

Abstract
The past decade has been one of rapid innovation in genome-editing technology. The opportunity now exists for investigators to manipulate virtually any gene in a diverse range of cell types and organisms with targeted nucleases designed with sequence-specific DNA-binding domains. The rapid development of the field has allowed for highly efficient, precise, and now cost-effective means by which to generate human and animal models of disease using these technologies. This review will outline the recent development of genome-editing technology, culminating with the use of CRISPR-Cas9 to generate novel mammalian models of disease. While the road to using this same technology for treatment of human disease is long, the pace of innovation over the past five years and early successes in model systems build anticipation for this prospect.
The emergence of genome-editing technology
The classical method for gene modification is homologous recombination. This approach has been widely used in mouse embryonic stem cells to generate germline knockout or knockin mice (1, 2). A disadvantage is that it typically takes more than a year to generate a genetically modified mouse using the standard approach. Furthermore, similar attempts at using homologous recombination in human cells have proven to be far more challenging, and alternative approaches to knock down gene expression, such as antisense oligonucleotides and short interfering RNAs, have instead become standard. However, these approaches only transiently reduce gene expression, and the effect is usually incomplete and can often affect off-target genes (3, 4). These shortcomings have fueled the demand for more effective methods of gene modification.
A new wave of technology that is variously termed “gene editing,” “genome editing,” or “genome engineering” has emerged to address this demand by giving investigators the ability to precisely and efficiently introduce a variety of genetic alterations into mammalian cells, ranging from knockin of single nucleotide variants to insertion of genes to deletion of chromosomal regions. We describe the key advantages and disadvantages of the three most popular genome-editing tools. This description is not meant to be a comprehensive review of the work leading to the development of the tools, but rather to give readers a working knowledge of the tools and the ability to select among the tools for desired tasks.
Zinc finger nucleases
Zinc finger nucleases (ZFNs) are increasingly being used in academia and industrial research for a variety of purposes ranging from the generation of animal models to human therapies (5). ZFNs are fusion proteins comprising an array of site-specific DNA-binding domains — adapted from zinc finger–containing transcription factors — attached to the endonuclease domain of the bacterial FokI restriction enzyme. Each zinc finger domain recognizes a 3- to 4-bp DNA sequence, and tandem domains can potentially bind to an extended nucleotide sequence (typically with a length that is a multiple of 3, usually 9 bp to 18 bp) that is unique within a cell’s genome.
To cleave a specific site in the genome, ZFNs are designed as a pair that recognizes two sequences flanking the site, one on the forward strand and the other on the reverse strand. Upon binding of the ZFNs on either side of the site, the pair of FokI domains dimerize and cleave the DNA at the site, generating a double-strand break (DSB) with 5′ overhangs (5). Cells repair DSBs using either (a) nonhomologous end joining (NHEJ), which can occur during any phase of the cell cycle, but occasionally results in erroneous repair, or (b) homology-directed repair (HDR), which typically occurs during late S phase or G2 phase when a sister chromatid is available to serve as a repair template (Figure 1).
Figure 1. Repair of DSBs.

With the creation of each DSB, two DNA repair processes proceed in concert. HDR results in high-fidelity repair using a template strand. If desired, an exogenous oligonucleotide sequence can be introduced to achieve site-specific mutagenesis. NHEJ yields WT clones as well as clones with frameshift/indel mutations through its inherently more error-prone mechanism of repair.
The error-prone nature of NHEJ can be exploited to introduce frameshifts into the coding sequence of a gene, potentially knocking out the gene by a combination of two mechanisms: premature truncation of the protein and nonsense-mediated decay of the mRNA transcript, the latter of which is not always particularly efficient (Figure 1). Alternatively, HDR can be utilized to insert a specific mutation, with the introduction of a repair template containing the desired mutation flanked by homology arms (Figure 1). In response to a DSB in DNA, HDR utilizes another closely matching DNA sequence to repair the break. Mechanistically, HDR can proceed in the same fashion as traditional homologous recombination, using an exogenous double-stranded DNA vector as a repair template (6). It can also use an exogenous single-stranded DNA oligonucleotide (ssODN) as a repair template. For ssODNs, homology arms of as little as 20 bp can enable introduction of mutations into the genome (7–9). In either case, the efficiency can be sufficiently high such that antibiotic selection to identify correctly targeted clones is unnecessary (8, 10). If antibiotic selection is not used, then extra steps to remove the antibiotic resistance cassette from the genome using systems such as Cre-lox and Flp-FRT are unnecessary, in contrast to traditional homologous recombination.
Despite the advantages of genome editing with ZFNs, there are several potential disadvantages. It has not proven to be straightforward to assemble zinc finger domains to bind an extended stretch of nucleotides with high affinity (11). This has made it difficult for nonspecialists to routinely engineer ZFNs. To surmount this difficulty, an academic consortium has developed an open-source library of zinc finger components and protocols to perform screens to identify ZFNs that bind with high affinity to a desired sequence (12, 13); nonetheless, it can still take months for nonspecialists to obtain optimized ZFNs. Another potential disadvantage is that target site selection is limited — the open-source ZFN components can only be used to target binding sites every 200 bps in random DNA sequence (of note, commercial sources of ZFNs offer higher design densities, with the ability to target binding sites every 50 bps in random DNA sequences). While this may be a nonissue if an investigator seeks to knock out a gene, since a frameshift introduced anywhere in the early coding sequence of the gene can produce the desired result, it may present challenges if a particular site is required, e.g., to knock in a specific mutation. Since the introduction of the open-source platform, alternative platforms to engineer optimized ZFNs have emerged, each with varying degrees of speed, flexibility in site selection, and success rates (14–18).
Finally, a significant concern about the use of proteins designed to introduce DSBs into the genome is that they will do so not only at the desired site but also at off-target sites. In one study in which ZFNs were used for genome editing in human pluripotent stem cells, the investigators identified ten possible off-target genomic sites based on high-sequence similarity to the on-target site and found a single off-target mutation in the 184 clones assessed (19). Two subsequent studies of ZFNs seeking to identify potential off-target sites for several ZFN pairs revealed off-target events at numerous loci in a cultured human tumor cell line (20, 21). Thus, investigators should be cognizant of the possibility that ZFNs designed for a particular purpose may incur undesired off-target events at a low rate. One strategy to reduce off-target events is to use a pair of ZFNs that have distinct FokI domains that are obligate heterodimers (22–24). This prevents a single ZFN from binding to two adjacent off-target sites and generating a DSB; rather, the only way an off-target event could occur is if both ZFNs in a pair bind adjacently and thus allow the FokI dimer to form. Another strategy that has been demonstrated to reduce off-target events is the introduction of purified ZFN proteins into cells (17).
Transcription activator-like effector nucleases
The recent discovery of a class of proteins called transcription activator-like effectors (TALEs), exclusive to a group of plant pathogens, has led to the characterization of a novel DNA-binding domain, termed TALE repeats. The naturally occurring TALE repeats comprise tandem arrays with 10 to 30 repeats that bind and recognize extended DNA sequences (25). Each repeat is 33 to 35 amino acids in length, with two adjacent amino acids (termed the repeat-variable di-residue [RVD]) conferring specificity for one of the four DNA base pairs (26–30). Thus, there is a one-to-one correspondence between the repeats and the base pairs in the target DNA sequences. Elucidation of the RVD code has made it possible to create a new type of engineered site-specific nuclease that fuses a domain of TALE repeats to the FokI endonuclease domain, termed TAL effector nucleases (TALENs) (refs. 31–33 and Figure 2). TALENs are similar to ZFNs in that they can generate DSBs at a desired target site in the genome and so can be used to knock out genes or knock in mutations in the same way (Figure 1).
Figure 2. Binding specificity of ZFNs and TALENs.
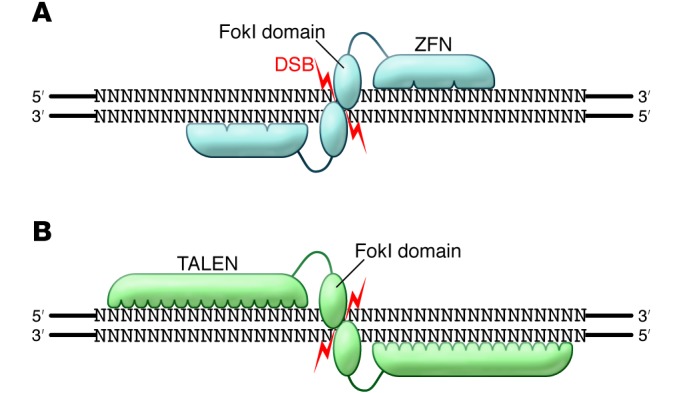
(A) The variable length ZFN DNA–binding domains bind to flanking DNA sequences and position their FokI nuclease domains such that they dimerize and generate a DSB between the binding sites. (B) Heterodimeric binding of TALENS, which like ZFNs, bind regions of variable length to generate DSB between binding sites.
In comparison with ZFNs, TALENs have turned out to be much easier to design. The RVD code has been employed to engineer many TALE repeat arrays that bind with high affinity to desired genomic DNA sequences; it appears that de novo–engineered TALE repeat arrays will bind to desired DNA sequences with high affinity at rates as high as 96% (32, 34, 35). TALENs can be designed and constructed in as short a time as two days and in as large a number as hundreds at a time (35, 36); indeed, a library with TALENs targeting all of the genes in the genome has been constructed (37).
One potential advantage over ZFNs is that the TALE repeat array can be easily extended to whatever length is desired. Whereas engineered ZFNs typically bind 9- to 18-bp sequences, TALENs are often built to bind 18-bp sequences or even longer, though recent evidence suggests that the use of larger TALENs may result in less specificity (38). Another advantage of TALENs over ZFNs is that there appear to be fewer constraints on site selection; in theory, there are multiple possible TALEN pairs available for each bp of a random DNA sequence (35).
As with ZFNs, off-target effects are a significant concern with TALENs. A study in which TALENs were used for genome editing in human pluripotent stem cells found low but measurable rates of mutagenesis at some of 19 possible off-target sites based on sequence similarity to the on-target sites (34). Although comparative data are scarce, one study found that for TALENs and ZFNs targeting the same site in the CCR5 gene, the TALENs produced fewer off-target mutations than the ZFNs at a highly similar site in the CCR2 gene (39). Furthermore, the ZFNs produced greater cell toxicity (i.e., inhibited their growth) when introduced into cells compared with the TALENs (however, it should be noted that ZFN versus TALEN protein concentrations were not normalized, and the ZFNs used in this study were of a design that was several years old, rather than being state-of-the-art ZFNs). As with ZFNs, TALENs with obligate heterodimer FokI domains are routinely used to minimize the possibility of off-target events. Recently, whole-genome sequencing studies of human pluripotent stem cell clones edited with TALENs showed that the overall off-target event rate is very low (40, 41).
A clear disadvantage of TALENs is their significantly larger size compared with ZFNs. The typical size for a cDNA encoding a TALEN is approximately 3 kb, whereas a cDNA encoding a ZFN is only approximately 1 kb. In principle, this makes it harder to deliver and express a pair of TALENs into cells compared with ZFNs, and the size of the TALENs makes them less attractive for therapeutic applications in which they must be delivered in viral vectors with limited cargo size (such as adenoassociated virus [AAV], with less than 5 kb) or as RNA molecules. Furthermore, the highly repetitive nature of the TALENs may impair their ability to be packaged and delivered by some viral vectors (42), though this can apparently be overcome by diversifying the coding sequences of the TALE repeats (43).
CRISPR-Cas9
The recent discovery of bacterial adaptive immune systems known as clustered regularly interspaced short palindromic repeats (CRISPR) and CRISPR-associated (Cas) systems has led to the newest set of genome-editing tools. CRISPR-Cas systems use a combination of proteins and short RNAs to target specific DNA sequences for cleavage. The bacteria collect “protospacers” from foreign DNA sequences (e.g., from bacteriophages), incorporate them into their genomes, and use them to express short guide RNAs, which can then be used by a CRISPR-Cas system to destroy any DNA sequences matching the protospacers.
In early 2013, four groups demonstrated that heterologous expression of a CRISPR-Cas system from Streptococcus pyogenes, comprising the Cas9 protein along with guide RNA(s) (either two separate RNAs, as found in bacteria, or a single chimeric RNA), in mammalian cells results in DSBs at target sites with (a) a 20-bp sequence matching the protospacer of the guide RNA and (b) an adjacent downstream NGG nucleotide sequence (termed the protospacer-adjacent motif [PAM]) (44–47). This occurs via the formation of a ternary complex in which Cas9 binds to the PAM in the DNA, then binds the nonprotospacer portion of the guide RNA, upon which the protospacer of the guide RNA hybridizes with one strand of the genomic DNA. Cas9 then catalyzes the DSB in the DNA at a position 3 bp upstream of the PAM (ref. 46 and Figure 3A).
Figure 3. Binding specificity of CRISPR-Cas9.

(A) With the most commonly used CRISPR-Cas9 system, a guide RNA recognizes and hybridizes a 20-bp protospacer in the genome. The DSB occurs at a site 3-bp upstream of the PAM sequence. (B) “Nickase” CRISPR-Cas9 bind to flanking DNA sequences and generate single-strand nicks that are the equivalent of a DSB. (C) Fusion proteins of catalytically dead CRISPR-Cas9 and FokI nuclease domains bind to flanking DNA sequences and position their FokI domains such that they dimerize and generate a DSB between binding sites.
In contrast to ZFNs and TALENs, which require recoding of proteins using large DNA segments (500–1500 bp) for each new target site, CRISPR-Cas9 can be easily adapted to target any genomic sequence by changing the 20-bp protospacer of the guide RNA, which can be accomplished by subcloning this nucleotide sequence into the guide RNA plasmid backbone. The Cas9 protein component remains unchanged. This ease of use for CRISPR-Cas9 is a significant advantage over ZFNs and TALENs, especially in generating a large set of vectors to target numerous sites (45) or even genome-wide libraries (48–51). Another potential advantage of CRISPR-Cas9 is the ability to multiplex, i.e., to use multiple guide RNAs in parallel to target multiple sites simultaneously in the same cell (44, 45). This makes it straightforward to mutate multiple genes at once or to engineer precise deletions in a genomic region, although it should be noted that simultaneous use of multiple ZFN or TALEN pairs can achieve the same outcomes (52).
With respect to site selection, CRISPR-Cas9 compares favorably with ZFNs and TALENs. With the most flexible version of the S. pyogenes CRISPR-Cas system, site selection is limited to 23-bp sequences on either strand that end in an NGG motif (the PAM for S. pyogenes Cas9), which occurs on average once every 8 bps (44). (Most CRISPR-Cas9 systems express the guide RNA from a plasmid using a RNA polymerase III promoter such as the U6 promoter, which requires a G in the first position, or the T7 promoter, which requires Gs in the first two positions; however, the G or Gs can simply be added to the 5′ end of the 20-nucleotide protospacer in the guide RNA as needed and thus do not affect site selection in the genome.) CRISPR-Cas systems from other species are starting to be employed in mammalian cells (44, 53, 54), and their versions of Cas9 have different PAM requirements, which allows for targeting of sites in the genome for which the S. pyogenes system is not optimal. For example, the canonical Neisseria meningitidis Cas9 PAM has been reported to be NNNNGATT, although it appears to be more tolerant of PAM variation than the S. pyogenes Cas9 (53, 54).
One disadvantage of CRISPR-Cas9 is the size of the Cas9 protein. The cDNA encoding S. pyogenes Cas9 is approximately 4.2 kb in size, making it somewhat larger than a TALEN monomer and much larger than a ZFN monomer (though both TALENs and ZFNs require dimerization, making their effective sizes larger). This size makes Cas9 challenging to deliver via viral vectors (which would additionally require a promoter and a polyadenylation sequence) or as an RNA molecule. The chimeric version of the guide RNA is only approximately 100 nucleotides in size, but it needs to be delivered in parallel with Cas9, either as a separate RNA molecule or via a DNA cassette with a separate promoter (typically the U6 promoter) with a size of approximately 500 bp. A lentivirus can just accommodate the S. pyogenes CRISPR-Cas9 system; AAV, with its cargo size limited to less than 5 kb, cannot accommodate it. Here again, the emerging availability of CRISPR-Cas systems from other species may prove helpful. For example, the cDNA encoding N. meningitidis Cas9 is approximately 3.2 kb in size and so should allow for delivery via AAV, which may be important for therapeutic applications.
Perhaps the biggest concern regarding CRISPR-Cas9 is the issue of off-target effects. It has recently been demonstrated that, although each nucleotide within the 20 nucleotide protospacer contributes to overall S. pyogenes Cas9 binding and specificity, single mismatches are often well tolerated, and multiple mismatches can sometimes be tolerated depending on their locations in the protospacer (55–58). Systematic analysis of the effect of alterations in the protospacer reveals an increasing tolerance for mismatches with increasing distance from the PAM. A number of studies in mammalian cells have documented off-target mutations occurring at significant rates at sites with sequence similarity to the on-target sites, occasionally rivaling or even surpassing mutagenesis at the on-target sites (55–60). These off-target effects, however, are guide RNA specific, and many guide RNAs have been reported for which no off-target activity is detectable (55–60). It has been posited that alternative CRISPR-Cas systems such as that from N. meningitidis may offer better targeting specificity by virtue of their longer protospacers (24 nucleotides for N. meningitidis) and longer PAMs. Experimental confirmation of improved specificity in mammalian cells remains to be shown. Early results with the N. meningitidis CRISPR-Cas9 system suggest that it may be less tolerant of mismatches in the protospacer compared with the S. pyogenes system (53).
Efforts to improve the specificity of CRISPR-Cas9 in mammalian cells are in progress. One strategy has been to use a mutant version of Cas9 that can only introduce a single-strand nick into the target DNA, rather than a DSB. Use of a pair of “nickase” CRISPR-Cas9 complexes with binding sites on opposite strands flanking the target site can produce the equivalent of a DSB with 5′ overhangs (Figure 3B), which is then repaired by NHEJ or HDR and can result in an on-target alteration. At an off-target site, a single-strand nick would be fixed by a different mechanism (base excision repair pathway) that is much less likely to result in a mutation. Because the likelihood of two nickases binding near each other elsewhere in the genome is very low, the off-target mutation rate should be dramatically reduced. Indeed, testing of this strategy in mammalian cells has demonstrated a reduction in off-target activity by up to three orders of magnitude with at most a modest reduction in on-target efficacy (57, 60, 61). Another strategy to reduce off-target effects is to reduce the length of the protospacer portion of the guide RNA, which makes it less tolerant of mismatches and thus can preserve the on-target efficacy while reducing off-target mutagenesis (62). A third successful strategy is to use a pair of proteins, each comprising a nuclease-dead Cas9 (that cannot cut DNA) fused to a FokI domain (Figure 3C); each Cas9 is targeted to either of two flanking sequences by a guide RNA, positioning the FokI domains to be able to dimerize and generate a DSB (63, 64). Although very large, these fusion proteins combine the most desirable properties of CRISPR-Cas9 and ZFNs/TALENs.
Genome editing in mammalian models
Although the creation of mouse lines with genetic alterations such as gene knockouts or conditional alleles has long been feasible with traditional homologous recombination employed in mouse embryonic stem cells, the last few years have seen the application of novel genome-editing tools for the generation of genetically modified mice with unprecedented ease and efficiency. Furthermore, these tools have made it possible to genetically modify animals for which embryonic stem cell lines are not widely available.
Initial studies of the efficacy of genome-editing tools in the mutagenesis of mammalian embryos were performed with rats. Inspired by studies in which injection of RNAs encoding ZFNs directly into the embryos of fruit flies and zebrafish yielded stable, heritable genomic alterations, injection of ZFN-encoding RNAs into one-cell rat embryos successfully generated monoallelic and biallelic frameshift mutations, resulting in gene knockout (65, 66). Numerous knockout rats have since been generated using this ZFN strategy. Subsequently, both TALENs and CRISPR-Cas9 have been used in similar fashion to generate knockout rats (67–69).
A particular advantage is that it is possible to obtain knockout animals in the first generation (assuming the targeted gene is not embryonic lethal), dramatically speeding up the time needed to do genetic studies in animals. Another advantage of this approach is that embryos from any of a variety of animal strains can be used; in the case of mice, there is no longer a restriction to a limited number of embryonic stem cell lines that necessitate backcrossing to an inbred strain of choice. Embryos from that inbred strain can be used to directly generate the knockout mice. Similarly, embryos from a strain that already carries genetic alterations can be used, relieving the need for many generations of interbreeding to obtain mice with multiple genetic alterations. The ability to perform multiplex gene targeting with CRISPR-Cas9 is also helpful in this regard.
All three engineered nucleases outlined above have proven effective at producing targeted mutations in mouse embryos (70–78). The efficiencies vary widely depending on the nuclease, target site in the genome, and amount of RNA injected. The most striking demonstration of efficiency has been with CRISPR-Cas9, with simultaneous targeting of both alleles of two genes in 80% of mice (76). CRISPR-Cas9 has also been used along with ssODNs or double-stranded DNA donor vectors in mouse embryos to knock in tags and fluorescent markers into endogenous gene loci and, most impressively, to generate conditional knockout mice in one step by simultaneously knocking in two loxP sites flanking an exon of a gene (77).
Finally, the high efficiencies of the genome-editing tools, particularly CRISPR-Cas9, have made it possible to generate targeted mutations in animals far beyond the reach of the traditional homologous recombination/embryonic stem cell approach. CRISPR-Cas9 technology has successfully generated modified organisms across the biologic spectrum, from sweet oranges to tilapia (79, 80). Both TALENs and CRISPR-Cas9 have now been used to generate genetically modified monkeys (81, 82), in each case targeting genes involved in human diseases. This is a remarkable accomplishment that suggests that there is no technical barrier to using genome-editing tools to modify human embryos, notwithstanding the profound social and ethical repercussions that would result if such attempts were to be made. On more secure ground will be attempts to use CRISPR-Cas9 for in vivo genome editing in adults to treat diseases. In an early proof of principle, a CRISPR-Cas9 plasmid and an ssODN were delivered into mouse liver via hydrodynamic injection, resulting in the correction of a patient-specific mutation in the Fah gene in a small proportion of hepatocytes (83). This resulted in the survival of mice that otherwise would have succumbed to liver failure from a disease analogous to type I tyrosinemia in humans.
Genome editing in human cells
To date, there have been a number of reports demonstrating the feasibility of performing genome editing in human pluripotent stem cells with ZFNs, TALENs, and CRISPRs (19, 34, 45, 61, 84–88). Genetically altered pluripotent stem cells offer the possibility of differentiating WT and mutant cell lines into whatever somatic cell type is desired, potentially giving new insights into disease pathophysiology. In one such study, the investigators generated induced pluripotent stem cells (iPSCs) from patients with Parkinson disease caused by the G2019S mutation of the LRRK2 gene as well as from control individuals (88). Upon differentiation into midbrain dopaminergic neurons, the cell lines displayed striking differences in whole-genome gene expression patterns, with clustering analysis showing that in some cases a patient line and a control line were more closely matched than lines generated from two different patients. Indeed, even iPSC lines generated from the same patient failed to cluster together, demonstrating the high degree of heterogeneity among iPSC lines. As an alternative approach, the investigators used ZFNs to correct the G2019S mutation in three of the patient-derived iPSC lines and to insert the mutation into a control iPSC line. They found that the matched sets of WT/mutant cell lines clustered together very closely, confirming the superiority of the genome-editing strategy for disease-modeling studies. The investigators consistently found that mutant neurons displayed less neurite outgrowth and more apoptosis in response to oxidative stress than matched WT neurons.
Other human cell types have proven to be quite amenable to genome editing. A therapeutic application that is currently in clinical trials is the use of ZFNs to disrupt the CCR5 gene in human T cells, thus rendering them impervious to HIV entry (89, 90). A similar intervention has been performed in human CD34+ hematopoietic stem/progenitor cells and may also be evaluated in clinical trials (91). In separate work, ZFNs have been used to insert a normal IL2RG gene into CD34+ hematopoietic stem/progenitor cells from a subject with X-linked severe combined immunodeficiency (92).
In another study, the investigators isolated intestinal stem cells from cystic fibrosis patients homozygous for the common Δ508 mutation in the CFTR gene (93). They used CRISPR-Cas9 targeting the site of the mutation, along with a double-stranded DNA donor vector, to correct one mutant allele (sufficient to “cure” the disease in this recessive disorder). They then used the mutant and corrected stem cells to create intestinal organoids in culture. Whereas the mutant organoids failed to respond to forskolin treatment by swelling, consistent with a lack of functional CFTR protein, the corrected organoids did respond by swelling, demonstrating a functional rescue.
The remarkable efficiency and ease of use of CRISPR-Cas9, where only 20 nucleotides in the guide RNA need be changed to retarget the nuclease, have led to the development of genome-wide CRISPR knockout libraries with the potential to knock out each of the genes in the genome. Several groups have performed proof-of-principle, genome-wide knockout screens in both human cells (48, 49, 51) and mouse cells (50). The results of the screens compared favorably with traditional genome-wide RNA interference screens, establishing a powerful new complementary approach to RNA interference to probe gene function in an unbiased fashion. Furthermore, CRISPR knockout libraries can potentially be used to target regions of interest in the noncoding genome (e.g., promoters, enhancers), enabling screens not possible with RNA interference.
Conclusion
The rapid development and improvement of genome-editing tools provide investigators with three well-characterized options for experiments as diverse as forward genetic screens to correction of pathogenic mutations in iPSC-derived human cells. ZFNs, TALENs, and CRISPRs can all generate site-specific DSBs with varying degrees of specificity and efficiency. The early uses of these systems have demonstrated remarkable new possibilities and allowed for the creation of model systems in a wide variety of organisms. With each iteration, the technology has improved, and the prospects for the study and treatment of human disease with genome editing have never been better.
Acknowledgments
This work was supported in part by a LaDue Fellowship in Cardiology (to R.M. Gupta) and a Sarnoff Scholar Award (to R.M. Gupta). Given the explosive nature of the genome-editing field in recent years, especially in the past year, we regret that due to space limitations we were unable to cite and describe many worthy studies, and we apologize for their omission.
Footnotes
Conflict of interest: The authors have declared that no conflict of interest exists.
Reference information:J Clin Invest. 2014;124(10):4154–4161. doi:10.1172/JCI72992.
References
- 1.Smithies O, Gregg RG, Boggs SS, Koralewski MA, Kucherlapati RS. Insertion of DNA sequences into the human chromosomal β-globin locus by homologous recombination. Nature. 1985;317(6034):230–234. doi: 10.1038/317230a0. [DOI] [PubMed] [Google Scholar]
- 2.Thomas KR, Capecchi MR. Site-directed mutagenesis by gene targeting in mouse embryo-derived stem cells. Cell. 1987;51(3):503–512. doi: 10.1016/0092-8674(87)90646-5. [DOI] [PubMed] [Google Scholar]
- 3.Qiu S, Adema CM, Lane T. A computational study of off-target effects of RNA interference. Nucleic Acids Res. 2005;33(6):1834–1847. doi: 10.1093/nar/gki324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Wang L, et al. CARM1 methylates chromatin remodeling factor BAF155 to enhance tumor progression and metastasis. Cancer Cell. 2014;25(1):21–36. doi: 10.1016/j.ccr.2013.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Urnov FD, Rebar EJ, Holmes MC, Zhang HS, Gregory PD. Genome editing with engineered zinc finger nucleases. Nat Rev Genet. 2010;11(9):636–646. doi: 10.1038/nrg2842. [DOI] [PubMed] [Google Scholar]
- 6.Rouet P, Smih F, Jasin M. Expression of a site-specific endonuclease stimulates homologous recombination in mammalian cells. Proc Natl Acad Sci U S A. 1994;91(13):6064–6068. doi: 10.1073/pnas.91.13.6064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Radecke S, Radecke F, Cathomen T, Schwarz K. Zinc-finger nuclease-induced gene repair with oligodeoxynucleotides: wanted and unwanted target locus modifications. Mol Ther. 2010;18(4):743–753. doi: 10.1038/mt.2009.304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Soldner F, et al. Generation of isogenic pluripotent stem cells differing exclusively at two early onset Parkinson point mutations. Cell. 2011;146(2):318–331. doi: 10.1016/j.cell.2011.06.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chen F, et al. High-frequency genome editing using ssDNA oligonucleotides with zinc-finger nucleases. Nat Methods. 2011;8(9):753–755. doi: 10.1038/nmeth.1653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Urnov FD, et al. Highly efficient endogenous human gene correction using designed zinc-finger nucleases. Nature. 2005;435(7042):646–651. doi: 10.1038/nature03556. [DOI] [PubMed] [Google Scholar]
- 11.Ramirez CL, et al. Unexpected failure rates for modular assembly of engineered zinc fingers. Nat Methods. 2008;5(5):374–375. doi: 10.1038/nmeth0508-374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Maeder ML, et al. Rapid “open-source” engineering of customized zinc-finger nucleases for highly efficient gene modification. Mol Cell. 2008;31(2):294–301. doi: 10.1016/j.molcel.2008.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Maeder ML, Thibodeau-Beganny S, Sander JD, Voytas DF, Joung JK. Oligomerized pool engineering (OPEN): an ‘open-source’ protocol for making customized zinc-finger arrays. Nat Protoc. 2009;4(10):1471–1501. doi: 10.1038/nprot.2009.98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sander JD, et al. Selection-free zinc-finger-nuclease engineering by context-dependent assembly (CoDA). Nat Methods. 2011;8(1):67–69. doi: 10.1038/nmeth.1542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gupta A, Christensen RG, Rayla AL, Lakshmanan A, Stormo GD, Wolfe SA. An optimized two-finger archive for ZFN-mediated gene targeting. Nat Methods. 2012;9(6):588–590. doi: 10.1038/nmeth.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bhakta MS, et al. Highly active zinc-finger nucleases by extended modular assembly. Genome Res. 2013;23(3):530–538. doi: 10.1101/gr.143693.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gaj T, Guo J, Kato Y, Sirk SJ, Barbas CF. Targeted gene knockout by direct delivery of zinc-finger nuclease proteins. Nat Methods. 2012;9(8):805–807. doi: 10.1038/nmeth.2030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kim HJ, Lee HJ, Kim H, Cho SW, Kim JS. Targeted genome editing in human cells with zinc finger nucleases constructed via modular assembly. Genome Res. 2009;19(7):1279–1288. doi: 10.1101/gr.089417.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hockemeyer D, et al. Efficient targeting of expressed and silent genes in human ESCs and iPSCs using zinc-finger nucleases. Nat Biotechnol. 2009;27(9):851–857. doi: 10.1038/nbt.1562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gabriel R, et al. An unbiased genome-wide analysis of zinc-finger nuclease specificity. Nat Biotechnol. 2011;29(9):816–823. doi: 10.1038/nbt.1948. [DOI] [PubMed] [Google Scholar]
- 21.Pattanayak V, Ramirez CL, Joung JK, Liu DR. Revealing off-target cleavage specificities of zinc-finger nucleases by in vitro selection. Nat Methods. 2011;8(9):765–770. doi: 10.1038/nmeth.1670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Doyon Y, et al. Enhancing zinc-finger-nuclease activity with improved obligate heterodimeric architectures. Nat Methods. 2011;8(1):74–79. doi: 10.1038/nmeth.1539. [DOI] [PubMed] [Google Scholar]
- 23.Szczepek M, Brondani V, Buchel J, Serrano L, Segal DJ, Cathomen T. Structure-based redesign of the dimerization interface reduces the toxicity of zinc-finger nucleases. Nat Biotechnol. 2007;25(7):786–793. doi: 10.1038/nbt1317. [DOI] [PubMed] [Google Scholar]
- 24.Miller JC, et al. An improved zinc-finger nuclease architecture for highly specific genome editing. Nat Biotechnol. 2007;25(7):778–785. doi: 10.1038/nbt1319. [DOI] [PubMed] [Google Scholar]
- 25.Bogdanove AJ, Voytas DF. TAL effectors: customizable proteins for DNA targeting. Science. 2011;333(6051):1843–1846. doi: 10.1126/science.1204094. [DOI] [PubMed] [Google Scholar]
- 26.Moscou MJ, Bogdanove AJ. A simple cipher governs DNA recognition by TAL effectors. Science. 2009;326(5959):1501. doi: 10.1126/science.1178817. [DOI] [PubMed] [Google Scholar]
- 27.Boch J, et al. Breaking the code of DNA binding specificity of TAL-type III effectors. Science. 2009;326(5959):1509–1512. doi: 10.1126/science.1178811. [DOI] [PubMed] [Google Scholar]
- 28.Morbitzer R, Romer P, Boch J, Lahaye T. Regulation of selected genome loci using de novo-engineered transcription activator-like effector (TALE)-type transcription factors. Proc Natl Acad Sci U S A. 2010;107(50):21617–21622. doi: 10.1073/pnas.1013133107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Streubel J, Blucher C, Landgraf A, Boch J. TAL effector RVD specificities and efficiencies. Nat Biotechnol. 2012;30(7):593–595. doi: 10.1038/nbt.2304. [DOI] [PubMed] [Google Scholar]
- 30.Cong L, Zhou R, Kuo YC, Cunniff M, Zhang F. Comprehensive interrogation of natural TALE DNA-binding modules and transcriptional repressor domains. Nat Commun. 2012;3:968. doi: 10.1038/ncomms1962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Christian M, et al. Targeting DNA double-strand breaks with TAL effector nucleases. Genetics. 2010;186(2):757–761. doi: 10.1534/genetics.110.120717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Miller JC, et al. A TALE nuclease architecture for efficient genome editing. Nat Biotechnol. 2011;29(2):143–148. doi: 10.1038/nbt.1755. [DOI] [PubMed] [Google Scholar]
- 33.Li T, et al. TAL nucleases (TALNs): hybrid proteins composed of TAL effectors and FokI DNA-cleavage domain. Nucleic Acids Res. 2011;39(1):359–372. doi: 10.1093/nar/gkq704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hockemeyer D, et al. Genetic engineering of human pluripotent cells using TALE nucleases. Nat Biotechnol. 2011;29(8):731–734. doi: 10.1038/nbt.1927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Reyon D, Tsai SQ, Khayter C, Foden JA, Sander JD, Joung JK. FLASH assembly of TALENs for high-throughput genome editing. Nat Biotechnol. 2012;30(5):460–465. doi: 10.1038/nbt.2170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cermak T, et al. Efficient design and assembly of custom TALEN and other TAL effector-based constructs for DNA targeting. Nucleic Acids Res. 2011;39(12):e82. doi: 10.1093/nar/gkr218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kim Y, et al. A library of TAL effector nucleases spanning the human genome. Nat Biotechnol. 2013;31(3):251–258. doi: 10.1038/nbt.2517. [DOI] [PubMed] [Google Scholar]
- 38.Guilinger JP, et al. Broad specificity profiling of TALENs results in engineered nucleases with improved DNA-cleavage specificity. Nat Methods. 2014;11(4):429–435. doi: 10.1038/nmeth.2845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mussolino C, Morbitzer R, Lutge F, Dannemann N, Lahaye T, Cathomen T. A novel TALE nuclease scaffold enables high genome editing activity in combination with low toxicity. Nucleic Acids Res. 2011;39(21):9283–9293. doi: 10.1093/nar/gkr597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Smith C, et al. Whole-genome sequencing analysis reveals high specificity of CRISPR/Cas9 and TALEN-based genome editing in human iPSCs. Cell Stem Cell. 2014;15(1):12–13. doi: 10.1016/j.stem.2014.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Suzuki K, et al. Targeted gene correction minimally impacts whole-genome mutational load in human-disease-specific induced pluripotent stem cell clones. Cell Stem Cell. 2014;15(1):31–36. doi: 10.1016/j.stem.2014.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Holkers M, et al. Differential integrity of TALE nuclease genes following adenoviral and lentiviral vector gene transfer into human cells. Nucleic Acids Res. 2013;41(5):e63. doi: 10.1093/nar/gks1446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yang L, et al. Optimization of scarless human stem cell genome editing. Nucleic Acids Res. 2013;41(19):9049–9061. doi: 10.1093/nar/gkt555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cong L, et al. Multiplex genome engineering using CRISPR/Cas systems. Science. 2013;339(6121):819–823. doi: 10.1126/science.1231143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mali P, et al. RNA-guided human genome engineering via Cas9. Science. 2013;339(6121):823–826. doi: 10.1126/science.1232033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Jinek M, Chylinski K, Fonfara I, Hauer M, Doudna JA, Charpentier E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science. 2012;337(6096):816–821. doi: 10.1126/science.1225829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Cho SW, Kim S, Kim JM, Kim JS. Targeted genome engineering in human cells with the Cas9 RNA-guided endonuclease. Nat Biotechnol. 2013;31(3):230–232. doi: 10.1038/nbt.2507. [DOI] [PubMed] [Google Scholar]
- 48.Wang T, Wei JJ, Sabatini DM, Lander ES. Genetic screens in human cells using the CRISPR-Cas9 system. Science. 2014;343(6166):80–84. doi: 10.1126/science.1246981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Shalem O, et al. Genome-scale CRISPR-Cas9 knockout screening in human cells. Science. 2014;343(6166):84–87. doi: 10.1126/science.1247005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Koike-Yusa H, Li Y, Tan EP, Velasco-Herrera Mdel C, Yusa K. Genome-wide recessive genetic screening in mammalian cells with a lentiviral CRISPR-guide RNA library. Nat Biotechnol. 2014;32(3):267–273. doi: 10.1038/nbt.2800. [DOI] [PubMed] [Google Scholar]
- 51.Zhou Y, et al. High-throughput screening of a CRISPR/Cas9 library for functional genomics in human cells. Nature. 2014;509(7501):487–491. doi: 10.1038/nature13166. [DOI] [PubMed] [Google Scholar]
- 52.Park CY, et al. Targeted inversion and reversion of the blood coagulation factor 8 gene in human iPS cells using TALENs. Proc Natl Acad Sci U S A. 2014;111(25):9253–9258. doi: 10.1073/pnas.1323941111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hou Z, et al. Efficient genome engineering in human pluripotent stem cells using Cas9 from Neisseria meningitidis. Proc Natl Acad Sci U S A. 2013;110(39):15644–15649. doi: 10.1073/pnas.1313587110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Esvelt KM, Mali P, Braff JL, Moosburner M, Yaung SJ, Church GM. Orthogonal Cas9 proteins for RNA-guided gene regulation and editing. Nat Methods. 2013;10(11):1116–1121. doi: 10.1038/nmeth.2681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Fu Y, et al. High-frequency off-target mutagenesis induced by CRISPR-Cas nucleases in human cells. Nat Biotechnol. 2013;31(9):822–826. doi: 10.1038/nbt.2623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hsu PD, et al. DNA targeting specificity of RNA-guided Cas9 nucleases. Nat Biotechnol. 2013;31(9):827–832. doi: 10.1038/nbt.2647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Mali P, et al. CAS9 transcriptional activators for target specificity screening and paired nickases for cooperative genome engineering. Nat Biotechnol. 2013;31(9):833–838. doi: 10.1038/nbt.2675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Pattanayak V, Lin S, Guilinger JP, Ma E, Doudna JA, Liu DR. High-throughput profiling of off-target DNA cleavage reveals RNA-programmed Cas9 nuclease specificity. Nat Biotechnol. 2013;31(9):839–843. doi: 10.1038/nbt.2673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Cradick TJ, Fine EJ, Antico CJ, Bao G. CRISPR/Cas9 systems targeting beta-globin and CCR5 genes have substantial off-target activity. Nucleic Acids Res. 2013;41(20):9584–9592. doi: 10.1093/nar/gkt714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Cho SW, et al. Analysis of off-target effects of CRISPR/Cas-derived RNA-guided endonucleases and nickases. Genome Res. 2014;24(1):132–141. doi: 10.1101/gr.162339.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ran FA, et al. Double nicking by RNA-guided CRISPR Cas9 for enhanced genome editing specificity. Cell. 2013;154(6):1380–1389. doi: 10.1016/j.cell.2013.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Fu Y, Sander JD, Reyon D, Cascio VM, Joung JK. Improving CRISPR-Cas nuclease specificity using truncated guide RNAs. Nat Biotechnol. 2014;32(3):279–284. doi: 10.1038/nbt.2808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Tsai SQ, et al. Dimeric CRISPR RNA-guided FokI nucleases for highly specific genome editing. Nat Biotechnol. 2014;32(6):569–576. doi: 10.1038/nbt.2908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Guilinger JP, Thompson DB, Liu DR. Fusion of catalytically inactive Cas9 to FokI nuclease improves the specificity of genome modification. Nat Biotechnol. 2014;32(6):577–582. doi: 10.1038/nbt.2909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Geurts AM, et al. Knockout rats via embryo microinjection of zinc-finger nucleases. Science. 2009;325(5939):433. doi: 10.1126/science.1172447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Mashimo T, et al. Generation of knockout rats with X-linked severe combined immunodeficiency (X-SCID) using zinc-finger nucleases. PLoS One. 2010;5(1):e8870. doi: 10.1371/journal.pone.0008870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Tesson L, et al. Knockout rats generated by embryo microinjection of TALENs. Nat Biotechnol. 2011;29(8):695–696. doi: 10.1038/nbt.1940. [DOI] [PubMed] [Google Scholar]
- 68.Li D, et al. Heritable gene targeting in the mouse and rat using a CRISPR-Cas system. Nat Biotechnol. 2013;31(8):681–683. doi: 10.1038/nbt.2661. [DOI] [PubMed] [Google Scholar]
- 69.Li W, Teng F, Li T, Zhou Q. Simultaneous generation and germline transmission of multiple gene mutations in rat using CRISPR-Cas systems. Nat Biotechnol. 2013;31(8):684–686. doi: 10.1038/nbt.2652. [DOI] [PubMed] [Google Scholar]
- 70.Carbery ID, et al. Targeted genome modification in mice using zinc-finger nucleases. Genetics. 2010;186(2):451–459. doi: 10.1534/genetics.110.117002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Cui X, Ji D, Fisher DA, Wu Y, Briner DM, Weinstein EJ. Targeted integration in rat and mouse embryos with zinc-finger nucleases. Nat Biotechnol. 2011;29(1):64–67. doi: 10.1038/nbt.1731. [DOI] [PubMed] [Google Scholar]
- 72.Meyer M, de Angelis MH, Wurst W, Kuhn R. Gene targeting by homologous recombination in mouse zygotes mediated by zinc-finger nucleases. Proc Natl Acad Sci U S A. 2010;107(34):15022–15026. doi: 10.1073/pnas.1009424107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Sung YH, et al. Knockout mice created by TALEN-mediated gene targeting. Nat Biotechnol. 2013;31(1):23–24. doi: 10.1038/nbt.2477. [DOI] [PubMed] [Google Scholar]
- 74.Wefers B, et al. Direct production of mouse disease models by embryo microinjection of TALENs and oligodeoxynucleotides. Proc Natl Acad Sci U S A. 2013;110(10):3782–3787. doi: 10.1073/pnas.1218721110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Shen B, et al. Generation of gene-modified mice via Cas9/RNA-mediated gene targeting. Cell Res. 2013;23(5):720–723. doi: 10.1038/cr.2013.46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Wang H, et al. One-step generation of mice carrying mutations in multiple genes by CRISPR/Cas-mediated genome engineering. Cell. 2013;153(4):910–918. doi: 10.1016/j.cell.2013.04.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Yang H, Wang H, Shivalila CS, Cheng AW, Shi L, Jaenisch R. One-step generation of mice carrying reporter and conditional alleles by CRISPR/Cas-mediated genome engineering. Cell. 2013;154(6):1370–1379. doi: 10.1016/j.cell.2013.08.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Wu Y, et al. Correction of a genetic disease in mouse via use of CRISPR-Cas9. Cell Stem Cell. 2013;13(6):659–662. doi: 10.1016/j.stem.2013.10.016. [DOI] [PubMed] [Google Scholar]
- 79.Jia H, Wang N. Targeted genome editing of sweet orange using Cas9/sgRNA. PLoS One. 2014;9(4):e93806. doi: 10.1371/journal.pone.0093806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Li M, et al. Efficient and heritable gene targeting in tilapia by CRISPR/Cas9. Genetics. 2014;197(2):591–599. doi: 10.1534/genetics.114.163667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Niu Y, et al. Generation of gene-modified cynomolgus monkey via Cas9/RNA-mediated gene targeting in one-cell embryos. Cell. 2014;156(4):836–843. doi: 10.1016/j.cell.2014.01.027. [DOI] [PubMed] [Google Scholar]
- 82.Liu H, et al. TALEN-mediated gene mutagenesis in rhesus and cynomolgus monkeys. Cell Stem Cell. 2014;14(3):323–328. doi: 10.1016/j.stem.2014.01.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Yin H, et al. Genome editing with Cas9 in adult mice corrects a disease mutation and phenotype. Nat Biotechnol. 2014;32(6):551–553. doi: 10.1038/nbt.2884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Lombardo A, et al. Gene editing in human stem cells using zinc finger nucleases and integrase-defective lentiviral vector delivery. Nat Biotechnol. 2007;25(11):1298–1306. doi: 10.1038/nbt1353. [DOI] [PubMed] [Google Scholar]
- 85.Zou J, et al. Gene targeting of a disease-related gene in human induced pluripotent stem and embryonic stem cells. Cell Stem Cell. 2009;5(1):97–110. doi: 10.1016/j.stem.2009.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Yusa K, Rad R, Takeda J, Bradley A. Generation of transgene-free induced pluripotent mouse stem cells by the piggyBac transposon. Nat Methods. 2009;6(5):363–369. doi: 10.1038/nmeth.1323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Sebastiano V, et al. In situ genetic correction of the sickle cell anemia mutation in human induced pluripotent stem cells using engineered zinc finger nucleases. Stem Cells. 2011;29(11):1717–1726. doi: 10.1002/stem.718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Reinhardt P, et al. Genetic correction of a LRRK2 mutation in human iPSCs links parkinsonian neurodegeneration to ERK-dependent changes in gene expression. Cell Stem Cell. 2013;12(3):354–367. doi: 10.1016/j.stem.2013.01.008. [DOI] [PubMed] [Google Scholar]
- 89.Perez EE, et al. Establishment of HIV-1 resistance in CD4+ T cells by genome editing using zinc-finger nucleases. Nat Biotechnol. 2008;26(7):808–816. doi: 10.1038/nbt1410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Tebas P, et al. Gene editing of CCR5 in autologous CD4 T cells of persons infected with HIV. N Engl J Med. 2014;370(10):901–910. doi: 10.1056/NEJMoa1300662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Holt N, et al. Human hematopoietic stem/progenitor cells modified by zinc-finger nucleases targeted to CCR5 control HIV-1 in vivo. Nat Biotechnol. 2010;28(8):839–847. doi: 10.1038/nbt.1663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Genovese P, et al. Targeted genome editing in human repopulating haematopoietic stem cells. Nature. 2014;510(7504):235–240. doi: 10.1038/nature13420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Schwank G, et al. Functional repair of CFTR by CRISPR/Cas9 in intestinal stem cell organoids of cystic fibrosis patients. Cell Stem Cell. 2013;13(6):653–658. doi: 10.1016/j.stem.2013.11.002. [DOI] [PubMed] [Google Scholar]