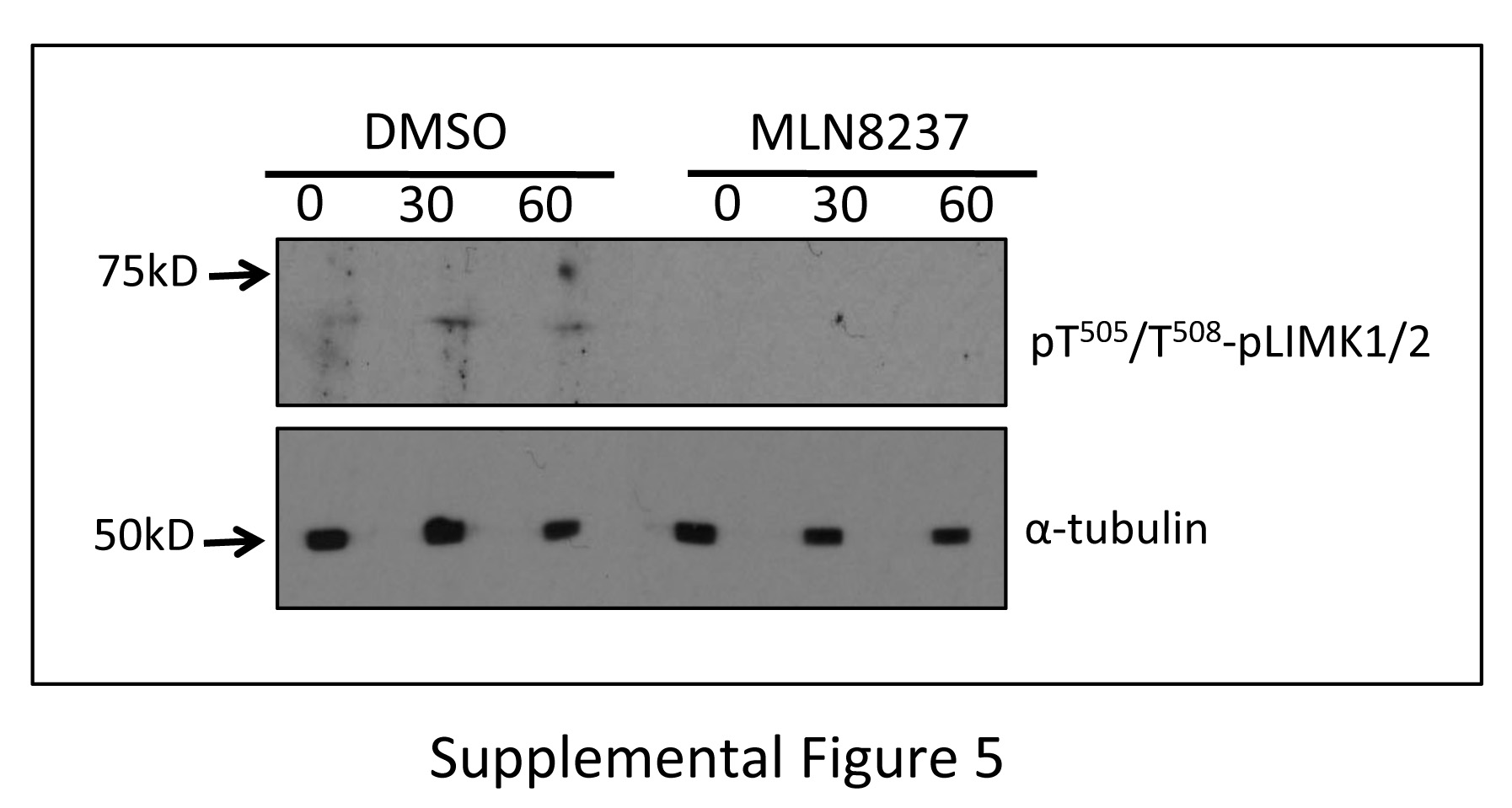
Recently, a bidirectional functional relationship between Aur-A and LIMK1 during mitosis has been demonstrated [8]. Earlier it was shown that LIMK1 phosphorylates Cofilin during mitosis [32]. To determine the contribution of LIMK1/2 in maintaining phospho-Cofilin levels during early mitotic phases, we examined the effect of the LIMK1/2 inhibitor BMS-5 on Cofilin phosphorylation by western blot analysis. We noted a significant reduction in phospho-Cofilin (pS3) levels in all time points in released M12 cells treated with BMS-5, which was further reduced to undetectable levels upon combination treatment of BMS-5 and MLN8237 (Fig. 7E). Because Aur-A phosphorylates and activates LIMK1 [8], we examined the activation status of LIMK1 during mitosis in cells treated with MLN8237. It could be noted from our results that phosphorylated LIMK1/2 was barely detectable in DMSO treated cells but was undetectable in MLN8237 treated cells (Supplemental Fig. 5). Since, low levels of pLIMK1/2 were detected in DMSO treated cells it is more likely that pLIMK1/2 is further lowered in MLN8237 treated cells rather than completely absent. Together, this data suggests that both LIMK and Aur-A participate in the regulation of Cofilin phosphorylation during mitosis.
4.0 Discussion
In this study, we show a novel interaction between Aur-A and Cofilin. Our study identified that Cofilin acts as a substrate of Aur-A, which phosphorylates Cofilin at multiple sites including S3, S8, and T25. Phosphorylation at S3 renders Cofilin inactive by blocking its binding to actin. Therefore, one role of Aur-A phosphorylation is to regulate the activity of Cofilin. Serine8 phosphorylation has been mentioned in two proteomics studies [33, 34] but has never been experimentally confirmed therefore, the consequence of this phosphorylation is unknown. Threonine25 phosphorylation has also been noted in a number of proteomics studies [34–36] including a mitotic phase proteomics study [37], but the function of this phosphorylation is also unknown.
In vitro phosphorylation of the CofilinS3A/S8A/T25A mutant suggested additional residues are phosphorylated by Aur-A. Because the C-terminal fragment of Cofilin (residues 90–166) was not phosphorylated by Aur-A, the additional phosphorylation sites most likely lie between amino acids 1–89. Two putative residues are S23 and S24 (RKSST) because they share a partial homology with the Aur-A phosphorylation motif ([K/N/R]-R-X-[pS/pT]-V) with a bias at the n+1 position. Interestingly the phosphorylation motif of Aur-A maintains that the n+1 position must not be a proline residue while T25 precedes a proline residue. Additionally, T63 and T70 may be phosphorylated by Aur-A but their phosphorylation would not have been detected by mass spectroscopy because the tryptic digestion would not have produced a peptide containing these residues. Serine41 and T88 are two residues that could have been the additional phosphorylation sites but were not detected by mass spectrometry.
Phosphorylation by Aur-A negatively regulates Cofilin activity as noted in actin polymerization assays. Phosphorylation at S3 inactivates Cofilin by preventing its ability to bind actin filaments. Therefore, reduced actin depolymerization by wild-type recombinant Cofilin incubated with His-Aur-A may be due to phosphorylation specifically at this site. Importantly, His-CofilinS3A activity was also noticeably reduced upon phosphorylation by His-Aur-A, while His-CofilinS3A/S8A/T25A retained a significantly higher level of activity when incubated with His-AurA. This data suggests that phosphorylation at residues in addition to S3 are involved in the regulation of the depolymerization activity of Cofilin. Since S8 is in close proximity to S3, phosphorylation at that site may result in a similar conformational change that would prevent binding to actin. Inhibition of Aur-A activity was also correlated with the reduced levels of F-actin in vivo. Taken together, it is likely that the alteration in F-actin by Aur-A was mediated through Cofilin.
Aur-A phosphorylation of Cofilin also influences intracellular localization of Cofilin. Wild type Cofilin-RFP and CofilinS3A/S8A/T25A-RFP showed distinct differences in subcellular localization. Cofilin has been reported to localize to the Golgi to aid in cargo sorting and fusion of carrier vesicles [38–40]. Aur-A may regulate Cofilin localization to this area through phosphorylation.
Our results also showed that Aur-A and Cofilin interact during mitosis and that this interaction is maintained during mitotic progression from prophase to telophase. However, the activation status of Cofilin through phosphorylation changes as cells progress through the mitotic phases. Phospho-Cofilin levels are at the peak when cells are mostly in prophase and metaphase but declined significantly as the cells start to progress to anaphase. It can be speculated that actin depolymerization is required as the spindles start to change shape and elongate during anaphase possibly through interaction with cortical actin. Interestingly, inhibition of Aur-A activity resulted in a sustained increase in phospho-Cofilin levels, which is counterintuitive of decreased phospho-Cofilin as a result of the inactivation of Aur-A. Importantly, MLN8237 treated cells showed a delayed progression of mitosis, as the majority of the cells are in prophase and only a small percentage of cells in metaphase. It can be speculated that inhibition of Aur-A activity induced mitotic delay is partly mediated by the failure of Cofilin-mediated depolymerization of actin. A differential localization of phosphomimic Cofilin at this stage is also indicative of altered actin dynamics. However, the question that we ask is how phospho-Cofilin levels increased upon inhibition of Aur-A kinase activity. We speculate that LIMK1 and SSH-1 phosphatase mediated Cofilin phosphorylation/dephosphorylation is responsible for the optimum phospho-Cofilin levels during mitosis. We have previously reported that Aur-A phosphorylates LIMK1 during mitosis, activating the protein and regulating its localization to the centrosomes [8]. An earlier report showed that Aur-A regulates SSH-1 expression, as Aur-A overexpression led to increased expression of SSH-1 and dephosphorylation of Cofilin in asynchronous cells [14]. Our data support this finding as SSH-1 expression was significantly reduced after treatment with MLN8237. Earlier studies indicated that the regulation of Cofilin phosphorylation is essential for cytokinesis as the accumulation of F-actin during late mitotic stages can cause cytokinesis defects leading to multinucleate cells [41]. Overexpression of LIMK1 also led to cytokinesis defects through enhanced F-actin via the inactivation of Cofilin [42]. Additionally, overexpression of a phosphatase inactive SSH1 mutant caused cytokinesis defects through increased levels of phospho-Cofilin and F-actin enhancement [32]. Our study demonstrate that the regulation of Cofilin phosphorylation is also important for progression of the early mitotic phases.
Based on these observations we propose a Cofilin phosphorylation model during mitosis (Fig. 10). In early mitotic phases, LIMK1 and Aur-A phosphorylate and inactivate Cofilin (Fig. 10D and A) while at the later stages SSH-1 inactivates LIMK1 by removing the phosphate group at T508 [43] (Fig. 10G), and additionally, dephosphorylates and activates Cofilin (Fig. 10F) [32]. Aur-A being a key regulator of early mitotic phases is participating in maintenance of phospho-Cofilin levels through the activation of LIMK1, and SSH-1 as a negative feedback loop (Fig. 10B and E), which possibly resulted in decreased phospho-Cofilin levels as cells start to progress to anaphase. It can be speculated that increased phosphorylation/inactivation of Cofilin following Inhibition of Aur-A activity by MLN8237 treatment may be linked with decreased levels of SSH-1 (Fig. 10E&F). Earlier studies showed that LIMK1 dependent phosphorylation of Cofilin is necessary for proper mitotic spindle orientation [28] and that treatment of cells with MLN8237 results in multipolar spindles and abnormal spindle morphology [8]. In conclusion, our data suggests that Aur-A functions in the early mitotic phases may be mediated, in part, through its control over Cofilin activity and actin polymerization.
Figure 10. Model of the Regulation of Cofilin Phosphorylation.

A: Cofilin, causing the inactivation of the protein leading to accumulation of F-actin. B: Aur-A phosphorylates LIMK1 at S307 priming it for full activation by phosphorylation at T508. C: Interaction with LIMK1 allows the activation of Aur-A through autophosphorylation at T288. D: LIMK1 phosphorylates Cofilin and inactivates it. E: Overexpression of Aur-A up regulates SSH-1. F: SSH-1 activates Cofilin in late mitosis through dephosphorylation leading to the depolymerization of F-actin. G: SSH-1 inactivates LIMK1 in late mitosis by dephosphorylation at T508.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Highlights.
Aurora A kinase phosphorylates Cofilin.
Aurora A regulates actin reorganization through Cofilin phosphorylation.
Aurora A maintains Cofilin phosphorylation status in early mitotic phases.
PhosphoCofilin level through the regulation of Slingshot expression.
Phosphorylated Cofilin shows differential localization in the early mitotic cells.
Acknowledgments
This work is supported by the research funding from the National Institute of Health (R15CA125681) and Department of Defense PCRP (PC041048) to RC.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
5.0. References
- 1.Hannak E, et al. Aurora-A kinase is required for centrosome maturation in Caenorhabditis elegans. J Cell Biol. 2001;155(7):1109–16. doi: 10.1083/jcb.200108051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Toji S, et al. The centrosomal protein Lats2 is a phosphorylation target of Aurora-A kinase. Genes Cells. 2004;9(5):383–97. doi: 10.1111/j.1356-9597.2004.00732.x. [DOI] [PubMed] [Google Scholar]
- 3.Conte N, et al. TACC1-chTOG-Aurora A protein complex in breast cancer. Oncogene. 2003;22(50):8102–16. doi: 10.1038/sj.onc.1206972. [DOI] [PubMed] [Google Scholar]
- 4.Mori D, et al. NDEL1 phosphorylation by Aurora-A kinase is essential for centrosomal maturation, separation, and TACC3 recruitment. Mol Cell Biol. 2007;27(1):352–67. doi: 10.1128/MCB.00878-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kufer TA, et al. Human TPX2 is required for targeting Aurora-A kinase to the spindle. J Cell Biol. 2002;158(4):617–23. doi: 10.1083/jcb.200204155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Koffa MD, et al. HURP is part of a Ran-dependent complex involved in spindle formation. Curr Biol. 2006;16(8):743–54. doi: 10.1016/j.cub.2006.03.056. [DOI] [PubMed] [Google Scholar]
- 7.Gruss OJ, Vernos I. The mechanism of spindle assembly: functions of Ran and its target TPX2. J Cell Biol. 2004;166(7):949–55. doi: 10.1083/jcb.200312112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ritchey L, et al. A functional cooperativity between Aurora A kinase and LIM kinase1: implication in the mitotic process. Cell Cycle. 2012;11(2):296–309. doi: 10.4161/cc.11.2.18734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hegarat N, et al. Aurora A and Aurora B jointly coordinate chromosome segregation and anaphase microtubule dynamics. The Journal of cell biology. 2011;195(7):1103–13. doi: 10.1083/jcb.201105058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Marumoto T, et al. Aurora-A kinase maintains the fidelity of early and late mitotic events in HeLa cells. J Biol Chem. 2003;278(51):51786–95. doi: 10.1074/jbc.M306275200. [DOI] [PubMed] [Google Scholar]
- 11.Li D, et al. Overexpression of oncogenic STK15/BTAK/Aurora A kinase in human pancreatic cancer. Clin Cancer Res. 2003;9(3):991–7. [PubMed] [Google Scholar]
- 12.Bischoff JR, et al. A homologue of Drosophila aurora kinase is oncogenic and amplified in human colorectal cancers. EMBO J. 1998;17(11):3052–65. doi: 10.1093/emboj/17.11.3052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Berdnik D, Knoblich JA. Drosophila Aurora-A is required for centrosome maturation and actin-dependent asymmetric protein localization during mitosis. Curr Biol. 2002;12(8):640–7. doi: 10.1016/s0960-9822(02)00766-2. [DOI] [PubMed] [Google Scholar]
- 14.Wang LH, et al. The mitotic kinase Aurora-A induces mammary cell migration and breast cancer metastasis by activating the Cofilin-F-actin pathway. Cancer Res. 2010;70(22):9118–28. doi: 10.1158/0008-5472.CAN-10-1246. [DOI] [PubMed] [Google Scholar]
- 15.Hirota T, et al. Aurora-A and an interacting activator, the LIM protein Ajuba, are required for mitotic commitment in human cells. Cell. 2003;114(5):585–98. doi: 10.1016/s0092-8674(03)00642-1. [DOI] [PubMed] [Google Scholar]
- 16.Moon W, Matsuzaki F. Aurora A kinase negatively regulates Rho-kinase by phosphorylation in vivo. Biochemical and biophysical research communications. 2013;435(4):610–5. doi: 10.1016/j.bbrc.2013.05.028. [DOI] [PubMed] [Google Scholar]
- 17.Uzbekov R, Kireyev I, Prigent C. Centrosome separation: respective role of microtubules and actin filaments. Biol Cell. 2002;94(4–5):275–88. doi: 10.1016/s0248-4900(02)01202-9. [DOI] [PubMed] [Google Scholar]
- 18.Whitehead CM, Winkfein RJ, Rattner JB. The relationship of HsEg5 and the actin cytoskeleton to centrosome separation. Cell Motil Cytoskeleton. 1996;35(4):298–308. doi: 10.1002/(SICI)1097-0169(1996)35:4<298::AID-CM3>3.0.CO;2-3. [DOI] [PubMed] [Google Scholar]
- 19.Thery M, et al. The extracellular matrix guides the orientation of the cell division axis. Nat Cell Biol. 2005;7(10):947–53. doi: 10.1038/ncb1307. [DOI] [PubMed] [Google Scholar]
- 20.Toyoshima F, Nishida E. Integrin-mediated adhesion orients the spindle parallel to the substratum in an EB1- and myosin X-dependent manner. EMBO J. 2007;26(6):1487–98. doi: 10.1038/sj.emboj.7601599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pelham RJ, Chang F. Actin dynamics in the contractile ring during cytokinesis in fission yeast. Nature. 2002;419(6902):82–6. doi: 10.1038/nature00999. [DOI] [PubMed] [Google Scholar]
- 22.Grigera PR, et al. Mass spectrometric analysis identifies a cortactin-RCC2/TD60 interaction in mitotic cells. Journal of proteomics. 2012;75(7):2153–9. doi: 10.1016/j.jprot.2012.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Arber S, et al. Regulation of actin dynamics through phosphorylation of cofilin by LIM-kinase. Nature. 1998;393(6687):805–9. doi: 10.1038/31729. [DOI] [PubMed] [Google Scholar]
- 24.Yang N, et al. Cofilin phosphorylation by LIM-kinase 1 and its role in Rac-mediated actin reorganization. Nature. 1998;393(6687):809–12. doi: 10.1038/31735. [DOI] [PubMed] [Google Scholar]
- 25.Toshima J, et al. Cofilin phosphorylation by protein kinase testicular protein kinase 1 and its role in integrin-mediated actin reorganization and focal adhesion formation. Mol Biol Cell. 2001;12(4):1131–45. doi: 10.1091/mbc.12.4.1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Toshima J, et al. Cofilin phosphorylation and actin reorganization activities of testicular protein kinase 2 and its predominant expression in testicular Sertoli cells. J Biol Chem. 2001;276(33):31449–58. doi: 10.1074/jbc.M102988200. [DOI] [PubMed] [Google Scholar]
- 27.Mizuno K. Signaling mechanisms and functional roles of cofilin phosphorylation and dephosphorylation. Cell Signal. 2013;25(2):457–69. doi: 10.1016/j.cellsig.2012.11.001. [DOI] [PubMed] [Google Scholar]
- 28.Kaji N, Muramoto A, Mizuno K. LIM kinase-mediated cofilin phosphorylation during mitosis is required for precise spindle positioning. J Biol Chem. 2008;283(8):4983–92. doi: 10.1074/jbc.M708644200. [DOI] [PubMed] [Google Scholar]
- 29.Davila M, et al. LIM kinase 1 is essential for the invasive growth of prostate epithelial cells: implications in prostate cancer. J Biol Chem. 2003;278(38):36868–75. doi: 10.1074/jbc.M306196200. [DOI] [PubMed] [Google Scholar]
- 30.Amano T, et al. Mitosis-specific activation of LIM motif-containing protein kinase and roles of cofilin phosphorylation and dephosphorylation in mitosis. J Biol Chem. 2002;277(24):22093–102. doi: 10.1074/jbc.M201444200. [DOI] [PubMed] [Google Scholar]
- 31.Wang LH, et al. The mitotic kinase Aurora-A induces mammary cell migration and breast cancer metastasis by activating the Cofilin-F-actin pathway. Cancer research. 2010;70(22):9118–28. doi: 10.1158/0008-5472.CAN-10-1246. [DOI] [PubMed] [Google Scholar]
- 32.Kaji N, et al. Cell cycle-associated changes in Slingshot phosphatase activity and roles in cytokinesis in animal cells. J Biol Chem. 2003;278(35):33450–5. doi: 10.1074/jbc.M305802200. [DOI] [PubMed] [Google Scholar]
- 33.Nagaoka R, Abe H, Obinata T. Site-directed mutagenesis of the phosphorylation site of cofilin: its role in cofilin-actin interaction and cytoplasmic localization. Cell Motil Cytoskeleton. 1996;35(3):200–9. doi: 10.1002/(SICI)1097-0169(1996)35:3<200::AID-CM3>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- 34.Wisniewski JR, et al. Brain phosphoproteome obtained by a FASP-based method reveals plasma membrane protein topology. J Proteome Res. 2010;9(6):3280–9. doi: 10.1021/pr1002214. [DOI] [PubMed] [Google Scholar]
- 35.Rigbolt KT, et al. System-wide temporal characterization of the proteome and phosphoproteome of human embryonic stem cell differentiation. Sci Signal. 2011;4 (164):rs3. doi: 10.1126/scisignal.2001570. [DOI] [PubMed] [Google Scholar]
- 36.Choudhary C, et al. Mislocalized activation of oncogenic RTKs switches downstream signaling outcomes. Mol Cell. 2009;36(2):326–39. doi: 10.1016/j.molcel.2009.09.019. [DOI] [PubMed] [Google Scholar]
- 37.Olsen JV, et al. Quantitative phosphoproteomics reveals widespread full phosphorylation site occupancy during mitosis. Sci Signal. 2010;3(104):ra3. doi: 10.1126/scisignal.2000475. [DOI] [PubMed] [Google Scholar]
- 38.Rosso S, et al. LIMK1 regulates Golgi dynamics, traffic of Golgi-derived vesicles, and process extension in primary cultured neurons. Mol Biol Cell. 2004;15(7):3433–49. doi: 10.1091/mbc.E03-05-0328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.von Blume J, et al. Actin remodeling by ADF/cofilin is required for cargo sorting at the trans-Golgi network. J Cell Biol. 2009;187(7):1055–69. doi: 10.1083/jcb.200908040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Salvarezza SB, et al. LIM kinase 1 and cofilin regulate actin filament population required for dynamin-dependent apical carrier fission from the trans-Golgi network. Mol Biol Cell. 2009;20(1):438–51. doi: 10.1091/mbc.E08-08-0891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Moulding DA, et al. Excess F-actin mechanically impedes mitosis leading to cytokinesis failure in X-linked neutropenia by exceeding Aurora B kinase error correction capacity. Blood. 2012;120(18):3803–11. doi: 10.1182/blood-2012-03-419663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yang X, et al. LATS1 tumour suppressor affects cytokinesis by inhibiting LIMK1. Nat Cell Biol. 2004;6(7):609–17. doi: 10.1038/ncb1140. [DOI] [PubMed] [Google Scholar]
- 43.Niwa R, et al. Control of actin reorganization by Slingshot, a family of phosphatases that dephosphorylate ADF/cofilin. Cell. 2002;108(2):233–46. doi: 10.1016/s0092-8674(01)00638-9. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.