

Abstract
Paroxysmal nocturnal hemoglobinuria (PNH) is a nonmalignant clonal disease of hematopoietic stem cells that is associated with hemolysis, marrow failure, and thrombophilia. PNH has been considered a monogenic disease that results from somatic mutations in the gene encoding PIGA, which is required for biosynthesis of glycosylphosphatidylinisotol-anchored (GPI-anchored) proteins. The loss of certain GPI-anchored proteins is hypothesized to provide the mutant clone with an extrinsic growth advantage, but some features of PNH argue that there are intrinsic drivers of clonal expansion. Here, we performed whole-exome sequencing of paired PNH+ and PNH– fractions on samples taken from 12 patients as well as targeted deep sequencing of an additional 36 PNH patients. We identified additional somatic mutations that resulted in a complex hierarchical clonal architecture, similar to that observed in myeloid neoplasms. In addition to mutations in PIGA, mutations were found in genes known to be involved in myeloid neoplasm pathogenesis, including TET2, SUZ12, U2AF1, and JAK2. Clonal analysis indicated that these additional mutations arose either as a subclone within the PIGA-mutant population, or prior to PIGA mutation. Together, our data indicate that in addition to PIGA mutations, accessory genetic events are frequent in PNH, suggesting a stepwise clonal evolution derived from a singular stem cell clone.
Introduction
Paroxysmal nocturnal hemoglobinuria (PNH), a prototypical disease of hematopoietic stem cells, is characterized by the clinical triad of intravascular hemolysis, thrombophilia, and bone marrow failure (1). While clonal, PNH has not been considered a malignancy. However, PNH does exhibit similarities to myelodysplastic syndrome (MDS), a chronic preleukemic myeloid neoplasm, including clonal hematopoiesis, persistence of an aberrant stem cell clone, and frequent derivation as a late complication of immune-mediated aplastic anemia (AA). The presence of a singular somatic mutation of the PIGA gene in hematopoietic stem cells leads to the defective biosynthesis of glycophosphatidylinositol (GPI) anchors, resulting in the deficiency and absence of GPI-anchored proteins on the cell surface, a hallmark of the PNH phenotype (2). These phenotypic features of affected PNH stem cells are believed to be responsible for an extrinsic growth advantage, which occurs in the context of immune-mediated hematopoietic suppression of hematopoiesis, as seen in AA (3). While immune privilege leading to clonal escape is plausible, it does not completely explain the evolution of PNH. Various observations suggest that intrinsic factors are also involved. For example, PNH often persists for many years after successful immunosuppression in AA, or PNH can present in a pure form without AA. Furthermore, the detection of tiny PIGA mutant clones in healthy individuals suggests a need for additional putative intrinsic factors that promote the expansion of GPI-deficient cells within the hematopoietic compartment (4, 5). These factors may include secondary genetic events such as somatic mutations. In support of this hypothesis, chromosomal abnormalities in the form of microdeletions involving the PIGA locus have been identified in a small proportion of cases of otherwise classical PNH (6, 7). Occasional clonal chromosomal abnormalities and somatic mutations including NRAS and JAK2 mutations have also been reported in PNH (8, 9), supporting the notion that PIGA mutations and additional somatic events correlate with each other and may be responsible for maintenance and expansion of the PNH clone.
Recently, the application of next-generation sequencing (NGS) to study malignant clonal diseases has revealed clonal architecture at a much higher level of complexity than previously anticipated, demonstrating both the stepwise acquisition of mutations and expansion of the most permissive subclones. In this study, we present data from whole-exome sequencing (WES) of clonal (GPI-deficient) and nonclonal cells from PNH patients to examine the mutational history of PNH. We hypothesized that the evolution of a PNH clone may be associated with additional somatic mutational events and that such events may be either of an ancestral or a facilitating nature. These additional somatic mutations, if present, may help to further clarify the mechanism of clonal expansion and persistence of the mutated PNH stem cell, as well as explain the clinical diversity of PNH and distinct behavior of the PNH clones. Our data suggest that PNH, a nonmalignant yet clonal disorder, displays clonal architecture in a manner similar to that of leukemia (10, 11).
Results
Mutational search using WES.
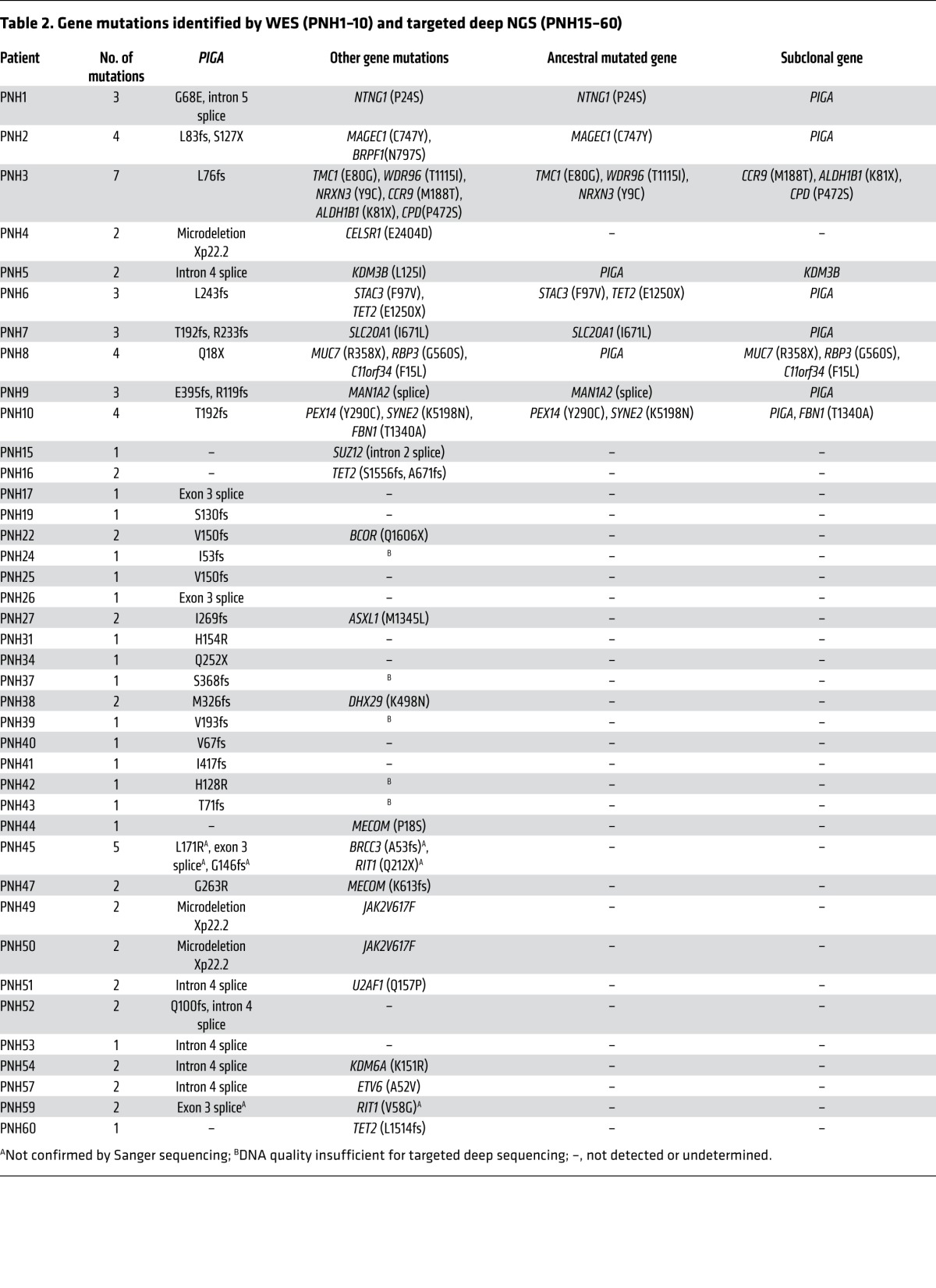
We analyzed a total of 60 patients with PNH (Table 1). After immunomagnetic sorting of wbc into PNH (CD59–) and non-PNH (CD59+) fractions, we verified purity by flow cytometry (Supplemental Table 1; supplemental material available online with this article; doi:10.1172/JCI74747DS1) and performed WES on 12 patients. Application of a bioanalytic algorithm designed to detect somatic mutations led to the selection of a total of 107 nonsilent alterations (98 were single nucleotide variants [SNVs] and 9 were indels). Through validation by Sanger sequencing and targeted deep NGS of candidate alterations (61 genes, Supplemental Table 2), we confirmed the presence of 38 somatic events in a total of 31 genes in PNH-derived DNA. The average coverage of exome sequencing and targeted deep NGS was 103 times and 316 times, respectively. Our stringent bioanalytic platform favored avoidance of false-negatives, and, consequently, the accuracy of the initial calling algorithm was 22% for SNVs and 89% for indels. In addition to 3 cases with Xp22.2 PIGA microdeletions, we identified a total of 31 missense, 8 nonsense, 26 frameshift, and 13 splice site mutations in cases analyzed by WES (n = 12) and targeted (n = 36) NGS sequencing (Table 2).
Table 1.
Patient characteristics
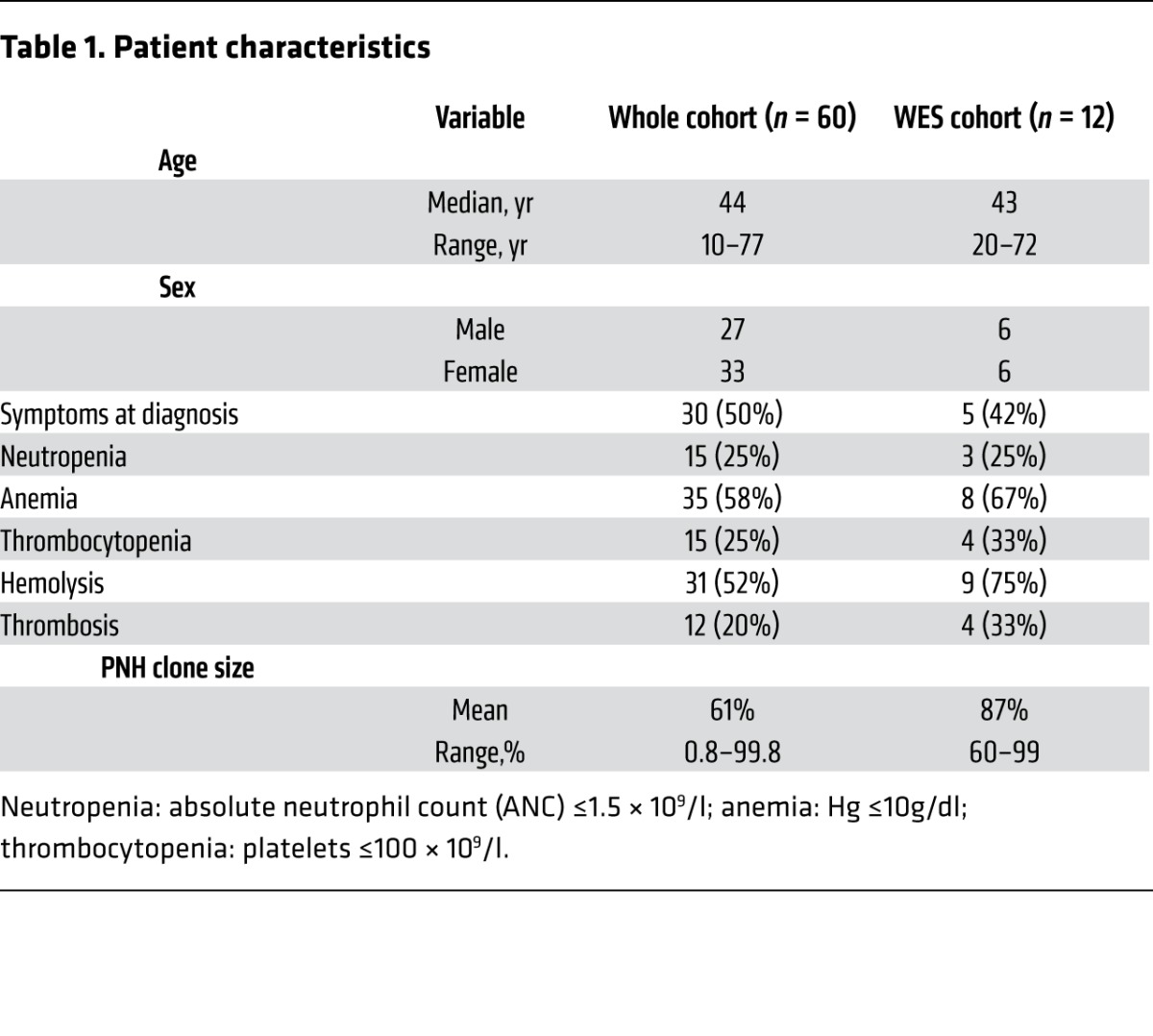
Table 2.
Gene mutations identified by WES (PNH1–10) and targeted deep NGS (PNH15–60)
PIGA mutations and deletions.
We detected somatic PIGA mutations (3 SNVs and 10 indels) in the PNH fractions of 9 of 12 cases analyzed by WES, while paired non-PNH CD59+ DNA samples were negative, although a small cross-contamination artifact was detected in some samples. For example, in PNH patient 5 (PNH5), we identified a single splice site mutation with a high variant allelic frequency in sorted PNH+ (CD59–) cells that was generally absent in normal (CD59+) cells (Figure 1A). To further explore the frequency of PIGA mutations in our cohort aside from the WES cases (n = 12), we used Sanger sequencing (n = 36) and targeted PIGA (n = 10) deep sequencing. Overall, PIGA mutations were detected in 60% (36 of 60) of patients (Table 2). As reported previously in the literature (12), some PNH patients harbor more than 1 PIGA mutation. In 14% (5 of 36) of PIGA-mutated cases, 2 independent mutations were found, and 1 case (PNH45) contained 3 mutations (Table 2 and representative patients depicted in Figure 1C and Supplemental Figure 1). In an index female PNH case (PNH1), the somatic nature of 2 PIGA mutations (p.G68E and intron 5 splice site) was confirmed, since deep sequencing indicated that both were confined to the PNH fraction (Figure 1C). The 2 mutations were located 431 nucleotides apart, and bacterial subcloning analysis demonstrated that they were not present in the same clone (Figure 1D). Two independent experiments involving Sanger sequencing of single colonies further validated the biclonal nature of the 2 PIGA mutations, as no colony contained both mutations (Figure 1E). Deep sequencing provided variant allelic frequencies (VAFs) of 18% (p.G68E) and 12% (intron 5 splice site) with a corresponding clonal size of 36% and 24%, respectively, as the mutations were heterozygous. Overall, in each of the 6 PNH cases with 2 or more PIGA mutations, we identified 2 (or 3) independent PNH clones with 1 unique PIGA mutation per clone, and all were codominant.
Figure 1. PIGA mutations can be either primary or secondary events.

The primary event (PNH5) is represented in A and B; the secondary event (PNH1) is represented in C–F. (A) Analysis of VAFs of the mutations identified in case PNH5 indicated that the KDM3B mutation was present at a lower frequency than the PIGA mutation, and both mutations were almost exclusively confined to the sorted PNH+ (CD59–) fraction. (B) Single-colony sequencing results confirmed that the PIGA and KDM3B mutations were present in the same cell population. (C) Deep sequencing VAFs for PIGA-1 (G68E), PIGA-2 (splice site), and NTNG1 (P24S) mutations, all of which were primarily present in the PNH fraction in the PNH1 case. (D) Bacterial subcloning and Sanger sequencing results demonstrated that the PIGA mutations in this case were independent, suggesting the presence of 2 separate PNH clones. (E) Single-colony sequencing further confirmed that 2 independent PNH clones were present and also suggested that the PIGA splice site mutation appeared to be a secondary event preceded by a NTNG1 mutation. (F) The combination of deep sequencing data with single-colony sequencing allowed for a representation of the clonal architecture in PNH1.
In total, 9 of 12 (75%) WES, 9 of 10 (90%) targeted deep sequencing, and 15 of 36 (42%) Sanger sequencing cases were positive for PIGA defects, including 3 nonsense, 21 frameshift, 5 missense, and 11 splice site mutations (Figure 2A). One WES case and an additional 2 cases contained a microdeletion involving the PIGA locus (delXp22.2, spanning an average of 559 kb, range 506–616 kb) as detected by SNP array. These microdeletions were confined to PNH cells and were not found in non-PNH fractions. In summary, we were able to detect either PIGA mutations or microdeletions in 60% (36 of 60) of patients studied, with 3% (1 of 36) triclonal, 14% (5 of 36) biclonal, 75% (27 of 36) monoclonal, and 8% (3 of 36) microdeletions (Figure 2B). We found no mutations or microdeletions in 40% (24 of 60) of the cases studied, with Sanger sequencing demonstrating a much lower detection rate than WES or targeted deep sequencing. Flow cytometric analysis of PNH clone size using Alexa 488 Proaerolysin Variant (FLAER) indicated that the only patient with 3 PIGA mutations also had a significant (>3%) wbc type II PNH clone (Figure 2C). Based on the evolution of PNH clone size as assessed longitudinally by flow cytometry in patients with at least a 4-year follow-up and a detected PIGA mutation (n = 16), the vast majority of patients tended to equilibrate at a clone size greater than 80% of all leukocytes regardless of the nature of the PIGA mutation (Figure 2D).
Figure 2. PIGA mutations and longitudinal analysis.

(A) Distribution of PIGA missense, frameshift, nonsense, splice site, and microdeletions. (B) Proportion and absolute number of patients with 1, 2, or 3 mutations as well as microdeletions and those patients with the PNH phenotype in which a PIGA mutation was not detected. (C) Representative flow cytometric plot quantifying the number of type I (normal), type II (intermediate GPI anchor loss), and type III (complete GPI anchor loss) cells. (D) Longitudinal flow cytometry quantifying the wbc PNH clone size in patients with approximately 4 years or more of follow-up (n = 16). Each time point corresponds to approximately 1 year.
Additional somatic mutations in PNH.
In addition to the pathognomonic PIGA mutations, 21 somatic mutations in 21 other genes were found in 83% (10 of 12) of cases tested by WES (Table 2). All of these mutations were confirmed by independent testing using both Sanger and targeted NGS. In contrast to what would be expected from a benign condition, the number of such novel gene mutations other than PIGA ranged between 0 and 6 per case, with an average of 2 additional events. In an index case of PNH with thrombocytosis (PNH1), we detected a novel somatic heterozygous mutation primarily in the PNH fraction in the NTNG1 gene (Figure 1C). While NTNG1 mutations or dysregulation of receptor ligand interactions have been described in various disorders including colorectal cancer (13–16), the p.P24S mutation has not been previously reported. Other somatic mutations discovered in PNH include those in TET2, MAGEC1, BRPF1, KDM3B, and STAC3 genes, all found in the PNH fraction and not in phenotypically normal cells (Table 2). All of these mutations were heterozygous without loss of heterozygosity (LOH) encompassing the affected gene locus. To assess whether these gene mutations were frequently recurrent in PNH, we screened an additional 36 cases by targeted deep sequencing, including a variety of genes (n = 61) that are frequently affected in MDS (Supplemental Table 2). In addition to 2 somatic homozygous JAK2 (p.V617F) mutations, both of which were present in PNH cases with microdeletions of Xp22.2, SUZ12, DHX29, MECOM, BCOR, U2AF1, ASXL1, BRCC3, ETV6, KDM6A, NTNG1, BRPF1, MAGEC1, CCR9, ALDH1B1, WDR96, TMC1, CPD, NRXN3, CELSR1, KDM3B, STAC3, SLC20A1, MUC7, RBP3, C11orf34, MAN1A2, PEX14, SYNE, and FBN1 mutations were found in a single PNH case each, with RIT1 and MECOM in 2 cases and TET2 in 3 cases, one of which harbored 2 different TET2 mutations (Table 2).
Clonal architecture.
In order to evaluate the clonal composition of each patient, we used single-colony sequencing assays, bacterial subcloning, and analysis of VAF data obtained from targeted deep sequencing. For example, in PNH5, a KDM3B mutation was also discovered, albeit at a lower frequency than that of the PIGA mutations (Figure 1A). Clonogenic assays were used to determine whether the mutations were independent (biclonal) or co-occuring (subclonal, Figure 1B). In this case, all colonies were either wild-type or had both mutations. Taken in context with allelic frequency data indicating that the KDM3B mutation was present at a lower frequency than the PIGA mutation, these results suggest that the PIGA mutation was the initial event.
Our first indication that a PIGA mutation may not always be the initial event in PNH came during our analysis of the WES data from PNH1. VAF of the NTNG1 mutation (18.3%) was slightly greater than that of either of the PIGA mutations (18% vs. 12%) detected (Figure 1C), prompting subcloning and single-colony sequencing experiments to determine the clonal composition of the observed mutations. These experiments confirmed the suspicion that the NTNG1 mutation preceded the PIGA intron 5 splice site mutation, as we observed a number of colonies with a NTNG1 mutation but without a PIGA mutation (Figure 1C). Furthermore, no colonies demonstrated the presence of a PIGA intron 5 splice site mutation in the absence of a concurrent NTNG1 mutation, leading to our current understanding of clonal architecture in this patient (Figure 1D), who clearly has 2 independent PIGA mutations, one of which evolved as a secondary event subsequent to the initial NTNG1 alteration.
As we continued our analysis of the data, it became evident that additional mutations further increase the complexity of clonal architecture in PNH. In an illustrative case (PNH8), we identified 1 PIGA mutation and an additional 3 somatic mutations (C11orf34, RBP3, MUC7; Figure 3A). Again, based on targeted deep NGS and single-colony sequencing (Figure 3B), we deduced that the PIGA mutation constituted an initial ancestral event followed by subclonal defects in 3 other genes (Figure 3C). The diversity of clonal architecture in PNH became apparent when more cases were analyzed. In PNH2, another case with 2 PIGA mutations, additional somatic events including MAGEC1 and BRPF1 mutations, were found and independently confirmed. Comparison of VAFs obtained through targeted deep NGS revealed that the corresponding clonal size for the MAGEC1 mutation was at least comparable to, if not larger than, that of the largest PIGA mutation, suggesting that the initial event may not have been a PIGA mutation, but indeed a MAGEC1 mutation (Supplemental Figure 1). In PNH9, we found that the initial event that may be either a malignant or a passenger mutation, again was not a PIGA mutation, but a novel mutation in MAN1A2, which was followed by the appearance of 2 PIGA mutations, thus creating 2 independent clones, both of which carried the original MAN1A2 mutation. In a somewhat similar case (PNH10, Supplemental Figure 2), somatic SYNE2 and PEX14 gene mutations were the initial events, followed by a PIGA frameshift mutation and an additional FBN1 mutation. Perhaps the most complex case, PNH3, demonstrated 7 somatic gene mutations that were all confined to the PNH fraction; single-colony sequencing confirmed that the PIGA mutation occurred after 2 other clonal mutations had already been acquired (TMC1 and WDR96) and, again, was not the initial event (Supplemental Figure 3).
Figure 3. PIGA mutation as the initial ancestral event in PNH8.

(A) A PIGA mutation together with 3 other somatic mutations (C11orf34, RBP3, and MUC7) were detected via WES at various allelic frequencies. All mutations were confined to the PNH fraction. Of note is that, in this case, the PIGA mutation was hemizygous, rendering the clone size equivalent to the allelic frequency. (B) Single-colony Sanger sequencing further confirmed the results obtained from deep sequencing and suggested that a PIGA mutation was the initial event. n = the number of colonies with the indicated mutations observed. Colonies that were not reproducible in independent experiments are not shown. (C) Proposed model of clonal architecture in this case based on VAFs and colony sequencing results.
In addition to the detection of concomitant mutations within the PNH clone, we found somatic events that predated the PIGA mutations and were present in both PNH and non-PNH myeloid fractions but not in germline DNA derived from T cells. These mutations included TET2, SUZ12, and JAK2. In PNH6, we observed concomitant TET2 and STAC3 mutations. However, mutant fractions were larger than those of the PIGA mutant, and the TET2 mutation was also present in the non-PNH fraction, which, in turn, was negative for STAC3 and PIGA mutations (Supplemental Figure 4). These results indicate that PNH, in this case, evolved as a subclone after a clonal TET2 mutation was acquired. However, in the bone marrow of another case (PNH3), we identified dysplastic changes along with trisomy 8 in 20 of 20 metaphases. FISH analysis resolved the origin of the trisomy 8, which was present only in the non-PNH fraction (data not shown). These results suggest that PNH in patient 3 evolved independently of the acquisition of trisomy 8.
Overall, our clonal analysis of 9 WES cases suggested that PIGA mutations were often acquired at a later stage (6 of 9 cases, Table 2), and mutations in other genes were the initial clonal events. Of note is that in a number of cases, the clonal composition showed significant overlap, preventing precise recapitulation of clonal hierarchy. In summary, our experiments indicate that the cohort can be stratified into 4 different scenarios (Figure 4): PIGA as the initial ancestral event accompanied by secondary mutations (Figure 4B, patients 5 and 8); PIGA as an event secondary to other mutations (Figure 4C, patients 1, 2, 6, 7, 9, and 10); PIGA as the lone mutation (Figure 4D, patients 17, 19, 25, 26, 31, 34, 40, 41, 52, and 53); and a PIGA mutation coexisting with other mutations responsible for the development of an MDS clone (Figure 4E, patient 3).
Figure 4. Mutational summary and clonal architecture scenarios.

(A) Cohort frequency of 4 different scenarios for the clonal architecture encountered in PNH: ancestral PIGA mutation (B), secondary PIGA mutation (C), PIGA mutation as an isolated genetic event (D), and PIGA mutation leading to a PNH clone that independently coexists with an MDS clone (E).
Clinical correlations.
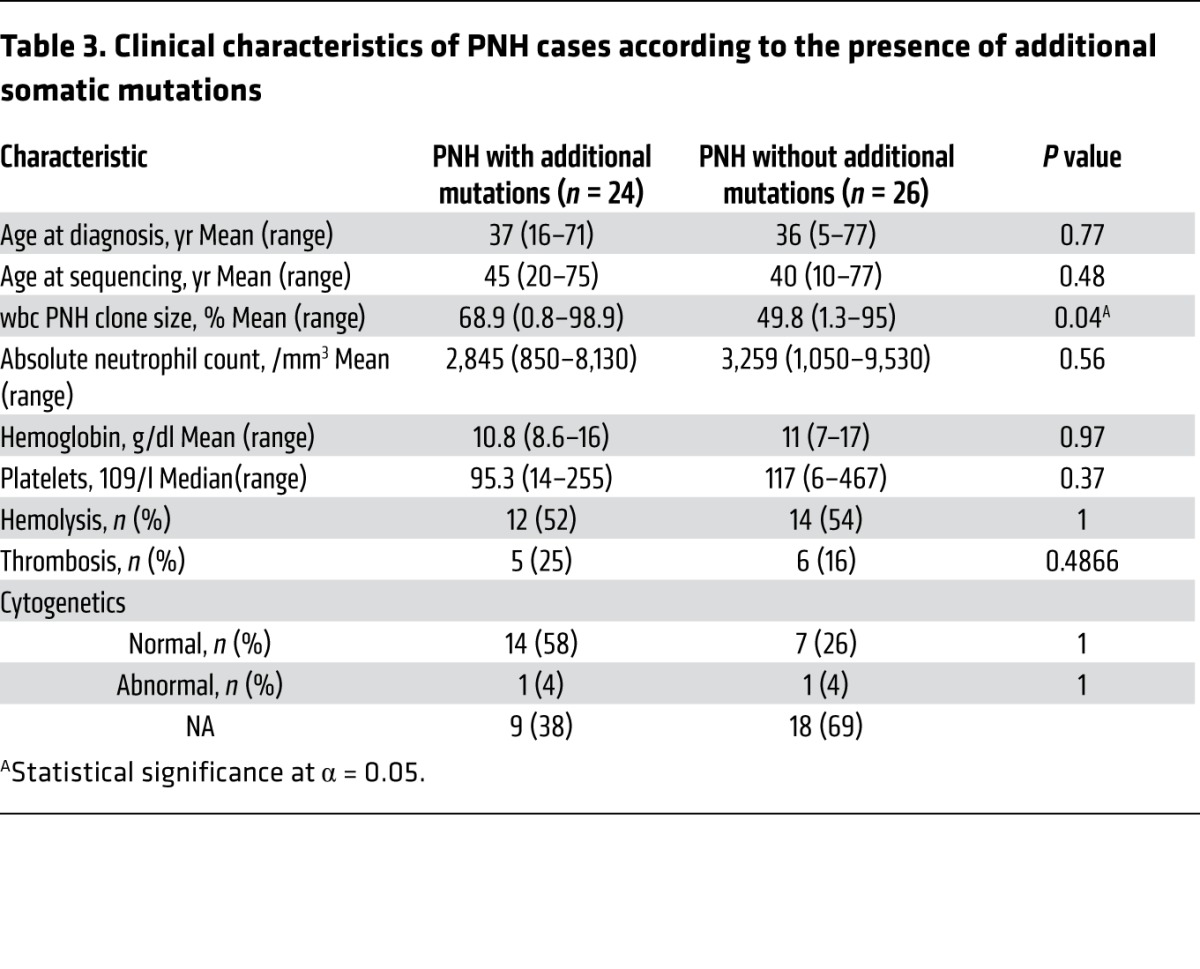
The discovery of additional somatic mutations in PNH prompted further investigation into the potential clinical impact these mutations may have. Analysis of various relevant clinical parameters with regard to the presence (n = 24) or absence (n = 26) of an additional mutation largely failed to find significant differences (Table 3). However, the presence of an additional mutation beyond PIGA was associated with a larger wbc PNH clone size as assessed by flow cytometry (P = 0.04; Table 3). Furthermore, based on the instructive discovery that PNH45 harbored 3 PIGA mutations (triclonal PNH) and a relatively large PNH type II wbc population (Figure 2C), we hypothesized that the presence of more than 1 PIGA mutation leads to the presence of both type II and type III PNH cells. Indeed, patients with more than 1 PIGA mutation had a higher incidence of a type II wbc population of greater than 3% than those with only 1 mutation (4 of 5 vs. 4 of 12, respectively; P = 0.048, data not shown).
Table 3.
Clinical characteristics of PNH cases according to the presence of additional somatic mutations
Discussion
One of the discoveries of cancer NGS projects was the realization of genetic heterogeneity due to a combination of somatic mutations and the complexity of clonal architecture reflective of the sequential acquisition of genetic defects (11, 17–21). While general principles of clonal dynamics have been derived from the study of leukemia, they have not been explored in nonmalignant diseases such as PNH. The results of our study lead to a conceptual understanding of this disease: PNH is subject to clonal dynamics and selection forces similar to those observed in hematopoietic neoplasms. Consequently, in PNH, additional clonal and subclonal mutations corroborate with the pathognomonic PIGA mutation responsible for the prevailing cell phenotype. An important conclusion of this new concept is that the nature and composition of additional clonal somatic mutations may modify the behavior of the PIGA clone and have the potential to explain various distinct clinical courses seen in individual PNH patients. Thus, the analogy to MDS is apparent with multiple mutations that arise within the clonal population and undergo clonal selection along with PIGA mutations. Furthermore, the mutational events discovered in our research are not unique to PNH, but overlap significantly with the spectrum of mutations seen in typical MDS. Many of these mutations, or other mutations in the same genes, have been identified as key drivers of clonal evolution in MDS or cancer, including U2AF1 (22, 23), TET2 (2, 24), MAGEC1 (25), BRPF1 (26), NRXN3 (27), KDM3B (28), SLC20A1 (29), MUC7 (30), PEX14 (31), FBN1 (32), SUZ12 (33), ASXL1 (34), BCOR (35), DHX29 (36), MECOM (37), RIT1 (38), and JAK2 V617F (39). In some ways, PNH appears genetically similar to, but pathogenetically distinct from, MDS. While additional mutations have been described in PNH (6), our results illustrate for the first time in a large PNH cohort that genetic clonal selection and evolution may be operative not only in malignant conditions, but also in otherwise benign hematologic diseases.
Our stringent bioanalytic approach has led to effective exclusion of germline variants based on comparatively similar high VAFs in both experimental and control fractions. If variant reads that derived from clonal mutations were present in the CD59+ cell population due to contamination, their VAF was low and was correspondingly high in the PNH+ population. As such, this would be incompatible with germline alterations, which would be expected to oscillate around 50% of reads in both fractions. With this in mind, all of the mutational events we report in this article are exclusively somatic. While we found that some mutations had similar VAFs in both fractions, they were clearly somatic, since the VAF was much lower than would be expected in heterozygous germline variants. In such situations, we verified the absence of the mutation in the CD3+ fraction and concluded that the PIGA mutation arose in the context of a preexisting clone. Future prospective studies would benefit from the use of nonhematopoietic tissue to clarify the mutational spectrum in PNH, as it is possible that some pre-PIGA mutations were excluded due to their presence in the entire hematopoietic compartment.
Our study, while showing that intrinsic somatic factors may contribute to clonal expansion, is also consistent with the immune selection theory of the evolution of PNH. Multiple independent clones characterized by PIGA mutations illustrate that a growth advantage may promote selection of several privileged clones, which in the process of disease may be further enabled by subsequent somatic events or by primordial passenger events in the ancestral stem cell affected by PIGA mutations. Phenotypically, this intraclonal diversity may not be easily distinguishable, although we have identified a significant relationship between the presence of both type II and type III PNH cells in patients with more than 1 PIGA mutation, supporting previous research (40). Nevertheless, our results suggest that deep targeted NGS of PIGA may have ancillary diagnostic potential in PNH, including quantitating the clonal size and composition of aberrant cells, yet flow cytometry remains the most effective diagnostic method, as we failed to detect PIGA mutations in 3 of 12 cases by WES, 1 of 10 by targeted deep sequencing, and 21 of 36 by Sanger sequencing. Other genes in the biosynthetic pathway, including all other PIG genes such as PIGT (41), were examined in WES cases, but no lesions were found, raising the possibility that intronic mutations may lead to the loss of GPI anchors on the cell surface. Furthermore, the high failure rate of Sanger sequencing could be due to a number of reasons including small clone size, intronic mutations, or micro/large deletions only detectable by SNP array.
At the onset of our study, we postulated several theoretical possibilities as to the clonal architecture of PNH: (a) PIGA gene defects are the initial ancestral events, and other mutations are acquired subsequently, similar to the situation in MDS; (b) other initial somatic events are followed by PIGA mutations constrained within and completely overlapping the PNH clone, suggesting that the initial non-PIGA events are passenger mutations; (c) secondary PIGA mutations arise in the context of other clonogenic mutations that are present in both PIGA mutant and wild-type cells; and (d) both myelodysplastic and PNH clones independently coexist. While PIGA mutations appear to be the initial event in many cases, we identified concomitant somatic mutations in a large proportion of PNH patients. In such cases, subsequent subclonal events can occur and are similar to those present in more aggressive hematologic malignancies. In cases in which the preceding clonal mutation was found but was limited to the PNH fraction, one could suggest that a passenger mutation was clonally “fixed” by the subsequent PIGA event. However, in some cases, permissive leukemogenic effects appeared to be instigated by mutations in JAK2, TET2, or STAC3, suggesting that these primary events arose, conveying an initial growth advantage, with a PIGA mutation as a subclone conveying an additional growth advantage. In particular, the presence of 2 cases with “myeloproliferative” JAK2 mutations suggests that the propensity toward clonal proliferation may be further modified by the presence of PIGA defects and that JAK2 mutations in concert with PIGA mutations lead to a markedly different phenotype than that of a myeloproliferative neoplasm (MPN). In addition, there were a significant number of cases in which PIGA was the lone mutation, although WES data suggest that this may be less frequent when entire exomes are analyzed.
Regardless, in many cases, the subclonal or clonal occurrence of associated mutations resembles the typical architecture of MDS (10). We and others have previously described additional somatic events in PNH patients demonstrating that even PIGA mutation–negative cases were clonal (6–8). The inability to detect significant recurrent additional mutations may be due to the relatively small cohort and surprising diversity discovered in this study. Nevertheless, our results suggest that an obligatory secondary event does not occur in PNH, yet PNH clonal expansions appear to be aided by the presence of additional mutations. Depending on the permissivity of additional genetic events, the PIGA mutant clone may expand quickly, smolder, or disappear. This heterogeneous scenario would also be consistent with a relative lack of correlation between immunosuppression and the size of the clone, which may be driven under some circumstances by additional subclonal events. Our results suggest that future investigations into PNH will offer the opportunity to examine the implications of hematopoiesis sustained primarily by 1 or 2 mutated stem cells, with many cases demonstrating an accumulation of additional mutations that may be due to an increase in the self-renewal burden placed on stem cells as a consequence of the disease.
Methods
Patients.
Bone marrow aspirates and/or blood samples were collected from 60 patients with PNH at the Cleveland Clinic. Diagnosis was confirmed and assigned according to the guidelines for the diagnosis and management of PNH (42).
Cytogenetics and SNP arrays.
Cytogenetic analysis was performed according to standard banding techniques based on 20 metaphases, if available. Technical details regarding sample processing for SNP array (SNP-A) assays were previously described (43, 44). Affymetrix 250K and 6.0 kits were used. A stringent algorithm was applied for the identification of SNP-A lesions. Patients with SNP-A lesions concordant with metaphase cytogenetics or typical lesions known to be recurrent required no further analysis. Changes reported in our internal or publicly available (Database of Genomic Variants; http://projects.tcag.ca/variation) copy number variation (CNV) databases were considered nonsomatic and excluded. Results were analyzed using CNAG version 3.0 (20) or Genotyping Console (Affymetrix). All other lesions were confirmed as somatic or germline by analysis of CD3-sorted cells (45).
WES.
WES was performed as previously reported (46). Briefly, tumor DNA was extracted from patients’ bone marrow or peripheral blood mononuclear cells. DNA was obtained from paired CD59-positive and -negative cells. Whole-exome capture was accomplished based on liquid-phase hybridization (SureSelect; Agilent Technologies) according to the manufacturer’s protocol. The SureSelect Human All Exon 50 Mb kit was used for 12 cases. The captured targets were subjected to massive sequencing using Illumina HiSeq 2000 according to the manufacturer’s instructions. The raw sequence data were processed through the in-house pipeline constructed for whole-exome analysis of paired cancer genomes at the Human Genome Center, Institute of Medical Science, The University of Tokyo, and are summarized in a previous report (46). Data processing was divided into 2 steps: (a) generation of a BAM file (http://samtools.sourceforge.net/) for paired normal and tumor samples for each case, and (b) detection of somatic SNVs and indels by comparing normal and tumor BAM files. Alignment of sequencing reads on hg19 was visualized using Integrative Genomics Viewer (IGV) software (http://www.broadinstitute.org/igv/) (47).
Targeted deep sequencing and Sanger sequencing.
We applied targeted exon sequencing to the validation of WES results as previously described (46, 48). Briefly, the number of reads containing SNVs and indels in both tumor and reference samples was determined using SAM tools, and the null hypothesis of equal allele frequencies in PNH and non-PNH samples was tested using the 2-tailed Fisher’s exact test. A variant was adopted as a candidate somatic mutation if it had a P value of less than 0.01. First, amplicons of each observation were subjected to Sanger sequencing by standard techniques on an ABI 3730xl DNA analyzer (Applied Biosystems). All mutations were confirmed by bidirectional sequencing and scored as pathogenic if not present in nonclonal, paired CD59–derived DNA. Then, deep NGS was applied to additional validation for variant allelic frequency and small clone detections. The amplicon libraries were generated according to an Illumina pair-end library protocol and subjected to deep sequencing on an Illumina MiSeq sequencer according to the standard protocol. For PIGA (exons 2–6) and KDM3B (exons 1–24), direct genomic sequencing was performed as previously described, together with deep sequencing, so that even very small mutated events would not be missed.
Single-cell colony culture.
Colony formation assays in semi-solid cultures were used to obtain colonies for single-colony sequencing. A total of 105 bone marrow cells from PNH patients were plated in 1 ml of methylcellulose supplemented with G-SCF, GM-SCF, and EPO cytokines (STEMCELL Technologies) as well as FBS in a 35-mm culture plate at 37°C with 5% CO2. After 7 to 10 days of culturing, individual colonies were removed, and DNA was extracted for sequencing.
Cell sorting.
Following lysis of rbc, immunomagnetic selection of cells was performed using anti-CD59 PE (Life Technologies) followed by anti-PE microbeads (Miltenyi Biotec). Samples were separated using LS Columns (Miltenyi Biotec), and purity was verified in each fraction by flow cytometry on a Beckman Coulter FC500.
Flow cytometry.
Whole blood was stained with antibodies against GlyA, CD15, CD24, CD55, CD59 (Beckman Coulter), CD59 (Life Technologies), and FLAER (Alexa 488 Proaerolysin Variant; Cedarlane Scientific) in various combinations to quantitate the number of GPI-deficient rbc and wbc in each patient. Samples were run on a Beckman Coulter FC500 or XL-MCL.
Accession codes.
WES results have been deposited in the Sequence Read Archive (SRA) public database (PRJNA254174).
Statistics.
JMP Pro 10 (SAS Institute Inc.) was used for statistical analysis. Either a Wilcoxon or Fisher’s exact test was performed to evaluate statistically significant differences between groups, with an α of 0.05.
Study approval.
Research was conducted according to protocols approved by the IRB of the Cleveland Clinic and in accordance with Declaration of Helsinki principles. Written informed consent was received from all patients prior to enrollment in the study.
Supplementary Material
Acknowledgments
This work was supported by grants from the NIH (RO1HL-082983, U54 RR019391, and K24 HL-077522, to J.P. Maciejewski); the Aplastic Anemia & MDS International Foundation (to J.P. Maciejewski and H. Makishima); the Robert Duggan Charitable Fund (to J.P. Maciejewski); Scott Hamilton CARES (to H. Makishima); the Ministry of Health, Labor and Welfare of Japan and KAKENHI (grants-in-aid 23249052, 22134006, and 21790907, to S. Ogawa); the Project for Development of Innovative Research on Cancer Therapeutics (P-DIRECT) (to S. Ogawa); and the Japan Society for the Promotion of Science (JSPS) through the Funding Program for World-Leading Innovative R&D on Science and Technology, initiated by the Council for Science and Technology Policy (CSTP) (to S. Ogawa). W. Shen was partly supported by the Jiangsu Health International Exchange Program (China).
Footnotes
Conflict of interest: The authors have declared that no conflict of interest exists.
Reference information:J Clin Invest. 2014;124(10):4529–4538. doi:10.1172/JCI74747.
References
- 1.Brodsky RA. Paroxysmal nocturnal hemoglobinuria: stem cells and clonality. Hematology Am Soc Hematol Educ Program. 2008:111–115. doi: 10.1182/asheducation-2008.1.111. [DOI] [PubMed] [Google Scholar]
- 2.Takeda J, et al. Deficiency of the GPI anchor caused by a somatic mutation of the PIG-A gene in paroxysmal nocturnal hemoglobinuria. Cell. 1993;73(4):703–711. doi: 10.1016/0092-8674(93)90250-T. [DOI] [PubMed] [Google Scholar]
- 3.Young NS, Calado RT, Scheinberg P. Current concepts in the pathophysiology and treatment of aplastic anemia. Blood. 2006;108(8):2509–2519. doi: 10.1182/blood-2006-03-010777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Maciejewski JP, Sloand EM, Sato T, Anderson S, Young NS. Impaired hematopoiesis in paroxysmal nocturnal hemoglobinuria/aplastic anemia is not associated with a selective proliferative defect in the glycosylphosphatidylinositol-anchored protein-deficient clone. Blood. 1997;89(4):1173–1181. [PubMed] [Google Scholar]
- 5.Araten DJ, Nafa K, Pakdeesuwan K, Luzzatto L. Clonal populations of hematopoietic cells with paroxysmal nocturnal hemoglobinuria genotype and phenotype are present in normal individuals. Proc Natl Acad Sci U S A. 1999;96(9):5209–5214. doi: 10.1073/pnas.96.9.5209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Inoue N, et al. Molecular basis of clonal expansion of hematopoiesis in 2 patients with paroxysmal nocturnal hemoglobinuria (PNH). Blood. 2006;108(13):4232–4236. doi: 10.1182/blood-2006-05-025148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.O’Keefe CL, et al. Deletions of Xp22.2 including PIG-A locus lead to paroxysmal nocturnal hemoglobinuria. Leukemia. 2011;25(2):379–382. doi: 10.1038/leu.2010.274. [DOI] [PubMed] [Google Scholar]
- 8.Mortazavi Y, Tooze JA, Gordon-Smith EC, Rutherford TR. N-RAS gene mutation in patients with aplastic anemia and aplastic anemia/ paroxysmal nocturnal hemoglobinuria during evolution to clonal disease. Blood. 2000;95(2):646–650. [PubMed] [Google Scholar]
- 9.Sugimori C, et al. Paroxysmal nocturnal hemoglobinuria and concurrent JAK2(V617F) mutation. Blood Cancer J. 2012;2(3):e63. doi: 10.1038/bcj.2012.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Walter MJ, et al. Clonal architecture of secondary acute myeloid leukemia. N Engl J Med. 2012;366(12):1090–1098. doi: 10.1056/NEJMoa1106968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ley TJ, et al. DNA sequencing of a cytogenetically normal acute myeloid leukaemia genome. Nature. 2008;456(7218):66–72. doi: 10.1038/nature07485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bessler M, Mason P, Hillmen P, Luzzatto L. omatic mutations and cellular selection in paroxysmal nocturnal haemoglobinuria. Lancet. 1994;343(8903):951–953. doi: 10.1016/S0140-6736(94)90068-X. [DOI] [PubMed] [Google Scholar]
- 13.Mazelin L, et al. Netrin-1 controls colorectal tumorigenesis by regulating apoptosis. Nature. 2004;431(7004):80–84. doi: 10.1038/nature02788. [DOI] [PubMed] [Google Scholar]
- 14.Arakawa H. Netrin-1 and its receptors in tumorigenesis. Nat Rev Cancer. 2004;4(12):978–987. doi: 10.1038/nrc1504. [DOI] [PubMed] [Google Scholar]
- 15.van Gils JM, et al. The neuroimmune guidance cue netrin-1 promotes atherosclerosis by inhibiting the emigration of macrophages from plaques. Nat Immunol. 2012;13(2):136–143. doi: 10.1038/ni.2205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cavard C, et al. Gene expression profiling provides insights into the pathways involved in solid pseudopapillary neoplasm of the pancreas. J Pathol. 2009;218(2):201–209. doi: 10.1002/path.2524. [DOI] [PubMed] [Google Scholar]
- 17.Burrell RA, McGranahan N, Bartek J, Swanton C. The causes and consequences of genetic heterogeneity in cancer evolution. Nature. 2013;501(7467):338–345. doi: 10.1038/nature12625. [DOI] [PubMed] [Google Scholar]
- 18.Meacham CE, Morrison SJ. Tumour heterogeneity and cancer cell plasticity. Nature. 2013;501(7467):328–337. doi: 10.1038/nature12624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cancer Genome Atlas Research Network Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature. 2008;455(7216):1061–1068. doi: 10.1038/nature07385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cancer Genome Atlas Research Network Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N Engl J Med. 2013;368(22):2059–2074. doi: 10.1056/NEJMoa1301689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Stransky N, et al. The mutational landscape of head and neck squamous cell carcinoma. Science. 2011;333(6046):1157–1160. doi: 10.1126/science.1208130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Graubert TA, et al. Recurrent mutations in the U2AF1 splicing factor in myelodysplastic syndromes. Nat Genet. 2011;44(1):53–57. doi: 10.1038/ng.1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Makishima H, et al. Mutations in the spliceosome machinery, a novel and ubiquitous pathway in leukemogenesis. Blood. 2012;119(14):3203–3210. doi: 10.1182/blood-2011-12-399774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ko M, et al. Impaired hydroxylation of 5-methylcytosine in myeloid cancers with mutant TET2. Nature. 2010;468(7325):839–843. doi: 10.1038/nature09586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Koh SS, et al. Differential gene expression profiling of primary cutaneous melanoma and sentinel lymph node metastases. Mod Pathol. 2012;25(6):828–837. doi: 10.1038/modpathol.2012.32. [DOI] [PubMed] [Google Scholar]
- 26.Shima H, et al. Bromodomain-PHD finger protein 1 is critical for leukemogenesis associated with MOZ-TIF2 fusion. Int J Hematol. 2014;99(1):21–31. doi: 10.1007/s12185-013-1466-x. [DOI] [PubMed] [Google Scholar]
- 27.Sun HT, Cheng SX, Tu Y, Li XH, Zhang S. FoxQ1 promotes glioma cells proliferation and migration by regulating NRXN3 expression. PLoS One. 2013;8(1):e55693. doi: 10.1371/journal.pone.0055693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kim JY, et al. KDM3B is the H3K9 demethylase involved in transcriptional activation of lmo2 in leukemia. Mol Cell Biol. 2012;32(14):2917–2933. doi: 10.1128/MCB.00133-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Liu L, Sanchez-Bonilla M, Crouthamel M, Giachelli C, Keel S. Mice lacking the sodium-dependent phosphate import protein, PiT1 (SLC20A1), have a severe defect in terminal erythroid differentiation and early B cell development. Exp Hematol. 2013;41(5):432–443. doi: 10.1016/j.exphem.2013.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Byrd JC, Bresalier RS. Mucins and mucin binding proteins in colorectal cancer. Cancer Metastasis Rev. 2004;23(1–2):77–99. doi: 10.1023/a:1025815113599. [DOI] [PubMed] [Google Scholar]
- 31.Cheung KJ, et al. High resolution analysis of follicular lymphoma genomes reveals somatic recurrent sites of copy-neutral loss of heterozygosity and copy number alterations that target single genes. Genes Chromosomes Cancer. 2010;49(8):669–681. doi: 10.1002/gcc.20780. [DOI] [PubMed] [Google Scholar]
- 32.Summers KM, et al. Experimental and bioinformatic characterisation of the promoter region of the Marfan syndrome gene, FBN1. Genomics. 2009;94(4):233–240. doi: 10.1016/j.ygeno.2009.06.005. [DOI] [PubMed] [Google Scholar]
- 33.Liu C, et al. SUZ12 is involved in progression of non-small cell lung cancer by promoting cell proliferation metastasis. Tumour Biol. 2014;35(6):6073–6082. doi: 10.1007/s13277-014-1804-5. [DOI] [PubMed] [Google Scholar]
- 34.Abdel-Wahab O, Dey A. The ASXL-BAP1 axis: new factors in myelopoiesis, cancer and epigenetics. Leukemia. 2013;27(1):10–15. doi: 10.1038/leu.2012.288. [DOI] [PubMed] [Google Scholar]
- 35.Panagopoulos I, et al. Fusion of the ZC3H7B and BCOR genes in endometrial stromal sarcomas carrying an X;22-translocation. Genes Chromosomes Cancer. 2013;52(7):610–618. doi: 10.1002/gcc.22057. [DOI] [PubMed] [Google Scholar]
- 36.Parsyan A, et al. The helicase protein DHX29 promotes translation initiation, cell proliferation, and tumorigenesis. Proc Natl Acad Sci U S A. 2009;106(52):22217–22222. doi: 10.1073/pnas.0909773106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Roy S, et al. BCR-ABL1 tyrosine kinase sustained MECOM expression in chronic myeloid leukaemia. Br J Haematol. 2012;157(4):446–456. doi: 10.1111/j.1365-2141.2012.09078.x. [DOI] [PubMed] [Google Scholar]
- 38.Gomez-Segui I, et al. Novel recurrent mutations in the RAS-like GTP-binding gene RIT1 in myeloid malignancies. Leukemia. 2013;27(9):1943–1946. doi: 10.1038/leu.2013.179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kralovics R, et al. A gain-of-function mutation of JAK2 in myeloproliferative disorders. N Engl J Med. 2005;352(17):1779–1790. doi: 10.1056/NEJMoa051113. [DOI] [PubMed] [Google Scholar]
- 40.Rollinson S, Richards S, Norfolk D, Bibi K, Morgan G, Hillmen P. Both paroxysmal nocturnal hemoglobinuria (PNH) type II cells and PNH type III cells can arise from different point mutations involving the same codon of the PIG-A gene. Blood. 1997;89(8):3069–3071. [PubMed] [Google Scholar]
- 41.Krawitz PM, et al. A case of paroxysmal nocturnal hemoglobinuria caused by a germline mutation and a somatic mutation in PIGT. Blood. 2013;122(7):1312–1315. doi: 10.1182/blood-2013-01-481499. [DOI] [PubMed] [Google Scholar]
- 42.Parker C, et al. Diagnosis and management of paroxysmal nocturnal hemoglobinuria. Blood. 2005;106(12):3699–3709. doi: 10.1182/blood-2005-04-1717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Maciejewski JP, Tiu RV, O’Keefe C. Application of array-based whole genome scanning technologies as a cytogenetic tool in haematological malignancies. Br J Haematol. 2009;146(5):479–488. doi: 10.1111/j.1365-2141.2009.07757.x. [DOI] [PubMed] [Google Scholar]
- 44.Gondek LP, et al. Chromosomal lesions and uniparental disomy detected by SNP arrays in MDS, MDS/MPD, and MDS-derived AML. Blood. 2008;111(3):1534–1542. doi: 10.1182/blood-2007-05-092304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Tiu RV, et al. New lesions detected by single nucleotide polymorphism array-based chromosomal analysis have important clinical impact in acute myeloid leukemia. J Clin Oncol. 2009;27(31):5219–5226. doi: 10.1200/JCO.2009.21.9840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yoshida K, et al. Frequent pathway mutations of splicing machinery in myelodysplasia. Nature. 2011;478(7367):64–69. doi: 10.1038/nature10496. [DOI] [PubMed] [Google Scholar]
- 47.Robinson JT, et al. Integrative genomics viewer. Nat Biotechnol. 2011;29(1):24–26. doi: 10.1038/nbt.1754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Makishima H, et al. Somatic SETBP1 mutations in myeloid malignancies. Nat Genet. 2013;45(8):942–946. doi: 10.1038/ng.2696. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.