

Abstract
Binge eating afflicts approximately 5% of US adults, though effective treatments are limited. Here, we showed that estrogen replacement substantially suppresses binge-like eating behavior in ovariectomized female mice. Estrogen-dependent inhibition of binge-like eating was blocked in female mice specifically lacking estrogen receptor-α (ERα) in serotonin (5-HT) neurons in the dorsal raphe nuclei (DRN). Administration of a recently developed glucagon-like peptide-1–estrogen (GLP-1–estrogen) conjugate designed to deliver estrogen to GLP1 receptor–enhanced regions effectively targeted bioactive estrogens to the DRN and substantially suppressed binge-like eating in ovariectomized female mice. Administration of GLP-1 alone reduced binge-like eating, but not to the same extent as the GLP-1–estrogen conjugate. Administration of ERα-selective agonist propylpyrazole triol (PPT) to murine DRN 5-HT neurons activated these neurons in an ERα-dependent manner. PPT also inhibited a small conductance Ca2+-activated K+ (SK) current; blockade of the SK current prevented PPT-induced activation of DRN 5-HT neurons. Furthermore, local inhibition of the SK current in the DRN markedly suppressed binge-like eating in female mice. Together, our data indicate that estrogens act upon ERα to inhibit the SK current in DRN 5-HT neurons, thereby activating these neurons to suppress binge-like eating behavior and suggest ERα and/or SK current in DRN 5-HT neurons as potential targets for anti-binge therapies.
Introduction
Binge eating is defined as the ingestion of a large amount of food in a brief period of time and is characterized by the preferential choice of highly palatable food. Binge eating, afflicting approximately 5% of US adults (1), is a central trait of major eating disorders, such as the binge-eating/purging type anorexia nervosa (AN), bulimia nervosa (BN), binge-eating disorder (BED), and other eating disorders not otherwise specified (EDNOS) (2, 3). Binge eating constitutes a significant public health concern by virtue of its strong associations with other medical and psychiatric disorders, most notably obesity and depression (1). Development of more effective treatments for binge eating is urgently needed.
Binge eating is more prevalent in women than in men, and women with binge eating often suffer from menstrual irregularity, presumably due to impaired functions of ovarian hormones (e.g., estrogens) (4–6). Interestingly, circulating 17β-estradiol levels are inversely associated with binge eating in women (7–11). Consistently, removal of endogenous ovarian hormones by ovariectomy (OVX) potentiates binge eating in female rats (12), whilereplacement with 17β-estradiol and progesterone in OVX rats suppresses binge eating (13). While these data suggest that hormone replacement therapy could be used to suppress binge-like eating, at least in women, the clinical application of ovarian hormones, e.g., estrogens, is limited by their gynecological and tumor-promoting actions. Therefore, better understanding the mechanisms by which ovarian hormones regulate binge-eating behavior may facilitate development of more selective hormone-based therapies that effectively inhibit binge eating but cause few or no side effects in humans.
Impaired brain serotonin (5-HT) signaling has been implicated in the development of binge eating (14–16). Interestingly, drugs that increase brain 5-HT content, such as fluoxetine, have been shown to suppress binge eating in humans (17–22). Notably, d-fenfluramine, a drug that increases 5-HT content by stimulating synaptic release of 5-HT and blocking its reuptake (23), effectively reduces the number of binge episodes in patients (24, 25). These findings suggest that brain 5-HT neurons play an important role in the regulation of binge-eating behavior.
Emerging evidence suggests that estrogens directly act on brain 5-HT neurons. For example, estrogen receptors, including estrogen receptor-α (ERα), are abundantly expressed in the dorsal raphe nuclei (DRN) (26), where most 5-HT neurons are located. Further, estrogen replacement increases neural activities in the DRN (27, 28). Based on these, we hypothesized that estrogens directly act through DRN 5-HT neurons to inhibit binge eating.
To test this hypothesis, we first examined whether estrogens regulate binge-like eating behavior in female mice. Then, using a genetic mouse model lacking ERα only in 5-HT neurons in the DRN, we tested to determine whether ERα in DRN 5-HT neurons is required to mediate the effects of estrogens on binge-like eating behavior. We further tested the effects of a newly developed glucagon-like peptide-1–estrogen (GLP-1–estrogen) conjugate on binge-like eating behavior in female mice. Finally, we tested to determine whether a small conductance Ca2+-activated K+ (SK) current mediates estrogenic actions on 5-HT neural activities and on binge-like eating behavior.
Results
17β-Estradiol inhibits binge-like eating in OVX female mice.
Czyzyk et al. recently showed that mice subjected to an intermittent high-fat diet (HFD) exposure paradigm display recurring binge-like eating behavior compared with mice subjected to a continuous HFD exposure paradigm (29). To test the effects of estrogens on binge-like eating behavior, female C57BL/6 mice received bilateral OVX, followed by s.c. implantations of pellets containing 17β-estradiol (0.5 μg/d for 60 days, OVXE) or containing vehicle (OVXV). After a 7-day recovery from the surgery, OVXV and OVXE female mice were subjected to intermittent HFD exposure or continuous HFD exposure for 1 week (as detailed in Methods). On the day to assess binge-like eating behavior (2 weeks after the surgeries), intake of HFD and chow was measured for 2.5 hours (from 11:00 am to 1:30 pm). First, OVXE significantly decreased HFD intake compared with OVXV treatment in mice exposed to intermittent HFD, while in mice exposed to continuous HFD, HFD intake was minimal and comparable between OVXE and OVXV groups (Figure 1A). Chow intake was minimal in all 4 groups (Figure 1B). In particular, OVXE significantly increased chow intake compared with OVXV in mice exposed to intermittent HFD, while such a difference was not observed between OVXV and OVXE mice exposed to continuous HFD (Figure 1B). In addition, we showed that body weights in all 4 groups were comparable on the binge assessment day (Figure 1C), suggesting that effects of estrogens on binge-like eating behavior were not confounded by different body weights. Together, these results indicate that estrogen replacement in OVX female mice effectively suppresses binge-like eating behavior.
Figure 1. Estrogen replacement inhibits binge-like eating in female mice.

(A and B) C57BL/6 female mice (12 weeks) were ovariectomized and implanted with s.c. 17β-estradiol pellets (0.5 μg/d for 60 days; OVXE) or vehicle pellets (OVXV). After a 7-day recovery, mice were subjected to intermittent HFD exposure or continuous HFD exposure for 1 week, as described in Methods. At the beginning of the next week, HFD and chow diet were provided to cages at 11:00 am, and 2.5-hour HFD intake (A) and chow intake (B) was measured. (C) Body weight was measured on the same day when binge-like behavior was assessed. n = 6–7/group. Results are shown as mean ± SEM. **P < 0.01; ***P < 0.001 between OVXV and OVXE mice in 2-way ANOVA analyses followed by post hoc Bonferroni’s test.
Importantly, we demonstrated that female mice exposed to either continuous or intermittent HFD displayed similar anxiety levels and locomotor activities measured in the light-dark test and the elevated plus maze (EPM) test (Supplemental Figures 1 and 2; supplemental material available online with this article; doi:10.1172/JCI74726DS1). Similarly, the forced swim test did not detect significant differences in depression levels in mice exposed to continuous or intermittent HFD (Supplemental Figure 3). These results suggest that binge-like behavior we observed in the “intermittent” group is unlikely to be associated with different anxiety and/or depression levels.
ERα in 5-HT neurons mediates inhibitory effects of estrogens on binge-like eating.
We used double immunofluorescence to examine colocalization of ERα and 5-HT in female C57BL/6 mouse brains. We found that about 40% of 5-HT neurons in the DRN coexpress ERα; about 90% of ERα neurons in this region are 5-HTergic (Figure 2A). None to few 5-HT neurons in the median raphe nuclei or caudal raphe nuclei (MRN or CRN) coexpress ERα (Figure 2, B and C). These results raise the possibility that ERα expressed by DRN 5-HT neurons may be involved in the estrogenic effects on binge-like eating behavior.
Figure 2. 5-HT neurons express ERα.
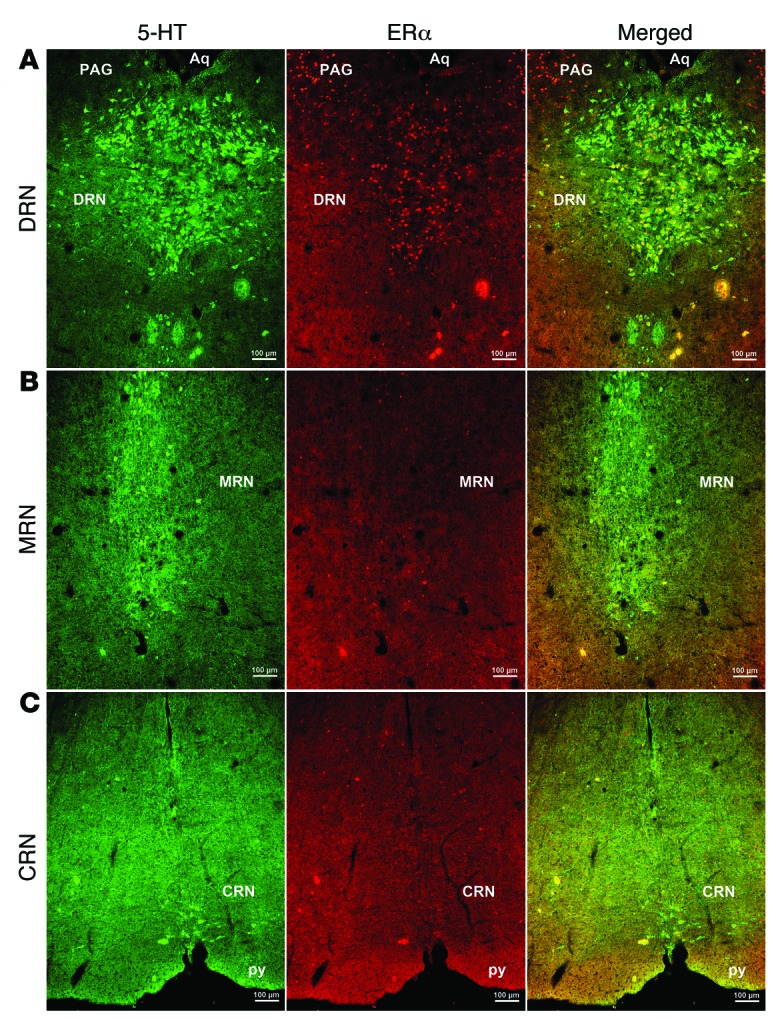
Representative immunofluorescent images for 5-HT (left, green) and ERα (middle, red) in coronal mouse brain sections containing the DRN (A), MRN (B), and CRN (C). Yellow neurons in the right panels indicate 5-HT neurons that coexpress ERα. Scale bars: 100 μm. Aq, aqueduct; PAG, periaqueductal gray; py, pyramidal tract.
In order to specifically target 5-HT neurons in functional mice, we first validated a Tph2-CreER mouse line in which expression of tamoxifen-inducible Cre recombinase is driven by tryptophan hydroxylase 2 (Tph2) promoter. To this end, Tph2-CreER mice were crossed to Rosa26-tdTOMATO mice to generate Tph2-CreER Rosa26-tdTOMATO mice. Upon tamoxifen induction (3 mg/injections, i.p., twice, 24 hours apart), the strong red fluorescence (TOMATO) was observed in the DRN, MRN, and CRN. Using double immunofluorescence for 5-HT (green) and TOMATO staining (red), we confirmed that all TOMATO-positive neurons are exclusively 5-HT neurons (Supplemental Figure 4, A–C). Importantly, no TOMATO signals were observed in other non–5-HT regions, including the arcuate nucleus (ARH), the ventromedial hypothalamic nucleus (VMH), and the paraventricular nucleus of the hypothalamus (PVH) (Supplemental Figure 5, A and B). In addition, as a negative control, we checked TOMATO staining in Tph2-CreER Rosa26-tdTOMATO mice without tamoxifen injections. No TOMATO signal was detected in these mice (data not shown), indicating that there is no leakage of Cre activity in Tph2-CreER mice.
To test whether ERα expressed by 5-HT neurons is required to mediate the inhibitory effects of estrogens on binge-like eating behavior, we crossed Tph2-CreER to Esr1fl/fl mice (30). This cross generated Esr1fl/fl (WT) and Esr1fl/fl Tph2-CreER (KO) mice. We validated that in KO mice (after tamoxifen induction), ERα was selectively deleted from the DRN, but ERα in other non–5-HT regions (including ARH and VMH) was intact (Figure 3, A–D). Both WT and KO mice (after tamoxifen induction) were divided into 2 groups to receive OVXV or OVXE treatment and then subjected to intermittent HFD exposure to induce binge-like eating behavior. We showed that in WT mice, OVXE treatment significantly suppressed binge-like eating behavior (measured by 2.5-hour HFD intake) compared with that in OVXV mice (Figure 3E). In contrast, both OVXV and OVXE KO mice showed comparable binge-like eating behavior (Figure 3E). In addition, we found that WT and KO female mice had comparable body weight, fat mass, and lean mass when binge-like eating behavior was assessed (Figure 3F). No difference in plasma orexin A levels was observed among these 4 groups (Figure 3G). These results indicate that the inhibitory effects of estrogens on binge-like eating behavior in female mice are largely mediated by ERα expressed by DRN 5-HT neurons.
Figure 3. ERα in 5-HT neurons mediates estrogenic actions to inhibit binge-like eating in female mice.

(A–D) Representative immunohistochemistry for ERα from female WT (A and C) and KO (B and D) mice (after tamoxifen inductions). Scale bars: 100 μm. 3V, third ventricle; ME, median eminence. (E) WT and KO mice received tamoxifen inductions at 8 weeks of age (3 mg/injections, i.p., 24 hours apart). At 24 weeks of age, mice were ovariectomized and implanted with s.c. 17β-estradiol pellets (0.5 μg/d for 60 days; OVXE) or vehicle pellets (OVXV). After a 7-day recovery, mice were subjected to intermittent HFD exposure for 1 week, as described in Methods. At the end of that week, HFD and chow diet were provided to cages at 11:00 am, and 2.5-hour HFD intake was measured. n = 7–10/group. Results are shown as mean ± SEM. ***P < 0.001, between OVXV and OVXE mice in 2-way ANOVA analyses followed by post hoc Bonferroni’s tests. (F) Body weight, fat mass, and lean mass of WT and KO mice measured when binge-like behavior was assessed. n = 16 or 18/group. Results are shown as mean ± SEM. (G) Plasma orexin A measured in WT and KO mice after assessment of binge-like eating behavior. n = 6–7/group. Results are shown as mean ± SEM.
GLP-1–estrogen targets the DRN and inhibits binge-like eating.
A stable GLP-1–estrogen conjugate was recently developed to preferentially deliver estrogens to GLP-1 receptor–enriched regions, but does not release estrogens into the circulation (31). GLP-1–estrogen has been shown to produce profound estrogen-mediated metabolic benefits in animals, while the common side effects associated with estrogen therapy (e.g., reproductive endocrine toxicity and breast cancer) are avoided (31). Interestingly, the DRN expresses abundant GLP-1 receptors (32). Further, we showed that injections of GLP-1–estrogen (4 μg/kg, s.c., 2 hours) significantly increased expression of tripartite motif–containing protein 25 (Trim25, a known estrogen target) in the DRN of OVX female C57BL/6 mice (Figure 4A). A similar increase in Trim25 mRNA was also observed in OVXE mice (Figure 4A). Importantly, GLP-1 (4 μg/kg) did not alter Trim25 mRNA in the DRN (Figure 4A). These results indicate that GLP-1–estrogen conjugate delivers bioactive estrogens to the DRN in addition to other sites (e.g., the hypothalamus) (31) and therefore raise the possibility that GLP-1–estrogen may act upon ERα in the DRN to inhibit binge-like eating behavior. To test this, we examined binge-like eating behaviors in OVX WT female mice 30 minutes after they received injections of saline, GLP-1, or GLP-1–estrogen. Interestingly, we observed that GLP-1 (4 μg/kg, s.c.) significantly reduced binge-like eating behavior (measured by 2.5-hour HFD intake) compared with saline treatment, by about 33% (Figure 4B). Importantly, GLP-1–estrogen (4 μg/kg, s.c.) produced a significantly more robust inhibition on binge-like eating behavior, by about 66% (Figure 4B), confirming that a portion of the effects induced by this conjugate stem from estrogens. In addition, we showed that effects of GLP-1 and GLP-1–estrogen on binge-like eating behavior were comparable in OVX female mice with ERα selectively deleted in the DRN (Figure 4B), indicating that estrogen-mediated inhibition on binge-like eating behavior requires ERα expressed by 5-HT neurons. As expected, the inhibitory effects induced by GLP-1 alone were not affected in KO mice (Figure 4B), since GLP-1’s effects are presumably mediated by GLP-1 receptors. Given that GLP-1–estrogen has been shown to avoid the common side effects associated with estrogens, our results provided the experimental basis that this newly developed conjugate, or its modified forms, could be used to treat binge eating, at least in women.
Figure 4. GLP-1–estrogen inhibits binge-like eating in female mice partly through estrogenic actions on ERα in 5-HT neurons.

(A) Real-time RT-PCR analyses for Trim25 mRNAs in the DRN from female OVX mice receiving s.c. implantation of placebo pellets (OVXV), 17β-estradiol pellets (0.5 μg/d for 2 weeks; OVXE), single injection of GLP-1 (4 μg/kg, s.c., 2 hours; OVX + GLP-1), or GLP-1–estrogen (4 μg/kg, s.c., 2 hours; OVX + GLP-1–estrogen). n = 4–6/group. Results are shown as mean ± SEM. *P < 0.05 and **P < 0.01 vs. OVXV; #P < 0.05 vs. OVX+GLP-1 in 1-way ANOVA analyses followed by post hoc Bonferroni’s tests. (B) WT and KO mice (24 weeks) were ovariectomized. After a 7-day recovery, mice were subjected to intermittent HFD exposure for 1 week, as described in Methods. At the end of that week, mice received s.c. injections of saline, GLP-1 (4 μg/kg), or GLP-1–estrogen (4 μg/kg) at 10:30 am, and HFD and chow diet were provided to cages at 11:00 am; 2.5-hour HFD intake was measured. n = 8 or 9/group. Results are shown as mean ± SEM. *P < 0.05; **P < 0.01; ***P < 0.001 in 1-way ANOVA analyses followed by post hoc Bonferroni’s tests.
Basic electrophysiological properties of identified DRN 5-HT neurons from WT and KO mice.
In order to delineate the cellular mechanisms by which ERα signals regulate 5-HT neurons, we crossed the Rosa26-tdTOMATO allele to both Tph2-CreER and Esr1fl/fl Tph2-CreER mice. This generated Tph2-CreER Rosa26-tdTOMATO (WT) and Esrfl/fl Tph2-CreER Rosa26-tdTOMATO (KO) mice. We used these mice (after tamoxifen induction) to perform electrophysiological recordings in identified 5-HT neurons (Figure 5, A–E). We first assessed the basic electrophysiological properties of identified DRN 5-HT neurons from WT and KO mice. Thus, passive membrane properties, including input resistance and resting membrane potential (RM), were determined by a series of 1-second hyperpolarizing current pulses from –160 pA to 0 pA, with steps of 20 pA (as an inset in Supplemental Figure 6). Input resistance was determined by the slope of a linear regression fitted line of a current-voltage (I-V) curve. RM was measured as the membrane voltage in the absence of current injection. The input resistances of 5-HT neurons from KO mice (493.90 ± 2.90 M⁄, n = 18) were higher than those in WT mice (401.30 ± 1.74 M⁄, n = 26, P < 0.05 by t test, Supplemental Figure 6, A–C). There was no significant difference in RM of DRN 5-HT neurons between WT and KO mice (WT: –41.97 ± 1.527 mV, n = 26; KO: –43.82 ± 2.65 mV, n = 18, P > 0.05 by t test, Supplemental Figure 6C). Further, we examined the characteristics of tonic or phasic firing in 5-HT neurons by observing action potential (AP) behavior of these neurons after direct depolarizing current injections ranged from 0 to 300 pA for 1 second (as an inset in Supplemental Figure 7A). We found that 84.38 % (27/32) WT and 77.78% (14/18) KO 5-HT neurons fired tonically (P > 0.05 by χ2 test, Supplemental Figure 7, A–E). In addition, we examined the characteristics of spontaneous or evoked firing in 5-HT neurons. We found that a larger proportion of 5-HT neurons fired spontaneously in KO mice compared with WT mice (WT: 59.38%, 19/32 vs. KO: 100%, 18/18, P < 0.05 by χ2 test, Supplemental Figure 8A). Finally, AP properties, including firing frequency, the amplitude of AP, the AP duration at 50% (APD50), and the amplitude of after-hyperpolarization potential (AHPA) in spontaneous firing neurons were analyzed. No significant differences were detected in these parameters in the DRN 5-HT neurons between WT and KO mice (Supplemental Figure 8, B–E).
Figure 5. An ERα agonist activates DRN 5-HT neurons.

(A–C) Brightfield (A), fluorescent with FRITC filter (B), and with FITC filter (C) illuminations of a targeted DRN 5-HT neuron. Scale bars: 10 μm. (D and E) Post hoc identification of the recorded neuron within the DRN in the fixed brain slice. Scale bars: 100 μm (D); 20 μm (E). (F–H) Representative traces before and after PPT treatment in 5-HT neurons from WT mice (F), in 5-HT neurons from KO mice (G), and in WT 5-HT neurons preincubated with 100 nM apamin for 2 hours (H). Upper panels: repetitive firing traces; values at the start of each trace are RMs. Lower panels: 3 continuous APs zoomed in from the upper-panel repetitive firing traces; values at the bottom of each trace are AHPA. (I) Cell numbers of 5-HT neurons with (responsive) or without (irresponsive) RM depolarization (> 1 mV) by PPT in various groups. ***P < 0.001. (J) Magnitude of depolarization induced by PPT in various groups. n = 9–18/group. Results are shown as mean ± SEM. **P < 0.01; ***P < 0.001. (K) Changes in firing rate induced by PPT in various groups. n = 7–18/group. Results are shown as mean ± SEM. ***P < 0.001. (L) Changes in AHPA induced by PPT in various groups. n = 7–18/group. Results are shown as mean ± SEM. ***P < 0.001.
Effects of the ERα agonist on DRN 5-HT neurons.
We showed that treatment with propylpyrazole triol (PPT) (100 nM), a selective ERα agonist, depolarized the RM (> 1 mV) in a proportion of 5-HT neurons from WT mice (53.13%, 17/32, Figure 5F), which is largely consistent with our earlier observation that only 40% of 5-HT neurons in the DRN coexpress ERα. Depending on the magnitude of PPT-induced depolarization, we defined all WT 5-HT neurons as “responsive neurons” (> 1 mV) or “irresponsive neurons” (≤ 1 mV). Lack of PPT’s effects in the irresponsive neurons was presumably because these neurons did not express ERα. Supporting this possibility, we showed that the majority of 5-HT neurons from KO mice (which lack ERα selectively in 5-HT neurons) were not depolarized by PPT (Figure 5G). Indeed, only 5.56% of(1/18) 5-HT neurons from KO mice responded to PPT with a depolarization (> 1 mV), which was significantly less frequent than in WT mice (KO: 5.56%, 1/18 in KO vs. WT: 53.13%, 17/32; P < 0.001 by χ2 test, Figure 5I). Further, the magnitude of PPT-induced depolarization in responsive neurons was significantly higher than those in irresponsive neurons and in 5-HT neurons from KO mice (P < 0.001 by 1-way ANOVA, Figure 5J). We further analyzed properties of AP in 5-HT neurons with spontaneous firing. Thus, we showed that PPT robustly increased firing rates of responsive neurons from WT mice, while this effect was significantly abolished in irresponsive neurons from WT mice and in 5-HT neurons from KO mice (P < 0.001 by 1-way ANOVA, Figure 5K). Finally, we also observed that PPT robustly decreased AHPA in all responsive neurons from WT mice, but not in irresponsive neurons from WT mice or in 5-HT neurons from KO mice (P < 0.001 by 1-way ANOVA, Figure 5L). Together, these results indicate that PPT activates DRN 5-HT neurons via ERα-dependent mechanisms.
The ERα agonist activates DRN 5-HT neurons partly by inhibiting an apamin-sensitive SK current.
The small conductance SK currents have been implicated in the regulation of firing properties and AHP of neurons (33). Thus, we assessed effects of PPT on SK-like currents in 5-HT neurons, using a published voltage clamp protocol (Figure 6A and ref. 34). We found that this protocol induced robust currents in WT 5-HT neurons, which were abolished by acute perfusion of apamin (100 nM, 8 minutes, Figure 6B). These data confirmed that these currents are apamin-sensitive SK-like currents. Further, we showed that PPT inhibited the SK-like currents in WT responsive neurons, but such PPT effects were absent in irresponsive neurons from WT mice, in 5-HT neurons from KO mice, and in WT 5-HT neurons preincubated with 100 nM apamin for 2 hours (P < 0.05 by 1-way ANOVA, Figure 6, C–F). Collectively, these results indicate that PPT, via acting upon ERα, inhibits an apamin-sensitive SK-like current in DRN 5-HT neurons.
Figure 6. Estrogens inhibit SK-like currents in DRN 5-HT neurons.

(A) A voltage clamp protocol to induce SK-like currents. (B) Representative traces for SK-like currents recorded from a 5-HT neuron before (black) and after (red) apamin perfusion. SK currents were shown as an outward tail current following step depolarization of Vh. (C–E) Representative traces before (black) and after (red) PPT treatment from responsive 5-HT neurons in WT brain slice (C), from 5-HT neurons in KO brain slice (D), from 5-HT neurons in WT slices preincubated with apamin (100 nM, 2 hours) (E). (F) PPT-induced changes in SK-like current in WT responsive neurons, in WT irresponsive neurons, in KO 5-HT neurons, or in WT 5-HT neurons preincubated with apamin (100 nM for 2 hours). n = 7–13/group. Results are shown as mean ± SEM. ***P < 0.001. (G) Effects of intra-DRN preinjections of saline or apamin (50 nM, 0.5 μl) on binge-like eating (2.5-hour HFD intake) in OVXV or OVXE WT and KO female mice. n = 5/group. Results are shown as mean ± SEM. **P < 0.01. (H) Intake of 0.1 M NaCl solution in OVX WT female mice receiving intra-DRN injections of saline or apamin (50 nM, 0.5 μl) in the CTA tests. n = 4/group. Results are shown as mean ± SEM.
To further determine whether the inhibition of the SK-like current contributes to PPT-induced activation of DRN 5-HT neurons, we assessed effects of PPT on WT 5-HT neurons after preincubation of apamin (2 hours, 100 nM). After apamin preincubation, the majority of WT 5-HT neurons were not depolarized by PPT (responsive neurons in untreated WT: 53.13%, 17/32; responsive neurons in apamin-treated WT: 12.5%, 2/16; P < 0.001 by χ2 test, Figure 5I). The magnitude of PPT-induced depolarization in RM was abolished in apamin-treated 5-HT neurons (P < 0.01 by 1-way ANOVA, Figure 5J). Similarly, apamin also significantly abolished PPT’s effects on firing rate (P < 0.001 by 1-way ANOVA, Figure 5K) and AHPA (P < 0.001 by 1-way ANOVA, Figure 5L). These results indicate that PPT activates DRN 5-HT neurons at least partly by inhibiting the apamin-sensitive SK-like current.
Local inhibition of the SK current in the DRN suppresses binge-like eating.
To test whether the SK current in 5-HT neurons is physiologically relevant for binge-like eating behavior, female mice received stereotaxic injections of apamin or vehicle into the DRN 30 minutes prior to measurement of binge-like eating behavior. We found that, compared with vehicle-treated OVXV WT mice, OVXV WT mice receiving intra-DRN injections of apamin (50 nM, 0.5 μl) showed significantly suppressed binge-like eating behavior, recapitulating responses seen in vehicle-treated OVXE WT mice (Figure 6G). Similarly, intra-DRN injections of apamin significantly suppressed binge-like eating behavior in OVXV KO mice (Figure 6G), suggesting that effects of SK current inhibition are likely downstream of ERα signals. Finally, in the conditioned taste-aversion tests, we showed that intra-DRN injections of apamin (50 nM, 0.5 μl) in OVX WT mice did not alter solution intake compared with saline injections (Figure 6H), suggesting that the inhibitory effects of apamin on binge-like eating behavior were not due to adverse effects per se.
Discussion
One major finding of these studies is that OVXE WT mice showed substantially decreased binge-like eating behavior compared with OVXV WT mice. This observation is in line with earlier reports that OVX rats show potentiated binge-like eating compared with gonad-intact female rats (12), while replacement with 17β-estradiol and progesterone suppresses binge-like eating in female OVX rats (13). Here, we further pinpointed that estrogen replacement alone can efficiently suppress binge-like eating behavior in female mice. Importantly, we assessed the binge-like eating behavior 2 weeks after OVXV and OVXE treatment. At this time, OVXV mice and OVXE mice had comparable body weights. Therefore, the decreased binge-like eating behavior we observed in OVXE mice was not confounded by body weight divergence or the associated hormonal signals (e.g., orexin A). In addition, we showed that during the 2.5-hour period when binge-like eating behavior was assessed, OVXE did not significantly alter food intake in female mice being exposed to “continuous” HFD. These observations argue that the effects of estrogens on binge-like eating behavior we observed were independent of the well-known anorexigenic effects of estrogens on basal feeding behavior (35).
Lower circulating estradiol levels have been associated with increased binge eating in women (7–11). Thus, our finding that estrogen replacement suppresses binge-like eating behavior in female mice suggests that estrogen replacement therapy could be used in ameliorating binge symptoms in women, especially in those after menopause or with menstrual irregularity (4–6). However, due to the broad spectrum of estrogenic actions through multiple estrogen receptors, current estrogen replacement therapy is often associated with detrimental side effects, such as breast cancer. One solution for this dilemma is to better understand where and how estrogens act to inhibit binge-like behavior.
Using electrophysiology combined with a genetic mouse model lacking ERα only in 5-HT neurons, we showed that estrogens, acting via ERα, depolarize DRN 5-HT neurons and stimulate their firing activities. Notably, impaired brain 5-HT signaling has been implicated in the development of binge eating. For example, binge patients are found to have increased 5-HT uptake in the brain and consequently decreased 5-HT content in the cerebrospinal fluid (14). In addition, effects of 5-HT precursor L-tryptophan (as measured by prolactin secretion) are significantly blunted in binge patients, presumably due to dysfunctions of 5-HT receptors and/or 5-HT synthesizing enzyme (TPH2) (14). On the other hand, drugs that enhance brain 5-HT signals, such as fluoxetine and d-Fenfluramine, have been shown to suppress binge eating in patients (17–22, 24, 25). Together, these results raise the possibility that estrogens may act upon ERα in 5-HT neurons to inhibit binge-like eating. This notion is further supported by our observations that selective deletion of ERα from 5-HT neurons substantially blocked effects of OVXE treatment to suppress binge-like eating behavior in female mice. Collectively, our results pinpointed ERα expressed by DRN 5-HT neurons as one key site that mediates estrogenic actions to inhibit binge-like eating behavior.
A recently developed stable GLP-1–estrogen conjugate uses GLP-1 as a “carrier” to deliver estrogens preferentially to GLP-1 receptor–enriched regions (31). GLP-1–estrogen, like 17β-estradiol, produces robust estrogen-mediated biological functions (e.g., lowering body weight) in mice (31). Importantly, GLP-1–estrogen does not cause common side effects associated with 17β-estradiol (e.g., reproductive endocrine toxicity and breast cancer), presumably because this conjugate does not deliver estrogens to the reproductive organs and breast tissue (31). Notably, body weight–lowering effects of GLP-1–estrogen conjugate are blunted in ERα-KO mice (lacking ERα globally), indicating that the conjugate acts at least partly via ERα (31). Further, estrogen-mediated body weight loss is blocked in mice lacking GLP-1 receptors only in the brain (31), suggesting that this conjugate acts, at least partly, in the brain regions that express both GLP-1 receptors and estrogen receptors to produce body weight–lowering effects. Interestingly, the DRN expresses abundant GLP-1 receptors (32) and ERα (26). This raises the possibility that GLP-1–estrogen can deliver estrogens to the DRN. Supporting this notion, we demonstrated that GLP-1–estrogen, at a very low dose (4 μg/kg), stimulated expression of a known estrogen target in the DRN. Remarkably, this effect of a single dose of GLP-1–estrogen was equivalent to that of chronic 17β-estradiol pellet supplementation in OVXE mice, suggesting that the GLP-1–estrogen conjugate can efficiently deliver bioactive estrogens to the DRN. More importantly, we showed that the single injection of GLP-1–estrogen substantially suppressed binge-like eating behavior in OVX female mice. Notably, effects of GLP-1–estrogen were more robust than those of GLP-1 alone, indicating that a portion of effects of GLP-1–estrogen on binge-like eating behavior are mediated by estrogens. Importantly, we showed that these estrogen-mediated inhibitory effects on binge-like eating behavior were blocked in OVX female mice with ERα selectively deleted in 5-HT neurons. Together, these results indicate that GLP-1–estrogen delivers bioactive estrogens to the DRN to inhibit binge-like eating behavior partly through acting upon ERα in DRN 5-HT neurons. Thus, our results provide the proof-of-concept that ERα expressed by DRN 5-HT neurons is a suitable target for the treatment of binge eating.
Of course, a portion of effects of GLP-1–estrogen on binge-like eating behavior stems from GLP-1, as GLP-1 alone also inhibits binge-like eating behavior in OVX mice. It is well established that GLP-1 exerts potent anorexigenic effects to suppress basal feeding behavior (36). Recent clinical studies revealed that patients with binge eating have lower circulating GLP-1 levels compared with healthy subjects (37, 38). Here, we showed that GLP-1 inhibits binge-like eating behavior, suggesting that GLP-1 could also be used to treat binge eating in humans. Future studies are therefore warranted to further delineate the acting sites and the mechanisms underlying the GLP-1 effects on binge eating. Nevertheless, the potent inhibitory effects of GLP-1–estrogen conjugate we observed in OVX female mice indicate that this conjugate could be used to ameliorate binge symptoms in women, especially in those after menopause or with menstrual irregularity.
We further explored the molecular mechanisms by which estrogen-ERα signals regulate 5-HT neural activities. We made an interesting observation that all 5-HT neurons depolarized by PPT (the ERα agonist) exhibit reduced AHPA, phenomena that were not seen in 5-HT neurons that did not respond to PPT or those with ERα deleted. Consistent with the notion that AHP is at least partly constituted by SK currents (33), we further showed that the PPT failed to suppress AHPA in 5-HT neurons when SK currents were preinhibited by apamin. In addition, using a voltage clamp protocol to directly examine SK-like currents, we demonstrated that PPT inhibited an apamin-sensitive SK-like current in 5-HT neurons, which was blocked in 5-HT neurons with ERα deletion. Importantly, inhibition of the SK current (by apamin) blocked PPT-induced excitation in 5-HT neurons. Collectively, our results suggest that estrogens act upon ERα to inhibit the SK channels, which in turn leads to reduced AHPA and consequently increased excitability of 5-HT neurons.
These electrophysiological results led us to hypothesize that modulations on the SK current in the DRN may regulate binge-like eating behavior in mice. This is supported by the observation that intra-DRN injections of apamin suppressed binge-like eating behavior in OVXV WT mice, recapitulating effects of estrogen replacement in OVXE WT mice. Further, we found that the inhibitory effects of apamin on binge-like eating were not affected by selective deletion of ERα from 5-HT neurons, arguing that the SK channel is downstream of ERα signals in 5-HT neurons. These results clearly indicate that pharmacological inhibition of SK currents in the DRN is sufficient to inhibit binge-like eating behavior in mice without endogenous estrogens or ERα.
While the current study focused on ERα as one mediator of estrogens on binge-like eating behavior, our data do not exclude the possibility that other estrogen receptors, e.g., estrogen receptor-β (ERβ), may also be involved in the effects of estrogens. Although it has been reported that stimulation of ERβ in the brain does not regulate basal food intake (39–41), effects of ERβ signals on binge-like eating behavior cannot be excluded. Indeed, abundance of ERβ is expressed in the DRN (26). Thus, future studies are warranted to explore the functions of ERβ in DRN neurons (or other brain regions) in the context of binge-like eating.
In summary, our data support a model that estrogens act upon ERα expressed by DRN 5-HT neurons to inhibit the SK current, which in turn leads to activation of 5-HT neurons and ultimately inhibition of binge-like eating behavior in female mice. Thus, we identified ERα and SK current in DRN 5-HT neurons as potential targets for anti-binge therapies. Indeed, we provided evidence that a newly developed GLP-1–estrogen conjugate can preferentially target this DRN ERα population to potently inhibit binge-like eating behavior in female mice.
Methods
Mice.
We crossed Tph2-CreER (Jackson Laboratory, #016584) transgenic mice and Esr1fl/fl mice (30). This cross produced KO mice (those that are homozygous for Esr1fl/fl and also carry the Tph2-CreER transgene) and WT mice (those that are homozygous for Esr1fl/fl, but do not carry the Tph2-CreER transgene). At 8 weeks of age, female KO mice received i.p. injections of tamoxifen (3 mg/mouse, twice, 24 hours apart) to induce Cre activity and therefore delete ERα only in 5-HT neurons. Female WT also received the same tamoxifen injections to rule out any possible effects of tamoxifen itself. Weekly body weight of these mice was monitored from weaning till the end of the study.
In parallel, we also crossed the Rosa26-tdTOMATO allele onto Tph2-CreER mice or Esr1fl/fl Tph2-CreER mice. This cross produced Tph2-CreER Rosa26-tdTOMATO (WT) and Esr1fl/fl Tph2-CreER Rosa26-tdTOMATO (KO) mice. Upon tamoxifen inductions similar to those described above, both mice expressed TOMATO exclusively in 5-HT neurons, and KO mice had Esr1 (ERα) deleted specifically in 5-HT neurons. These mice were used for electrophysiology recordings.
All the breeders have been backcrossed to C57BL/6 background for more than 12 generations. In addition, some C57BL/6 mice were purchased from the mouse facility of Baylor College of Medicine. Mice were housed in a temperature-controlled environment in groups of 2 to 5 at 22ÚC to 24ÚC using a 12-hour light/12-hour dark cycle. The mice were fed standard chow (6.5% fat, #2920; Harlan-Teklad) until training and assessment of binge-like eating behavior. Water was provided ad libitum.
Histology.
We used dual immunofluorescence to examine the colocalization of ERα and 5-HT in mouse brain. Briefly, C57BL/6 female mice were perfused with 10% formalin, and brain sections were cut at 25 μm. The sections were incubated at room temperature in primary goat anti–5-HT antibody (1:5,000, #20079; Immunostar) overnight, followed by the secondary donkey anti-goat Alexa Fluor 488 (1:500; #A-11055; Invitrogen) for 1.5 hours. Then, the sections were incubated in the primary rabbit anti-ERα antibody (1:10,000, #06-935; Millipore) overnight, followed by secondary donkey anti-rabbit Alexa Fluor 594 (1:500; #A-21207; Invitrogen) for 1.5 hours. Slides were coverslipped with DAPI-containing mounting media (H1500; Vector Laboratories). Fluorescence images were analyzed using a Leica DM5500 fluorescence microscope with OptiGrid structured illumination configuration.
Similarly, we performed immunofluorescence for 5-HT in Tph2-CreER Rosa26-tdTOMATO mice to confirm colocalization of TOMATO and 5-HT. Briefly, mice (after tamoxifen inductions) were perfused with 10% formalin, and brain sections were cut at 25 μm. The sections were incubated at room temperature in primary goat anti–5-HT antibody (1:5000, #20079; Immunostar) overnight, followed by the secondary donkey anti-goat Alexa Fluor 488 (1:500, #A-11055; Invitrogen) for 1.5 hours. Slides were coverslipped with DAPI-containing mounting media (H1500; Vector Laboratories) and analyzed using a Leica DM5500 fluorescence microscope with OptiGrid structured illumination configuration. TOMATO signals were observed directly with a red fluorescence channel. As a negative control, TOMATO signals were also examined in Tph2-CreER Rosa26-tdTOMATO mice without tamoxifen induction.
We also performed immunohistochemistry for ERα in WT and KO mice to validate selective deletion of ERα in 5-HT neurons. Briefly, mice (after tamoxifen inductions) were perfused with 10% formalin, and brain sections were cut at 25 μm. The sections were incubated at room temperature in the primary rabbit anti-ERα antibody (1:20,000; Upstate) overnight followed by biotinylated anti-rabbit secondary antibody (1:1,000; Vector) for 2 hours. Sections were then incubated in the avidin-biotin complex (1:500, ABC; Vector Elite Kit) and incubated in 0.04% 3, 3′-diaminobenzidine and 0.01% hydrogen peroxide. After dehydration through graded ethanol, the slides were then immersed in xylene and coverslipped. Images were analyzed using a brightfield Leica microscope.
Training and assessment of binge-like eating behavior.
We used the published protocol (29) to train and assess binge-like eating behavior in mice. Briefly, mice were randomly assigned into “intermittent” or “continuous” group. “Intermittent” mice were exposed to both regular chow pellets (6.5% fat, #2920; Harlan) and HFD pellets (40% fat, TD.95217; Harlan) for 48 hours (from Monday 11:00 am to Wednesday 11:00 am) and then exposed to only chow for the rest of the week. On the binge assessment day (Monday of the 2nd week), HFD was given back to cages at 11:00 am, and HFD and chow intake were measured for 2.5 hours (from 11:00 am to 1:30 pm). The “continuous” group was exposed to chow and HFD for the entire study, and HFD and chow intake were measured for 2.5 hours at the same time as in the “intermittent” group. Mice were housed in their original home cages for the entire training and study period. These cages (width: 7.25 inch, length: 11.5 inch, height: 5 inch; # RC71U-UD; Alternative Design) were made of polysulfone, with a gridded metal top holding a water bottle and pellet diets. A metal board was vertically inserted into the food holder to separate chow and HFD pellets.
Binge-like eating behavior in OVXV and OVXE mice.
Female littermates were anesthetized with isoflurane. As previously described (42, 43), bilateral OVX was performed, followed by s.c. implantations of a pellet containing 17β-estradiol (0.5 μg/d for 60 days, OVXE) or containing vehicle (OVXV). These pellets were purchased from Innovative Research of America. After a 7-day recovery, mice were subjected to the intermittent or continuous (as control) exposure to HFD for 1 week in order to induce binge-like eating behavior. Two weeks after the surgery, binge-like eating behavior was assessed as detailed above. Body weight and food intake were monitored every other day during the entire study period. Body composition was determined using quantitative magnetic resonance on the same day when binge-like behavior was assessed. After assessment of binge-like eating behavior, mice were deeply anesthetized with isoflurane, and blood was collected through cardiac aspiration. Plasma was obtained by centrifugation, and plasma orexin A was measured with a mouse orexin A ELISA kit (MBS815052; MyBioSource).
Effects of GLP-1–estrogen on binge-like eating behavior in OVX female mice.
First, to determine whether stable GLP-1–estrogen delivers bioactive estrogens into mouse DRN, we examined effects of GLP-1–estrogen on expression of Trim25 (a known estrogen target) in the DRN. To this end, female C57BL/6 mice (12 weeks of age) received OVX surgery as described above. After a 7-day recovery, these mice received s.c. injections of GLP-1 (4 μg/kg) or GLP-1–estrogen (4 μg/kg). Two hours after injections, mice were sacrificed and the DRN was quickly microdissected and stored at –80ÚC. As control groups, another cohort of female C57BL/6 mice (12 weeks of age) received OVXV or OVXE surgery as described above. After a 7-day recovery, the DRN was quickly microdissected and stored at –80ÚC. As described previously (44), total mRNA was isolated using TRIzol Reagent (Invitrogen) according to the manufacturer’s protocol, and reverse transcription reactions were performed from 2 μg of total mRNA using a High-Capacity cDNA Reverse Transcription Kits (Invitrogen). Samples were amplified on a CFX384 Real-Time System (Bio-Rad) using SsoADV SYBR Green Supermix (Bio-Rad). Correct melting temperatures for all products were verified after amplification. Results were normalized against the expression of housekeeping gene cyclophilin. Primer sequences were as follows: cyclophilin, forward: TGGAGAGCACCAAGACAGACA; cyclophilin, reverse: TGCCGGAGTCGACAATGAT; Trim25, forward: TGATGTGGCTGTGCATGATA; Trim25, reverse: AAGACCTGCTCCCCTACGAC.
Further, we tested to determine whether GLP-1–estrogen can inhibit binge-like eating behavior via acting upon ERα in 5-HT neurons. To this end, WT and KO female mice received tamoxifen inductions as described above at 8 weeks of age. At 24 weeks of age, these WT and KO mice were anesthetized with isoflurane and received bilateral OVX surgery. After a 7-day recovery period, mice were subjected to the 1-week intermittent HFD exposure to induce binge-like eating behavior as described above. On the binge assessment day, vehicle (saline), GLP-1 (4 μg/kg), or GLP-1–estrogen (4 μg/kg) was s.c. injected at 10:30 am, followed by assessment of binge-like eating behavior (11:00 am to 1:30 pm). The doses of GLP-1 and GLP-1–estrogen were chosen based on the previous report (31) and our preliminary studies.
Effects of intra-DRN injections on binge-like eating behavior in OVX female mice.
WT and KO female mice received tamoxifen induction as described above at 8 weeks of age. At 24 weeks of age, mice were anesthetized with i.p. injections of the ketamine/xylazine cocktail (100 mg/kg ketamine and 10 mg/kg xylazine) and received OVXV or OVXE treatment as described above. During the same period under anesthesia, an indwelling microinjection cannula was stereotaxically inserted to target the DRN (midline, 4.36 mm posterior and 3.1 ventral to the bregma). After a 7-day recovery, mice were subjected to the 1-week intermittent HFD exposure to induce binge-like eating behavior as described above. On the binge assessment day, vehicle (saline) or apamin (50 nM, 0.5 μl, # STA-200; Alomone Labs) was injected into the DRN at 10:30 am, followed by assessment of binge-like eating behavior (11:00 am to 1:30 pm). The dose of apamin was chosen based on preliminary studies.
Conditioned taste-aversion tests.
To rule out the possibility that intra-DRN injections of apamin inhibit binge-like eating behavior through nonspecific adverse effects, we performed conditioned taste aversion (CTA) tests as described in the literature (45). Briefly, C57BL/6 female mice (12 weeks of age) were bilaterally ovariectomized, and during the same period under anesthesia, an indwelling cannula was inserted to target the DRN as described above. After a 7-day recovery, these mice were housed individually and acclimated to intra-DRN injections (saline solutions) and a 2-hour daily water supply (3:00 pm–5:00 pm) over 8 days. On a test day, mice were given access to 0.2 m NaCl solution for 2 hours (3:00 pm–5:00 pm) in the same type of bottle with which they were usually presented with water. Immediately after exposure to the NaCl solution, mice received intra-DRN injections of saline or apamin (50 nM, 0.5 μl). The amount of solution intake was measured. The same tests were repeated in another 5 trials every other day.
Anxiety tests (light-dark test and EPM).
To determine whether intermittent HFD exposure causes anxiety in mice, an independent cohort of C57BL/6 female mice (12 weeks) were subjected to intermittent or continuous HFD exposure for 1 week as described above. Binge-like eating behavior was confirmed in the “intermittent” HDF group in the second week. At 11:00 am on Monday of the third week (the same time when mice would be tested for binge-like eating behavior), these mice were subjected to the light-dark tests using published protocol (46). Briefly, the test consisted of a polypropylene chamber (44 × 21 × 21 cm) unequally divided into a larger, brightly illuminated open compartment (clear polypropylene) and a smaller, dark compartment (in dark polypropylene), connected by a small opening. Mice were placed in the illuminated chamber and allowed to move freely between the 2 chambers for 10 minutes. The latency to enter the light and dark chambers, the time spent in the chambers, the total number of transitions, and distance traveled in each chamber was measured using the VersaMax Animal Activity Monitoring System (AccuScan Instruments Inc.) and analyzed. Transfer of all 4 paws of an experimental animal from one chamber to the other was considered as 1 transition event.
At 11:00 am on Tuesday of the third week, these mice were subjected to the EPM using published protocol (46). The EPM was constructed of Plexiglas, with 2 open arms (30 × 5 cm) and 2 enclosed black arms (30 × 5 × 15 cm) at an elevation of 50 cm above the floor. The arms of the maze form a cross, with the 2 open arms facing each other. The maze was cleaned with 70% ethanol solution after each session and allowed to dry between the sessions. Anxiety-like behavior was measured by placing the mice in the center of the junction of the arms of the maze facing an open arm, and the behavior was analyzed for 10 minutes. The number of entries into the open and closed arms, the time spent exploring the open and closed arms, and the distance traveled were recorded and analyzed using the ANY-maze software (Stoelting Co.). The changes in anxiety-like behavior were calculated by dividing the number of entries into the open arms by the total number of entries into all 4 arms (open to total ratio for entries) or by dividing the amount of time spent in the open arms by the amount of time spent in all 4 arms (OTR for time). The time spent in the center platform not exploring any of the arms was not included in these calculations.
Forced swim tests.
To determine whether the intermittent HFD exposure causes depression in mice, an independent cohort of C57BL/6 mice was subjected to the intermittent or continuous HFD exposure for 1 week as described above. Binge-like eating behavior was confirmed in the intermittent HDF group in the second week. At 11:00 am on Monday of the third week (the same time when mice would be tested for binge-like eating behavior), these mice were subjected to the forced swim tests using published protocol (47–49). Briefly, the mice were individually placed into a glass cylinder (25 cm tall × 10 cm diameter) containing 8 cm of water, maintained at 23°C to 25°C for 6 minutes. The mouse movement was digitally recorded from the side using a camcorder. Water in the cylinder was replaced after each recording. The starting and total time of immobility for each mouse was recorded. Immobility is defined as the absence of active floating with only minimal movements needed to keep head above water. The immobility was scored every 30 seconds for the last 4 minutes. During each 30 seconds, 1 immobility score was recorded if the immobility time was more than 10 seconds. Data from mice that had difficulty keeping heads above water were excluded from analyses. All the videos were blindly rated by a different experimenter.
Electrophysiology.
Mice were deeply anesthetized with isoflurane and transcardially perfused (50) with a modified ice-cold artificial cerebral spinal fluid (aCSF) in: 10 mM NaCl, 25 mM NaHCO3, 195 mM sucrose, 5 mM glucose, 2.5 mM KCl, 1.25 mM NaH2PO4, 2 mM sodium pyruvate, 0.5 mM CaCl2, and 7 mM MgCl2) (51). The mice were then decapitated, and the entire brain was removed. Brains was quickly sectioned in ice-cold aCSF solution (126 mM NaCl, 2.5 mM KCl, 1.2 mM MgCl2, 2.4 mM CaCl2, 1 mM NaH2PO4, 11.1 mM glucose, and mM 21.4 NaHCO3) (52) saturated with 95% O2 and 5% CO2. Coronal sections containing the DRN (270 μm) were cut with a Microm HM 650V vibratome (Thermo Scientific) and then preincubated in the aCSF (52) at 34°C for at least 1 hour before recording.
Whole-cell patch clamp recordings were performed in the target neurons in the DRN visually identified by an upright microscope (Eclipse FN-1; Nikon) equipped with IR-DIC optics (×40 NIR; Nikon). Signals were processed using Multiclamp 700B amplifier (Axon Instruments), sampled using Digidata 1440A, and analyzed offline on a PC with pCLAMP 10.3 (Axon Instruments). The slices were bathed in oxygenated aCSF (52) (32°C–34°C) at a flow rate of approximately 2 ml/min. Patch pipettes with resistances of 3 to 5 M⁄ were filled with solution containing 126 mM K gluconate, 10 mM NaCl, 10 mM EGTA, 1 mM MgCl2, 2 mM Na-ATP, and 0.1 mM Mg-GTP (adjusted to pH 7.3 with KOH) (34). Voltage clamp was used to record SK-like currents. Current clamp was engaged to test neural firing, RM, and input resistance. Based on previous reports and our preliminary data, PPT (100 nM, perfusion up to 6 minutes at 2 ml/min; Sigma-Aldrich, H6036) (53), acute apamin perfusion (100 nM, perfusion upon to 8 minutes at 2 ml/min), and chronic apamin incubation (100 nM, incubation about 2 hours) (34) were used. Alexa Fluor 488 (0.01 mM) was included in the pipette solution to trace the recorded neurons. After recordings, slices were fixed with 4% formalin overnight and mounted onto slides. Cells were then visualized with the a Leica DM5500 fluorescence microscope to identify post hoc the anatomical location of the recorded neurons in the DRN.
Statistics.
The data are presented as mean ± SEM. Statistical analyses were performed using GraphPad Prism. Most data were analyzed by 1- or 2-way ANOVA, followed by post hoc Bonferroni’s tests. Comparisons between 2 groups were analyzed by t tests. Numbers of responsive neurons in each group were analyzed by χ2 tests. P < 0.05 was considered statistically significant.
Study approval.
Care of all animals and procedures were approved by the Baylor College of Medicine Institutional Animal Care and Use Committee.
Supplementary Material
Acknowledgments
This work was supported by grants from the NIH (R01DK093587, R00DK085330, and P30 DK079638-03 to Y. Xu; T32CA059268 to S. Khan; HD62512 to S.K. Mani; and R01DK092605 to Q. Tong), by NSF IOS1424017 (to S.K. Mani), by the American Diabetes Association (to Y. Xu), by Marcadia Biotech (to B. Yang and R. DiMarchi), by the Klarman Family Foundation (to Y. Xu), by the Naman Family Fund for Basic Research (to Y. Xu), by the Curtis Hankamer Basic Research Fund (to Y. Xu), and the American Heart Association (to Q. Tong). Measurements of body composition were performed in the Mouse Metabolic Research Unit (MMRU) at the USDA/ARS Children’s Nutrition Research Center, Baylor College of Medicine, which is supported by funds from the USDA ARS (www.bcm.edu/cnrc/mmru). The authors acknowledge the expert assistance of Firoz Vohra and the MMRU Core Director, Marta Fiorotto. The behavioral tests (e.g., light-dark test, EPM test) were performed at the Mouse Neurobehavior Core, Baylor College of Medicine, which is supported by NIH P30HD024064.
Footnotes
Conflict of interest: Bin Yang and Richard DiMarchi received support from Marcadia Biotech.
Reference information:J Clin Invest. 2014;124(10):4351–4362. doi:10.1172/JCI74726.
Yuxin Feng’s present address is: Combined Blood and Marrow Transplantation Program, Spectrum Health, Grand Rapids, Michigan, USA.
References
- 1.Hudson JI, Hiripi E, Pope HG, Pope HG, Jr, Kessler RC. The prevalence and correlates of eating disorders in the National Comorbidity Survey Replication. Biol Psychiatry. 2007;61(3):348–358. doi: 10.1016/j.biopsych.2006.03.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mitchell JE, Mussell MP. Comorbidity and binge eating disorder. Addict Behav. 1995;20(6):725–732. doi: 10.1016/0306-4603(95)00095-X. [DOI] [PubMed] [Google Scholar]
- 3.Patrick L. Eating disorders: a review of the literature with emphasis on medical complications and clinical nutrition. Altern Med Rev. 2002;7(3):184–202. [PubMed] [Google Scholar]
- 4.Whitaker AH. An epidemiological study of anorectic and bulimic symptoms in adolescent girls: implications for pediatricians. Pediatr Ann. 1992;21(11):752–759. doi: 10.3928/0090-4481-19921101-10. [DOI] [PubMed] [Google Scholar]
- 5.Verri A, Nappi RE, Cecchini AP, Vallero E, Luzi S, Zara C. Eating disorders and Axis I psychiatric comorbidity in amenorrheic women. Int J Eat Disord. 1998;24(2):137–146. doi: 10.1002/(sici)1098-108x(199809)24:2<137::aid-eat3>3.0.co;2-f. [DOI] [PubMed] [Google Scholar]
- 6.Poyastro Pinheiro A, et al. Patterns of menstrual disturbance in eating disorders. Int J Eat Disord. 2007;40(5):424–434. doi: 10.1002/eat.20388. [DOI] [PubMed] [Google Scholar]
- 7.Lester NA, Keel PK, Lipson SF. Symptom fluctuation in bulimia nervosa: relation to menstrual-cycle phase and cortisol levels. Psychol Med. 2003;33(1):51–60. doi: 10.1017/s0033291702006815. [DOI] [PubMed] [Google Scholar]
- 8.Gladis MM, Walsh BT. Premenstrual exacerbation of binge eating in bulimia. Am J Psychiatry. 1987;144(12):1592–1595. doi: 10.1176/ajp.144.12.1592. [DOI] [PubMed] [Google Scholar]
- 9.Price WA, Torem MS, DiMarzio LR. Premenstrual exacerbation of bulimia. Psychosomatics. 1987;28(7):378–379. doi: 10.1016/S0033-3182(87)72511-0. [DOI] [PubMed] [Google Scholar]
- 10.Edler C, Lipson SF, Keel PK. Ovarian hormones and binge eating in bulimia nervosa. Psychol Med. 2007;37(1):131–141. doi: 10.1017/S0033291706008956. [DOI] [PubMed] [Google Scholar]
- 11.Klump KL, Keel PK, Culbert KM, Edler C. Ovarian hormones and binge eating: exploring associations in community samples. Psychol Med. 2008;38(12):1749–1757. doi: 10.1017/S0033291708002997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Klump KL, Suisman JL, Culbert KM, Kashy DA, Keel PK, Sisk CL. The effects of ovariectomy on binge eating proneness in adult female rats. Horm Behav. 2011;59(4):585–593. doi: 10.1016/j.yhbeh.2011.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yu Z, Geary N, Corwin RL. Ovarian hormones inhibit fat intake under binge-type conditions in ovariectomized rats. Physiol Behav. 2008;95(3):501–507. doi: 10.1016/j.physbeh.2008.07.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jimerson DC, Lesem MD, Kaye WH, Hegg AP, Brewerton TD. Eating disorders and depression: is there a serotonin connection? Biol Psychiatry. 1990;28(5):443–454. doi: 10.1016/0006-3223(90)90412-U. [DOI] [PubMed] [Google Scholar]
- 15.Monteleone P, Brambilla F, Bortolotti F, Maj M. Serotonergic dysfunction across the eating disorders: relationship to eating behaviour, purging behaviour, nutritional status and general psychopathology. Psychol Med. 2000;30(5):1099–1110. doi: 10.1017/S0033291799002330. [DOI] [PubMed] [Google Scholar]
- 16.Steiger H. Eating disorders and the serotonin connection: state, trait and developmental effects. J Psychiatry Neurosci. 2004;29(1):20–29. [PMC free article] [PubMed] [Google Scholar]
- 17.Sabine EJ, Yonace A, Farrington AJ, Barratt KH, Wakeling A. Bulimia nervosa: a placebo controlled double-blind therapeutic trial of mianserin. Br J Clin Pharmacol. 1983;15(suppl 2):195S–202S. doi: 10.1111/j.1365-2125.1983.tb05866.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pope HG, Pope HG, Jr, Hudson JI, Jonas JM, Yurgelun-Todd D. Bulimia treated with imipramine: a placebo-controlled, double-blind study. Am J Psychiatry. 1983;140(5):554–558. doi: 10.1176/ajp.140.5.554. [DOI] [PubMed] [Google Scholar]
- 19.Mitchell JE, Groat R. A placebo-controlled, double-blind trial of amitriptyline in bulimia. J Clin Psychopharmacol. 1984;4(4):186–193. [PubMed] [Google Scholar]
- 20.Walsh BT, Stewart JW, Roose SP, Gladis M, Glassman AH. Treatment of bulimia with phenelzine. A double-blind, placebo-controlled study. Arch Gen Psychiatry. 1984;41(11):1105–1109. doi: 10.1001/archpsyc.1983.01790220095015. [DOI] [PubMed] [Google Scholar]
- 21.Hughes PL, Wells LA, Cunningham CJ, Ilstrup DM. Treating bulimia with desipramine. A double-blind, placebo-controlled study. Arch Gen Psychiatry. 1986;43(2):182–186. doi: 10.1001/archpsyc.1986.01800020092012. [DOI] [PubMed] [Google Scholar]
- 22.Barlow J, Blouin J, Blouin A, Perez E. Treatment of bulimia with desipramine: a double-blind crossover study. Can J Psychiatry. 1988;33(2):129–133. doi: 10.1177/070674378803300211. [DOI] [PubMed] [Google Scholar]
- 23.Rowland NE, Carlton J. Neurobiology of an anorectic drug: fenfluramine. Prog Neurobiol. 1986;27(1):13–62. doi: 10.1016/0301-0082(86)90011-0. [DOI] [PubMed] [Google Scholar]
- 24.Stunkard A, Berkowitz R, Tanrikut C, Reiss E, Young L. d-fenfluramine treatment of binge eating disorder. Am J Psychiatry. 1996;153(11):1455–1459. doi: 10.1176/ajp.153.11.1455. [DOI] [PubMed] [Google Scholar]
- 25.Blouin AG, Blouin JH, Perez EL, Bushnik T, Zuro C, Mulder E. Treatment of bulimia with fenfluramine and desipramine. J Clin Psychopharmacol. 1988;8(4):261–269. [PubMed] [Google Scholar]
- 26.Merchenthaler I, Lane MV, Numan S, Dellovade TL. Distribution of estrogen receptor alpha and beta in the mouse central nervous system: in vivo autoradiographic and immunocytochemical analyses. J Comp Neurol. 2004;473(2):270–291. doi: 10.1002/cne.20128. [DOI] [PubMed] [Google Scholar]
- 27.Dalmasso C, Amigone JL, Vivas L. Serotonergic system involvement in the inhibitory action of estrogen on induced sodium appetite in female rats. Physiol Behav. 2011;104(3):398–407. doi: 10.1016/j.physbeh.2011.04.029. [DOI] [PubMed] [Google Scholar]
- 28.Robichaud M, Debonnel G. Oestrogen and testosterone modulate the firing activity of dorsal raphe nucleus serotonergic neurones in both male and female rats. J Neuroendocrinol. 2005;17(3):179–185. doi: 10.1111/j.1365-2826.2005.01292.x. [DOI] [PubMed] [Google Scholar]
- 29.Czyzyk TA, Sahr AE, Statnick MA. A model of binge-like eating behavior in mice that does not require food deprivation or stress. Obesity (Silver Spring). 2010;18(9):1710–1717. doi: 10.1038/oby.2010.46. [DOI] [PubMed] [Google Scholar]
- 30.Feng Y, Manka D, Wagner KU, Khan SA. Estrogen receptor-alpha expression in the mammary epithelium is required for ductal and alveolar morphogenesis in mice. Proc Natl Acad Sci U S A. 2007;104(37):14718–14723. doi: 10.1073/pnas.0706933104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Finan B, et al. Targeted estrogen delivery reverses the metabolic syndrome. Nat Med. 2012;18(12):1847–1856. doi: 10.1038/nm.3009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gu G, Roland B, Tomaselli K, Dolman CS, Lowe C, Heilig JS. Glucagon-like peptide-1 in the rat brain: Distribution of expression and functional implication. J Comp Neurol. 2013;521(10):2235–2261. doi: 10.1002/cne.23282. [DOI] [PubMed] [Google Scholar]
- 33.Adelman JP, Maylie J, Sah P. Small-conductance Ca2+-activated K+ channels: form and function. Annu Rev Physiol. 2012;74:245–269. doi: 10.1146/annurev-physiol-020911-153336. [DOI] [PubMed] [Google Scholar]
- 34.Pagadala P, et al. Loss of NR1 subunit of NMDARs in primary sensory neurons leads to hyperexcitability and pain hypersensitivity: involvement of Ca(2+)-activated small conductance potassium channels. J Neurosci. 2013;33(33):13425–13430. doi: 10.1523/JNEUROSCI.0454-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Asarian L, Geary N. Estradiol enhances cholecystokinin-dependent lipid-induced satiation and activates estrogen receptor-alpha-expressing cells in the nucleus tractus solitarius of ovariectomized rats. Endocrinology. 2007;148(12):5656–5666. doi: 10.1210/en.2007-0341. [DOI] [PubMed] [Google Scholar]
- 36.Drucker DJ. Minireview: the glucagon-like peptides. Endocrinology. 2001;142(2):521–527. doi: 10.1210/endo.142.2.7983. [DOI] [PubMed] [Google Scholar]
- 37.Dossat AM, Bodell LP, Williams DL, Eckel LA, Keel PK. Preliminary examination of glucagon-like peptide-1 levels in women with purging disorder and bulimia nervosa. Int J Eat Disord. doi: 10.1002/eat.22264. [published online ahead of print March 4, 2014]. doi: 10.1002/eat.22264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Naessen S, Carlstrom K, Holst JJ, Hellstrom PM, Hirschberg AL. Women with bulimia nervosa exhibit attenuated secretion of glucagon-like peptide 1, pancreatic polypeptide, and insulin in response to a meal. Am J Clin Nutr. 2011;94(4):967–972. doi: 10.3945/ajcn.111.014837. [DOI] [PubMed] [Google Scholar]
- 39.Santollo J, Wiley MD, Eckel LA. Acute activation of ER alpha decreases food intake, meal size, and body weight in ovariectomized rats. Am J Physiol Regul Integr Comp Physiol. 2007;293(6):R2194–R2201. doi: 10.1152/ajpregu.00385.2007. [DOI] [PubMed] [Google Scholar]
- 40.Santollo J, Katzenellenbogen BS, Katzenellenbogen JA, Eckel LA. Activation of ERα is necessary for estradiol’s anorexigenic effect in female rats. Horm Behav. 2010;58(5):872–877. doi: 10.1016/j.yhbeh.2010.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Heine PA, Taylor JA, Iwamoto GA, Lubahn DB, Cooke PS. Increased adipose tissue in male and female estrogen receptor-α knockout mice. Proc Natl Acad Sci U S A. 2000;97(23):12729–12734. doi: 10.1073/pnas.97.23.12729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Xu Y, et al. Distinct hypothalamic neurons mediate estrogenic effects on energy homeostasis and reproduction. Cell Metab. 2011;14(4):453–465. doi: 10.1016/j.cmet.2011.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhu L, et al. Steroid receptor coactivator-1 mediates estrogenic actions to prevent body weight gain in female mice. Endocrinology. 2013;154(1):150–158. doi: 10.1210/en.2012-2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Xu Y, et al. PI3K signaling in the ventromedial hypothalamic nucleus is required for normal energy homeostasis. Cell Metab. 2010;12(1):88–95. doi: 10.1016/j.cmet.2010.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cannon CM, Scannell CA, Palmiter RD. Mice lacking dopamine D1 receptors express normal lithium chloride-induced conditioned taste aversion for salt but not sucrose. Eur J Neurosci. 2005;21(9):2600–2604. doi: 10.1111/j.1460-9568.2005.04077.x. [DOI] [PubMed] [Google Scholar]
- 46.Oyola MG, et al. Anxiolytic effects and neuroanatomical targets of estrogen receptor-β (ERβ) activation by a selective ERβ agonist in female mice. Endocrinology. 2012;153(2):837–846. doi: 10.1210/en.2011-1674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Flaisher-Grinberg S, Einat H. A possible utilization of the mice forced swim test for modeling manic-like increase in vigor and goal-directed behavior. J Pharmacol Toxicol Methods. 2009;59(3):141–145. doi: 10.1016/j.vascn.2009.03.003. [DOI] [PubMed] [Google Scholar]
- 48.Pechnick RN, Chesnokova VM, Kariagina A, Price S, Bresee CJ, Poland RE. Reduced immobility in the forced swim test in mice with a targeted deletion of the leukemia inhibitory factor (LIF) gene. Neuropsychopharmacology. 2004;29(4):770–776. doi: 10.1038/sj.npp.1300402. [DOI] [PubMed] [Google Scholar]
- 49.Poleszak E, et al. NMDA/glutamate mechanism of antidepressant-like action of magnesium in forced swim test in mice. Pharmacol Biochem Behav. 2007;88(2):158–164. doi: 10.1016/j.pbb.2007.07.018. [DOI] [PubMed] [Google Scholar]
- 50.Hill JW, et al. Acute effects of leptin require PI3K signaling in hypothalamic proopiomelanocortin neurons in mice. J Clin Invest. 2008;118(5):1796–1805. doi: 10.1172/JCI32964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ren H, et al. FoxO1 target Gpr17 activates AgRP neurons to regulate food intake. Cell. 2012;149(6):1314–1326. doi: 10.1016/j.cell.2012.04.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Pinto S, et al. Rapid rewiring of arcuate nucleus feeding circuits by leptin. Science. 2004;304(5667):110–115. doi: 10.1126/science.1089459. [DOI] [PubMed] [Google Scholar]
- 53.Merlo S, Frasca G, Canonico PL, Sortino MA. Differential involvement of estrogen receptor alpha and estrogen receptor beta in the healing promoting effect of estrogen in human keratinocytes. J Endocrinol. 2009;200(2):189–197. doi: 10.1677/JOE-08-0442. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.