

Abstract
We recently showed that germline transmission of mitochondrial DNA mutations via the oocyte cause aggravation of aging phenotypes in prematurely aging mtDNA mutator (PolgAmut/mut) mice. We discovered that 32% of these mice also exhibit stochastic disturbances of brain development, when maternal mtDNA mutations were combined with homozygosity for the PolgA mutation, leading to de novo somatic mtDNA mutations. Surprisingly, we also found that maternally transmitted mtDNA mutations can cause mild premature aging phenotypes also in mice with a wild-type nuclear DNA background. We now report that in addition to the early onset of aging phenotypes, these mice, burdened only by low levels of mtDNA mutations transmitted via the germline, also exhibit reduced longevity. Our data thus demonstrate that low levels of maternally inherited mtDNA mutations when present during development can affect both overall health and lifespan negatively.
The accumulation of mitochondrial DNA (mtDNA) mutations resulting in mitochondrial dysfunction has been heavily implicated in the aging process as well as various age-related disorders and diseases1,2,3,4. Replication of the mitochondrial genome continues in mitotic and meiotic cells, as well as in non-dividing cells, with an ~10-fold higher mutation rate than nuclear DNA1,2,3,4,5. Thus, mutations can occur in the maternal germline and be transmitted to offspring. Despite the presence of protective mechanisms that eliminate deleterious mtDNA mutations6,7, evidence indicates inheritability of low levels of heteroplasmy in humans8,9; however, the influence of such mutations on health and lifespan has been largely unclear.
To determine the extent to which inherited mtDNA mutations may contribute to the rate of aging, we designed a series of mouse mutants and previously demonstrated that germline mtDNA mutations can induce and augment aging phenotypes10. We also unexpectedly found that a combination of inherited and somatic mtDNA mutations cause stochastic brain malformations. These results suggest that starting life with healthy mitochondria might be important for the maintenance of health during aging. Interestingly, a recent study by Shen et al.11 supports this notion. The authors found that genetic mutations and environmental factors early in life of C. elegans affected the day-3 mitoflash frequency, which they found to predict lifespan. This suggests that the rate of aging may be set early in life before reproduction ends. We now present evidence to demonstrate that the presence of low levels of germline-transmitted mtDNA mutations during development can have life-long consequences not only by causing premature aging phenotypes, but also by shortening lifespan.
Results
Knock-in mice expressing a proofreading deficient version of the nucleus-encoded catalytic subunit of mtDNA polymerase-γ (PolgA)12 were used to test the extent to which inherited mtDNA mutations can contribute to the aging process. Homozygous PolgAmut/mut mice (mtDNA mutator mice) develop high levels of point mutations (20–30 mutations per mtDNA molecule) and linear deletions (~25% of total mtDNA). The mtDNA mutator mice show many signs of premature aging seen in humans, including reduced lifespan (~42–43 wks), alopecia, weight loss, anemia, sarcopenia, loss of subcutaneous fat, reduced fertility, impaired hearing13, and osteoporosis. Elevated brain lactate levels14,15 and reduced cytochrome c oxidase (COX) activity10,12,14,15,16,17 have also been reported in all similar models of mtDNA mutator mice12,15,18.
Using heterozygous PolgAwt/mut mice, we created a series of inbred mutant mice (Fig. 1) since it has been shown that low levels of mtDNA mutations are transmitted through the maternal germline19. Heterozygotes were intercrossed to generate Type I, Type II, and Type III mice, all with inherited mtDNA mutations. For mice lacking germline mtDNA mutations, male heterozygotes were crossed with female wild-type mice to generate Type IV and Type V animals.
Figure 1. Breeding scheme to generate wild-type variants.

(a) Mice heterozygous for the mtDNA mutator allele (PolgAwt/mut) were intercrossed to generate Type I (PolgAwt/wt), Type II (PolgAwt/mut), and Type III (PolgAmut/mut) mice, all with inherited germline mtDNA mutations from their heterozygous (PolgAwt/mut) mother. Type II (PolgAwt/mut) and Type III (PolgAmut/mut) mice also formed de novo somatic mtDNA mutations. (b) Male Type II (PolgAwt/mut) mice were crossed with female wild-type mice to generate Type IV (PolgAwt/wt) and Type V (PolgAwt/mut) mice, both without inherited mtDNA mutations. Schematic adapted from Ross et al. 2013.
Using these mutant mice, we recently reported10 that ongoing mtDNA mutagenesis in the maternal germline causes anticipation of fecundity phenotypes when intercrossing Type II mice, including decreasing litter sizes, decreasing total number of litters per female, and decreasing proportions of mtDNA mutator mice (Type III) born. An additional 17% reduction in longevity and anticipation of aging phenotypes, such as reduced body size and weight, kyphosis, alopecia, anemia, and organ enlargement, also were found in homozygous mtDNA mutator mice (Type III) born with inherited mtDNA mutations, as compared to mtDNA mutator mice born to Type II mothers with re-introduced wild-type mtDNA. Stochastic brain malformations, mostly microscopic and occasionally macroscopic, were only observed in ~32% of Type III mtDNA mutator mice, but not in Type I, II, IV, or V mice, or in mtDNA mutator mice born to Type II mothers with re-introduced wild-type mtDNA. Thus, the presence of inherited mtDNA mutations in combination with somatic mtDNA mutagenesis was required to cause disturbances of brain development.
Surprisingly, we found that even low levels of maternally transmitted mtDNA mutations can induce premature aging phenotypes in Type I mice with a completely wild-type nuclear genome, including alopecia, kyphosis, reduced body size, lower body weight, and decreased spontaneous rearing. However, longevity of such animals was not known, a critical outstanding question in terms of supporting a mitochondrial role in aging. We have now followed the lifespan of these mice and have discovered that not only do germline-only mtDNA mutations affect the onset and progression of aging phenotypes in Type I mice, but that longevity is also reduced ~30% (P<0.0001). We observed that wild-type mice derived from the standard breeding (Type I) had a reduced mean lifespan of 100.2 weeks (n = 13) (Fig. 2, black line), whereas wild-type mice, whose mothers lacked germline mtDNA mutations, had a mean lifespan of 141.1 weeks (n = 25) (Fig. 2, red line).
Figure 2. Longevity in mice with wild-type nuclear genomes is shortened by germline inherited mtDNA mutations.
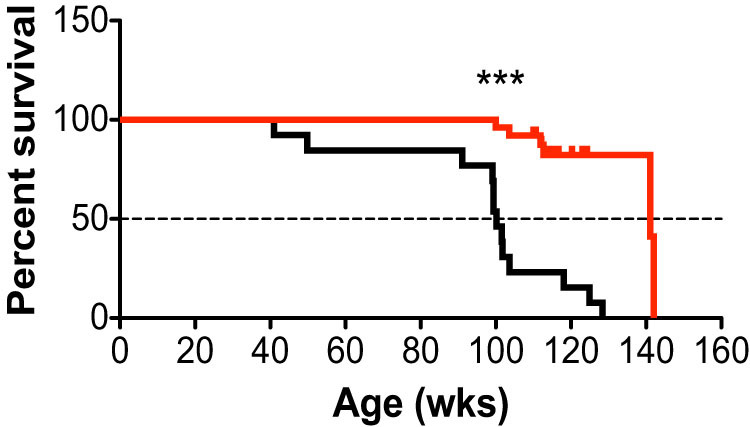
Type I wild-type mice (both males and females) obtained from standard intercrosses of PolgAwt/mut mice (black line, n = 13) with maternally transmitted mtDNA mutations have a significantly reduced lifespan (Χ2(1) = 24.4), compared to Type IV wild-type mice (PolgAwt/wt) with no experimentally induced mtDNA mutations (red line, n = 25). Median survival decreased from 141.1 wks to 100.2 wks of age in Type I mice. Significances were determined by the Mantel–Cox test, *** P<0.0001.
Discussion
The mechanisms underlying the relationship between mtDNA mutation load and the process of aging are not well understood. Deficient oxidative phosphorylation and loss of ATP for cellular processes would lead to loss of oxidative phosphorylation enzymes, particularly Complex I, an elevated NADH/NAD+ ratio, and reduced levels of NAD+ (also see Gomes et al.20). NAD+ is required as a substrate for generating ADP-ribose monomers, needed for the generation of PARP (Poly (ADP-ribose) polymerase) which, in turn, is critical for the identification of single-strand DNA breaks and the signaling to activate enzymatic DNA repair. There is an extensive literature on how the accumulation of DNA damage with time eventually exceeds repair capacity, leading to manifestations of aging pathophysiology in many organ systems21,22,23. Relative reduction of NAD+ may thus underlie many age-related problems (see review by Hipkiss24). In addition, reduced levels of NAD+ may lead to reduced activity of Sirtuin 1, which may further exacerbate age-related organ pathology25.
A second hypothesis about aging involves the accumulation of free radicals and reactive oxygen species (ROS)3,26. However, there is little evidence for ROS accumulation in the mtDNA mutator mouse model18,27,28,29,30. Finally, there are studies suggesting that stem cells play a “renewal” role in adult organisms and that a deficiency of stem cell-mediated renewal may be part of the aging process. Indeed, work from our own lab on abnormalities of brain development10 and from others on hematopoietic stem cells31 suggest stem cells are particularly vulnerable to the mitochondrial dysfunction in mtDNA mutator mice. Interestingly, losses in DNA repair processes in mice are reported to limit the ability of hematopoietic stem cells to self-renew with age32.
Our previous and present findings allow us to conclude that inherited mtDNA mutations alone or in combination with somatic mtDNA mutations, augments the rate of aging and shortens lifespan. These results also provide additional evidence for the hypothesis that certain determinants of aging are present prior to conception and during development. It would be interesting to understand if the rate of aging, determined early during life, can be altered. Recent studies suggest that aging phenotypes might be completely reversible with NAD+ treatment20 and with forced exercise using similar mtDNA mutator mice33. These studies, together with our findings, might thus suggest that the rate of aging, even in the presence of inherited predispositions for age-related health issues, might be responsive to environmental factors. Our results demonstrate that low levels of maternally transmitted mtDNA mutations present during development constitute an important phenotypic determinant, acting on both overall fitness and lifespan.
Methods
Animals
All mice were on an inbred C57BL/6NCrl nuclear background. The study was performed in accordance with guidelines from the Federation of the European Laboratory Animal Science (FELASA). Experiments were approved by the animal welfare ethics committee and in accordance with Swedish law. Mice were fed ad libitum (R34, Lactamin/Lantmännen, Stockholm, Sweden), had free access to water, and were kept on a 12:12 h light:dark cycle at 22–23°C.
Statistical analysis
Statistical analyses were performed using appropriate software (GraphPad Software, GraphPad Software, San Diego, CA), with an alpha level of 0.05.
Author Contributions
J.M.R. and G.C. performed breeding, collected data, and prepared the figures. J.M.R., B.J.H., and L.O. wrote the paper.
Acknowledgments
The Swedish Brain Foundation (JMR), Swedish Brain Power (LO, JMR), the Swedish Society for Medical Research (GC), the Foundation for Geriatric Diseases at Karolinska Institutet (GC), Karolinska Institutet Research Foundations (GC), Loo och Hans Ostermans Foundation for Medical Research (GC), ERC Advanced Investigator grant (322744; LO), the Swedish Research Council (K2012-62X-03185-42-4; LO), and the Karolinska Distinguished Professor Award (LO). We thank Karin Pernold and Anna Lindberg for technical support.
References
- Corral-Debrinski M. et al. Mitochondrial DNA deletions in human brain: regional variability and increase with advanced age. Nat. Genet. 2, 324–329 (1992). [DOI] [PubMed] [Google Scholar]
- Cortopassi G. A. & Arnheim N. Detection of a specific mitochondrial DNA deletion in tissues of older humans. Nucleic Acids Res. 18, 6927–6933 (1990). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harman D. The biologic clock: the mitochondria? J Am Geriatr Soc 20, 145–147 (1972). [DOI] [PubMed] [Google Scholar]
- Müller-Höcker J. Cytochrome-c-oxidase deficient cardiomyocytes in the human heart--an age-related phenomenon. A histochemical ultracytochemical study. Am. J. Pathol. 134, 1167–1173 (1989). [PMC free article] [PubMed] [Google Scholar]
- Brown W. M., George M. & Wilson A. C. Rapid evolution of animal mitochondrial DNA. Proc. Natl. Acad. Sci. U.S.A. 76, 1967–1971 (1979). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan W. et al. A mouse model of mitochondrial disease reveals germline selection against severe mtDNA mutations. Science 319, 958–962 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart J. B., Freyer C., Elson J. L. & Larsson N.-G. Purifying selection of mtDNA and its implications for understanding evolution and mitochondrial disease. Nat. Rev. Genet. 9, 657–662 (2008). [DOI] [PubMed] [Google Scholar]
- Payne B. A. I. et al. Universal heteroplasmy of human mitochondrial DNA. Hum. Mol. Genet. 22, 384–390 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li M. et al. Detecting heteroplasmy from high-throughput sequencing of complete human mitochondrial DNA genomes. Am. J. Hum. Genet. 87, 237–249 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross J. M. et al. Germline mitochondrial DNA mutations aggravate ageing and can impair brain development. Nature 501, 412–415 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen E.-Z. et al. Mitoflash frequency in early adulthood predicts lifespan in Caenorhabditis elegans. Nature (2014) 10.1038/nature13012. [DOI] [PubMed] [Google Scholar]
- Trifunovic A. et al. Premature ageing in mice expressing defective mitochondrial DNA polymerase. Nature 429, 417–423 (2004). [DOI] [PubMed] [Google Scholar]
- Niu X., Trifunovic A., Larsson N.-G. & Canlon B. Somatic mtDNA mutations cause progressive hearing loss in the mouse. Exp. Cell Res. 313, 3924–3934 (2007). [DOI] [PubMed] [Google Scholar]
- Ross J. M. et al. High brain lactate is a hallmark of aging and caused by a shift in the lactate dehydrogenase A/B ratio. Proc. Natl. Acad. Sci. U.S.A. 107, 20087–20092 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mito T. et al. Mitochondrial DNA mutations in mutator mice confer respiration defects and B-cell lymphoma development. PLoS One 8, e55789 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vermulst M. et al. DNA deletions and clonal mutations drive premature aging in mitochondrial mutator mice. Nat. Genet. 40, 392–394 (2008). [DOI] [PubMed] [Google Scholar]
- Ross J. M. Visualization of Mitochondrial Respiratory Function using Cytochrome C Oxidase/Succinate Dehydrogenase (COX/SDH) Double-labeling Histochemistry. J Vis Exp (2011) 10.3791/3266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kujoth G. C. et al. Mitochondrial DNA mutations, oxidative stress, and apoptosis in mammalian aging. Science 309, 481–484 (2005). [DOI] [PubMed] [Google Scholar]
- Ameur A. et al. Ultra-Deep Sequencing of Mouse Mitochondrial DNA: Mutational Patterns and Their Origins. PLoS Genet. 7, e1002028 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomes A. P. et al. Declining NAD(+) induces a pseudohypoxic state disrupting nuclear-mitochondrial communication during aging. Cell 155, 1624–1638 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamilton M. L. et al. Does oxidative damage to DNA increase with age? Proc. Natl. Acad. Sci. U.S.A. 98, 10469–10474 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swain U. & Subba Rao K. Study of DNA damage via the comet assay and base excision repair activities in rat brain neurons and astrocytes during aging. Mech. Ageing Dev. 132, 374–381 (2011). [DOI] [PubMed] [Google Scholar]
- Vogel H., Lim D. S., Karsenty G., Finegold M. & Hasty P. Deletion of Ku86 causes early onset of senescence in mice. Proc. Natl. Acad. Sci. U.S.A. 96, 10770–10775 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hipkiss A. R. Mitochondrial dysfunction, proteotoxicity, and aging: causes or effects, and the possible impact of NAD+-controlled protein glycation. Adv. Clin. Chem. 50, 123–150 (Elsevier, 2010). [PubMed] [Google Scholar]
- Cerutti R. et al. NAD(+)-dependent activation of Sirt1 corrects the phenotype in a mouse model of mitochondrial disease. Cell Metab. 19, 1042–1049 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harman D. Aging: a theory based on free radical and radiation chemistry. J Gerontol 11, 298–300 (1956). [DOI] [PubMed] [Google Scholar]
- Trifunovic A. et al. Somatic mtDNA mutations cause aging phenotypes without affecting reactive oxygen species production. Proc. Natl. Acad. Sci. U.S.A. 102, 17993–17998 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai D.-F. et al. Age-dependent cardiomyopathy in mitochondrial mutator mice is attenuated by overexpression of catalase targeted to mitochondria. Aging Cell 9, 536–544 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hiona A. et al. Mitochondrial DNA mutations induce mitochondrial dysfunction, apoptosis and sarcopenia in skeletal muscle of mitochondrial DNA mutator mice. PLoS One 5, e11468 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Logan A. et al. In vivo levels of mitochondrial hydrogen peroxide increase with age in mtDNA mutator mice. Aging Cell 13, 765–768 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahlqvist K. J. et al. Somatic progenitor cell vulnerability to mitochondrial DNA mutagenesis underlies progeroid phenotypes in Polg mutator mice. Cell Metab. 15, 100–109 (2012). [DOI] [PubMed] [Google Scholar]
- Rossi D. J. et al. Hematopoietic stem cell quiescence attenuates DNA damage response and permits DNA damage accumulation during aging. Cell cycle 6, 2371–2376 (2007). [DOI] [PubMed] [Google Scholar]
- Safdar A. et al. Endurance exercise rescues progeroid aging and induces systemic mitochondrial rejuvenation in mtDNA mutator mice. Proc. Natl. Acad. Sci. U.S.A. 108, 4135–4140 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]