

Cells are continuously faced with life-and-death decisions based on their ability to handle stressful situations. One indicator of stress is the accumulation of unfolded proteins within the ER, which induces a transcriptional cascade aimed at increasing the folding capacity of the ER. If the burden is too great and homeostasis cannot be restored, the response shifts from damage control to the induction of apoptotic pathways. This unfolded protein response (UPR) is conserved among all eukaryotes, and dysfunction in this pathway underlies many human diseases, including diabetes and cancer. The 2014 Albert Lasker Basic Medical Research award honors Kazutoshi Mori of Kyoto University and Peter Walter of the UCSF (Figure 1) for their contributions toward unraveling the pathways involved in mediating the complex cellular response to ER stress.
Figure 1. The recipients of the 2014 Albert Lasker Basic Medical Research Award.

Kazutoshi Mori (left) of Kyoto University and Peter Walter (right) of the UCSF have been selected for their work identifying key players of the UPR.
A simple question
In the 1970s, the identification of a set of proteins that were induced in response to viral transformation set the stage for understanding heat shock-independent cellular stress responses. These particular proteins were constitutively present in cells and notably increased in response to glucose deprivation (1) or agents that block post-translational glycosylation, such as tunicamycin. Based on this apparent glucose-dependent regulation, they became known as glucose-regulated proteins (GRPs). One of the most strongly induced proteins, GRP78, was found to localize to the ER, and soon after, researchers realized that GRP78 was the same as a previously identified protein, the immunoglobulin heavy chain binding protein (BiP). BiP was known to function as a chaperone and bind both nascent and malformed peptides in the ER, leading to the hypothesis that the presence of unfolded or misfolded proteins underlies induction of the GRPs, and not glucose deprivation per se. To test this hypothesis, Yasunori Kozutsumi, as part of a team led by Mary-Jane Gething and Joe Sambrook at the University of Texas Southwestern Medical Center at Dallas, transformed mammalian cells with mutant forms of influenza hemagglutin (HA) and revealed that HA molecules that were unable to leave the ER induced transcription of the genes encoding BiP and other known GRPs. Moreover, the same transcriptional response that was induced by the accumulation of mutant HA in the ER was recapitulated with tunicamycin treatment (2). The revelation by Kozutsumi and colleagues that the accumulation of unfolded or misfolded proteins in the ER results in a distinct transcriptional response gave rise to a simple question. “How are signals from the ER relayed to the nucleus?” Two of the scientists that provided answers to this question had backgrounds in chemistry and left their home countries to join influential cell biology labs in the United States.
The draw of cell biology
After finishing his undergraduate degree in chemistry in his native Germany, Peter Walter was encouraged to study abroad in the United States. After an unsuccessful Fulbright Scholarship application, Walter was accepted as an exchange student at Vanderbilt University as part of the master’s program in organic chemistry. As Walter recounted to the JCI, “I was adopted into the lab of Thomas Harris, where I studied the synthesis of an alkaloid produced by a fungus that grows on red clover in Tennessee. When cows ingest the fungus it causes them to slobber.” During his time at Vanderbilt, Walter found himself in a one-on-one meeting with Nobel laureate Stanford Moore, who at the time was on the Board of Trustees at Vanderbilt and a professor at The Rockefeller University. “I can’t recall how a lowly student, such as myself, ended up having a one-on-one meeting with Dr. Moore, but he strongly encouraged me to apply to the doctorate program at Rockefeller.” Walter applied and was put on the waiting list before finally being accepted. After initially being turned down, Walter would eventually join Günter Blobel’s lab, a massively transformative experience in his career. “It was a small lab at the time that was just incredibly interactive and energetic, with many different projects going on. Many of Günter’s ideas were highly contested and controversial, and he was a fighter out there for his ideas, with so much passion and conviction. However, he was also always capable of listening to data and changing his opinion as the landscape changed. It is a wonderful thing to have a goal and march toward it, then as the work progresses you make new discoveries and change the paradigm.” During his time in the Blobel lab, Walter became interested in how proteins are targeted to specific cellular locations throughout the cells and was part of the team that identified the signal recognition particle (SRP), which directs nascent peptides of secreted proteins to the ER (3). Walter continued the work on intracellular targeting in his own lab at the UCSF.
Kazutoshi Mori always knew that he wanted to be a scientist and recently told the JCI, “Although there was no atmosphere of science in my family, it was my dream since childhood to be a PhD scientist. My interest was driven by the comics, which I loved to read, such as Mighty Atom (aka Astro Boy) and Mach GoGoGo (aka Speed Racer). I found a bright future in science and technology. I was also affected by the movie Modern Times by Charlie Chaplin.” As an undergraduate, Mori was more interested in chemistry and physics than biology; however, this would change after reading a newspaper article describing the work of Nobel laureate Susumu Tonegawa. “Before that time, I felt like there were no basic rules in biology and that I had to learn everything by heart. I was amazed by the fact that the genetic code is conserved from E. coli to humans, namely biology has a basic rule!” As a graduate student, under the tutelage of Ikuo Yamashina and Toshisuke Kawasaki, Mori analyzed the biological role of mannan-binding protein, a liver protein that binds to high-mannose-type carbohydrates in the ER. After working for a time as an instructor in the lab of Dr. Kyozo Hayashi at Gifu Pharmaceutical University, using biochemical methods to investigate a cancer cell–secreted factor, Mori took a huge risk and decided to go to the United States to study molecular biology, which had piqued his interest. After several futile applications, Yasunori Kozutsumi, a former colleague from the Yamashina lab, suggested that Mori apply to work with Mary-Jane Gething and Joe Sambrook. “Very fortunately, they accepted me. Mary-Jane and Joe gave me freedom, encouraged me, and were excited about my results.”
A simple screen provides exciting answers
Prior to Mori’s arrival at the University of Texas Southwestern, the Gething and Sambrook lab had determined that yeast have a homologous response to unfolded proteins in the ER as well a BiP homolog (encoded by KAR2) (4), allowing this response to be evaluated in a genetically tractable model. Mori set forth to understand the regulatory elements within the KAR2 promoter and identified three distinct elements that independently mediated constitutive, heat shock–induced, and UPR-induced KAR2 expression (5). The UPR element (UPRE) within the KAR2 promoter turned out to be a 22-bp sequence that when localized upstream of a gene promotes expression of the gene in response to ER stress.
At UCSF, Peter Walter had set up his lab and was greatly influenced by his fellow colleagues. As he told the JCI, “UCSF was a different environment. There was a lot more communication between labs compared to what I had experienced at Rockefeller. There was a lot more discussion and thought about genetics and structure. Ira Herskowitz strongly promoted genetic approaches in yeast, while Bob Stroud promoted the use of crystallography, which were both useful for the continuation of the work on SRP and protein translocation. At the same time, two graduate students joined the lab and wanted to do something a bit different.”
The revelation that the UPR was conserved between mammals and yeast and the identification of the UPRE in the KAR2 promoter provided the tools necessary to unravel how the ER communicates its stress level to the nucleus. Both Mori, as part of the Gething and Sambrook lab, and Jeff Cox and Caroline Shamu, Walter’s adventurous graduate students, set up yeast genetic screens to identify mediators of the UPR. The genetic screens were simple but elegant: generate a yeast strain harboring lacZ, which produces a colorimetric reporter when transcribed, downstream of a UPRE-containing promoter, mutagenize the strain, induce the UPR, and screen for mutants unable to induce lacZ in response to ER stress. Mutants with an altered UPR would appear white on a plate, while mutants with a normal UPR would appear blue. The independent screens both hit the same target, the gene encoding Ire1p (6, 7). Ire1p had been identified previously in a screen for yeast genes able to complement inositol auxotrophy and, based on sequence homology, was proposed to be a protein kinase (8). Mori et al. and Cox et al. both confirmed that Ire1p is a transmembrane serine/threonine kinase and proposed that Ire1p is well poised to transmit signals from the lumen of the ER to the cytoplasm (6, 7).
As a kinase, it was assumed that Ire1p would transmit the unfolded protein signal via phosphorylation of a downstream target, ultimately activating a transcription factor. The chase was on to identify the downstream transcription factor responsible for transcription of UPR-induced genes. Walter and his group moved forward with a high-copy suppressor screen, which would identify genes able to overcome the defective UPR response in Ire1p mutants. After the identification of Ire1p in 1993, Mori was ready to return to Japan and secured a position at the HSP Research Institute. As Mori recounted, “My new director, Dr. Takashi Yura, who had just retired from Kyoto University (where he investigated heat shock response in E. coli), allowed me to continue working on the UPR. The next target was the transcription factor specific to yeast UPR. Because the HSP Research Institute was focused on transcription factors, Takashi advised me not to employ a multicopy suppressor screen and encouraged me to devise a method to obtain the transcription factor directly. I finally came up with the idea to employ one-hybrid screening, which would be the most important moment in my career.” In 1996, Mori and Walter published papers identifying Hac1p as the transcription factor responsible for the UPR (9, 10). It was now important to determine how Ire1p activates Hac1p-dependent transcription.
Unconventional pathways
Unlike many other transcription factors that are activated by kinase cascades, Hac1p was not constitutively present in cells and waiting for the the input signal from Ire1p. Instead, Hac1p expression was unexpectedly determined to be regulated by splicing of HAC1 mRNA (10, 11). Walter described to the JCI his reaction to the first time that his graduate student showed him the data, “We couldn’t show that Hac1p was phosphorylated when the UPR was activated, it was only produced when the response was on. Hac1p could have been degraded when it is not needed or only made when it was needed, so Jeff ran a simple Northern blot to look at the changes of HAC1 mRNA abundance following induction of the response. These experiments led to the discovery that the abundance of HAC1 mRNA didn’t change, but there was a new form that appeared. This turned out to be the spliced form. It’s just amazing. The first time Jeff showed this to me, my response was, ‘Do it again, and don’t degrade the RNA.’ He was so proud when he proved me wrong. It was an incredibly exciting time.”
Walter and his group initially proposed that the translation of the unspliced full-length HAC1 mRNA produced an unstable protein that was rapidly targeted for degradation by the ubiquitin/proteasome system. Mori and his team also found that HAC1 mRNA is spliced following UPR induction; however, they determined that unspliced HAC1 mRNA is not translated due to the presence of an internal intron (11). Therefore, UPR-induced splicing of HAC1 mRNA is required for translation of this transcription factor. The next step for Walter was determining how HAC1 splicing was mediated. Walter and his group set up a screen that revealed that HAC1 splicing does not involve the spliceosome and requires a tRNA ligase (12). Mori and his group identified the sequences within the HAC1 transcript that were targeted by the UPR-associated endonuclease (13). Subsequently, another graduate student in Walter’s lab, Carmela Sidrauski, unveiled the surprising result that Ire1p itself is the molecule that specifically cuts HAC1 mRNA and that tRNA ligase then joins the severed exons to complete the splicing reaction (14). Together, the work from both the Mori and Walter groups in the 1990s revealed that in yeast the accumulation of unfolded proteins in the ER induces Ire1p-dependent splicing of constitutively expressed HAC1 mRNA. This splicing event allows translation of Hac1p, which not only induces its own expression but also activates transcription of numerous target genes involved in the UPR. The screens and experiments designed both by the Walter lab and Mori lab led to convergent discoveries, with both groups hot on the others’ heels. Walter reflected on this dynamic, “Having two labs pursuing the same thing provides great motivation and immediate validation and extension of your work. I think the synergy between our groups helped move the field forward rapidly and cohesively.”
A 3-pronged approach for handling ER stress
After focusing much of his early career on the UPR in yeast, Mori decided it was time to switch systems and began to investigate the UPR in mammalian systems. As Mori told the JCI, “Because the goal of HSP Research Institute was to develop drugs using the heat shock response and UPR, I decided to extend my work to mammalian cells.” As he had done previously, Mori turned to the promoters of UPR-induced genes, specifically those encoding GRPs, to identify the mammalian transcription factors that mediate the UPR. As Mori explained, “Many homeostatic GRP-encoding genes are simultaneously induced when the UPR is activated. To do so, their promoter regions must contain common cis-acting elements, as in yeast; however, no one had identified such elements in mammals. It had also been noted that the ER stress-responsive promoters of mammalian GRP genes contain multiple CCAAT motifs. Aligning these CCAAT and neighboring sequences, Hiderou Yoshida, now professor at Hyogo Prefectural University, in my group noticed that a structural motif of CCAAT-NNNNNNNNN-CCACG is present in all GRP promoters. We named this the ER stress response element (ERSE). Discovery of ERSE was a major breakthrough, because we could start identifying mammalian UPR-specific transcription factors.”
Using the ERSE and one-hybrid screening, Mori’s group would identify a mammalian Hac1p homolog, x-box–binding protein 1 (XBP1), and another transcription factor, ATF6, as mediators of the UPR in mammalian cells (15). Unlike XBP1, which is regulated by UPR-induced splicing (16), Mori and colleagues determined that ATF6 was constitutively present in cells as a 90-kDa protein and processed into a 50-kDa protein following UPR induction. Mori’s group also demonstrated that full-length ATF6 localizes to the ER and that, upon induction of the UPR, ATF6 is cleaved, and this processed form is targeted to the nucleus, in which it induces transcription (17). ATF6 shares the same activation mechanisms as the SREBPs, and like this class of transcription factors, ATF6 traffics from the ER to the Golgi apparatus following induction of the UPR, in which it is cleaved by the same proteases that process SREBPs (18).
A third branch of the UPR was identified in the late 1990s by groups led by David Ron and Ron Wek, respectively (19, 20). This branch involves activation of protein kinase RNA-like endoplasmic reticulum kinase (PERK), which localizes to the ER and is very similar to IRE1 but lacks an RNase domain. As part of the UPR, PERK phosphorylates the translation initiation factor eIF2α, resulting in a global reduction of translation in the cell, reducing total protein burden in the ER; however, phosphorylated eIF2α promotes translation of a few select mRNAs, including those coding for the transcription factor ATF4. ATF4 induces expression of genes involved in amino acid metabolism and oxidative stress resistance (21, 22). Among the ATF4-regulated genes is the gene encoding the transcription factor CHOP, which induces expression of genes involved in apoptosis (23).
The three branches of the UPR in mammalian systems (Figure 2) provide multiple avenues for checks and balances, which allow cells to determine the best course of action when faced with an abundance of unfolded proteins within the ER (24, 25). While many of the pathways that regulate the UPR are known, the exact signals that are detected by and activate IRE1, ATF6, and PERK remain to be determined, although there is mounting evidence that unfolded proteins are indeed the ligand for Ire1p in yeast (see ref. 26 for a review). Moreover, it is still not clear how these three pathways integrate within mammalian cells to coordinate restoration of homeostasis or tip the balance toward apoptosis. It has been proposed that the three branches serve to elicit a temporal response to ER stress, which allows an initial cell-protective response aimed at relieving the protein burden in the ER. This initial wave is followed by the transcriptional activation of genes encoding ER chaperones. If the signals that mediate these responses are not turned off, the cell initiates apoptotic pathways (24, 25).
Figure 2. Simplified schematic of the three pathways that mediate the UPR in mammalian cells.

Under homeostatic conditions, transmembrane proteins IRE1, ATF6, and PERK reside within the ER membrane, and their lumenal domains bind the protein-folding chaperone BiP. The accumulation of unfolded proteins in the ER triggers alterations of these proteins that result in transcriptional and translational changes in the cell that either deal with the excess unfolded proteins in the ER or promote apoptosis. Upon activation, PERK phosphorylates the translation initiation factor eIF2α, which reduces global translation but promotes translation of the transcription factor ATF4. Activation of IRE1 results in oligomerization, autophosphorylation, and RNA splicing activity. IRE1 specifically splices XBP1 mRNA, which is then relegated and translated. XBP1 then induces transcription of genes involved in the UPR. In response to unfolded proteins, ATF6 is targeted to the Golgi apparatus via vesicular transport. Once in the Golgi, AFT6 is cleaved by S1P and S2P, allowing the cytoplasmic portion of the protein to enter the nucleus and mediate transcription of UPR-associated genes.
The future unfolds
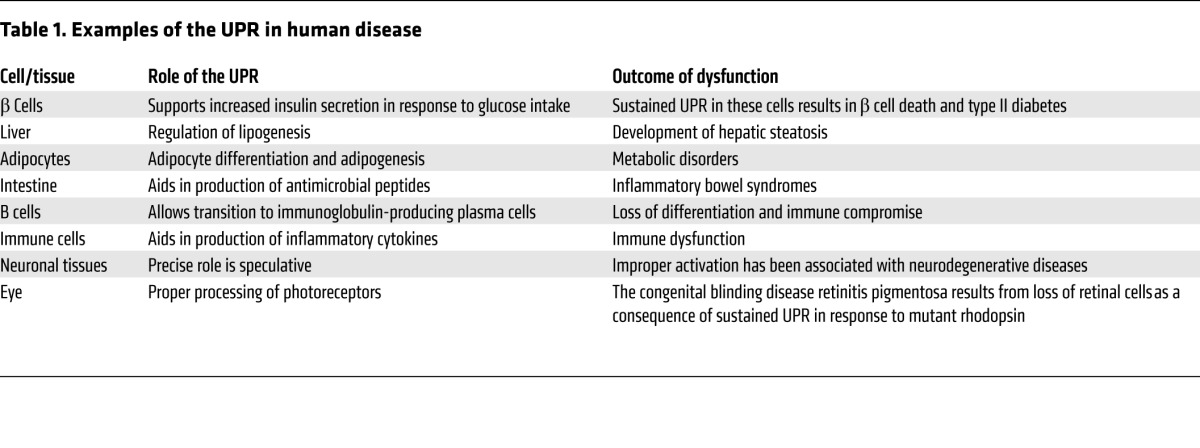
Mori and Walter both began studying the UPR because of their interest in basic cell biology. Peter Walter credits much of his success to the mentors, students, postdocs, and collaborators with whom he has worked through the years. Walter is also especially grateful for his status as a Howard Hughes Investigator for giving him the freedom to focus on his research pursuits. Kazutoshi Mori is also thankful for those that have helped him throughout his career, especially his colleagues at Kyoto University, who have been supportive of his research program. In talking with the JCI, both have expressed their delight in the mounting evidence that their initial studies in yeast have translated to humans. Many diseases and infections (See Table 1 for examples) have now been shown to involve UPR dysfunction or continuous activation of the system, which tips the response toward death in stressed populations, such as insulin-secreting pancreatic β cells, the loss of which underlies diabetes (see refs. 27, 28 for reviews). Animal models have been informative in trying to tease apart the specific roles of different components of the UPR in the development of disease. Moreover, these models have demonstrated that targeting the UPR may provide therapeutic benefit for some diseases, including inhibition of tumor cell growth (see ref. 29 for review). Walter told the JCI, “I’m very excited. I started out as a chemist and then moved into a yeast model system, and it is really very rewarding to see how the early work relates to potential clinical application. It would be a wonderful thing if our research would lead to insights that help people in a tangible way.” Mori agreed with this sentiment, “When Peter and I were analyzing the mechanism of yeast UPR, we hoped that this system also operated in humans. The revelation that the UPR plays very important roles in humans has been a dream come true.”
Table 1.
Examples of the UPR in human disease
Footnotes
Reference information:J Clin Invest. 2014;124(10):4138–4142. doi:10.1172/JCI78419.
References
- 1.Shiu RP, Pouyssegur J, Pastan I. Glucose depletion accounts for the induction of two transformation-sensitive membrane proteinsin Rous sarcoma virus-transformed chick embryo fibroblasts. Proc Natl Acad Sci U S A. 1977;74(9):3840–3844. doi: 10.1073/pnas.74.9.3840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kozutsumi Y, Segal M, Normington K, Gething M-J, Sambrook J. The presence of malfolded proteins in the endoplasmic reticulum signals the induction of glucose-regulated proteins. Nature. 1988;332(6163):462–464. doi: 10.1038/332462a0. [DOI] [PubMed] [Google Scholar]
- 3.Walter P, Blobel G. Purification of a membrane-associated protein complex required for protein translocation across the endoplasmic reticulum. Proc Natl Acad Sci U S A. 1980;77(12):7112–7116. doi: 10.1073/pnas.77.12.7112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Normington K, Kohno K, Kozutsumi Y, Gething MJ, Sambrook J. S. cerevisiae encodes an essential protein homologous in sequence and function to mammalian BiP. Cell. 1989;57(7):1223–1236. doi: 10.1016/0092-8674(89)90059-7. [DOI] [PubMed] [Google Scholar]
- 5.Mori K, Sant A, Kohno K, Normington K, Gething MJ, Sambrook JF. A 22 bp cis-acting element is necessary and sufficient for the induction of the yeast KAR2 (BiP) gene by unfolded proteins. EMBO J. 1992;11(7):2583–2593. doi: 10.1002/j.1460-2075.1992.tb05323.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mori K, Ma W, Gething MJ, Sambrook J. A transmembrane protein with a cdc2+/CDC28-related kinase activity is required for signaling from the ER to the nucleus. Cell. 1993;74(4):743–756. doi: 10.1016/0092-8674(93)90521-q. [DOI] [PubMed] [Google Scholar]
- 7.Cox JS, Shamu CE, Walter P. Transcriptional induction of genes encoding endoplasmic reticulum resident proteins requires a transmembrane protein kinase. Cell. 1993;73(6):1197–1206. doi: 10.1016/0092-8674(93)90648-a. [DOI] [PubMed] [Google Scholar]
- 8.Nikawa JI, Yamashita S. IRE1 encodes a putative protein kinase containing a membrane-spanning domain and is required for inositol phototrophy in Saccharomyces cerevisiae. Mol Microbiol. 1992;6(11):1441–1446. doi: 10.1111/j.1365-2958.1992.tb00864.x. [DOI] [PubMed] [Google Scholar]
- 9.Mori K, Kawahara T, Yoshida H, Yanagi H, Yura T. Signalling from endoplasmic reticulum to nucleus: transcription factor with a basic-leucine zipper motif is required for the unfolded protein-response pathway. Genes Cells. 1996;1(9):803–817. doi: 10.1046/j.1365-2443.1996.d01-274.x. [DOI] [PubMed] [Google Scholar]
- 10.Cox JS, Walter P. A novel mechanism for regulating activity of a transcription factor that controls the unfolded protein response. Cell. 1996;87(3):391–404. doi: 10.1016/s0092-8674(00)81360-4. [DOI] [PubMed] [Google Scholar]
- 11.Kawahara T, Yanagi H, Yura T, Mori K. Endoplasmic reticulum stress-induced mRNA splicing permits synthesis of transcription factor Hac1p/Ern4p that activates the unfolded protein response. Mol Biol Cell. 1997;8(10):1845–1862. doi: 10.1091/mbc.8.10.1845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sidrauski C, Cox JS, Walter P. tRNA ligase is required for regulated mRNA splicing in the unfolded protein response. Cell. 1996;87(3):405–413. doi: 10.1016/s0092-8674(00)81361-6. [DOI] [PubMed] [Google Scholar]
- 13.Kawahara T, Yanagi H, Yura T, Mori K. Unconventional splicing of HAC1/ERN4 mRNA required for the unfolded protein response. Sequence-specific and non-sequential cleavage of the splice sites. J Biol Chem. 1998;273(3):1802–1807. doi: 10.1074/jbc.273.3.1802. [DOI] [PubMed] [Google Scholar]
- 14.Sidrauski C, Walter P. The transmembrane kinase Ire1p is a site-specific endonuclease that initiates mRNA splicing in the unfolded protein response. Cell. 1997;90(6):1031–1039. doi: 10.1016/s0092-8674(00)80369-4. [DOI] [PubMed] [Google Scholar]
- 15.Yoshida H, Haze K, Yanagi H, Yura T, Mori K. Identification of the cis-acting endoplasmic reticulum stress response element responsible for transcriptional induction of mammalian glucose-regulated proteins. Involvement of basic leucine zipper transcription factors. J Biol Chem. 1998;273(50):33741–33749. doi: 10.1074/jbc.273.50.33741. [DOI] [PubMed] [Google Scholar]
- 16.Yoshida H, Matsui T, Yamamoto A, Okada T, Mori K. XBP1 mRNA is induced by ATF6 and spliced by IRE1 in response to ER stress to produce a highly active transcription factor. Cell. 2001;107(7):881–891. doi: 10.1016/s0092-8674(01)00611-0. [DOI] [PubMed] [Google Scholar]
- 17.Haze K, Yoshida H, Yanagi H, Yura T, Mori K. Mammalian transcription factor ATF6 is synthesized as a transmembrane protein and activated by proteolysis in response to endoplasmic reticulum stress. Mol Biol Cell. 1999;10(11):3787–3799. doi: 10.1091/mbc.10.11.3787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ye J, et al. ER stress induces cleavage of membrane-bound ATF6 by the same proteases that process SREBPs. Mol Cell. 2000;6(6):1355–1364. doi: 10.1016/s1097-2765(00)00133-7. [DOI] [PubMed] [Google Scholar]
- 19.Shi Y, et al. Identification and characterization of pancreatic eukaryotic initiation factor 2 alpha-subunit kinase, PEK, involved in translational control. Mol Cell Biol. 1998;18(12):7499–7509. doi: 10.1128/mcb.18.12.7499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Harding HP, Zhang Y, Ron D. Protein translation and folding are coupled by an endoplasmic-reticulum-resident kinase. Nature. 1999;397(6716):271–274. doi: 10.1038/16729. [DOI] [PubMed] [Google Scholar]
- 21.Harding HP, et al. An integrated stress response regulates amino acid metabolism and resistance to oxidative stress. Mol Cell. 2003;11(3):619–633. doi: 10.1016/s1097-2765(03)00105-9. [DOI] [PubMed] [Google Scholar]
- 22.Harding HP, et al. Regulated translation initiation controls stress-induced gene expression in mammalian cells. Mol Cell. 2000;6(5):1099–1108. doi: 10.1016/s1097-2765(00)00108-8. [DOI] [PubMed] [Google Scholar]
- 23.Wang XZ, et al. Identification of novel stress-induced genes downstream of chop. EMBO J. 1998;17(13):3619–3630. doi: 10.1093/emboj/17.13.3619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mori K. Signalling pathways in the unfolded protein response: development from yeast to mammals. J Biochem. 2009;146(6):743–750. doi: 10.1093/jb/mvp166. [DOI] [PubMed] [Google Scholar]
- 25.Walter P, Ron D. The unfolded protein response: from stress pathway to homeostatic regulation. Science. 2011;334(6059):1081–1086. doi: 10.1126/science.1209038. [DOI] [PubMed] [Google Scholar]
- 26.Gardner BM, Pincus D, Gotthardt K, Gallagher CM, Walter P. Endoplasmic reticulum stress sensing in the unfolded protein response. Cold Spring Harb Perspect Biol. 2013;5(3):a013169. doi: 10.1101/cshperspect.a013169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kaufman RJ. Orchestrating the unfolded protein response in health and disease. J Clin Invest. 2002;110(10):1389–1398. doi: 10.1172/JCI16886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wang S, Kaufman RJ. The impact of the unfolded protein response on human disease. J Cell Biol. 2012;197(7):857–867. doi: 10.1083/jcb.201110131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hetz C, Chevet E, Harding HP. Targeting the unfolded protein response in disease. Nat Rev Drug Discov. 2013;12(9):703–719. doi: 10.1038/nrd3976. [DOI] [PubMed] [Google Scholar]