

Abstract
Antibiotics have been a cornerstone of innovation in the fields of public health, agriculture, and medicine. However, recent studies have shed new light on the collateral damage they impart on the indigenous host-associated communities. These drugs have been found to alter the taxonomic, genomic, and functional capacity of the human gut microbiota, with effects that are rapid and sometimes persistent. Broad-spectrum antibiotics reduce bacterial diversity while expanding and collapsing membership of specific indigenous taxa. Furthermore, antibiotic treatment selects for resistant bacteria, increases opportunities for horizontal gene transfer, and enables intrusion of pathogenic organisms through depletion of occupied natural niches, with profound implications for the emergence of resistance. Because these pervasive alterations can be viewed as an uncoupling of mutualistic host-microbe relationships, it is valuable to reconsider antimicrobial therapies in the context of an ecological framework. Understanding the biology of competitive exclusion, interspecies protection, and gene flow of adaptive functions in the gut environment may inform the design of new strategies that treat infections while preserving the ecology of our beneficial constituents.
The effects of antibiotics on the indigenous microbes of the gut have been the subject of extensive study ever since the discovery of these drugs in the 1940s and their subsequent commercial production. Yet nearly all of this work has concerned the effects of individual antibiotics on individual, cultivated strains of bacteria in the laboratory, or on specific species of bacteria cultivated from antibiotic-exposed hosts. Additionally, most of these studies have employed relatively high concentrations of these drugs in comparison to their typical concentrations in naturally occurring microbial communities and focused on pathogenic bacterial species. As a result, much of our understanding about the effects of antibiotics is skewed toward mechanisms of killing and specific resistance genotypes and phenotypes in the context of a narrow subset of the gut microbiota in isolation from the rest of the community.
Over the past decade or so, the study of antibiotics and the gut microbiota has taken a decidedly more ecological and system-wide perspective. Ecological principles and molecular approaches are now prominently featured in research laboratories and in the clinical workplace. As the microbial community and its local ecosystem have become a unit of study, we review here recent work that examines the effects of antibiotics on the human gut bacterial community, highlighting the implications for lateral transfer of resistance genes.
Antibiotics: an ecological perspective
Antibiotic resistance genes are ubiquitous in present day natural environments. In part, this reflects the widespread human use of large quantities of antibiotics over the past 70 years and the strong selective pressures this use has exerted, including the dissemination of resistant organisms. However, the resistance genes themselves are ancient. DNA sequences predicted to encode β-lactamases, the tetracycline resistance determinant TetM, and vancomycin resistance proteins have been recovered from 30,000-year-old permafrost sediment in northern British Columbia (1). Although these sequences are related to those of present-day resistance genes, they were significantly divergent. But in at least one case (the VanA ligase), the gene expressed a protein with the expected function. Bacterial isolates that have been recovered from ancient, protected sites, including one site that was allegedly isolated for more than 4 million years (2), have been shown to express multi-drug resistance. The presence of these genes well before modern use of the related antibiotics by humans strongly suggests that these antibiotics were widely distributed in the environment and that resistance genes have been circulating for at least thousands of years. Modern use of high doses of antibiotics enriches for resistance genes and, from a host ecological perspective, may lead to an uncoupling of mutualistic relationships that have evolved over long periods of time among gut microbiota and the human host (3, 4).
Low concentrations of antibiotics at levels believed to be present in natural environments such as soil elicit nonlethal responses that lead to altered physiology and behavior (5–7); thus, antibiotics and a wide variety of small molecules that display growth-inhibitory properties at high concentrations may have evolved long ago to serve as interspecies signaling molecules. These molecules mediate species-species interactions that shape the structure of and confer stability to naturally occurring microbial communities. The diversity and number of small molecules and potential antibiotics produced by gut commensal bacteria are almost certainly far greater than previously recognized. “Genome-mining” approaches have revealed a number of novel, potent small molecules from both environmental and commensal bacteria, including some new peptidyl nucleoside antibiotics (8, 9). With the development of exploratory bioinformatics tools and expression screening techniques, previously studied strains have yielded new antibacterial molecules of substantial interest (10, 11); prior screening efforts failed to identify these molecules, in large part because they were not expressed under routine laboratory growth conditions (12).
Microbial community-wide effects of antibiotics
For several decades, a variety of studies have been conducted in order to characterize the effect of antibiotics on specific strains and species, and the emergence of drug resistance, within the gut bacterial communities. In recent years, efforts have begun to turn to the effects of antibiotics on the overall taxonomic composition of the fecal microbiota, and on the abundance and diversity of antibiotic resistance genes. Still relatively ignored are the effects on clinically relevant, community-wide properties of the gut microbiota and on host response. However, one of the earliest described features of the effect of antibiotics on the gut was the loss of colonization resistance (i.e., the loss of “competitive exclusion,” refs. 13, 14). The loss was manifested by the much greater ease of colonization and disease caused by Salmonella immediately following antibiotic treatment. Both resource competition and direct interference competition play roles in the resistance of the intact microbiota to colonization by pathogens. Indirect factors include the induction of multiple innate immune response pathways and effector molecules (15). Antibiotics disrupt community structure sufficiently to cause large-scale disturbances in resources and species-species interactions. Recent work in mice suggests that antibiotics lead to an increase in the abundance of host-derived free sialic acid in the gut, which can then be utilized by opportunistic pathogens such as Salmonella typhimurium and Clostridium difficile to enhance their growth (16).
In general, studies of the effects of antibiotics on gut community taxonomic composition have found diminished levels of bacterial diversity, stereotypic declines and expansions in the relative abundances of certain taxa, some degree of recovery in most individuals but persistent effects in others, and antibiotic- and individual host–specific effects. Antibiotics with strong and broad activity against anaerobes, for example clindamycin, have typically caused the longest-lasting effects on gut community composition (17–19). Jakobsson studied the impact of seven days of clarithromycin, metronidazole, and omeprazole on the pharyngeal and fecal taxonomic composition, and found broad taxonomic compositional effects with rapid but only partial recovery in some cases and persistent effects at least four years after exposure (20).
Ciprofloxacin, which has relatively little activity against standard cultivated anaerobes, has profound effects on the gut microbiota composition. Dethlefsen et al. found that five days of ciprofloxacin influenced the abundance of about a third of the bacterial taxa in the gut and decreased taxonomic richness within days of initial exposure (21). Some responses among the human subjects were conserved (such as abrupt decreases in bacterial diversity and depletion of many of the Ruminococcaceae), while others were individualized (such as the degree or timing of community composition recovery following each of the ciprofloxacin exposures). Nearly complete recovery was seen in most cases by four weeks after exposure, although some compositional effects lasted for six months. There were no symptoms or signs in these subjects, suggesting the importance of functional redundancy within the microbiota (22). A second identical ciprofloxacin exposure six months after the first caused similar acute effects but was associated with less complete recovery in some cases (23). As ecologists have noted in other ecosystems, compounded (e.g., antibiotic) disturbances can lead to “ecological surprises” (24).
Efforts to explore the effect of antibiotics on other microbiota-associated host phenotypes are underway. Cho et al. have shown that early-life exposure of mice to subtherapeutic doses of antibiotics, in addition to altering the composition of the intestinal microbiota, also increased total and relative body fat mass, bone density, and intestinal microbiota production of short-chain fatty acids and altered hepatic fatty acid metabolism (25). Whether and to what degree these effects occur in children, and the degree to which these effects are dependent on other microbial, host, and environmental factors remains to be seen.
The antibiotic resistance reservoir of the microbiome
The pervasive effects of antibiotics on the population structure in the gut are paralleled by their alteration of the genomic capacity of gut microbial communities. Treatment of bacterial populations with antibiotics in the laboratory selects and enriches for resistant strains and species (26, 27), and studies of antibiotics and resistance in vivo, while still nascent, have yielded similar findings (Figure 1). In the Jakobsson study, patients treated with a clarithromycin-containing combination antibiotic regimen for H. pylori–associated peptic ulcers exhibited 1,000-fold increases in the ermB resistance gene, which encodes the macrolide target–modifying RNA methylase, immediately following their course of treatment. Although subjects did not receive additional antibiotics throughout the course of the study, their gut microbiota continued to harbor comparable levels of the resistance genes four years later (20). Subtherapeutic doses also appear to result in the enrichment of resistance genes. The use of ASP40 growth-enhancing antibiotics (a cocktail of chlortetracycline, sulfamethazine, and penicillin) in swine feed produced significant increases in abundance and diversity of multiple resistance genes after only three days of exposure. Genes conferring resistance to classes of drugs that were not present in the feed source, such as aminoglycoside resistance genes, were also enriched, providing evidence for the role of antibiotics in promoting resistance to unrelated classes of drugs in commensal microbes (28).
Figure 1. Microbial community-wide effects of antibiotics on the human gut microbiota.
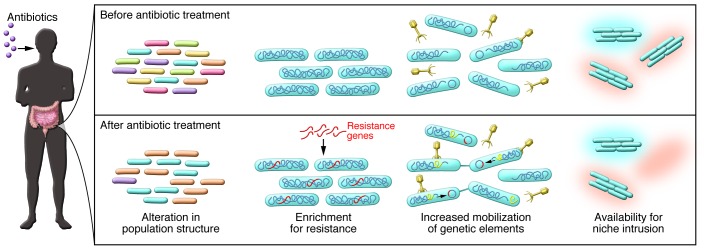
Antibiotic treatment alters the population structure of the indigenous microbiota, reducing bacterial diversity and redistributing member composition in both transient and persistent effects. Changes to the highly co-evolved microbial community architecture lead to changes in resource availability and species-species interactions, opening niches available for pathogenic intrusion and leading to the loss of colonization resistance. Antibiotics also select for antibiotic-resistant community members, enriching the presence of resistance genes in the microbiome. Treatment with antibiotics promotes the transfer of genetic information among bacteria by increasing conjugation, phage transduction, and plasmid mobility, primarily through the activation of cellular stress responses.
The development of antibiotic resistance in the gut microbiota has been most commonly studied using genomic and metagenomic approaches or alternatively by tracking the phenotypes of particular species (29, 30). As of yet, the dynamics of the evolution of resistance at the community level remain unclear, as do the mechanisms that sustain the microbiota in the face of antibiotic stress. Identifying the source of this resistance is central to understanding the emergence of resistance. Resistance can arise spontaneously due to the mutability of bacterial genomes (31), or it can emerge from the transfer of genetic material from one cell to another (Figures 1 And 2). As a further example of the intrinsic and natural role of drug resistance in microbial ecosystems (1, 32), the human microbiome has recently been found to serve as an impressive reservoir of antibiotic resistance genes. Colonization of the intestine by resistant bacteria can occur within three days of birth (33); additionally, resistance phenotypes have been documented in remote human communities with limited exposure to antibiotics (34).
Figure 2. Mechanisms for the acquisition of resistance genes.
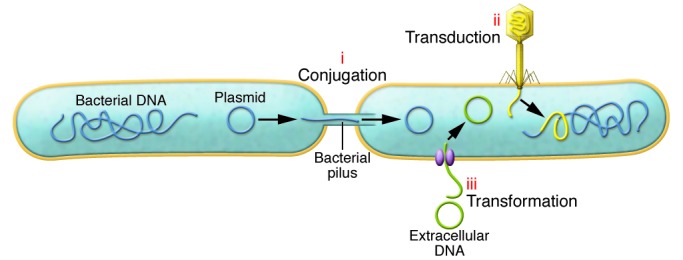
Bacteria exchange genetic information with one another using horizontal routes of conjugation, phage transduction, and natural transformation. In conjugation (i), donor and recipient cells are physically connected through the formation of a transient bridge (pilus), and DNA copied from one cell flows to the next. Cells can transfer plasmid DNA, integrative conjugative elements (chromosomally encoded gene clusters with autonomous conjugation machinery), or chromosomal DNA through high frequency of recombination mediated by F plasmids. Phages or bacterial viruses serve as vehicles for bacterial gene transfer (ii) by transducing DNA from one host cell to another. During lysis, phages can inadvertently package bacterial DNA, either randomly incorporating pieces of the bacterial genome into phage particles (generalized transduction) or taking up bacterial DNA positioned near the phage integration site (specialized transduction). Upon lysogenic infection of a new host, genetic material can be maintained in the genome by homologous recombination or site-specific integration. In the process of natural transformation (iii), certain bacterial species can take up free DNA from the environment using membrane protein complexes. While some species exhibit competence during phases of their life cycle, others respond to extracellular cues to initiate DNA uptake.
Once present in the community, selective pressures and opportunities to mitigate the costs of antibiotic resistance (35) likely contribute to its maintenance by the ecosystem. A study that described culturable microbes from over 600 individuals revealed that the majority of these individuals harbored gut communities in which at least 10% of members were resistant to a single antibiotic, and over a third carried microbes that exhibited multidrug resistance phenotypes (36). These subjects came from rural, urban, and hospital environments, and although resistance to certain drugs was detected at higher frequency in the hospitalized group, resistance to most antibiotics was present at similar levels in all cohorts. In a separate study, fecal bacterial populations from two individuals were studied using culture-independent and culture-dependent approaches (37). At least 20% of isolates cultivated from each individual were resistant to 10 different drugs, comprising a range of antibiotic classes and bacteriostatic and bactericidal drugs. Genomic inserts captured from total community DNA further revealed an expansive repertoire of coding sequences providing resistance functions in the human microbiome. Because the majority of host-associated bacteria are recalcitrant to current culture techniques, this metagenomic perspective provides a less biased assessment of the prevalence of resistance genes across the breadth of the intestinal microbial community.
Horizontal gene transfer in the gut environment
The reservoir of antibiotic resistance determinants in the microbiome presents an available cache of genetic information for rapid innovation by members of the gut community using mechanisms of gene exchange. Bacteria are capable of transferring genetic information to one another using the horizontal routes of conjugation, phage transduction, and natural transformation (ref. 38 and Figure 2). Intercellular movement of DNA is further facilitated by its intracellular mobilization, such as by transposons and mobile integrons. These elements can translocate to plasmids and integrating conjugative elements, facilitating their accessibility in the collective bacterial metagenome. Mobilized traits offer bacteria the capacity for niche expansion and functional diversification by enabling the sampling of innovations perfected by coexisting microbes (39). Because of the intrinsic evolvability and mechanisms for replication of horizontally acquired genetic material, such material can constitute up to 15% to 20% of prokaryotic genomes (40).
Recent work has indicated that gene transfer is common in the gut. A broad assessment of lateral gene transfer events between bacteria from a multitude of environments showed that human-associated bacteria are 25 times more likely to exchange genetic material than bacteria from other environments, and that closely related human-associated bacteria exhibit horizontal gene transfer (HGT) in 20% to 40% of all pairwise bacterial comparisons (41). This expansive and highly connected network for gene exchange may be a consequence of the unique circumstances of microbial inhabitation of the gut. While particular species have evolved specialized functions in the intestinal ecosystem, microbiota share similar goals in their mutualistic relationships with their hosts and are subject to the same selective pressures that constrain viability in the human gut. For example, mechanical forces in the gastrointestinal tract and turnover of mucosal epithelial cells threaten displacement, requiring that bacteria associate with the intestinal mucosa and mucus or engage in other physical interactions. Because the human immune system responds to microbial cues and regulates community composition, bacterial subversion and cooperation are not uncommon as means for maintaining homeostatic conditions for occupancy (42). The potential fragility of the population structure in the face of perturbation (21, 23, 43) suggests that functional redundancy among community members is an important feature of their co-evolved relationship with the host.
As these physical and functional forces may promote information sharing through HGT, the accessibility of conduits for gene flow further suggests a mechanistic basis for the abundance of HGT in the gut. The intestinal ecosystem boasts the highest density of microbes of all known environments, with 1011–1012 cells per gram of gastrointestinal content (44); opportunities for HGT by conjugation are afforded by the intimate proximity of microorganisms. In addition to their role in forming intercellular mating bridges, bacterial pili promote cell adhesion, aiding in colonization of the gastrointestinal tract. Dual functionality of this cellular apparatus may influence chances for conjugation in this setting. Host immune system activity can also increase opportunities for mating events between similar species by affecting population blooms (45). A metagenomic study of microbiota from Japanese individuals provided support for these conjugation-conducive features, revealing that a conjugative transposon family (Tn1549-like CTns) was highly prevalent across individuals and abundant within individual metagenomes, accounting for almost 1% of all predicted genes (46).
Phage, another means for gene transfer, can have a profound influence on the lifestyles of environmental microbes (47), and early analyses of viral interactions in the gut show that phages may similarly affect the indigenous microbiota. A metagenomic analysis of viruses from twin pairs and their mothers revealed that individuals harbored vastly different viral populations, but intrapersonal variability of virotypes was low, and phage populations were consistent over the year-long period of investigation (48). This stability intimated lysogeny, and in corroboration most of the phages identified were temperate and integrase genes were abundantly annotated in viral metagenomes. Clustered regularly interspaced short palindromic repeat (CRISPR) elements identified in the microbial community, hallmarks of phage resistance, did not match corresponding individuals’ viral sequences, suggesting that the gut environment utilizes phage as vehicles for HGT rather than as predatory agents.
Despite our current knowledge regarding routes for HGT in the gut environment, it is unclear which members of the community are actively participating in these various mechanisms. For example, the normal human microbiota includes naturally transformable bacterial species (49, 50), but many of these are low-abundance pathogens and their role in contributing to the exchange of information in this environment is not well understood. Furthermore, microbiome annotations of two individuals sampled as part of the MetaHit consortium showed that several conjugative protein orthologs were in the top 80th percentile of represented protein families, yet over 50% of these sequences were harbored by species of low representation (51).
The mobility of antibiotic resistance genes encoded in the microbiome
Because of their acute fitness advantage in a range of environments, antibiotic resistance genes are frequently encoded on mobile elements. With an estimated 1% to 3% of the developed world undergoing antibiotic treatment on a daily basis (3), the dynamic environment of the human intestine benefits (and may suffer in some cases) from metagenome accessibility of these genes on conjugative elements, viral particles, and plasmids (46, 52–54). Interestingly, in a study of genomic variation in over 200 individuals, conjugative elements with resistance functions were found to have the highest SNP density of all annotatable genes (55). Accordingly, in addition to introducing new genes, HGT can also be a source of variability at those loci, highlighting the evolutionary plasticity associated with mechanisms of gene transfer (56).
Mobility of resistance is compounded by the tendency of antibiotics to facilitate transfer of genetic information (Figure 1). DNA synthesis–inhibiting antibiotics have been shown to induce interspecies exchange of integrating conjugative elements encoding multidrug resistance (57). In vivo experiments have demonstrated increased conjugation between the gut species Enterococcus faecalis and Listeria monocytogenes in mice administered tetracycline in their drinking water (58). Antibiotic treatment is known to enhance phage mobility through activation of the bacterial DNA-damage response, which has cross-talk with phage regulation (59), and recent work has shown that antibiotic treatment in vivo increases the connectivity of phage-bacterial networks, thus potentiating microbial access to the phage metagenome (60).
Intestinal microbes are important to their hosts in part due to their role in community-mediated exclusion of pathogenic bacteria. As mentioned earlier, commensals prevent pathogen invasion by outcompeting virulent microbes for space and nutrients (3) and by inducing host defenses of the colonic epithelium to actively protect against infections of the intestine (61, 62). Antibiotic-mediated disruption of the highly organized population structure of the gut environment can reduce these defenses and open new niches for occupation (Figure 1). Exacerbated by the increased availability and mobility of resistance genes resulting from antibiotic treatment, co-localization of pathogenic and commensal communities offers opportunities for the transfer of resistance to virulent species.
Pathogen accessibility of the human microbiome is exemplified by the USA300 community-acquired strain of methicillin-resistant Staphylococcus aureus (MRSA), which obtained a gene cluster from the skin commensal Staphylococcus epidermis that provides functionality for improved colonization of host sites (63). Additionally, substantial evidence exists for the movement of contextually relevant genetic information between biomes. In one example, specialized polysaccharide degradation genes from marine bacteria were identified in microbiota of Japanese individuals, likely as a consequence of bacterial interactions facilitated by seaweed consumption by humans over hundreds of years (64). Also of note, functional metagenomic screening provided evidence for the exchange of genes conferring protection to multiple classes of antibiotics, between soil bacteria and common human pathogens (65). On a macroscopic level, transfer of antibiotic resistance genes across environmental boundaries takes place more frequently than within a given environment (41), illustrating the facile movement of adaptive functions between ecologically diverse bacteria. However, the transfer of resistant genes between microbiota and pathogen gene pools has not yet been definitively confirmed. Culture-independent investigation of the reservoir of resistance genes in the human microbiome revealed that functional sequences exhibited low homology to resistance genes of known pathogens (37). Accordingly, uncharacterized barriers may hinder resistance transfer between commensal and pathogenic communities and promote compartmentalization of the resistome.
Although antibiotic treatment poses significant implications for the dissemination of resistance genes, much has yet to be discerned about the mechanisms that govern gene flow in in vivo environments and the functional context in which this information transfer occurs. For example, HGT induced by antibiotic treatment may improve the capacity of microbiota to endure stress perturbation and maintain its contribution to host function as a consequence of increased connectivity of the microbiome. Carbohydrate-active enzymes, which promote bacterial survival in the gut environment and provide energy to the host, are transferred extensively across diverse phylogenies of commensal bacteria, and it is surmised that horizontal dispersal of these genes enables convergent community function to endure shared challenges faced in the dynamic gut ecosystem (66). Antibiotic treatment of mice has been shown to result in the enrichment of phage-encoded carbohydrate-active enzymes, raising the possibility that genomic reservoirs augmented by increased gene transfer, such as the phage metagenome, may serve to buffer the gut environment during stress by allowing bacteria to store and access functional elements that aid in niche recolonization (60). Given the complex and co-evolved relationship of the host and its associated communities, HGT in the human gut may drive innovation and evolution of functions benefiting the host.
Unanswered questions
The complexity of the disturbances arising from antibiotic treatment highlights the importance for developing augmented or alternative antimicrobial therapies that minimize consequences to the microbiome. Antibiotic-induced functional and phylogenetic alterations reveal the interdependence of gut microbiota in maintaining colonization of the intestinal space and its contribution to host function. This appreciation for gut-associated bacteria as a connected community in communication with its environment offers an ecosystem framework with which to devise new lines of inquiry regarding therapeutic modulation of the gut environment.
Although the microbiome represents a permeable network for gene flow, it remains unclear how other inter-organism interactions are utilized to mediate protection in the face of antibiotic perturbation. Microbiotas may recruit other ecosystem members to regain their homeostatic composition, such as phages and the host immune system; furthermore, some species may protect others from antibiotic stress, such as through cell signaling and heterogeneously organized biofilms (67, 68). Bacteria have been shown to exhibit cooperative mechanisms of resistance when confronted with antibiotic treatment in vitro (26). Host-microbiota mutualism may promote altruistic sharing of public goods that directly benefit individual species in order to maintain the community architecture that sustains the population as a whole. Understanding the extent to which these mechanisms occur in the gut environment may inform strategies to preemptively induce resilience of the gut community and preserve its function during perturbation.
While inter-organism interactions may contribute to the robustness of the gut environment, the population structure creates a foundation for stability, and specific features of this structure likely buffer against community disturbances. What is the basis for competitive exclusion of foreign bacteria from the gut environment, and what specific alterations to the composition enable pathogenic intrusion? As mentioned above, recent work suggests that increased nutrient availability in the absence of certain scavenging commensals depleted by antibiotic treatment contributes to pathogenic proliferation (16). The pursuit of investigations along these lines might enable the informed design of biologically inspired therapeutics, such as consortia-based solutions. Multi-organism cocktails may potentially rebalance the community more effectively than antibiotics, while reducing adverse consequences to the intestinal ecosystem and opportunities for resistance. In addition to treating infections, community-based therapeutics might also be used to reboot dysbiotic ecosystems; however, one of the challenges with restoration ecology is that exogenously delivered communities are not always maintained and effects can be transient (69). A modification of synthetic communities to exploit the selfish nature of genes with HGT, such as loading constituents to encode conjugative-based elements or broad host range plasmids, may allow imparted functions to endure beyond elimination of the delivered community. Phage-based therapeutics might also be used to distribute functions to the microbiome, perhaps engineered to carry genes that aid in recolonization of the gut environment or genes that encode functions depleted due to sustained ecosystem damage.
As antibiotics are currently our main line of defense against bacterial infections, it will be important to take advantage of insights from the emergence of resistance in vivo to innovate strategies to curb the spread of this resistance. Bacteria in the gut environment contribute to a microbiome that is highly accessible and replete with resistance functions readily able to transcend ecological boundaries. This is further complicated by the long-term persistence of resistance carriage and the propensity for diverse resistance elements to be co-mobilized, raising important implications for the unchecked evolution of cross-resistance. However, it remains unclear what governs the dissemination of resistance into clinical environments and whether specific features exist to sequester the resistome from pathogen access. The flow of this information has unfavorable consequences for the gut community, as resistant pathogens serve as more venerable adversaries in competing for inhabitation of the host. An identification and understanding of evolutionarily advantageous, context-specific regulation of HGT might help to constrain the transmission of potent genetic information and mitigate deleterious consequences for the human host. Because the microbiota uses HGT to preserve functional capacity following stress perturbation, it will be equally important to better understand how beneficial gene flow occurs such that it can be maintained and promoted. As we continue to elucidate the resounding impact of environmental disturbances on the gut microbiome, efforts to preserve the ecology of native commensal and mutualistic community members will be critical to human health and the evolution of improved relationships with our microbial companions.
Acknowledgments
The authors wish to thank members of their laboratories for valuable input and intellectual discourse. We wish to acknowledge the large number of relevant and important studies that we were unable to mention or discuss in this Review. S.R. Modi and J.J. Collins are supported by the Defense Threat Reduction Agency grant HDTRA1-14-1-0006 and the Howard Hughes Medical Institute. D.A. Relman is supported by grants from NIH (AI112401, GM099534, DE023113) and Office of Naval Research (N000141010233) and by the Thomas C. and Joan M. Merigan Endowment at Stanford University.
Footnotes
Conflict of interest: The authors have declared that no conflict of interest exists.
Reference information:J Clin Invest. 2014;124(10):4212–4218. doi:10.1172/JCI72333.
References
- 1.D’Costa VM, et al. Antibiotic resistance is ancient. Nature. 2011;477(7365):457–461. doi: 10.1038/nature10388. [DOI] [PubMed] [Google Scholar]
- 2.Bhullar K, et al. Antibiotic resistance is prevalent in an isolated cave microbiome. PLoS One. 2012;7(4):e34953. doi: 10.1371/journal.pone.0034953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Costello EK, Stagaman K, Dethlefsen L, Bohannan BJ, Relman DA. The application of ecological theory toward an understanding of the human microbiome. Science. 2012;336(6086):1255–1262. doi: 10.1126/science.1224203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dethlefsen L, McFall-Ngai M, Relman DA. An ecological and evolutionary perspective on human-microbe mutualism and disease. Nature. 2007;449(7164):811–818. doi: 10.1038/nature06245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Davies J, Ryan KS. Introducing the parvome: bioactive compounds in the microbial world. ACS Chem Biol. 2012;7(2):252–259. doi: 10.1021/cb200337h. [DOI] [PubMed] [Google Scholar]
- 6.Finley RL, et al. The scourge of antibiotic resistance: the important role of the environment. Clin Infect Dis. 2013;57(5):704–710. doi: 10.1093/cid/cit355. [DOI] [PubMed] [Google Scholar]
- 7.Kohanski MA, DePristo MA, Collins JJ. Sublethal antibiotic treatment leads to multidrug resistance via radical-induced mutagenesis. Mol Cell. 2010;37(3):311–320. doi: 10.1016/j.molcel.2010.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kersten RD, et al. A mass spectrometry-guided genome mining approach for natural product peptidogenomics. Nat Chem Biol. 2011;7(11):794–802. doi: 10.1038/nchembio.684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Walsh CT, Zhang W. Chemical logic and enzymatic machinery for biological assembly of peptidyl nucleoside antibiotics. ACS Chem Biol. 2011;6(10):1000–1007. doi: 10.1021/cb200284p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Liu WT, Kersten RD, Yang YL, Moore BS, Dorrestein PC. Imaging mass spectrometry and genome mining via short sequence tagging identified the anti-infective agent arylomycin in Streptomyces roseosporus. J Am Chem Soc. 2011;133(45):18010–18013. doi: 10.1021/ja2040877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yamanaka K, et al. Direct cloning and refactoring of a silent lipopeptide biosynthetic gene cluster yields the antibiotic taromycin A. Proc Natl Acad Sci U S A. 2014;111(5):1957–1962. doi: 10.1073/pnas.1319584111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Challis GL. Genome mining for novel natural product discovery. J Med Chem. 2008;51(9):2618–2628. doi: 10.1021/jm700948z. [DOI] [PubMed] [Google Scholar]
- 13.Bohnhoff M, Miller CP. Enhanced susceptibility to Salmonella infection in streptomycin-treated mice. J Infect Dis. 1962;111:117–127. doi: 10.1093/infdis/111.2.117. [DOI] [PubMed] [Google Scholar]
- 14.Miller CP, Bohnhoff M, Rifkind D. The effect of an antibiotic on the susceptibility of the mouse’s intestinal tract to Salmonella infection. Trans Am Clin Climatol Assoc. 1957;68:51–55. [PMC free article] [PubMed] [Google Scholar]
- 15.Buffie CG, Pamer EG. Microbiota-mediated colonization resistance against intestinal pathogens. Nat Rev Immunol. 2013;13(11):790–801. doi: 10.1038/nri3535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ng KM, et al. Microbiota-liberated host sugars facilitate post-antibiotic expansion of enteric pathogens. Nature. 2013;502(7469):96–99. doi: 10.1038/nature12503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jernberg C, Lofmark S, Edlund C, Jansson JK. Long-term ecological impacts of antibiotic administration on the human intestinal microbiota. ISME J. 2007;1(1):56–66. doi: 10.1038/ismej.2007.3. [DOI] [PubMed] [Google Scholar]
- 18.Jernberg C, Lofmark S, Edlund C, Jansson JK. Long-term impacts of antibiotic exposure on the human intestinal microbiota. Microbiology. 2010;156(pt 11):3216–3223. doi: 10.1099/mic.0.040618-0. [DOI] [PubMed] [Google Scholar]
- 19.Jernberg C, Sullivan A, Edlund C, Jansson JK. Monitoring of antibiotic-induced alterations in the human intestinal microflora and detection of probiotic strains by use of terminal restriction fragment length polymorphism. Appl Environ Microbiol. 2005;71(1):501–506. doi: 10.1128/AEM.71.1.501-506.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jakobsson HE, Jernberg C, Andersson AF, Sjolund-Karlsson M, Jansson JK, Engstrand L. Short-term antibiotic treatment has differing long-term impacts on the human throat and gut microbiome. PLoS One. 2010;5(3):e9836. doi: 10.1371/journal.pone.0009836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dethlefsen L, Huse S, Sogin ML, Relman DA. The pervasive effects of an antibiotic on the human gut microbiota, as revealed by deep 16S rRNA sequencing. PLoS Biol. 2008;6(11):e280. doi: 10.1371/journal.pbio.0060280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Naeem S, Li SB. Biodiversity enhances ecosystem reliability. Nature. 1997;390:507–509. doi: 10.1038/37348. [DOI] [Google Scholar]
- 23.Dethlefsen L, Relman DA. Incomplete recovery and individualized responses of the human distal gut microbiota to repeated antibiotic perturbation. Proc Natl Acad Sci U S A. 2011;108(suppl 1):4554–4561. doi: 10.1073/pnas.1000087107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Paine RT, Tegner MJ, Johnson EA. Compounded perturbations yield ecological surprises. Ecosystems. 1998;1(6):535–545. doi: 10.1007/s100219900049. [DOI] [Google Scholar]
- 25.Cho I, et al. Antibiotics in early life alter the murine colonic microbiome and adiposity. Nature. 2012;488(7413):621–626. doi: 10.1038/nature11400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lee HH, Molla MN, Cantor CR, Collins JJ. Bacterial charity work leads to population-wide resistance. Nature. 2010;467(7311):82–85. doi: 10.1038/nature09354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Toprak E, Veres A, Michel JB, Chait R, Hartl DL, Kishony R. Evolutionary paths to antibiotic resistance under dynamically sustained drug selection. Nat Genet. 2012;44(1):101–105. doi: 10.1038/ng.1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Looft T, et al. In-feed antibiotic effects on the swine intestinal microbiome. Proc Natl Acad Sci U S A. 2012;109(5):1691–1696. doi: 10.1073/pnas.1120238109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lofmark S, Jernberg C, Jansson JK, Edlund C. Clindamycin-induced enrichment and long-term persistence of resistant Bacteroides spp. and resistance genes. J Antimicrob Chemother. 2006;58(6):1160–1167. doi: 10.1093/jac/dkl420. [DOI] [PubMed] [Google Scholar]
- 30.Sjolund M, Wreiber K, Andersson DI, Blaser MJ, Engstrand L. Long-term persistence of resistant Enterococcus species after antibiotics to eradicate Helicobacter pylori. Ann Intern Med. 2003;139(6):483–487. doi: 10.7326/0003-4819-139-6-200309160-00011. [DOI] [PubMed] [Google Scholar]
- 31.Bagel S, Hullen V, Wiedemann B, Heisig P. Impact of gyrA and parC mutations on quinolone resistance, doubling time, and supercoiling degree of Escherichia coli. Antimicrob Agents Chemother. 1999;43(4):868–875. doi: 10.1128/aac.43.4.868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dantas G, Sommer MO, Oluwasegun RD, Church GM. Bacteria subsisting on antibiotics. Science. 2008;320(5872):100–103. doi: 10.1126/science.1155157. [DOI] [PubMed] [Google Scholar]
- 33.Koenig JE, et al. Succession of microbial consortia in the developing infant gut microbiome. Proc Natl Acad Sci U S A. 2011;108(suppl 1):4578–4585. doi: 10.1073/pnas.1000081107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bartoloni A, et al. High prevalence of acquired antimicrobial resistance unrelated to heavy antimicrobial consumption. J Infect Dis. 2004;189(7):1291–1294. doi: 10.1086/382191. [DOI] [PubMed] [Google Scholar]
- 35.Andersson DI, Hughes D. Antibiotic resistance and its cost: is it possible to reverse resistance? Nat Rev Microbiol. 2010;8(4):260–271. doi: 10.1038/nrmicro2319. [DOI] [PubMed] [Google Scholar]
- 36.Levy SB, Marshall B, Schluederberg S, Rowse D, Davis J. High frequency of antimicrobial resistance in human fecal flora. Antimicrob Agents Chemother. 1988;32(12):1801–1806. doi: 10.1128/AAC.32.12.1801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sommer MO, Dantas G, Church GM. Functional characterization of the antibiotic resistance reservoir in the human microflora. Science. 2009;325(5944):1128–1131. doi: 10.1126/science.1176950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Frost LS, Leplae R, Summers AO, Toussaint A. Mobile genetic elements: the agents of open source evolution. Nat Rev Microbiol. 2005;3(9):722–732. doi: 10.1038/nrmicro1235. [DOI] [PubMed] [Google Scholar]
- 39.Ochman H, Lawrence JG, Groisman EA. Lateral gene transfer and the nature of bacterial innovation. Nature. 2000;405(6784):299–304. doi: 10.1038/35012500. [DOI] [PubMed] [Google Scholar]
- 40.Koonin EV, Makarova KS, Aravind L. Horizontal gene transfer in prokaryotes: quantification and classification. Annu Rev Microbiol. 2001;55:709–742. doi: 10.1146/annurev.micro.55.1.709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Smillie CS, Smith MB, Friedman J, Cordero OX, David LA, Alm EJ. Ecology drives a global network of gene exchange connecting the human microbiome. Nature. 2011;480(7376):241–244. doi: 10.1038/nature10571. [DOI] [PubMed] [Google Scholar]
- 42.Ley RE, Peterson DA, Gordon JI. Ecological and evolutionary forces shaping microbial diversity in the human intestine. Cell. 2006;124(4):837–848. doi: 10.1016/j.cell.2006.02.017. [DOI] [PubMed] [Google Scholar]
- 43.David LA, et al. Diet rapidly and reproducibly alters the human gut microbiome. Nature. 2014;505(7484):559–563. doi: 10.1038/nature12820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.O’Hara AM, Shanahan F. The gut flora as a forgotten organ. EMBO Rep. 2006;7(7):688–693. doi: 10.1038/sj.embor.7400731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Stecher B, et al. Gut inflammation can boost horizontal gene transfer between pathogenic and commensal Enterobacteriaceae. Proc Natl Acad Sci U S A. 2012;109(4):1269–1274. doi: 10.1073/pnas.1113246109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kurokawa K, et al. Comparative metagenomics revealed commonly enriched gene sets in human gut microbiomes. DNA Res. 2007;14(4):169–181. doi: 10.1093/dnares/dsm018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rohwer F, Thurber RV. Viruses manipulate the marine environment. Nature. 2009;459(7244):207–212. doi: 10.1038/nature08060. [DOI] [PubMed] [Google Scholar]
- 48.Reyes A, et al. Viruses in the faecal microbiota of monozygotic twins and their mothers. Nature. 2010;466(7304):334–338. doi: 10.1038/nature09199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Human Microbiome Project Consortium Structure, function diversity of the healthy human microbiome. Nature. 2012;486(7402):207–214. doi: 10.1038/nature11234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Johnsborg O, Eldholm V, Havarstein LS. Natural genetic transformation: prevalence, mechanisms and function. Res Microbiol. 2007;158(10):767–778. doi: 10.1016/j.resmic.2007.09.004. [DOI] [PubMed] [Google Scholar]
- 51.Arumugam M, et al. Enterotypes of the human gut microbiome. Nature. 2011;473(7346):174–180. doi: 10.1038/nature09944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Fancello L, Desnues C, Raoult D, Rolain JM. Bacteriophages and diffusion of genes encoding antimicrobial resistance in cystic fibrosis sputum microbiota. J Antimicrob Chemother. 2011;66(11):2448–2454. doi: 10.1093/jac/dkr315. [DOI] [PubMed] [Google Scholar]
- 53.Ghosh TS, Gupta SS, Nair GB, Mande SS. In silico analysis of antibiotic resistance genes in the gut microflora of individuals from diverse geographies and age-groups. PLoS One. 2013;8(12):e83823. doi: 10.1371/journal.pone.0083823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Pallecchi L, et al. Population structure and resistance genes in antibiotic-resistant bacteria from a remote community with minimal antibiotic exposure. Antimicrob Agents Chemother. 2007;51(4):1179–1184. doi: 10.1128/AAC.01101-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Schloissnig S, et al. Genomic variation landscape of the human gut microbiome. Nature. 2013;493(7430):45–50. doi: 10.1038/nature11711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kunz BA, Glickman BW. The infidelity of conjugal DNA transfer in Escherichia coli. Genetics. 1983;105(3):489–500. doi: 10.1093/genetics/105.3.489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Beaber JW, Hochhut B, Waldor MK. SOS response promotes horizontal dissemination of antibiotic resistance genes. Nature. 2004;427(6969):72–74. doi: 10.1038/nature02241. [DOI] [PubMed] [Google Scholar]
- 58.Doucet-Populaire F, Trieu-Cuot P, Dosbaa I, Andremont A, Courvalin P. Inducible transfer of conjugative transposon Tn1545 from Enterococcus faecalis to Listeria monocytogenes in the digestive tracts of gnotobiotic mice. Antimicrob Agents Chemother. 1991;35(1):185–187. doi: 10.1128/AAC.35.1.185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Goerke C, Koller J, Wolz C. Ciprofloxacin and trimethoprim cause phage induction and virulence modulation in Staphylococcus aureus. Antimicrob Agents Chemother. 2006;50(1):171–177. doi: 10.1128/AAC.50.1.171-177.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Modi SR, Lee HH, Spina CS, Collins JJ. Antibiotic treatment expands the resistance reservoir and ecological network of the phage metagenome. Nature. 2013;499(7457):219–222. doi: 10.1038/nature12212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Brandl K, et al. Vancomycin-resistant enterococci exploit antibiotic-induced innate immune deficits. Nature. 2008;455(7214):804–807. doi: 10.1038/nature07250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Fukuda S, et al. Bifidobacteria can protect from enteropathogenic infection through production of acetate. Nature. 2011;469(7331):543–547. doi: 10.1038/nature09646. [DOI] [PubMed] [Google Scholar]
- 63.Diep BA, et al. Complete genome sequence of USA300, an epidemic clone of community-acquired meticillin-resistant Staphylococcus aureus. Lancet. 2006;367(9512):731–739. doi: 10.1016/S0140-6736(06)68231-7. [DOI] [PubMed] [Google Scholar]
- 64.Hehemann JH, Correc G, Barbeyron T, Helbert W, Czjzek M, Michel G. Transfer of carbohydrate-active enzymes from marine bacteria to Japanese gut microbiota. Nature. 2010;464(7290):908–912. doi: 10.1038/nature08937. [DOI] [PubMed] [Google Scholar]
- 65.Forsberg KJ, Reyes A, Wang B, Selleck EM, Sommer MO, Dantas G. The shared antibiotic resistome of soil bacteria and human pathogens. Science. 2012;337(6098):1107–1111. doi: 10.1126/science.1220761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Lozupone CA, et al. The convergence of carbohydrate active gene repertoires in human gut microbes. Proc Natl Acad Sci U S A. 2008;105(39):15076–15081. doi: 10.1073/pnas.0807339105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Vega NM, Allison KR, Khalil AS, Collins JJ. Signaling-mediated bacterial persister formation. Nat Chem Biol. 2012;8(5):431–433. doi: 10.1038/nchembio.915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Vega NM, Allison KR, Samuels AN, Klempner MS, Collins JJ. Salmonella typhimurium intercepts Escherichia coli signaling to enhance antibiotic tolerance. Proc Natl Acad Sci U S A. 2013;110(35):14420–14425. doi: 10.1073/pnas.1308085110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Lemon KP, Armitage GC, Relman DA, Fischbach MA. Microbiota-targeted therapies: an ecological perspective. Sci Transl Med. 2012;4(137):137rv135. doi: 10.1126/scitranslmed.3004183. [DOI] [PMC free article] [PubMed] [Google Scholar]