

Abstract
The encapsulation of living mammalian cells within a semi-permeable hydrogel matrix is an attractive procedure for many biomedical and biotechnological applications, such as xenotransplantation, maintenance of stem cell phenotype and bioprinting of three-dimensional scaffolds for tissue engineering and regenerative medicine. In this review, we focus on naturally derived polymers that can form hydrogels under mild conditions and that are thus capable of entrapping cells within controlled volumes. Our emphasis will be on polysaccharides and proteins, including agarose, alginate, carrageenan, chitosan, gellan gum, hyaluronic acid, collagen, elastin, gelatin, fibrin and silk fibroin. We also discuss the technologies commonly employed to encapsulate cells in these hydrogels, with particular attention on microencapsulation.
Keywords: cell encapsulation, natural polymers, microencapsulation
1. Introduction
Cell encapsulation technologies aim at entrapping viable and functional cells within a semi-permeable matrix. A suitable matrix must be biocompatible, it must support cell survival and therefore it must be permeable to oxygen, to the incoming nutrients and to the outgoing toxic metabolites. Other properties may depend on the specific application. For example, encapsulation techniques have been investigated for xenotransplantation to treat endocrine diseases, such as diabetes [1], anaemia [2] and restricted growth [3]. The transplanted cells must be shielded from the host immune system to avoid the use of immunosuppressant drugs that lead to undesirable side effects [4,5]. Therefore, the matrix should have a fine-tuned porosity to block antibodies and T-cells while permitting the passage of the incoming signalling molecule and the outgoing response. Furthermore, it should elicit a minimal host reaction and have a low degradation kinetics to prolong the protection of cells. For applications other than immunoprotection, the properties of the matrix may be very different. For applications directed towards the repair or regeneration of tissue, biodegradability of the matrix could be desirable. When the matrix degrades, the entrapped cells may proliferate and create their own extracellular matrix in place of the artificial one used to entrap them. Given the different applications of this technology, many biomaterials have been proposed as entrapping matrix to fulfil the specific requirements. Suitable materials for cell encapsulation should mimic the extracellular matrix and should be processed under conditions compatible with the presence of cells. Many of these materials are naturally derived polymers that form hydrogels. Hydrogels are highly hydrated materials composed of hydrophilic polymers that are cross-linked to form three-dimensional networks. They are soft, highly porous structures that once implanted induce a low protein adsorption because of the low interfacial tension with the surrounding fluids [6]. Hydrogels derived from natural materials have a similar structure to the extracellular matrix of many human tissues. They are made of polymers similar to the biological macromolecules engineered by nature to perform specific functions in a demanding environment. Some of them are abundant (e.g. marine sources [7]) and furthermore they can often be processed under mild conditions compatible with cell survival [8].
Once processed and formed, the hydrogel will exhibit different transport properties depending on its structure, chemical composition and the degree of cross-linking. This aspect is crucial in determining the ability of cells to obtain nutrients and remove waste that directly influences cell survival or death. In fact, the hydrogel matrix can act as a mechanical and/or chemical barrier towards incoming and outgoing molecules. When the barrier acts as a mechanical obstacle, there is an upper size limit of the solute that can pass through the matrix. The corresponding molecular weight cut-off can be quantified by assessing the permeation of a tracer molecule (e.g. a fluorescent dye using confocal microscopy) into the matrix or by assessing outside the matrix the presence of molecules produced by cells. When the barrier is chemical, the passage of molecules smaller than the molecular weight cut-off may be hindered or inhibited. Oxygen, signalling molecules, cell nutrients and metabolites should be able to diffuse through the matrix without being blocked. Furthermore, cell survival inside the matrix depends on permeability for nutrient supply and removal of metabolites, generating different gradients of concentration along the distance from the border. Cells that are far from the border of the matrix will receive a lower amount of nutrients in a given time. This means that a low mass transfer rate of nutrients may be enough for cells next to the external border but may not be for the inner cells. Also, if high cell densities of entrapped cells are needed, the mass transfer rate of nutrients should also be higher to sustain them. These observations suggest that microbeads with high surface area to volume ratio may perform better in providing the right conditions to support viable cells [9]. Typically, the upper size limit for these beads is considered to be 400 µm in diameter, two times the maximum diffusion distance of oxygen and nutrients from blood vessels to cells [1]. Microbeads, in fact, guarantee a higher exchange rate of substances between cells and the surrounding environment than standard size beads [10]. The smaller size also contributes to the mechanical properties of the beads—they are less prone to breakage and, as such, elicit a milder pericapsular reaction once implanted [11]. It has been reported that smaller beads can lead to more inadequately encapsulated cells that protrude from the beads [12]. However there is also strong evidence that microsize beads may be more biocompatible than standard size beads [13,14]. For these reason many groups have developed different microencapsulation techniques that will be the major focus of this review. Some of the properties of the microcapsules in relation to the encapsulation of cells are outlined in figure 1.
Figure 1.

A scheme outlining the properties of the microcapsules for the encapsulation of cells.
In the first part of this review, we discuss some selected naturally derived polymers that can be processed under mild conditions and that show potential for cell encapsulation (table 1). The gel phase can be formed by the reversible effect of external stimuli (temperature, ionic strength) or by permanent cross-linking [40]. We discuss their structure, their biodegradability [41,42] and their sol–gel mechanism. In the second part of this review, we describe some of the methods for cell encapsulation that allow the fabrication of cell-laden beads and capsules in the micrometric range.
Table 1.
Summary of some properties of the materials discussed in this review with references on their use for cell encapsulation. ECM, extracellular matrix.
| name | source | type | gelation | biodegradability | references |
|---|---|---|---|---|---|
| agarose | seaweed | polysaccharide | thermal | no | [15–17] |
| carrageenan | seaweed | polysaccharide | thermal/ionotropic | no | [18,19] |
| alginate | seaweed | polysaccharide | ionotropic | no | [1,20,21] |
| chitosan | crustaceans | polysaccharide | ionotropic | yes | [22,23] |
| gellan gum | seaweed | polysaccharide | thermal/ionotropic | not clear | [24,25] |
| hyaluronic acid | ECM | polysaccharide | thermal/photo upon chemical modification | yes | [26–28] |
| collagen | ECM | protein | thermal/pH variation | yes | [29,30] |
| gelatin | ECM/collagen | protein | thermal | yes | [31,32] |
| elastin | ECM/synthetic | protein | thermal | yes | [33–35] |
| fibrin | blood | protein | enzymatic | yes | [36,37] |
| silk fibroin | silk | protein | thermal/pH variation | dependsa | [38,39] |
aDepends on the processing technique, e.g. β-sheet content.
2. Gelling mechanism
The entrapment of cells in a hydrogel usually starts by suspending cells in a water-based solution of a hydrogel precursor, the sol flowing phase. The suspension then undergoes a transition to the gel non-flowing phase by physical, chemical or biochemical processes (generally referred to as ‘hardening’ in this review). Every passage of this process must be compatible with cell survival and should induce a minimal stress to cells. This means that, during the entire process, the environmental conditions should be as close as possible to the physiological conditions. Sometimes, however, it is not possible to comply with these requirements (e.g. photo-cross-linkable polymers). In these cases, it is important to note that the toxicity of a particular treatment is related not only to its nature and intensity, but also to the time of exposure [43]. In this review, we mainly discuss polymers that undergo a sol–gel transition in the presence of ions, when irradiated by light or when the temperature changes. See figure 2 for a general scheme of these methodologies.
Figure 2.

A simplified scheme of the three common gelling mechanisms. (a) Thermal gelation: due to a change in temperature the polymer molecules rearrange from random coil to helix, then the helices assemble in clusters joined together by the untwined regions. (b) Ionic cross-linking: the sections of the polymer backbone carrying the charge bind with ions (circles) of opposite charge. (c) Chemical cross-linking (photo-cross-linking): the photoinitiator molecules (green particles) in solution form radicals when irradiated by UV and cross-link the polymer chains.
Thermoresponsive polymers for cell encapsulation are polymers that form gels by a change in temperature. Some polymers form gel when heated above the transition temperature (e.g. elastin and to some extent collagen), while others form gels when cooled (e.g. gelatin, agarose). Polymers that form gel when cooled present an upper critical solution temperature (UCST) above which the water and the polymer are miscible. By decreasing the temperature below the UCST, the polymer becomes more hydrophobic and insoluble forming a gel. In contrast, polymers that form gels upon heating present a lower critical solution temperature (LCST) below which they are miscible with water. Most of natural thermoresponsive polymers, in contrast with the synthetic ones, exhibit a UCST and form gels by cooling warm water-based solutions [44,45]. When a polymer is dissolved in water, three types of interaction are possible [46]: polymer–polymer, polymer–water and water–water. The change in temperature makes the polymer–water interaction unfavourable, causing, for example, the transition from random coil to helix to minimize the exposure of the macromolecule to water. The transition of polymers that present an LCST is driven by a change in the entropy of water (hydrophobic effect), while for polymers presenting a UCST the transition is driven by a change in enthalpy [47]. Thermoresponsive polymers that are suitable for cell encapsulation should have a sol–gel transition temperature around physiological temperature.
Some polymers instead form gels in the presence of electrically charged species. These polymers are polyelectrolytes and carry a net charge along their backbone. When polyelectrolytes are combined with multi-valent cations of opposite sign they cross-link forming insoluble complexes. The complexes are insoluble because the charged groups, responsible for the solubility of the polymer in water, are mutually shielded [48]. Commonly used natural polymers are negatively charged (alginate, hyaluronic acid, carrageenan), mainly because of the presence of carboxyl or sulfate groups along the chain. The influence of the charge type and density on cellular response is not fully understood. However, many negatively charged biomaterials do not induce a strong inflammatory response in contrast to positively charged polymers that tend to attract inflammatory cells [49].
Ionically and thermally cross-linked hydrogels are physical gels formed by ionic or secondary forces. These gels can be dissolved under appropriate conditions such as a reverse temperature change or when in contact with chelating agents to remove the cross-linking ions. Other gels are formed by chemically irreversible cross-linking. Hydrogels formed by light irradiation are an important family of chemically cross-linked hydrogels for cell encapsulation. The photo-cross-linking process involves the presence of photoinitiator compounds whose chemical nature determines the reaction rate and the wavelength of absorption [50]. When irradiated by light, typically in the UV range, the photoinitiators form free radicals that react with functional groups of the polymer backbone forming intermolecular bonds. Biopolymers must be chemically modified to be photo-cross-linkable, typically by introducing functional groups such as acrylates. Another family of chemically cross-linked hydrogels that has recently received a lot of attention is enzymatically cross-linked hydrogels [51]. These gels are inspired by the natural cross-linking reactions occurring in our body such as the formation of blood clots by the enzyme transglutaminase. The enzymatic reaction is catalysed at neutral pH and physiological temperature. Different enzymes can lead to the formation of gels with different properties [52] and the reaction can be controlled by controlling the activity of the enzyme [53].
3. Materials
3.1. Carbohydrates
3.1.1. Agarose
Agarose is a polysaccharide derived from the cell wall of a group of red algae (Rhodophyceae), including Gelidium and Gracilaria [54]. The plant is harvested and agarose is extracted after a series of purification and homogenization steps [55]. The main structure of agarose consists of alternating units of β-d-galactopyranose and 3,6-anhydro-α-l-galactopyranose. Agarose extracted from different sources can have different chemical compositions; for example, sulfates can be found instead of the hydroxyl groups with a variable degree of substitution. Agarose is a responsive polymer and its aqueous solutions undergo a sol–gel transition upon cooling. Above the sol–gel temperature, agarose exhibits a random-coil conformation in solution, and upon cooling the structure changes to a double helix. Some of the helices then aggregate and the hydrogen bonds between structural water and galactose stabilize the structure (figure 2a) [56]. The gelling temperature depends on the concentration of the solution, the average molecular weight of the polymer and its structure. For this reason, there is a wide range of commercially available agarose, characterized by different gel strengths and sol–gel transition temperatures. Some of them can be used for cell encapsulation since their sol–gel transition occurs at around 37°C. The thermal sol–gel transition of agarose is reversible and presents a marked thermal hysteresis, which is a wide temperature difference between gelling and liquefaction [57].
The average pore size of agarose hydrogels and, as a consequence, the mass transport properties are influenced by the concentration of the polymer in solution and the settling temperature. An increase in concentration results in tightly packed helices that translate to a decrease in pore size [58]. For a Bio-Rad Certified low-melt agarose, Narayanan et al. [59] measured an average pore size of 600 nm for a concentration of 1% w/v decreasing to 100 nm or less when the concentration was 3%. A decrease in settling temperature results in gel with smaller pores and higher elastic modulus (compression test). For example, Aymard et al. [60] showed a decrease in elastic modulus for a type I-A agarose (Sigma, 36°C gelling temperature) from 78 kPa for samples cured at 5°C to 53 kPa for samples cured at 35°C.
Agarose does not provide adhesion motifs to cells and does not allow interaction between adherent cells and the entrapping matrix [61]. However, it can be supplemented with adhesion molecules of the extracellular matrix, such as fibronectin [62] or RGD soluble peptide [63].
Agarose is not biodegradable—it can only be degraded by specific bacteria, not mammals. It can be degraded in vitro by agarases, which are classified according to their cleavage pattern into three types: α-agarase, β-agarase and β-porphyranase [64–66].
Agarose hydrogels have been extensively investigated for cartilage repair in vitro [67]. They support chondrocytes in culture for up to six weeks [68] and gels embedded into the cells can be placed in a bioreactor that mimics dynamic physiological loading shortly after encapsulation [69]. However, the poor biodegradability of agarose inhibits the spontaneous repair process in vivo probably as a result of the foreign body reaction to this material [70].
3.1.2. Carrageenan
Carrageenan is a water-soluble anionic polysaccharide derived from the Rhodophyceae red algae by alkali extraction. Carrageenan is a galactan, like agarose, and it consists of repeat sequences of β-d-galactose and α-d-galactose with variable proportions of sulfate groups. In carrageenan, the β-galactose is d while in agarose it is l. Commercially available carrageenan can be divided into three families based on the position and number of sulfate groups: κ-(kappa), ι-(iota) and λ-(lambda) carrageenan carrying 1, 2 and 3 sulfate groups, respectively [71]. Aqueous solutions of κ- and ι-carrageenan can reversibly form hydrogels in the presence of cations, while λ-carrageenan does not undergo a sol–gel transition. In fact, carrageenan in solution has a random-coil conformation and upon cooling κ- and ι-conformation becomes a double helix with the sulfate groups pointing outwards, while the higher sulfate content of λ-carrageenan inhibits the formation of the helicoidal structure [72]. The positive cations in solution neutralize the charge of the sulfate groups allowing a tighter aggregation of the helices [73]. Divalent cations are effective in promoting the formation of strong hydrogels for κ- and ι-carrageenan while monovalent ions are particularly effective on κ-carrageenan. Hydrogels prepared using ι-carrageenan are softer and more deformable than those prepared using κ-carrageenan [74]. Rochas et al. [75] measured a compression Young's modulus of 1 kPa for 1.2% w/v ι-carrageenan hardened overnight with a 0.25 M KCl solution, while Young's modulus of κ-carrageenan was 10 kPa. They also showed [76] that an increase in Young's modulus increases the molarity of the hardening bath and the concentration of the polymer in solution.
Despite the extensive and documented use of this polymer as an inducer of chronic and acute inflammation [77–79], there is no consensus on its effect on the inflammatory response of the host when newly developed purified carrageenan is used [80]. Also, both ι- and κ-carrageenan hydrogels have been used for cell encapsulation by ionic cross-linking [18] and by the formation of complexes with polycations such as chitosan [81].
Similar to alginate the degradation of carrageenan hydrogels is driven by the exchange of ions with the surrounding fluids, while only some bacteria produce the enzymes that can cleave the polymer chain [82].
3.1.3. Alginate
Alginate is a polysaccharide, a polyanionic linear block copolymer containing blocks of (1,4)-linked β-d-mannuroic (M block) and α-l-guluronic (G block) acids [83]. It is extracted from many different species of brown seaweed and is also produced by two kinds of bacteria, Pseudomonas and Azotobacter [84]. When derived from seaweed, the crop is cleaned and then alginate is extracted with a solution of a sodium salt followed by precipitation [85].
Alginate is a commonly used polymer for encapsulation of therapeutic agents [86] and ever since the first successful microencapsulation of pancreatic islets was reported by Lim & Sun [33], it has become the most studied material for encapsulation of living cells [1,87]. When multi-valent cations (e.g. Ca2+) are added to a water-based alginate solution, they bind adjacent alginate chains forming ionic interchain bridges (figure 2b) that cause a fast sol–gel transition compatible with the survival of the entrapped cells. It is generally assumed that cations bind preferably to the G blocks of the chains but relatively recent studies also suggest that the M block (in particular, the alternating MG block) has an active role in cross-linking the polymer chains [88]. In alginate, a naturally occurring biomaterial, the relative ratio between the G and M blocks is not constant and depends on the seaweed from which it is extracted. The G blocks provide rigidity to the polymeric structure and the mechanical properties of alginates are influenced by the ratio of G and M blocks, and as expected high G alginates result in the formation of stronger gels in compression [89] and tension tests [90]. Alginates can form polyelectrolyte complexes in the presence of polycations such as poly-l-lysine or chitosan. Poly-l-lysine has been widely used to coat the alginate beads as a way of controlling their molecular weight cut-off. A positively charged cation may be immunogenic and attract host inflammatory cells [49,91]. For this reason, another external alginate coating is often added to the beads to form the so-called ‘alginate–polylysine–alginate’ (APA) system. However, developments in the characterization of APA capsules [92] suggest that these capsules are not multi-layered; instead they consist of an inner calcium-alginate core covered by one single external layer of a poly-l-lysine and alginate blend. The binding strength of the initial poly-l-lysine layer depends on the relative ratio of the G and M blocks in the alginate core. Poly-l-lysine does not bind tightly to alginates with a high content of G blocks because, in contrast to M blocks, they do not allow complete interaction with the polycation. When these capsules are implanted or incubated they induce a stronger response than capsules without poly-l-lysine [93,94]. Alginates can also be combined with other biopolymers to improve the biological response of the host. Such studies were recently performed using high-throughput methodologies for the evaluation of the in vitro [95] and in vivo [96] response to different combinations of biomaterials. Furthermore, alginate does not provide cell adhesion motifs, but it can be conjugated with RGD peptides to improve cell adhesion [97].
Alginate is characterized by a wide pore size distribution (5–200 nm) with the most open structure found in alginates with high G content [98,99]. The permeability of alginate is strongly influenced by the concentration and nature of the hardening ions; higher concentrations of ions create tighter structures (especially in the outer part of the gel in direct contact with the hardening bath) and as a consequence decrease the diffusion rate of large molecules outside the gel [100,101]. Instead, when the hardening bath consists of salts with low solubility in water (e.g. CaCo3) the structure that is formed is more uniform and the hydrogel has higher mechanical stability [102]. Furthermore, it should be noted that, as most of the proteins are negatively charged at pH 7, they do not easily diffuse into the gel while they diffuse out more quickly than expected [98].
The kinetics of dissolution of the alginate gel in vivo depends on the G/M composition and it is driven by the exchange of cross-linking ions with monovalent cations of the surrounding fluids. Once unbounded, the polymer chain cannot be degraded by the biological activity of the host. Alginate can be degraded by alginase but this enzyme is not present in mammals. Alginate degradation, however, can be manipulated by chemical modifications of the chain. A slight oxidation of alginate using sodium peroxide leads to a polymer that degrades in aqueous medium without interfering significantly in its ability to form gels [103–105]. The degradation kinetics of alginate can also be tuned by controlling its molecular weight distribution by gamma irradiation [106]. The dissolution of the alginate gel in vitro can be obtained by the exchange of ions with a buffer (e.g. phosphate-buffered saline without calcium and magnesium) or by using chelating agents such as EDTA or sodium citrate. Being non-adhesive and having been characterized by mild dissolution methods, alginate hydrogels have recently been used to manufacture switchable hydrogels, in which alginate is mixed with cell adhesive matrices such as collagen. In these constructs, alginate acts as a self-renewal permissive substrate for human pluripotent stem cells and, when needed, it can be removed from the matrix permitting the differentiation of cells on the collagen substrate [107,108].
Alginate is by far the most studied material for cell encapsulation [87], and it has been adopted for many biomedical applications. Alginate has historically been used as a protective barrier to enhance cell therapies, for immunoprotection of pancreatic islets [1,33], treatment of brain tumours [109], treatment of anaemia [2] and cryopreservation [110].
3.1.4. Chitosan
Chitosan is a polysaccharide derived from chitin. Chitin is a naturally occurring polymer synthesized by many natural species as a structural component of their exoskeleton [111]. Chitin consists of N-acetyl-β-d-glucosamine chains arranged in semi-crystalline structures. Chitin is chemically similar to cellulose, another structural polysaccharide, with a hydroxyl position at C-2 being replaced by an acetylamide group [112]. Crabs and shrimp shells are the most common sources for commercially available chitin. To obtain chitin from the shells, an acid treatment is needed to dissolve the minerals followed by purification with an alkaline treatment to remove the proteins retained from the surrounding tissue [113,114]. The alkaline treatment can also remove some acetyl groups bound to the amine [115]. When enough acetyl groups are removed (50% deacetylation or more) the polymer is called chitosan. In acidic solutions, the primary amine is protonated and chitosan becomes a soluble, positively charged polyelectrolyte [116]. As a polycation, chitosan has extensively been used in a layer-by-layer technique, which allows the build-up of multi-layered thin films made of polymers of opposite charge [117,118]. In fact, chitosan can easily form polyelectrolyte complexes with other polyanions such as alginate [119], pectin [120], elastin [121] or even DNA [122]. For example, chitosan/alginate multi-layers have been used as an alternative to poly-l-lysine to coat alginate beads containing cells, producing capsules the core of which could be liquefied [123,124]. The ability of chitosan to form complexes with polyanions is influenced by the degree of deacetylation because by removing acetyl groups more free amine groups can participate in the formation of the complex [125]. The degree of deacetylation influences the physical, mechanical and biological properties of chitosan. For example, chitin can be degraded in vivo, by the activity of lysozymes up to the formation of amino-sugars [126]. According to Tomihata & Ikada [127], the rate of degradation decreases with the degree of deacetylation, showing a marked reduction after 70 mol% of deacetylation and being very low for fully deacetylated chitosan. This is in agreement with the results of Freier et al. [128], who also reported prolonged degradation for fully deacetylated chitosan. Furthermore, they reported a good adhesion and proliferation for dorsal root ganglion neurons for fully deacetylated chitosan that decreased proportionally with the degree of deacetylation, which was very low at 50%. Also, Chatelet et al. [129] reported that the degree of deacetylation has no significant influence on the in vitro cytocompatibility of chitosan films for keratinocytes and fibroblasts but plays an important role in the cell adhesion and proliferation of these two cell lines. Fibroblasts adhere twice as strongly to chitosan as to keratinocytes. However, while the rate of proliferation of keratinocytes decreased proportionally with the degree of deacetylation, the fibroblasts did not proliferate regardless of the number of acetyl groups present. The authors suggested that the high adhesion affinity of fibroblasts for chitosan materials could alter their growth.
The alkalinization of chitosan acidic solution up to a pH compatible with the presence of cells (around 7) leads to the formation of gel precipitates. When the protonated amine that induces electrostatic repulsion among the chains is neutralized, a three-dimensional network forms due to hydrogen bonding and hydrophobic interactions [130]. With the addition of phosphate salts in solution, chitosan becomes soluble at physiological pH and becomes thermoresponsive, forming a gel upon heating to physiological temperature [131,132]. Chitosan can also be chemically modified to improve its solubility at neutral pH or its complexation properties without affecting its biocompatibility and biodegradability. These modifications typically involve the covalent binding of a molecule through graft polymerization of the free amine groups on deacetylated units or the hydroxyl groups on the C3 and C6 carbons on acetylated or deacetylated units [133,134]. A typical example is graft polymerization with moieties bearing carboxylic groups, such as carboxymethyl. This modification increases the solubility of chitosan up to physiological pH without affecting its cationic character [135]. Another example is chitosan grafted with polyethylene glycol or temperature responsive chains, becoming a water-soluble thermoresponsive polymer that forms gels at physiological temperature [136,137].
3.1.5. Gellan gum
Gellan gum is a polysaccharide derived from the microbial fermentation product of Sphingomonas elodea. A pure culture of S. elodea is inoculated in a medium of glucose, nitrogen and inorganic salts. When the glucose is exhausted by the biological activity the broth is refined and the gum is collected after precipitation with alcohol [138]. Gellan gum is a linear anionic polysaccharide composed of tetrasaccharide repeating units (1,3-β-d-glucose, 1,4-β-d-glucuronic acid, 1,4-bβ-d-glucose, 1,4-α-l-rhamnose) [139,140]. Gellan gum is available in two isoforms, an acylated form that produces soft hydrogels and a deacylated form that produces hard and brittle gels. Both forms are in random-coil conformation at high temperature and upon cooling there is a transition to double-helix conformation [141]. The double-helices self-assemble in clusters of anti-parallel structures called junction zones which are joined together in a three-dimensional structure by untwined regions of the polymer chain. The presence of cations in solution is needed to obtain a stable hydrogel. Without cations the negatively charged carboxyl side groups repel each other hindering the aggregation of the helices. Monovalent cations in solution can electrically shield the carboxyl group by a tighter aggregation of the helices while divalent cations, in addition to their screening effect, can bind together two carboxyl groups producing stronger gels [142,143]. These requirements for the sol–gel transition (temperature, presence of cations) are compatible with a physiological environment and as such the sol–gel transition of gellan gum is compatible with cell entrapment and survival. Furthermore, the carboxyl group of gellan gum can be used for chemical modifications to improve its mechanical properties and stability in vivo. For example, when modified with methacrylates, the hydrogel obtained is photo-cross-linkable and presents a higher stability because of the covalent bonds between the chains [144].
Ferris & in het Panhuis [145] have characterized the mechanical properties of Gelzan, a commercial formulation of gellan gum. They showed an increase in elastic modulus in compression from 100 to 400 kPa by increasing the concentration of the polymer in solution from 0.5% to 1.5%. The number of adherent cells (L-929 mouse fibroblasts) decreased following an opposite trend and reached a minimum for gellan gum of 1.5%. They also showed an initial increase in elastic modulus from 250 to 350 kPa by increasing the concentration of calcium ions in the hardening bath from 5 to 10 mM. The elastic modulus then decreased back to 200 kPa by further increasing the concentration of ions to 15 mM. The number of adherent cells followed a similar trend and was maximum for the gellan gum hardened with a solution of 10 mM.
Some authors have conducted experiments aimed at characterizing the degradation of this hydrogel in vitro and in vivo [146,147]. The degradation rate of the gel is influenced by the number of acyl groups; in particular, hydrogels made of low acyl gellan gums degrade more slowly than the high acyl forms [148]. Furthermore, gellan gum is degraded by the enzyme galactomannanase [149].
3.1.6. Hyaluronic acid
Hyaluronic acid is a polysaccharide present in all living organisms and is found in most connective tissues [150]. It is a glycosaminoglycan synthesized by membrane-bound hyaluronan synthases, which distinguishes it from other glycosaminoglycans that are produced in the Golgi apparatus. Hyaluronic acid can be obtained from many tissues by extraction or enzymatic digestion and it can also be produced by bacteria. Different sources and extraction protocols can lead to preparations of hyaluronic acid characterized by similar molecular weight but different content of endotoxins and contaminating proteins, leading to a different behaviour in in vivo and in vitro experiments [151,152]. Hyaluronic acid is a linear anionic polysaccharide comprising 1,3-β-d-glucuronic acid and 1,4-β-N-acetyl-d-glucosamine, a structure conserved in all mammals [153]. It is a hydrophilic polymer that can form highly viscous solutions at low concentrations [154]; it is widely used as a lubricant and in preventing post-surgical adhesions [155]. The molecule of hyaluronic acid in solution is stiffened by a combination of the chemical structure of the disaccharide, internal hydrogen bonds and interactions with the solvent [156]. The polymer has to be chemically modified to form a hydrogel. This procedure may involve the modification of the carboxyl or the hydroxyl group by esterification and cross-linking with, for example, glutaraldehyde, carbodiimide and divinyl sulfone [157]. For cell encapsulation, the polymer can be treated with methacrylic anhydride to obtain methacrylated hyaluronic acid, a photo-cross-linkable polymer (figure 2c) [158]. Seidlits et al. [159] showed that the mechanical properties of methacrylated hyaluronic acid can be tuned to be close to the natural central neural tissues. The compressive modulus depended, as expected, on the initial degree of methacrylation (DM) and ranged from 3 to 10 kPa for a 1% w/v hyaluronic acid with a molecular weight of 1500 kDa. They also observed that the diffusion of small molecules (DAPI) was inversely related to the DM. Bencherif et al. [160] observed a monotonic increase in the shear modulus as the DM increased with a variation less pronounced for highly methacrylated hydrogels. The hydrogel with a DM of 14% had a shear modulus of 15 kPa, increasing up to 30 kPa for 30% DM and 60 kPa for 90% DM. They measured the influence of the concentration of the polymer on the shear modulus for a hyaluronic acid with 32% DM, ranging from 22 kPa for a 2% w/v hyaluronic acid up to 70 kPa for 10% w/v. They did not observe substantial differences in cell attachment and proliferation on hydrogels with different DM.
Hyaluronic acid does not favour cell adhesion but it can be further modified to incorporate adhesion motifs such as RGD [161]. Hyaluronic acid is used for the encapsulation of all those cell lines whose extracellular matrix is rich in glycosaminoglycans or hyaluronic acid itself. For example, hyaluronic acid has been studied extensively for cartilage tissue engineering, demonstrating its ability to support chondrogenic differentiation of mesenchymal stem cells and the formation of cartilaginous matrix [162]. Another important field of application of this polymer is neural tissue engineering since hyaluronic acid has an important role in the development of the central neural system, in nerve regeneration, in astrocyte activation and proliferation after a spinal cord injury [163].
Hyaluronic acid is biodegradable in mammals. It is rapidly degraded by hyaluronidase, β-glucuronidase and β-N-acetyl-glucosaminidase up to the formation of low molecular weight hyaluronic acid and oligosaccharides that enter the glycolytic pathway [150].
Being naturally present in most connective tissue, hyaluronic acid is commonly used for the encapsulation of cells whose extracellular matrix is rich in hyaluronic acid such as chondrocytes [164].
3.2. Proteins
3.2.1. Collagen and gelatin
Collagen is the most abundant protein in humans and the main component of the extracellular matrix of many tissues. It is mostly synthesized by fibroblasts and osteoblasts [165]. Among the existing collagen types, collagen type I is the most abundant and is extracted from tissues (ligaments, skin) by enzymatic and acid treatments [166]. Collagen proteins consist of a unique triple helix extending over a large portion of the molecule. The helices assemble in complex supramolecular structures. Every third amino acid of the chain of the helix is glycine. Glycine, a very small amino acid, occupies the centre of the helix allowing a tight packing of the three chains. About one-third of the remaining amino acids are proline and hydroxyproline and have their side chains pointing outwards from the helix [167]. Collagen type I is typically dissolved in diluted acid. Collagen self-assembles to form a hydrogel when the solution is neutralized (e.g. with NaOH) and heated to physiological temperature [168]. For cell encapsulation, cells can be mixed with the neutralized collagen solution and the suspension can then be moved to an incubator. Collagen contains some adhesion motifs as RGD (Arg-Gly-Asp) an important tripeptide for the interaction between a variety of cells and the extracellular matrix. Collagen can be rapidly biodegraded in mammals via collagenases and metalloproteinases to yield the corresponding amino acids. The rate of degradation can be controlled by enzymatic treatment or chemical cross-linking [169]. Because collagen is the main component of the extracellular matrix of many tissues it has found many potential applications for tissue engineering [170] such as cartilage repair [171], mesenchymal stem cell-based therapies [172], bone regeneration [173], etc. Collagen is the most common clinically used biomaterial for skin repair [174].
The triple helix of collagen can be broken down into single-strain molecules to obtain gelatin [175]. Two different types of gelatin can be obtained by treating collagen: one is the result of the hydrolysis of the amide groups of asparagine and glutamine into carboxyl groups, while the other is the result of an acid treatment. The carboxyl groups make gelatin negatively charged [176]. Gelatin is thermoresponsive, undergoing a reversible sol–gel transition by cooling a water-based solution of the polymer below 35°C. The hydrogel can be liquefied by heating it to physiological temperature. This property has been exploited to fabricate hydrogels with an inner gelatin core that ‘melts' once placed in physiological conditions [31] or to fabricate porous cell-laden scaffolds with the gelatin beads acting as porogen [177]. Gelatin can be chemically modified to encapsulate cells so that it does not liquefy when placed at physiological temperature. For example, methacrylate groups can be added to the side chains of gelatin, resulting in photo-cross-linkable gelatin–methacrylamide [178].
3.2.2. Fibrin
Fibrin is a major component of blood clots and a key regulator of wound healing. Fibrin is a polymer similar to collagen formed by the enzymatic polymerization of the protein fibrinogen in the presence of thrombin [179]. Fibrinogen is a water-soluble glycoprotein of 340 kDa, comprising two sets of three polypeptide chains that are linked together by disulfide bonds. Fibrinogen is synthesized primarily in the liver, it is present in the human blood and its concentration increases after a trauma. Fibrinogen can be isolated by precipitation starting from the plasma of autologous blood, for example, by a series of freezing and thawing cycles or by using chemicals aimed at decreasing the solubility of the protein [180]. Thrombin catalyses the cleavage of fibrinopeptides leading to fibrin monomers [181]. Cleavage of the fibrinopeptides occurs in the central N-terminal part of the fibrinogen exposing the ‘A-’ and ‘B-knobs’ binding sites [182]. The knobs interact with the ‘A-’ and ‘B-holes’ present at the end of the fibrinogen molecule forming insoluble fibrin fibres. The branching of the fibres results in a three-dimensional fibrin network. Finally, in the presence of calcium the transglutaminase factor XIIIa cross-links and stabilizes the structure. Factor XIIIa is derived from the thrombin-mediated cleavage of factor XIII, a transglutaminase that can promote stem cell adhesion and proliferation [183]. The formation of the gel and its mechanical properties are influenced by the concentration of fibrinogen and thrombin [184]. Lower concentrations of thrombin lead to more compact gels, with thicker fibres and better mechanical properties. Furthermore, when fibrin is used as the matrix to encapsulate stem cells, the concentration of thrombin and fibrinogen can influence their proliferation rate and their differentiation. The results from Catelas et al. [185] show that formulations containing a lower concentration of fibrinogen (5 mg ml−1) can support human mesenchymal stem cell growth, while a higher concentration (50 mg ml−1) has an increased potential for their differentiation into osteoblasts. In mammals, fibrin can degrade rapidly owing to the presence of proteolytic enzymes (fibrinolysis). In fact, during wound healing, fibrin is gradually degraded and replaced by mature extracellular matrix. The degradation kinetic of fibrin can be controlled by protease inhibitors such as aprotinin [186,187].
A cell suspension in fibrinogen and thrombin can be injected to form a fibrin hydrogel in situ. This particular approach has found many applications in tissue engineering, specifically for cell therapies to enhance cell retention after transplant. There are many examples in the literature of these injectable systems not only for restoring the myocardium after myocardial infarction [188–190] but also for other applications such as bone [191] or muscle regeneration [36].
3.2.3. Elastin
Elastic fibres are present in all those tissues that require the recovery of their initial shape after deformation, such as skin, ligaments and blood vessels [192]. The main components of elastic fibres are elastin and microfibrils. Elastin is a protein formed by the polymerization of water-soluble tropoelastin in a process called elastogenesis. The tropoelastin proteins are synthesized by cells and they consist of hydrophobic and cross-linking hydrophilic domains. The hydrophobic domains mainly consist of valine, glycine and proline, while lysine and alanine are the main components of the hydrophilic domains [193]. The tropoelastin proteins are secreted by cells; outside the wall they self-assemble in a process called coacervation, following a precise patterning of mostly alternating hydrophobic and hydrophilic sequences [194]. The tropoelastin aggregates are then deposited and cross-linked onto microfibrillar templates forming elastin [195].
Natural elastin can be extracted from tissues by harsh alkaline treatments leading to a poor yield. Typically, elastin-like polypeptides (ELPs) are used, which are synthetic naturally inspired polymers using human elastin sequences as their building blocks [196]. ELPs are expressed from a plasmid-borne gene in Escherichia coli [197]. The polypeptides obtained by the purification of the cell lysates are macromolecules constituted by large monomers that are repeated a few times. For this reason and to underline their engineered nature, Rodríguez-Cabello et al. [198] proposed the term elastin-like recombinamer (ELR). The most studied members of the ELR family are based on the VPGVG pentapeptide, the most abundant sequence in natural human elastin. Other ELRs have been synthesized by substituting the fourth amino acid with other natural amino acids except proline. The ELRs are thermally responsive and they undergo a reversible sol–gel transition upon heating. Under the transition temperature, they are in random-coil conformation and upon heating the chain folds hydrophobically forming a regular, ordered β-spiral structure stabilized by hydrophobic interactions. The transition temperature is influenced by the molecular weight, the concentration in solution and the amino acid composition but can be easily controlled to be between room and body temperature. Furthermore, biodegradation sequences [199,200] or specific cell adhesion motifs can be added to the polymer chain [201,202]. For example, a negatively charged ELR containing the RGD motif was produced by Costa et al. [203] to fabricate microcapsules with a layer-by-layer technique using chitosan as a polycation.
3.2.4. Silk fibroin
Silkworm silk, namely silk produced by Bombyx mori, is a filament made from two different proteins, sericin (the outer coating, about 25%) and fibroin. For biomedical applications, sericin is removed by a process called degumming, typically by boiling silk in an aqueous solution of sodium carbonate. The primary structure of the protein is the recurrent amino acid sequence (Gly-Ser-Gly-Ala-Gly-Ala)n forming a light chain (MW 25 kDa) and a heavy chain (MW 350 kDa) linked by disulfide bonds. The fibroin spun by the silkworm consists of β-sheet secondary structures arranged so that the methyl groups and hydrogen groups of opposing sheets interact [204,205]. This water-insoluble secondary structure, known as silk II, is stabilized by strong hydrogen bonds and van der Waals forces. Solvents with high ionic strength and high concentration of salts (e.g. lithium bromide) are needed to break down the hydrogen bonds and obtain a water-soluble random-coil conformation known as silk I. The solution is then dialysed against distilled water for 3 days to remove the salt. The concentration of the protein in solution can then be increased by dialysis against polyethylene glycol [206]. Hydrogels can be prepared by fibroin solutions in water; this process can occur in mild conditions permitting the encapsulation of cells. The water-soluble silk I is metastable, and fibroin chains tend to aggregate forming stable β-sheet structures [207]. The sol–gel transition process is not fully understood but is known to be influenced by mechanical stresses, the concentration of the protein, temperature, pH and concentration of salts in solution [208]. For example, Wang et al. [38] successfully encapsulated cells into a fibroin gel by sonicating fibroin aqueous solutions at different concentrations. The gel forming of fibroin can occur under mild conditions, permitting the encapsulation of cells. After sonication, the liquid solutions were mixed with cells and the suspensions were moved to an incubator where they formed hydrogels. The authors stated that several physical factors related to sonication, including local temperature increase, mechanical/shear forces and increased air–liquid interfaces, affect the process of rapid gelation of silk fibroin (0.5–2 h). Kim et al. [207] characterized the influence of temperature, Ca2+ and K+ concentrations, and pH on the sol–gel transition of fibroin. They showed a decrease in gelation time at 37°C from 30 days to one week by increasing the concentration of the polymer in solution from 2% to 20% and that the presence of ions could shorten the time needed for the sol–gel transition even more. They also measured the compressive modulus of the gels at 37°C, which increased from a few kPa for fibroin 4% up to 4 MPa for fibroin 16%. They also observed a decreased average pore size for the gels made from higher concentration of polymer. Fibroin hydrogels can also be prepared with low cytotoxic chemical cross-linkers such as genipin. Sun & Incitti [209] characterized genipin cross-linked fibroin hydrogels formed after 48 h of incubation at 37°C. The long incubation time was necessary to obtain a high cross-linking degree (about 93%) needed for the formation of the gel, since genipin preferentially reacts with lysine and arginine which are present in fibrin only in low percentages. The hydrogel obtained had a storage modulus of 41 kPa and supported the long-term proliferation of pluripotent cells.
Fibroin undergoes a slow proteolytic degradation and the protein is broken down into smaller polypeptides and free amino acids [210]. The rate of degradation depends on the β-sheet content and is related to the preparation methods of the hydrogel [211].
4. Methods
The materials and the hardening strategies should be combined with adequate technologies to encapsulate cells. This section will explore methods that allow the production of hydrogels with controlled size, often in the form of capsules or particles in the micrometre range (table 2). Most of these capsules or particles are spherical since there is a wide range of applicability of spherical-based devices for tissue engineering and other biomedical applications [216].
Table 2.
A comparison of microencapsulation techniques.
| name | scalability | control on size | minimum size (d in µm) | size dispersion | best gelling mechanism |
|---|---|---|---|---|---|
| extrusion | medium/good | medium | 80 [212] | medium/low | fast/ionotropic |
| lithography | medium | very good | 50 [26] | low | all |
| emulsion | very good | low | 10 [213] | high/medium | thermal |
| microfluidic | medium | very good | 50 [214] | low | all |
| bioprinting | medium/low | very good | 100 [37] | low | all (more complex with thermal) |
| superhydrophobic surfaces | low | very good | 1000 [215] | low | all |
4.1. Extrusion
The easiest method to encapsulate cells into hydrogel beads is by gravitational dripping. A suspension of hydrogel precursor and cells is extruded through a small tube (i.e. a needle), the drop grows and when it reaches a critical mass it freely falls into a suitable hardening bath. According to Poncelet et al. [217], the final diameter of the capsule is influenced by the density of the solution, the surface tension of the drop and the diameter of the pendant droplet neck (approximately the external diameter of the needle). This method usually leads to capsules above 1 mm diameter and because of the impact onto the bath they are not always spherical. Pereira et al. [24] applied this method to suspensions of immortalized mouse lung fibroblasts and gellan gum with two different degrees of acylation, using a phosphate buffer as the hardening agent. The beads had a tear shape and there was a significant difference in diameter between the beads obtained using the two polymers due to the different properties of the solutions. They did not observe any substantial difference in cytotoxicity and cell viability up to 72 h.
When the extrusion is made directly into a hardening bath without any air gap, the interfacial shear forces on the drop at the tip of the needle inhibit its growth. This technique produces fibres and is called wet spinning. Popa et al. [218] compared the two techniques showing that beads fabricated by dripping were at least two times bigger than wet spun fibres. The hydrogels were used to encapsulate ATD5 cells into alginate–carrageenan hydrogels. This mixture of hydrocolloids is interesting since the two polymers share a similar gelling mechanism. In fact, they both cross-link in the presence of positively charged cations and a single bath of CaCl2−KCl can be an adequate hardening agent for the mixture.
Smaller beads can be obtained by applying different methods aimed at ‘breaking’ the jet. One of these methods is by coaxial air flow, where a compressed gas is forced around the extruding droplet and the shear stresses applied by the gas cause the droplet to detach before it would fall due to gravity. Higher gas flows produce smaller beads while higher viscosities of the solution have a stabilizing effect resulting in bigger beads [219]. Using this encapsulator, Zhou & Xu [220] successfully encapsulated human umbilical cord mesenchymal stem cells in a blend of oxidized alginate and fibrin. The microbeads (several hundred micrometres) showed a quicker degradation rate and a higher cell density than pure and oxidized alginate. This device has also been used by Mazzitelli et al. [221], who adopted this technique to encapsulate IB3-1 cells in alginate using a bath of barium chloride as the hardening agent. The microencapsulation procedure did not alter the viability of the encapsulated cells and the cells could still be induced to pro-inflammatory responses after treatment with tumour necrosis factor-α. A coaxial encapsulator can also be used with a liquid instead of a gas to break the drop. Sakai et al. [222] used such a device to entrap cells in a multi-layered capsule made of phenolic-derived alginate and cellulose. The extruding liquid was a suspension of the polymers, horseradish peroxidase and cells. In this case, the outer fluid was liquid paraffin that contained hydrogen peroxide to allow the enzymatic cross-linking of the cell suspension.
There are other ways to break the drop and create microcapsules, one of which is the vibrational encapsulator, characterized by Mazzitelli et al. [212]. This system is based on the principle that laminar liquid jets break up into equally sized droplets by applying a vibration to the extrusion nozzle. By using low flow rates and high frequency of vibration, it is possible to obtain beads up to 100 µm. They were able to produce calcium-alginate gel beads with a very narrow size distribution and excellent morphological characteristics. The microcapsules did not alter the morphology, viability and functional properties of neonatal porcine islets.
Prüsse et al. [223] have extensively characterized the JetCutter, a droplet generator that can be used with highly viscous fluids. This technology is based on a mechanical cut of a liquid jet by rotating cutting wires. The droplet size mainly depends on the diameter of the nozzle, the rotation and frequency of the cutting tool, the number and the diameter of the wires. The diameter of the beads produced by this technology ranges from 200 µm up to a few millimetres. With the JetCutter, Schwinger et al. [20] successfully encapsulated murine fibroblasts into alginate/poly-l-lysine complexes, showing that encapsulated cells were able to survive in culture for extended periods of time with unchanged rates of proliferation and preserved morphology.
Smaller beads can also be obtained by applying an electrical potential to a metallic extruder (figure 3). The technique, commonly referred to as bio-electrospray, consists in spraying a polymer solution pumped through a needle connected to a high-voltage generator [225]. The solution at the tip of the needle reacts to the presence of the electrical charge by accumulating charges of opposite sign on its surface. The resulting Coulombic repulsive forces create stresses on the liquid surface and deform it into a conical shape known as the Taylor cone [226]. If the electric field is sufficiently high, this electrostatic stress overcomes the surface tension at the apex of the liquid cone and a jet of drops is formed to expel the excess of surface charge (Rayleigh limit) [227]. Once the jetting condition is initiated the size of the drop is independent of the voltage applied and the distance from the target anode. Gasperini et al. [21] used this technique starting from a suspension of B50 mouse cells and alginate. The cell-laden droplets of the jet were collected in a calcium chloride bath acting as the hardening agent. They showed that, by increasing the concentration of cells in the suspension, higher applied voltages were needed to initiate the jetting condition. However, once the condition was initiated there were no substantial differences in the diameter of the beads, which ranged from 200 µm up to few millimetres. Cells did remain viable up to 30 days after encapsulation but they showed a different behaviour from control cells once released from the beads and cultured on a tissue culture plate. Cells migrated to form clusters leaving large portions of completely cell-free tissue culture plate and were not able to reach confluence.
Figure 3.
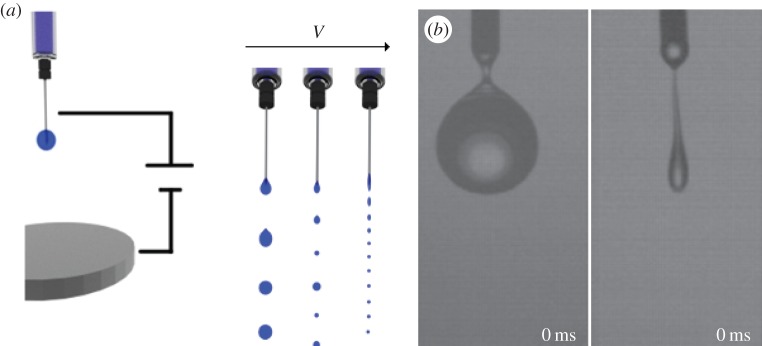
(a) Scheme of the bio-electrospray encapsulation. A potential is applied to the metallic needle of a syringe placed above a metallic plate connected to the ground. When the potential applied is zero the drops freely fall from the needle as in gravitational dripping. By increasing the potential, there is a transition to a jetting mechanism and small mono-sized beads are obtained. (b) Real images of dripping (left) and jetting (right) modes. Adapted from Xie & Wang [224] with permission (Copyright © 2007, Elsevier).
4.2. Lithography
Lithography techniques for cell encapsulation can be divided into soft lithography and photolithography [228] (figure 4). Soft lithography is a strategy based on self-assembly and replica moulding for carrying out micro- and nanofabrication [229]. In this technique, a polymer solution is poured or spin-coated onto a master and then is cross-linked, obtaining a rubbery replica. The replica can contain channels that can be filled with a suspension of a hydrogel precursor and cells. The channels are usually filled by pressing the replica onto the cell suspension. This technique was adopted by Khademhosseini et al. [26] to encapsulate NIH-3T3 in photo-cross-linkable hyaluronic acid using a polydimethylsiloxane (PDMS) replica. They filled the voids in the replica by pressing it onto the cell suspension, then the replica was moved onto an acrylated glass substrate and exposed to UV. After 6 h, about 85% of the cells were viable and after 5 days cells were able to emerge from the hydrogel.
Figure 4.

(a) Soft lithography approach to cell encapsulation. A mould is placed onto a cell suspension and the microvolumes are filled by capillary forces. The mould, transparent to UV, is moved onto a clean surface and placed under UV. The microgels that form can be detached from the mould. (b) Photolithography. A mask is placed on the cell suspension and then is irradiated by light for cross-linking. Microgels are formed and the rest of the suspension can be washed away.
In photolithography, a mask is placed on a suspension made of cells and a photo-cross-linkable hydrogel precursor. The suspension is then irradiated by light, typically in the UV range. The area of the suspension accessible by light cross-links forming a hydrogel while the uncross-linked portion of the suspension can be washed away. The hydrogels obtained in this way are thin and their heights can be increased by repeating the process, thus obtaining multi-layer structures [230]. Using this technique, Shin et al. [231] encapsulated NIH-3T3 cells into hybrid hydrogels of photo-cross-linkable gelatin and graphene oxide on glass slides coated with polyethylene glycol diacrylate. They optimized the photo-cross-linking process by increasing the time of exposure when a higher concentration of graphene oxide was present in the gel. The hybrid hydrogels demonstrated tunable mechanical strength and enhanced electrical properties. The gels supported cellular spreading with improved viability and proliferation.
4.3. Emulsion
The preparation of spherical objects by emulsion-based techniques has been widely used in the pharmaceutical industry [232]. Encapsulation by emulsion is usually obtained by dispersing a hydrogel precursor into a non-miscible phase. Surfactants can be used to stabilize the emulsion and to obtain smaller drops (microemulsion) [233]. When the dispersion reaches equilibrium, the polymer drops are hardened according to the sol–gel mechanism of the hydrogel. Luan & Iwata [16] adopted this technique to encapsulate rat islets in agarose microbeads carrying complement receptor-1, a potent inhibitor of complement activation pathways. A suspension of islets and agarose was mixed with liquid paraffin at 40°C. The tube containing the emulsion was then immersed in an ice bath for 3 min to harden the agarose beads. After adding buffer to the tube and stirring at 4°C, the beads could be collected and the complement receptor was immobilized on their surface. It was shown that agarose microbeads functionalized with the complement receptor contributed to the marked prolongation of islet graft survival in rat-to-mouse xenotransplantation compared with plain agarose microbeads. Batorsky et al. [234] used a collagen–agarose hydrogel to encapsulate adult mesenchymal stem cells using PDMS as a non-miscible phase. This mixture of polymers is challenging since both polymers present a sol–gel transition at physiological temperature but with an opposite behaviour: one is a hydrogel when the other is a fluid. Collagen was neutralized at 4°C with NaOH at 37°C followed by mixing it with agarose at 60°C. The mixture was first added into the emulsification bath at 37°C, stirred for 6 min and then it was cooled with an ice bath for 30 min. They obtained beads from 30 to 150 μm in diameter, containing viable cells up to 8 days. They also showed that by increasing the relative concentration of collagen in the mixture, as expected, cells were more spread due to the affinity for collagen.
4.4. Microfluidics
Microfluidics is a technique dealing with the handling of fluids in microenvironments, such as microchannels where the flow of fluids is generally laminar. The flow is characterized by low Reynolds numbers [235], meaning that it is dominated by viscous stresses with negligible inertia effects. The laminar flow allows a fine control over the characteristics of the microdrop [236]. The generation of the microdrops usually involves the formation of emulsions of the polymer droplets in a non-miscible continuous phase. The generation of individual drops through microfluidics can be seen as a bottom-up approach to emulsification, compared with standard emulsification techniques being the top-down approach. In this bottom-up approach, a suspension of hydrogel precursor and cells is injected into a microchannel and the droplets are formed when the suspension intersects the continuous phase coming from other inlets. The intersection between the channels can have different geometries, with the T-junction [214,237] and the flow-focusing [238,239] being the most common (figure 5).
Figure 5.
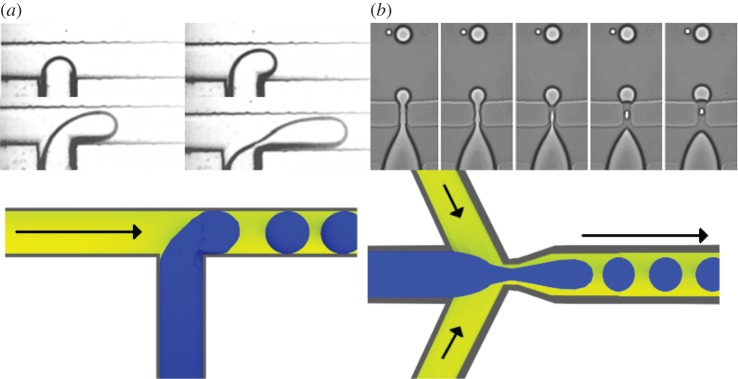
Microfluidic encapsulation devices, real images and schemes showing the hydrogel precursor and the continuous phase. (a) T-junction, hydrogel precursor from the bottom channel (adapted from Fu et al. [240] with permission. Copyright © 2010, Elsevier). (b) The flow-focus, hydrogel precursor form the left channel (adapted from Anna et al. [239] with permission. Copyright © 2003, AIP Publishing LLC).
When two channels merge at a right angle the junction is called a T-junction. In this set-up, the continuous phase flows in the bigger main channel while the cell suspension is injected into the orthogonal inlet. The suspension coming from the orthogonal inlet penetrates the continuous phase and the droplet begins to grow. The flow intensity and the pressure gradient in the main channel bend the drop towards the direction of the flow until it breaks. Its size is influenced by the size of the microchannel and the flow intensity of the two phases. In the flow-focusing set-up, the continuous phase flows in side channels while the cell suspension flows in an inner channel between them. At the junction, the continuous phases force the cell suspension through an orifice located downstream of the inner channel. The inner fluid then breaks forming a droplet. The beads can be hardened (e.g. by UV or thermal gelation) inside or outside the microfluidic chip. Sakai et al. [31] proposed a flow-focusing microfluidic device able to fabricate cell-enclosing gelatin capsules in a two-step process. First, they fabricated microparticles of less than 200 µm and then used the same device to coat the particles with gelatin incorporating phenolic hydroxyl groups. After cross-linking the gelatin coating via a horseradish peroxidase catalysed reaction, a hollow-core structure was obtained by liquefying the inner unmodified gelatin after incubation at 37°C. Jun et al. [241] reported an encapsulation technique that produces microfibres and is based on a flow-focusing device using a cross-linking agent as the continuous phase. In this experiment, they used a suspension of pancreatic islets in an alginate–collagen solution for the inner channel and delivered a calcium chloride solution through the outer channels. In this way, they obtained a coaxial flux of cell suspension and hardening agent inside the cylindrical outlet. The extruded fibres were transplanted and the entrapped islets survived for more than four weeks. The fibres did not degrade and successfully protected the cells from the host immune system. They hypothesized a synergistic effect between alginate hydrogel (acting as an immunological barrier) and collagen (supporting the viability of the cells).
4.5. Bioprinting
Bioprinting is a rapid prototyping technology that consists in the computer-aided layer-by-layer deposition of cells [17,242]. Recently, many bioprinting techniques have been developed, the working principle of which is often similar to standard printers for documents. In conventional printers, the ink is transferred from a reservoir or a substrate and deposited onto the paper by a piezoelectric/thermal head or a laser beam. The paper is engineered to absorb the ink. Similarly in some bioprinters, a suspension of cells, called bio-ink, is deposited by the printer onto a specific substrate, called bio-paper. The bio-paper is often engineered specifically for the bio-ink (to cross-link the bio-ink or to be cross-linked by it). Some bioprinters can be seen as an encapsulating device on top of a positioning system; in fact, bioprinting is often referred to as a bottom-up approach using microdrops as building blocks [243].
Cui & Boland [37] adopted this technique to print cell-laden fibrin scaffolds. In this case, they used bio-ink of thrombin and human microvascular endothelial cells that was printed using a modified commercial printer for documents. The cells could proliferate and connect to each other to form a confluent lining along the fibrin scaffold after 21 days of culture. Bioprinting technologies rely on a high-resolution placing system, and this allows unusual encapsulation methods. For example, it is possible to deposit a hydrogel precursor and then dispense a cell suspension on the exact same spot. Lee et al. [29] successfully encapsulated cells in collagen by depositing collagen type I on top of a substrate coated with sodium carbonate, acting as a pH-altering cross-linking agent. Then the droplets of cells in culture medium were dispensed on the partially cross-linked collagen layer to be lodged inside. Finally, the hydrogel was cross-linked by a ultrafine mist of calcium carbonate solution. More recently, Gasperini et al. [244] used a bio-electrospray deposition system to print cell-laden alginate scaffolds on top of a gelatin-enriched biopaper. Using this hydrogel as biopaper, it was possible to cross-link the alginate upon contact onto the substrate and print samples up to 3 mm thick. Billiet et al. [245] used a commercial bioprinter to fabricate cell-laden constructs using a photo-cross-linkable gelatin. They adapted the printer to obtain a homogeneous temperature during the extrusion of the liquid gelatin and with a Peltier cell they cooled the deposition substrate below the sol–gel temperature. Furthermore, by optimizing the printing and UV hardening processes they obtained highly ordered three-dimensional scaffolds with a high viability of entrapped cells.
4.6. Superhydrophobic surfaces
A drop of water placed onto a superhydrophobic surface will maintain a spherical shape. Cells can be encapsulated by dripping onto a superhydrophobic surface a suspension of cells in a water-based hydrogel precursor [215]. Then the drops, almost completely surrounded by air (or another desired atmosphere), are hardened over the hydrophobic surface maintaining the spherical geometry. This technique permits the preparation of a large range of particulate systems under mild conditions avoiding any loss of the cargo during the processing (encapsulation efficiency very close to 100%). Moreover, there is a good control over the size of the particles dictated by the volume of the dispensed suspension. Lima et al. [22] adopted this technique using a suspension of thermoresponsive chitosan (solution of chitosan and glycerophosphate) and cells. The drops were placed on a superhydrophobic surface of polystyrene and a hydrogel complex was formed by the addition of sodium tripolyphosphate. Upon heating, at 37°C a second sol–gel transition could be observed, predicting a hardening effect when exposed to physiological conditions. Cells could remain viable up to 7 days. Using this method, mesenchymal stem cells were encapsulated in alginate beads containing fibronectin. Cells exhibited good viability and osteogenic capability. Moreover, in vivo tests showed that these particles could stimulate bone regeneration in cranial defects [246]. More sophisticated layered particles could be obtained by the sequential deposition of hydrogel precursors over previously hardened particles allowing the distribution of cells or drugs in compartments with a radial arrangement [247]. Future developments on this technology should include dispensing systems able to place smaller volumes of hydrogel precursors over adequate superhydrophobic surfaces to obtain particles smaller than 800 µm.
4.7. Comments
Extrusion techniques range from simple semi-automatic extrusions of hydrogel precursors into a hardening bath to more complex patented systems. Generally, these techniques are best suited for fast gelling hydrogels, such as alginate. With these polymers, the surface of the droplet quickly cross-links once in contact with the hardening agent and the hydrogel coating that is formed stabilizes the shape of the drop. The liquid core can then be hardened by keeping the capsule in the bath, allowing the hardening agent to diffuse inside the capsule. The most interesting extrusion techniques are those able to fabricate gels in the micrometre range. Although these techniques are able to produce beads with a relatively good size distribution, it is not trivial to predict the final size of the beads. If we take as an example the bio-electrospray process and alginate, the beads are formed from a suspension of a shear thinning non-Newtonian fluid and viscous particles (cells) under the effect of an electric field. The final size of the droplets depends on the diameter of the needle, the electrical and rheological properties of the suspension, the flux of the suspension and the applied voltage. General qualitative information can be drawn but it is difficult to predict quantitatively the influence of these parameters on the final outcome. A similar observation can be made for other extrusion techniques. Techniques other than extrusion, such as lithography, have a better control on size, meaning that the final dimension of the bead can be easily predicted. In fact, in soft lithography the droplet is physically constrained inside a mould of predefined dimensions while in photolithography the dimensions of the microgel are determined by the dimensions of a photoresistant mask. Lithography techniques, specifically soft lithography, rely on the production of masters or masks that involves technologies which are not available to all research facilities, while extrusion or emulsion techniques rely on much more common equipment. The same can be said for microfluidic techniques since the microfluidic chip is usually fabricated by lithography or advanced micromachining. In microfluidics, there is also an added layer of complexity represented by the flow of the fluid in the microchannels. The chip has to be properly designed to accommodate inlets, outlets and the junctions where the droplet is formed by the intersection of the fluids. Furthermore, pumps are needed to control the flux of fluids inside the channels. However, once the chip set-up is optimized, microfluidic techniques allow an easier continuous encapsulation of cells with minimal human intervention. While photolithography techniques are restricted to photo-cross-linkable polymers, lithography and microfluidics are really versatile and allow the encapsulation of cells with basically all the polymers (and their modifications) discussed in this review. In soft lithography, once the fluid has filled the channel, it is trivial to put it in contact with hardening agents, change its temperature or irradiate it by UV. Similarly, in microfluidics an inlet can carry the cross-linking agent, the chip can be heated or cooled and it can be manufactured with materials transparent to UV such as PDMS, a common material to manufacture microfluidic chips.
Bioprinting might be the more complex technique to encapsulate cells. It is based on a deposition/encapsulation system which is coupled to an accurate positioning system. Even if complex, bioprinting can rely on cheap but accurate machines developed for the computer market (document printers and their controlling drivers). These printers make this technology appealing in the face of little investment. Furthermore, they are colour printers and usually contain four reservoirs (red, green, blue and black) to allow, for example, the encapsulation of different kinds of cells suspended into different hydrogel precursors. These systems are particularly complex encapsulating machines but this complexity is justified by the possibility to design drop-by-drop arbitrary structures that translate directly into cell-laden scaffolds. On the opposite side are emulsion techniques, which are simple, based on common equipment and can be easily scaled to produce a high number of cell-laden beads. These techniques can also rely on a well-established industrial technology developed for cosmetics and pharmaceuticals. The droplets obtained by emulsion typically have a wider size distribution than those obtained by other techniques. However, water-in-oil emulsion can be stabilized using surfactants (i.e. Tween and Span) that may help in narrowing the distribution and controlling the size of the droplets. Also in this case, as with extrusion, predicting the final dimension of the beads is not trivial. The system has to be carefully designed by choosing the surfactants and their relative amount in the suspension, considering the specific hydrogel precursor and the apolar phase used. The relative concentration of each component is usually established empirically. When thermal treatment is used to harden the droplets careful attention should be given to the kinetics of the sol–gel transition. Typically during this treatment, the suspension of droplets and non-miscible phase is quickly cooled (e.g. with an ice bath) to induce the sol–gel transition. The thermal treatment should be long enough to obtain the hydrogel but not too long to cause thermal shock to cells.
5. Conclusion
Since the first cell encapsulation by Lim & Sun [33], a notable amount of research has been aimed at optimizing the materials and process selection. Many authors have proposed materials and methods tailored for specific applications that range from the encapsulation for xenotransplantation to the production of scaffolds. Among these materials, an important part is represented by naturally derived polymers owing to their inherent similarity to the extracellular matrix and mild processing conditions. In this review, we described how some of these authors have combined the knowledge of specific polymers and their sol–gel mechanism with specific processing conditions compatible with the presence of cells. These experiments could be a model to further develop and improve this technology that could have a huge impact in the field of biotechnology and other biomedical applications.
Funding statement
This work was supported by European Research Council grant agreement ERC-2012-ADG 20120216-321266 for project ComplexiTE.
References
- 1.de Vos P, Faas MM, Strand B, Calafiore R. 2006. Alginate-based microcapsules for immunoisolation of pancreatic islets. Biomaterials 27, 5603–5617. ( 10.1016/j.biomaterials.2006.07.010) [DOI] [PubMed] [Google Scholar]
- 2.Murua A, Orive G, Hernández RM, Pedraz JL. 2009. Xenogeneic transplantation of erythropoietin-secreting cells immobilized in microcapsules using transient immunosuppression. J. Control. Release 137, 174–178. ( 10.1016/j.jconrel.2009.04.009) [DOI] [PubMed] [Google Scholar]
- 3.Chang P. 1999. The in vivo delivery of heterologous proteins by microencapsulated recombinant cells. Trends Biotechnol. 17, 78–83. ( 10.1016/S0167-7799(98)01250-5) [DOI] [PubMed] [Google Scholar]
- 4.Braun F, Behrend M. 2007. Drugs that act on the immune system: immunosuppressive and immunostimulatory drugs. In Side effects of drugs annual, 29th edition, vol. 29, ch. 38 (ed. Aronson JK.), pp. 424–479. Amsterdam, The Netherlands: Elsevier. [Google Scholar]
- 5.Taylor AL, Watson CJE, Bradley JA. 2005. Immunosuppressive agents in solid organ transplantation: mechanisms of action and therapeutic efficacy. Crit. Rev. Oncol. Hematol. 56, 23–46. ( 10.1016/j.critrevonc.2005.03.012) [DOI] [PubMed] [Google Scholar]
- 6.Uludag H, De Vos P, Tresco PA. 2000. Technology of mammalian cell encapsulation. Adv. Drug Delivery Rev. 42, 29–64. ( 10.1016/S0169-409X(00)00053-3) [DOI] [PubMed] [Google Scholar]
- 7.Silva TH, et al. 2012. Materials of marine origin: a review on polymers and ceramics of biomedical interest. Int. Mater. Rev. 57, 276–306. ( 10.1179/1743280412Y.0000000002) [DOI] [Google Scholar]
- 8.Malafaya PB, Silva GA, Reis RL. 2007. Natural-origin polymers as carriers and scaffolds for biomolecules and cell delivery in tissue engineering applications. Adv. Drug Delivery Rev. 59, 207–233. ( 10.1016/j.addr.2007.03.012) [DOI] [PubMed] [Google Scholar]
- 9.Schrezenmeir J, Kirchgessner J, Gerö L, Kunz LA, Beyer J, Mueller-Klieser W. 1994. Effect of microencapsulation on oxygen distribution in islets organs. Transplantation 57, 1308–1314. ( 10.1097/00007890-199405150-00003) [DOI] [PubMed] [Google Scholar]
- 10.Sakai S, Kawabata K, Ono T, Ijima H, Kawakami K. 2005. Development of mammalian cell-enclosing subsieve-size agarose capsules (<100 μm) for cell therapy. Biomaterials 26, 4786–4792. ( 10.1016/j.biomaterials.2004.11.043) [DOI] [PubMed] [Google Scholar]
- 11.Ma X, Vacek I, Sun A. 1994. Generation of alginate-poly-l-lysine-alginate (APA) biomicrocapsules: the relationship between the membrane strength and the reaction conditions. Artif. Cells Blood Substit. Biotechnol. 22, 43–69. ( 10.3109/10731199409117399) [DOI] [PubMed] [Google Scholar]
- 12.De Vos P, De Haan B, Pater J, Van Schilfgaarde R. 1996. Association between capsule diameter, adequacy of encapsulation, and survival of microencapsulated rat islet allografts. Transplantation 62, 893–899. ( 10.1097/00007890-199610150-00004) [DOI] [PubMed] [Google Scholar]
- 13.Sakai S, Mu C, Kawabata K, Hashimoto I, Kawakami K. 2006. Biocompatibility of subsieve-size capsules versus conventional-size microcapsules. J. Biomed. Mater. Res. A 78, 394–398. ( 10.1002/jbm.a.30676) [DOI] [PubMed] [Google Scholar]
- 14.Robitaille R, Pariseau JF, Leblond FA, Lamoureux M, Lepage Y, Hallé JP. 1999. Biocompatibility of smaller versus standard microcapsules. J. Biomed. Mater. Res. 44, 116–120. ( 10.1002/(SICI)1097-4636(199901)44:1<116::AID-JBM13>3.0.CO;2-9) [DOI] [PubMed] [Google Scholar]
- 15.Dang SM, Gerecht-Nir S, Chen J, Itskovitz-Eldor J, Zandstra PW. 2004. Controlled, scalable embryonic stem cell differentiation culture. Stem cells 22, 275–282. ( 10.1634/stemcells.22-3-275) [DOI] [PubMed] [Google Scholar]
- 16.Luan NM, Iwata H. 2012. Xenotransplantation of islets enclosed in agarose microcapsule carrying soluble complement receptor 1. Biomaterials 33, 8075–8081. ( 10.1016/j.biomaterials.2012.07.048) [DOI] [PubMed] [Google Scholar]
- 17.Norotte C, Marga FS, Niklason LE, Forgacs G. 2009. Scaffold-free vascular tissue engineering using bioprinting. Biomaterials 30, 5910–5917. ( 10.1016/j.biomaterials.2009.06.034) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rocha PM, Santo VE, Gomes ME, Reis RL, Mano JF. 2011. Encapsulation of adipose-derived stem cells and transforming growth factor-β 1 in carrageenan-based hydrogels for cartilage tissue engineering. J. Bioactive Compatible Polym. 26, 493–507. ( 10.1177/0883911511420700) [DOI] [Google Scholar]
- 19.Popa E, Reis R, Gomes M. 2012. Chondrogenic phenotype of different cells encapsulated in κ-carrageenan hydrogels for cartilage regeneration strategies. Biotechnol. Appl. Biochem. 59, 132–141. ( 10.1002/bab.1007) [DOI] [PubMed] [Google Scholar]
- 20.Schwinger C, Koch S, Jahnz U, Wittlich P, Rainov NG, Kressler J. 2008. High throughput encapsulation of murine fibroblasts in alginate using the JetCutter technology. J. Microencapsul. 19, 273–280. ( 10.1080/02652040110105328) [DOI] [PubMed] [Google Scholar]
- 21.Gasperini L, Maniglio D, Migliaresi C. 2013. Microencapsulation of cells in alginate through an electrohydrodynamic process. J. Bioactive Compatible Polym. 28, 413–425. ( 10.1177/0883911513501599) [DOI] [Google Scholar]
- 22.Lima AC, Correia CR, Oliveira MB, Mano JF. 2013. Sequential ionic and thermogelation of chitosan spherical hydrogels prepared using superhydrophobic surfaces to immobilize cells and drugs. J. Bioactive Compatible Polym. 29, 50–65. ( 10.1177/0883911513513660) [DOI] [Google Scholar]
- 23.Ramesh S, Rajagopal K, Vaikkath D, Nair PD, Madhuri V. 2014. Enhanced encapsulation of chondrocytes within a chitosan/hyaluronic acid hydrogel: a new technique. Biotechnol. Lett. 36, 1107–1111. ( 10.1007/s10529-014-1461-1) [DOI] [PubMed] [Google Scholar]
- 24.Pereira DR, et al. 2011. Development of gellan gum-based microparticles/hydrogel matrices for application in the intervertebral disc regeneration. Tissue Eng. C Methods 17, 961–972. ( 10.1089/ten.tec.2011.0115) [DOI] [PubMed] [Google Scholar]
- 25.Silva-Correia J, Zavan B, Vindigni V, Silva TH, Oliveira JM, Abatangelo G, Reis RL. 2013. Biocompatibility evaluation of ionic- and photo-crosslinked methacrylated gellan gum hydrogels: in vitro and in vivo study. Adv. Healthcare Mater. 2, 568–575. ( 10.1002/adhm.201200256) [DOI] [PubMed] [Google Scholar]
- 26.Khademhosseini A, Eng G, Yeh J, Fukuda J, Blumling J, III, Langer R, Burdick JA. 2006. Micromolding of photocrosslinkable hyaluronic acid for cell encapsulation and entrapment. J. Biomed. Mater. Res. A 79, 522–532. ( 10.1002/jbm.a.30821) [DOI] [PubMed] [Google Scholar]
- 27.Duan B, Hockaday LA, Kapetanovic E, Kang KH, Butcher JT. 2013. Stiffness and adhesivity control aortic valve interstitial cell behavior within hyaluronic acid based hydrogels. Acta Biomater. 9, 7640–7650. ( 10.1016/j.actbio.2013.04.050) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Shen Y-I, Abaci HE, Krupski Y, Weng L-C, Burdick JA, Gerecht S. 2014. Hyaluronic acid hydrogel stiffness and oxygen tension affect cancer cell fate and endothelial sprouting. Biomater. Sci. 2, 655–665. ( 10.1039/c3bm60274e) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lee W, Debasitis JC, Lee VK, Lee J-H, Fischer K, Edminster K, Park J-K, Yoo S-S. 2009. Multi-layered culture of human skin fibroblasts and keratinocytes through three-dimensional freeform fabrication. Biomaterials 30, 1587–1595. ( 10.1016/j.biomaterials.2008.12.009) [DOI] [PubMed] [Google Scholar]
- 30.Wang L, Rao RR, Stegemann JP. 2013. Delivery of mesenchymal stem cells in chitosan/collagen microbeads for orthopedic tissue repair. Cells Tissues Organs 197, 333–343. ( 10.1159/000348359) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sakai S, Ito S, Inagaki H, Hirose K, Matsuyama T, Taya M, Kawakami K. 2011. Cell-enclosing gelatin-based microcapsule production for tissue engineering using a microfluidic flow-focusing system. Biomicrofluidics 5, 13402 ( 10.1063/1.3516657) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ma S, Natoli M, Liu X, Neubauer MP, Watt FM, Fery A, Huck WTS. 2013. Monodisperse collagen-gelatin beads as potential platforms for 3D cell culturing. J. Mater. Chem. B 1, 5128 ( 10.1039/c3tb20851f) [DOI] [PubMed] [Google Scholar]
- 33.Lim F, Sun A. 1980. Microencapsulated islets as bioartificial endocrine pancreas. Science 210, 908–910. ( 10.1126/science.6776628) [DOI] [PubMed] [Google Scholar]
- 34.Bandiera A. 2011. Transglutaminase-catalyzed preparation of human elastin-like polypeptide-based three-dimensional matrices for cell encapsulation. Enzyme Microb. Technol. 49, 347–352. ( 10.1016/j.enzmictec.2011.06.012) [DOI] [PubMed] [Google Scholar]
- 35.Betre H, Ong SR, Guilak F, Chilkoti A, Fermor B, Setton LA. 2006. Chondrocytic differentiation of human adipose-derived adult stem cells in elastin-like polypeptide. Biomaterials 27, 91–99. ( 10.1016/j.biomaterials.2005.05.071) [DOI] [PubMed] [Google Scholar]
- 36.Liu J, Xu HHK, Zhou H, Weir MD, Chen Q, Trotman CA. 2013. Human umbilical cord stem cell encapsulation in novel macroporous and injectable fibrin for muscle tissue engineering. Acta Biomater. 9, 4688–4697. ( 10.1016/j.actbio.2012.08.009) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cui X, Boland T. 2009. Human microvasculature fabrication using thermal inkjet printing technology. Biomaterials 30, 6221–6227. ( 10.1016/j.biomaterials.2009.07.056) [DOI] [PubMed] [Google Scholar]
- 38.Wang X, Kluge JA, Leisk GG, Kaplan DL. 2008. Sonication-induced gelation of silk fibroin for cell encapsulation. Biomaterials 29, 1054–1064. ( 10.1016/j.biomaterials.2007.11.003) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Davis NE, Beenken-Rothkopf LN, Mirsoian A, Kojic N, Kaplan DL, Barron AE, Fontaine MJ. 2012. Enhanced function of pancreatic islets co-encapsulated with ECM proteins and mesenchymal stromal cells in a silk hydrogel. Biomaterials 33, 6691–6697. ( 10.1016/j.biomaterials.2012.06.015) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mano JF. 2008. Stimuli-responsive polymeric systems for biomedical applications. Adv. Eng. Mater. 10, 515–527. ( 10.1002/adem.200700355) [DOI] [Google Scholar]
- 41.Hutmacher DW. 2000. Scaffolds in tissue engineering bone and cartilage. Biomaterials 21, 2529–2543. ( 10.1016/S0142-9612(00)00121-6) [DOI] [PubMed] [Google Scholar]
- 42.Vert M, Li SM, Spenlehauer G, Guerin P. 1992. Bioresorbability and biocompatibility of aliphatic polyesters. J. Mater. Sci. 3, 432–446. ( 10.1007/BF00701240) [DOI] [Google Scholar]
- 43.Rozman KK, Doull J. 2000. Dose and time as variables of toxicity. Toxicology 144, 169–178. ( 10.1016/S0300-483X(99)00204-8) [DOI] [PubMed] [Google Scholar]
- 44.Jeong B, Kim SW, Bae YH. 2002. Thermosensitive sol-gel reversible hydrogels. Adv. Drug Delivery Rev. 54, 37–51. ( 10.1016/S0169-409X(01)00242-3) [DOI] [PubMed] [Google Scholar]
- 45.Roy D, Brooks WLA, Sumerlin BS. 2013. New directions in thermoresponsive polymers. Chem. Soc. Rev. 42, 7214–7243. ( 10.1039/c3cs35499g) [DOI] [PubMed] [Google Scholar]
- 46.Klouda L, Mikos AG. 2008. Thermoresponsive hydrogels in biomedical applications. Eur. J. Pharm. Biopharm. 68, 34–45. ( 10.1016/j.ejpb.2007.02.025) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ward M, Georgiou TK. 2011. Thermoresponsive polymers for biomedical applications. Polymers 3, 1215–1242. ( 10.3390/polym3031215) [DOI] [Google Scholar]
- 48.Bekturov EA, Bimedina LA. 1981. Interpolymer complexes. Adv. Polym. Sci. 41, 99–147. ( 10.1007/3-540-10554-9_11) [DOI] [Google Scholar]
- 49.Bhatia SR, Khattak SF, Roberts SC. 2005. Polyelectrolytes for cell encapsulation. Curr. Opin. Colloid Interface Sci. 10, 45–51. ( 10.1016/j.cocis.2005.05.004) [DOI] [Google Scholar]
- 50.Ferreira P, Coelho JFJ, Almeida JF, Gil MH. 2009. Photocrosslinkable polymers for biomedical applications. In Biomedical engineering—frontiers and challenges (ed. Fazel R.), pp. 55–74. Rijeka, Croatia: InTech. [Google Scholar]
- 51.Moreira Teixeira LS, Feijen J, van Blitterswijk CA, Dijkstra PJ, Karperien M. 2012. Enzyme-catalyzed crosslinkable hydrogels: emerging strategies for tissue engineering. Biomaterials 33, 1281–1290. ( 10.1016/j.biomaterials.2011.10.067) [DOI] [PubMed] [Google Scholar]
- 52.Chen T, Embree HD, Brown EM, Taylor MM, Payne GF. 2003. Enzyme-catalyzed gel formation of gelatin and chitosan: potential for in situ applications. Biomaterials 24, 2831–2841. ( 10.1016/S0142-9612(03)00096-6) [DOI] [PubMed] [Google Scholar]
- 53.Sofia S, Singh A, Kaplan D. 2002. Peroxidase-catalyzed crosslinking of functionalized polyaspartic acid polymers. J. Macromol. Sci. A 39, 1151–1181. ( 10.1081/MA-120014843) [DOI] [Google Scholar]
- 54.Fu XT, Kim SM. 2010. Agarase: review of major sources, categories, purification method, enzyme characteristics and applications. Mar. Drugs 8, 200–218. ( 10.3390/md8010200) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Meena R, Siddhanta A, Prasad K, Ramavat B, Eswaran K, Thiruppathi S, Ganesan M, Mantri V, Rao PS. 2007. Preparation, characterization and benchmarking of agarose from Gracilaria dura of Indian waters. Carbohydr. Polym. 69, 179–188. ( 10.1016/j.carbpol.2006.09.020) [DOI] [Google Scholar]
- 56.Lahaye M, Rochas C. 1991. Chemical structure and physico-chemical properties of agar. In International workshop on gelidium (eds Juanes JA, Santelices B, McLachlan JL.), pp. 137–148. Dordrecht, The Netherlands: Springer. [Google Scholar]
- 57.Indovina PL, Tettamanti E, Micciancio-Giammarinaro MS, Palma MU. 1979. Thermal hysteresis and reversibility of gel-sol transition in agarose-water systems. J. Chem. Phys. 70, 2841 ( 10.1063/1.437817) [DOI] [Google Scholar]
- 58.Pernodet N, Tinland B, Sadron IC, Pasteur C-UL. 1997. Pore size of agarose gels by atomic force microscopy. Electrophoresis 18, 55–58. ( 10.1002/elps.1150180111) [DOI] [PubMed] [Google Scholar]
- 59.Narayanan J, Xiong J-Y, Liu X-Y. 2006. Determination of agarose gel pore size: absorbance measurements vis a vis other techniques. J. Phys. Conf. Ser. 28, 83–86. ( 10.1088/1742-6596/28/1/017) [DOI] [Google Scholar]
- 60.Aymard P, Martin DR, Foster TJ, Clark AH, Norton IT. 2001. Influence of thermal history on the structural and mechanical properties of agarose gels. Biopolymers 59, 131–144. ( 10.1002/1097-0282(200109)59:3<131::AID-BIP1013>3.0.CO;2-8) [DOI] [PubMed] [Google Scholar]
- 61.Tang S, Yang W, Mao X. 2007. Agarose/collagen composite scaffold as an anti-adhesive sheet. Biomed. Mater. 2, S129–S134. ( 10.1088/1748-6041/2/3/S09) [DOI] [PubMed] [Google Scholar]
- 62.Karoubi G, Ormiston ML, Stewart DJ, Courtman DW. 2009. Single-cell hydrogel encapsulation for enhanced survival of human marrow stromal cells. Biomaterials 30, 5445–5455. ( 10.1016/j.biomaterials.2009.06.035) [DOI] [PubMed] [Google Scholar]
- 63.Guaccio A, Borselli C, Oliviero O, Netti PA. 2008. Oxygen consumption of chondrocytes in agarose and collagen gels: a comparative analysis. Biomaterials 29, 1484–1493. ( 10.1016/j.biomaterials.2007.12.020) [DOI] [PubMed] [Google Scholar]
- 64.Chi W-J, Chang Y-K, Hong S-K. 2012. Agar degradation by microorganisms and agar-degrading enzymes. Appl. Microbiol. Biotechnol. 94, 917–930. ( 10.1007/s00253-012-4023-2) [DOI] [PubMed] [Google Scholar]
- 65.Zhang L-M, Wu C-X, Huang J-Y, Peng X-H, Chen P, Tang S-Q. 2012. Synthesis and characterization of a degradable composite agarose/HA hydrogel. Carbohydr. Polym. 88, 1445–1452. ( 10.1016/j.carbpol.2012.02.050) [DOI] [Google Scholar]
- 66.Emans PJ, van Rhijn LW, Welting TJM, Cremers A, Wijnands N, Spaapen F, Voncken JW, Shastri VP. 2010. Autologous engineering of cartilage. Proc. Natl Acad. Sci. USA 107, 3418–3423. ( 10.1073/pnas.0907774107) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Benya P. 1982. Dedifferentiated chondrocytes reexpress the differentiated collagen phenotype when cultured in agarose gels. Cell 30, 215–224. ( 10.1016/0092-8674(82)90027-7) [DOI] [PubMed] [Google Scholar]
- 68.Hung CT, Lima EG, Mauck RL, Taki E, LeRoux MA, Lu HH, Stark RG, Guo X, Ateshian GA. 2003. Anatomically shaped osteochondral constructs for articular cartilage repair. J. Biomech. 36, 1853–1864. ( 10.1016/S0021-9290(03)00213-6) [DOI] [PubMed] [Google Scholar]
- 69.Mauck RL, Seyhan SL, Ateshian GA, Hung CT. 2002. Influence of seeding density and dynamic deformational loading on the developing structure/function relationships of chondrocyte-seeded agarose hydrogels. Ann. Biomed. Eng. 30, 1046–1056. ( 10.1114/1.1512676) [DOI] [PubMed] [Google Scholar]
- 70.Hunziker EB. 2002. Articular cartilage repair: basic science and clinical progress. A review of the current status and prospects. Osteoarthritis Cartilage 10, 432–463. ( 10.1053/joca.2002.0801) [DOI] [PubMed] [Google Scholar]
- 71.Hossain KS, Miyanaga K, Maeda H, Nemoto N. 2001. Sol-gel transition behavior of pure iota-carrageenan in both salt-free and added salt states. Biomacromolecules 2, 442–449. ( 10.1021/bm000117f) [DOI] [PubMed] [Google Scholar]
- 72.Campo VL, Kawano DF, Silva DBD, Carvalho I. 2009. Carrageenans: biological properties, chemical modifications and structural analysis—a review. Carbohydr. Polym. 77, 167–180. ( 10.1016/j.carbpol.2009.01.020) [DOI] [Google Scholar]
- 73.Yuguchi Y, Thu Thuy TT, Urakawa H, Kajiwara K. 2002. Structural characteristics of carrageenan gels: temperature and concentration dependence. Food Hydrocolloids 16, 515–522. ( 10.1016/S0268-005X(01)00131-X) [DOI] [Google Scholar]
- 74.Michel A-S, Mestdagh M, Axelos M. 1997. Physico-chemical properties of carrageenan gels in presence of various cations. Int. J. Biol. Macromol. 21, 195–200. ( 10.1016/S0141-8130(97)00061-5) [DOI] [PubMed] [Google Scholar]
- 75.Rochas C, Rinaudo M, Landry S. 1989. Relation between the molecular structure and mechanical properties of carrageenan gels. Carbohydr. Polym. 10, 8–10. ( 10.1016/0144-8617(89)90061-1) [DOI] [Google Scholar]
- 76.Rochas C, Rinaudo M, Landry S. 1990. Role of the molecular weight on the mechanical properties of kappa carrageenan gels. Carbohydr. Polym. 12, 255–266. ( 10.1016/0144-8617(90)90067-3) [DOI] [Google Scholar]
- 77.Di Rosa M. 1972. Biological properties of carrageenan. J. Pharm. Pharmacol. 24, 89–102. ( 10.1111/j.2042-7158.1972.tb08940.x) [DOI] [PubMed] [Google Scholar]
- 78.Loram LC, Fuller A, Fick LG, Cartmell T, Poole S, Mitchell D. 2007. Cytokine profiles during carrageenan-induced inflammatory hyperalgesia in rat muscle and hind paw. J. Pain 8, 127–136. ( 10.1016/j.jpain.2006.06.010) [DOI] [PubMed] [Google Scholar]
- 79.Yoo S, Han S, Park YS, Lee J-H, Oh U, Hwang SW. 2009. Lipoxygenase inhibitors suppressed carrageenan-induced Fos-expression and inflammatory pain responses in the rat. Mol. Cells 27, 417–422. ( 10.1007/s10059-009-0059-2) [DOI] [PubMed] [Google Scholar]
- 80.Popa EG, et al. 2014. Evaluation of the in vitro and in vivo biocompatibility of carrageenan-based hydrogels. J. Biomed. Mater. Res. A 2009, 1–11. ( 10.1002/jbm.a.35081) [DOI] [PubMed] [Google Scholar]
- 81.Luna SM, Gomes ME, Mano JF, Reis RL. 2010. Development of a novel cell encapsulation system based on natural origin polymers for tissue engineering applications. J. Bioactive Compatible Polym. 25, 341–359. ( 10.1177/0883911510372173) [DOI] [Google Scholar]
- 82.Guibet M, Colin S, Barbeyron T, Genicot S, Kloareg B, Michel G, Helbert W. 2007. Degradation of lambda-carrageenan by Pseudoalteromonas carrageenovora lambda-carrageenase: a new family of glycoside hydrolases unrelated to kappa- and iota-carrageenases. Biochem. J. 404 105–114. ( 10.1042/BJ20061359) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Rowley JA, Madlambayan G, Mooney DJ. 1999. Alginate hydrogels as synthetic extracellular matrix materials. Biomaterials 20, 45–53. ( 10.1016/S0142-9612(98)00107-0) [DOI] [PubMed] [Google Scholar]
- 84.Remminghorst U, Rehm BHA. 2006. Bacterial alginates: from biosynthesis to applications. Biotechnol. Lett. 28, 1701–1712. ( 10.1007/s10529-006-9156-x) [DOI] [PubMed] [Google Scholar]
- 85.Calumpong H, Maypa A, Magbanua M. 1999. Population and alginate yield and quality assessment of four Sargassum species in Negros Island, central Philippines. In Sixteenth International Seaweed Symposium, ch. 4 (eds Kain JM, Brown MT, Lahaye M.), pp. 211–215. Dordrecht, The Netherlands: Springer. [Google Scholar]
- 86.Goh CH, Heng PWS, Chan LW. 2012. Alginates as a useful natural polymer for microencapsulation and therapeutic applications. Carbohydr. Polym. 88, 1–12. ( 10.1016/j.carbpol.2011.11.012) [DOI] [Google Scholar]
- 87.Murua A, Portero A, Orive G, Hernández RM, de Castro M, Pedraz JL. 2008. Cell microencapsulation technology: towards clinical application. J. Control. Release 132, 76–83. ( 10.1016/j.jconrel.2008.08.010) [DOI] [PubMed] [Google Scholar]
- 88.Donati I, Holtan S, Mørch YA, Borgogna M, Dentini M, Skjåk-Braek G. 2005. New hypothesis on the role of alternating sequences in calcium-alginate gels. Biomacromolecules 6, 1031–1040. ( 10.1021/bm049306e) [DOI] [PubMed] [Google Scholar]
- 89.Mancini M, Moresi M, Rancini R. 1999. Mechanical properties of alginate gels: empirical characterization. J. Food Eng. 39, 369–378. ( 10.1016/S0260-8774(99)00022-9) [DOI] [Google Scholar]
- 90.Drury JL, Dennis RG, Mooney DJ. 2004. The tensile properties of alginate hydrogels. Biomaterials 25, 3187–3199. ( 10.1016/j.biomaterials.2003.10.002) [DOI] [PubMed] [Google Scholar]
- 91.Strand B, Ryan L, Veld P, Kulseng B, Rokstad A, Skjåk-Braek G, Espevik T. 2001. Poly-l-lysine induces fibrosis on alginate microcapsules via the induction of cytokines. Cell Transplant 10, 263–275. ( 10.3727/000000001783986800) [DOI] [PubMed] [Google Scholar]
- 92.Tam SK, Dusseault J, Polizu S, Ménard M, Hallé J-P, Yahia L. 2005. Physicochemical model of alginate-poly-L-lysine microcapsules defined at the micrometric/nanometric scale using ATR-FTIR, XPS, and ToF-SIMS. Biomaterials 26, 6950–6961. ( 10.1016/j.biomaterials.2005.05.007) [DOI] [PubMed] [Google Scholar]
- 93.Vos PD, Haan BD, Schilfgaarde RV. 1997. Effect of the alginate composition on the biocompatibility of alginate-polylysine microcapsules. Biomaterials 18, 273–278. ( 10.1016/S0142-9612(96)00135-4) [DOI] [PubMed] [Google Scholar]
- 94.Juste S, Lessard M, Henley N, Ménard M, Hallé J-P. 2005. Effect of poly-L-lysine coating on macrophage activation by alginate-based microcapsules: assessment using a new in vitro method. J. Biomed. Mater. Res. A 72, 389–398. ( 10.1002/jbm.a.30254) [DOI] [PubMed] [Google Scholar]
- 95.Salgado CL, Oliveira MB, Mano JF. 2012. Combinatorial cell-3D biomaterials cytocompatibility screening for tissue engineering using bioinspired superhydrophobic substrates. Integr. Biol. 4, 318–327. ( 10.1039/c2ib00170e) [DOI] [PubMed] [Google Scholar]
- 96.Oliveira MB, Ribeiro MP, Miguel SP, Neto AI, Coutinho P, Correia IJ, Mano J. In press. In vivo high-content evaluation of three-dimensional scaffolds biocompatibility. Tissue Eng. Part C, Methods. ( 10.1089/ten.TEC.2013.0738) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Yu J, Gu Y, Du KT, Mihardja S, Sievers RE, Lee RJ. 2009. The effect of injected RGD modified alginate on angiogenesis and left ventricular function in a chronic rat infarct model. Biomaterials 30, 751–756. ( 10.1016/j.biomaterials.2008.09.059) [DOI] [PubMed] [Google Scholar]
- 98.Smidsrod O, Skjak-Braek G. 1990. Alginate as immobilization matrix for cells. Trends Biotechnol. 8, 71–78. ( 10.1016/0167-7799(90)90139-O) [DOI] [PubMed] [Google Scholar]
- 99.Martinsen A, Skjak-Braek G, Smidsrod O. 1989. Alginate as immobilization material: I. Correlation between chemical and physical properties of alginate gel beads. Biotechnol. Bioeng. 33, 79–89. ( 10.1002/bit.260330111) [DOI] [PubMed] [Google Scholar]
- 100.Aslani P, Kennedy RA. 2006. Studies on diffusion in alginate gels. I. Effect of cross-linking with calcium or zinc ions on diffusion of acetaminophen. J. Control. Release 42, 75–82. ( 10.1016/0168-3659(96)01369-7) [DOI] [Google Scholar]
- 101.Tanaka H, Matsumura M, Veliky IA. 1984. Diffusion characteristics of substrates in Ca-alginate gel beads. Biotechnol. Bioeng. 26, 53–58. ( 10.1002/bit.260260111) [DOI] [PubMed] [Google Scholar]
- 102.Kuo CK, Ma PX. 2001. Ionically crosslinked alginate hydrogels as scaffolds for tissue engineering: Part 1. Structure, gelation rate and mechanical properties. Biomaterials 22, 511–521. ( 10.1016/S0142-9612(00)00201-5) [DOI] [PubMed] [Google Scholar]
- 103.Gomez C, Rinaudo M, Villar M. 2007. Oxidation of sodium alginate and characterization of the oxidized derivatives. Carbohydr. Polym. 67, 296–304. ( 10.1016/j.carbpol.2006.05.025) [DOI] [Google Scholar]
- 104.Bouhadir KH, Lee KY, Alsberg E, Damm KL, Anderson KW, Mooney DJ. 2001. Degradation of partially oxidized alginate and its potential application for tissue engineering. Biotechnol. Progress 17, 945–950. ( 10.1021/bp010070p) [DOI] [PubMed] [Google Scholar]
- 105.Boontheekul T, Kong H-J, Mooney DJ. 2005. Controlling alginate gel degradation utilizing partial oxidation and bimodal molecular weight distribution. Biomaterials 26, 2455–2465. ( 10.1016/j.biomaterials.2004.06.044) [DOI] [PubMed] [Google Scholar]
- 106.Kong HJ, Kaigler D, Kim K, Mooney DJ. 2004. Controlling rigidity and degradation of alginate hydrogels via molecular weight distribution. Biomacromolecules 5, 1720–1727. ( 10.1021/bm049879r) [DOI] [PubMed] [Google Scholar]
- 107.Gillette BM, Jensen JA, Wang M, Tchao J, Sia SK. 2010. Dynamic hydrogels: switching of 3D microenvironments using two-component naturally derived extracellular matrices. Adv. Mater. 22, 686–691. ( 10.1002/adma.200902265) [DOI] [PubMed] [Google Scholar]
- 108.Dixon JE, Shah DA, Rogers C, Hall S, Weston N, Parmenter CDJ, McNally D, Denning C, Shakesheff KM. 2014. Combined hydrogels that switch human pluripotent stem cells from self-renewal to differentiation. Proc. Natl Acad. Sci. USA 111, 5580–5585. ( 10.1073/pnas.1319685111) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Bhujbal SV, de Vos P, Niclou SP. 2014. Drug and cell encapsulation: alternative delivery options for the treatment of malignant brain tumors. Adv. Drug Delivery Rev. 67–68, 142–153. ( 10.1016/j.addr.2014.01.010) [DOI] [PubMed] [Google Scholar]
- 110.Gryshkov O, Pogozhykh D, Hofmann N, Glasmacher B. 2013. Cell encapsulation into alginate micro-capsules provides living cells with a mild environment during cryopreservation. Cryobiology 66, 352 ( 10.1016/j.cryobiol.2013.02.043) [DOI] [Google Scholar]
- 111.Rinaudo M. 2006. Chitin and chitosan: properties and applications. Progr. Polym. Sci. 31, 603–632. ( 10.1016/j.progpolymsci.2006.06.001) [DOI] [Google Scholar]
- 112.Ravi Kumar MN. 2000. A review of chitin and chitosan applications. Reactive Funct. Polym. 46, 1–27. ( 10.1016/S1381-5148(00)00038-9) [DOI] [Google Scholar]
- 113.Hayes M, Carney B, Slater J, Brück W. 2008. Mining marine shellfish wastes for bioactive molecules: chitin and chitosan–Part A: extraction methods. Biotechnol. J. 3, 871–877. ( 10.1002/biot.200700197) [DOI] [PubMed] [Google Scholar]
- 114.Liu XF, Guan YL, Yang DZ, Li Z, Yao KD. 2000. Antibacterial action of chitosan and carboxymethylated chitosan. J. Appl. Polym. Sci. 79, 1324–1335. [Google Scholar]
- 115.Percot A, Viton C, Domard A. 2003. Optimization of chitin extraction from shrimp shells. Biomacromolecules 4, 12–18. ( 10.1021/bm025602k) [DOI] [PubMed] [Google Scholar]
- 116.Rinaudo M, Pavlov G, Desbrie J. 1999. Influence of acetic acid concentration on the solubilization of chitosan. Polymer 40, 7029–7032. ( 10.1016/S0032-3861(99)00056-7) [DOI] [Google Scholar]
- 117.Martins GV, Mano JF, Alves NM. 2011. Dual responsive nanostructured surfaces for biomedical applications. Langmuir 27, 8415–8423. ( 10.1021/la200832n) [DOI] [PubMed] [Google Scholar]
- 118.Martins GV, Merino EG, Mano JF, Alves NM. 2010. Crosslink effect and albumin adsorption onto chitosan/alginate multilayered systems: an in situ QCM-D study. Macromol. Biosci. 10, 1444–1455. ( 10.1002/mabi.201000193) [DOI] [PubMed] [Google Scholar]
- 119.Costa NL, Sher P, Mano JF. 2011. Liquefied capsules coated with multilayered polyelectrolyte films for cell immobilization. Adv. Eng. Mater. 13, B218–B224. ( 10.1002/adem.201080138) [DOI] [Google Scholar]
- 120.Puga AM, Lima AC, Mano JF, Concheiro A, Alvarez-Lorenzo C. 2013. Pectin-coated chitosan microgels crosslinked on superhydrophobic surfaces for 5-fluorouracil encapsulation. Carbohydr. Polym. 98, 331–340. ( 10.1016/j.carbpol.2013.05.091) [DOI] [PubMed] [Google Scholar]
- 121.Costa RR, Testera AM, Arias FJ, Rodríguez-Cabello JC, Mano JF. 2013. Layer-by-layer film growth using polysaccharides and recombinant polypeptides: a combinatorial approach. J. Phys. Chem. B 117, 6839–6848. ( 10.1021/jp4028518) [DOI] [PubMed] [Google Scholar]
- 122.Liu W, Sun S, Cao Z, Zhang X, Yao K, Lu WW, Luk KDK. 2005. An investigation on the physicochemical properties of chitosan/DNA polyelectrolyte complexes. Biomaterials 26, 2705–2711. ( 10.1016/j.biomaterials.2004.07.038) [DOI] [PubMed] [Google Scholar]
- 123.Correia CR, Reis RL, Mano JF. 2013. Multilayered hierarchical capsules providing cell adhesion sites. Biomacromolecules 14, 743–751. ( 10.1021/bm301833z) [DOI] [PubMed] [Google Scholar]
- 124.Correia CR, Sher P, Reis RL, Mano JF. 2013. Liquified chitosan-alginate multilayer capsules incorporating poly(l-lactic acid) microparticles as cell carriers. Soft Matter 9, 2125–2130. ( 10.1039/c2sm26784e) [DOI] [Google Scholar]
- 125.Lee KY, Park WH, Ha WS. 1997. Polyelectrolyte complexes of sodium alginate with chitosan or its derivatives for microcapsules. J. Appl. Polym. Sci. 63, 425–432. ( 10.1002/(SICI)1097-4628(19970124)63:4<425::AID-APP3>3.0.CO;2-T) [DOI] [Google Scholar]
- 126.Pangburn S, Trescony P, Heller J. 1982. Lysozyme degradation of partially deacetylated chitin, its films and hydrogels. Biomaterials 3, 105–108. ( 10.1016/0142-9612(82)90043-6) [DOI] [PubMed] [Google Scholar]
- 127.Tomihata K, Ikada Y. 1997. In vitro and in vivo degradation of films of chitin and its deacetylated derivatives. Biomaterials 18, 567–575. ( 10.1016/S0142-9612(96)00167-6) [DOI] [PubMed] [Google Scholar]
- 128.Freier T, Koh HS, Kazazian K, Shoichet MS. 2005. Controlling cell adhesion and degradation of chitosan films by N-acetylation. Biomaterials 26, 5872–5878. ( 10.1016/j.biomaterials.2005.02.033) [DOI] [PubMed] [Google Scholar]
- 129.Chatelet C, Damour O, Domard A. 2001. Influence of the degree of acetylation on some biological properties of chitosan films. Biomaterials 22, 261–268. ( 10.1016/S0142-9612(00)00183-6) [DOI] [PubMed] [Google Scholar]
- 130.Chenite A, Buschmann M, Wang D, Chaput C, Kandani N. 2001. Rheological characterisation of thermogelling chitosan/glycerol-phosphate solutions. Carbohydr. Polym. 46, 39–47. ( 10.1016/S0144-8617(00)00281-2) [DOI] [Google Scholar]
- 131.Chenite A, et al. 2000. Novel injectable neutral solutions of chitosan form biodegradable gels in situ. Biomaterials 21, 2155–2161. ( 10.1016/S0142-9612(00)00116-2) [DOI] [PubMed] [Google Scholar]
- 132.Couto DS, Hong Z, Mano JF. 2009. Development of bioactive and biodegradable chitosan-based injectable systems containing bioactive glass nanoparticles. Acta Biomater. 5, 115–123. ( 10.1016/j.actbio.2008.08.006) [DOI] [PubMed] [Google Scholar]
- 133.Mano JF, et al. 2007. Natural origin biodegradable systems in tissue engineering and regenerative medicine: present status and some moving trends. J. R. Soc. Interface 4, 999–1030. ( 10.1098/rsif.2007.0220) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Jayakumar R, Prabaharan M, Reis R, Mano J. 2005. Graft copolymerized chitosan present status and applications. Carbohydr. Polym. 62, 142–158. ( 10.1016/j.carbpol.2005.07.017) [DOI] [Google Scholar]
- 135.Alves N, Mano J. 2008. Chitosan derivatives obtained by chemical modifications for biomedical and environmental applications. Int. J. Biol. Macromol. 43, 401–414. ( 10.1016/j.ijbiomac.2008.09.007) [DOI] [PubMed] [Google Scholar]
- 136.Bhattarai N, Matsen FA, Zhang M. 2005. PEG-grafted chitosan as an injectable thermoreversible hydrogel. Macromol. Biosci. 5, 107–111. ( 10.1002/mabi.200400140) [DOI] [PubMed] [Google Scholar]
- 137.Prabaharan M, Mano JF. 2006. Stimuli-responsive hydrogels based on polysaccharides incorporated with thermo-responsive polymers as novel biomaterials. Macromol. Biosci. 6, 991–1008. ( 10.1002/mabi.200600164) [DOI] [PubMed] [Google Scholar]
- 138.Gibson W, Sanderson G. 1997. Thickening and gelling agents for food. Boston, MA: Springer US. [Google Scholar]
- 139.Moorhouse R, Colegrove GT, Sanford PA, Baird JK, Kang KS. 1981. Solution properties of polysaccharides, vol. 150 of ACS Symposium Series. Washington, DC: American Chemical Society. [Google Scholar]
- 140.Jansson P-E, Lindberg B, Sandford PA. 1983. Structural studies of gellan gum, an extracellular polysaccharide elaborated by Pseudomonas elodea. Carbohydr. Res. 124, 135–139. ( 10.1016/0008-6215(83)88361-X) [DOI] [Google Scholar]
- 141.Oliveira JT, Martins L, Picciochi R, Malafaya PB, Sousa RA, Neves NM, Mano JF, Reis RL. 2010. Gellan gum: a new biomaterial for cartilage tissue engineering applications. J. Biomed. Mater. Res. A 93, 852–863. ( 10.1002/jbm.a.32574) [DOI] [PubMed] [Google Scholar]
- 142.Miyoshi E, Takaya T, Nishinari K. 1996. Rheological and thermal studies of gel-sol transition in gellan gum aqueous solutions. Carbohydr. Polym. 30, 109–119. ( 10.1016/S0144-8617(96)00093-8) [DOI] [Google Scholar]
- 143.Grasdalen H, Smidsrød O. 1987. Gelation of gellan gum. Carbohydr. Polym. 7, 371–393. ( 10.1016/0144-8617(87)90004-X) [DOI] [Google Scholar]
- 144.Silva-Correia J, Oliveira JMT, Caridade SG, Sousa RA, Mano JF, Reis RL. 2011. Gellan gum-based hydrogels for intervertebral disc tissue-engineering applications. J. Tissue Eng. Regener. Med. 5, 97–107. ( 10.1002/term.363) [DOI] [PubMed] [Google Scholar]
- 145.Ferris CJ, in het Panhuis M. 2009. Conducting bio-materials based on gellan gum hydrogels. Soft Matter 5, 3430–3437. ( 10.1039/b909795c) [DOI] [Google Scholar]
- 146.Coutinho DF, Sant SV, Shin H, Oliveira JT, Gomes ME, Neves NM, Khademhosseini A, Reis RL. 2010. Modified gellan gum hydrogels with tunable physical and mechanical properties. Biomaterials 31, 7494–7502. ( 10.1016/j.biomaterials.2010.06.035) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Oliveira JT, Santos TC, Martins L, Picciochi R, Marques AP, Castro AG, Neves NM, Mano JF, Reis RL. 2010. Gellan gum injectable hydrogels for cartilage tissue engineering applications: in vitro studies and preliminary in vivo evaluation. Tissue Eng. Part A 16, 343–353. ( 10.1089/ten.TEA.2009.0117) [DOI] [PubMed] [Google Scholar]
- 148.Lee H, Fisher S, Kallos MS, Hunter CJ. 2011. Optimizing gelling parameters of gellan gum for fibrocartilage tissue engineering. J. Biomed. Mater. Res. B Appl. Biomat. 98, 238–245. ( 10.1002/jbm.b.31845) [DOI] [PubMed] [Google Scholar]
- 149.Singh BN, Trombetta LD, Kim KH. 2004. Biodegradation behavior of gellan gum in simulated colonic media. Pharm. Dev. Technol. 9, 399–407. ( 10.1081/PDT-200035793) [DOI] [PubMed] [Google Scholar]
- 150.Ljrent C, Hospital RM, Laurent TC, Fraser JR. 1992. Hyaluronan. FASEB J. 6, 2397–2404. [PubMed] [Google Scholar]
- 151.Shiedlin A, et al. 2004. Evaluation of hyaluronan from different sources: Streptococcus zooepidemicus, rooster comb, bovine vitreous, and human umbilical cord. Biomacromolecules 5, 2122–2127. ( 10.1021/bm0498427) [DOI] [PubMed] [Google Scholar]
- 152.Kogan G, Soltés L, Stern R, Gemeiner P. 2007. Hyaluronic acid: a natural biopolymer with a broad range of biomedical and industrial applications. Biotechnol. Lett. 29, 17–25. ( 10.1007/s10529-006-9219-z) [DOI] [PubMed] [Google Scholar]
- 153.Chen WYJ, Abatangelo G. 1999. Functions of hyaluronan in wound repair. Wound Repair Regener. 7, 79–89. ( 10.1046/j.1524-475X.1999.00079.x) [DOI] [PubMed] [Google Scholar]
- 154.Balazs EA. 2004. Viscoelastic properties of hyaluronan and its therapeutic use. In Chemistry and biology of hyaluronan (eds Garg HG, Hales CA.), pp. 415–455. Amsterdam, The Netherlands: Elsevier. [Google Scholar]
- 155.Yeo Y, Highley CB, Bellas E, Ito T, Marini R, Langer R, Kohane DS. 2006. In situ cross-linkable hyaluronic acid hydrogels prevent post-operative abdominal adhesions in a rabbit model. Biomaterials 27, 4698–705. ( 10.1016/j.biomaterials.2006.04.043) [DOI] [PubMed] [Google Scholar]
- 156.Necas J, Bartosikova L, Brauner P, Kolar J. 2008. Hyaluronic acid (hyaluronan): a review. Synthesis 2008, 397–411. [Google Scholar]
- 157.Collins MN, Birkinshaw C. 2007. Comparison of the effectiveness of four different crosslinking agents with hyaluronic acid hydrogel films for tissue-culture applications. J. Appl. Polym. Sci. 104, 3183–3191. ( 10.1002/app.25993) [DOI] [Google Scholar]
- 158.Smeds KA, Grinstaff MW, Pfister-serres A, Miki D, Dastgheib K, Inoue M, Hatchell DL. 2001. Photocrosslinkable polysaccharides for in situ hydrogel formation. J. Biomed. Mater. Res. 54, 115–121. ( 10.1002/1097-4636(200101)54:1<115::AID-JBM14>3.0.CO;2-Q) [DOI] [PubMed] [Google Scholar]
- 159.Seidlits SK, Khaing ZZ, Petersen RR, Nickels JD, Vanscoy JE, Shear JB, Schmidt CE. 2010. The effects of hyaluronic acid hydrogels with tunable mechanical properties on neural progenitor cell differentiation. Biomaterials 31, 3930–3940. ( 10.1016/j.biomaterials.2010.01.125) [DOI] [PubMed] [Google Scholar]
- 160.Bencherif SA, Srinivasan A, Horkay F, Hollinger JO, Matyjaszewski K, Washburn NR. 2008. Influence of the degree of methacrylation on hyaluronic acid hydrogels properties. Biomaterials 29, 1739–1749. ( 10.1016/j.biomaterials.2007.11.047) [DOI] [PubMed] [Google Scholar]
- 161.Park YD, Tirelli N, Hubbell JA. 2003. Photopolymerized hyaluronic acid-based hydrogels and interpenetrating networks. Biomaterials 24, 893–900. ( 10.1016/S0142-9612(02)00420-9) [DOI] [PubMed] [Google Scholar]
- 162.Kim IL, Mauck RL, Burdick JA. 2011. Hydrogel design for cartilage tissue engineering: a case study with hyaluronic acid. Biomaterials 32, 8771–8782. ( 10.1016/j.biomaterials.2011.08.073) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Struve J, Maher PC, Li Y-Q, Kinney S, Fehlings MG, Kuntz C, Sherman LS. 2005. Disruption of the hyaluronan-based extracellular matrix in spinal cord promotes astrocyte proliferation. Glia 52, 16–24. ( 10.1002/glia.20215) [DOI] [PubMed] [Google Scholar]
- 164.Chung C, Burdick JA. 2009. Influence of three-dimensional hyaluronic acid microenvironments on mesenchymal stem cell chondrogenesis. Tissue Eng. Part A 15, 243–254. ( 10.1089/ten.tea.2008.0067) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Pachence JM. 1996. Collagen-based devices for soft tissue repair. J. Biomed. Mater. Res. 33, 35–40. ( 10.1002/(SICI)1097-4636(199621)33:1<35::AID-JBM6>3.0.CO;2-N) [DOI] [PubMed] [Google Scholar]
- 166.Ber S, Torun Köse G, Hasirci V. 2005. Bone tissue engineering on patterned collagen films: an in vitro study. Biomaterials 26, 1977–1986. ( 10.1016/j.biomaterials.2004.07.007) [DOI] [PubMed] [Google Scholar]
- 167.van der Rest M, Garrone R. 1991. Collagen family of proteins. FASEB J. 5, 2814–2823. [PubMed] [Google Scholar]
- 168.Xu T, Molnar P, Gregory C, Das M, Boland T, Hickman JJ. 2009. Electrophysiological characterization of embryonic hippocampal neurons cultured in a 3D collagen hydrogel. Biomaterials 30, 4377–4383. ( 10.1016/j.biomaterials.2009.04.047) [DOI] [PubMed] [Google Scholar]
- 169.Weadock KS, Miller EJ, Keuffel EL, Dunnl MG. 1996. Effect of physical crosslinking methods on collagen-fiber durability in proteolytic solutions. J. Biomed. Mater. Res. 32, 221–226. ( 10.1002/(SICI)1097-4636(199610)32:2<221::AID-JBM11>3.0.CO;2-M) [DOI] [PubMed] [Google Scholar]
- 170.Lee CH, Singla A, Lee Y. 2001. Biomedical applications of collagen. Int. J. Pharm. 221, 1–22. ( 10.1016/S0378-5173(01)00691-3) [DOI] [PubMed] [Google Scholar]
- 171.Sams A, Nixon A. 1995. Chondrocyte-laden collagen scaffolds for resurfacing extensive articular cartilage defects. Osteoarthritis Cartilage 3, 47–59. ( 10.1016/S1063-4584(05)80037-8) [DOI] [PubMed] [Google Scholar]
- 172.Chan BP, Hui TY, Yeung CW, Li J, Mo I, Chan GCF. 2007. Self-assembled collagen-human mesenchymal stem cell microspheres for regenerative medicine. Biomaterials 28, 4652–4666. ( 10.1016/j.biomaterials.2007.07.041) [DOI] [PubMed] [Google Scholar]
- 173.Sittinger M, Hutmacher DW, Risbud MV. 2004. Current strategies for cell delivery in cartilage and bone regeneration. Curr. Opin. Biotechnol. 15, 411–418. ( 10.1016/j.copbio.2004.08.010) [DOI] [PubMed] [Google Scholar]
- 174.MacNeil S. 2008. Biomaterials for tissue engineering of skin. Mater. Today 11, 26–35. ( 10.1016/S1369-7021(08)70087-7) [DOI] [Google Scholar]
- 175.Kuijpers AJ, Engbers GHM, Feijen J, De Smedt SC, Meyvis TKL, Demeester J, Krijgsveld J, Zaat SAJ, Dankert J. 1999. Characterization of the network structure of carbodiimide cross-linked gelatin gels. Macromolecules 32, 3325–3333. ( 10.1021/ma981929v) [DOI] [Google Scholar]
- 176.Young S, Wong M, Tabata Y, Mikos AG. 2005. Gelatin as a delivery vehicle for the controlled release of bioactive molecules. J. Control. Release 109, 256–274. ( 10.1016/j.jconrel.2005.09.023) [DOI] [PubMed] [Google Scholar]
- 177.Hwang CM, Sant S, Masaeli M, Kachouie NN, Zamanian B, Lee S-H, Khademhosseini A. 2010. Fabrication of three-dimensional porous cell-laden hydrogel for tissue engineering. Biofabrication 2, 035003 ( 10.1088/1758-5082/2/3/035003) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Van Den Bulcke A, Bogdanov B, De Rooze N, Schacht EH, Cornelissen M, Berghmans H. 2000. Structural and rheological properties of methacrylamide modified gelatin hydrogels. Biomacromolecules 1, 31–38. ( 10.1021/bm990017d) [DOI] [PubMed] [Google Scholar]
- 179.Mosesson MW. 2005. Fibrinogen and fibrin structure and functions. J. Thromb. Haemost. 3, 1894–1904. ( 10.1111/j.1538-7836.2005.01365.x) [DOI] [PubMed] [Google Scholar]
- 180.Oseni AO, Butler PE, Seifalian AM. 2013. Rapid production of autologous fibrin hydrogels for cellular encapsulation in organ regeneration. Methods Mol. Biol. (Clifton, N.J.) 1001, 145–152. ( 10.1007/978-1-62703-363-3_12) [DOI] [PubMed] [Google Scholar]
- 181.Weisel JW. 2005. Fibrinogen and fibrin. Adv. Protein Chem. 70, 247–299. ( 10.1016/S0065-3233(05)70008-5) [DOI] [PubMed] [Google Scholar]
- 182.Litvinov RI, Gorkun OV, Owen SF, Shuman H, Weisel JW. 2005. Polymerization of fibrin: specificity, strength, and stability of knob-hole interactions studied at the single-molecule level. Blood 106, 2944–2951. ( 10.1182/blood-2005-05-2039) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Ho ST, Cool SM, Hui JH, Hutmacher DW. 2010. The influence of fibrin based hydrogels on the chondrogenic differentiation of human bone marrow stromal cells. Biomaterials 31, 38–47. ( 10.1016/j.biomaterials.2009.09.021) [DOI] [PubMed] [Google Scholar]
- 184.Rowe SL, Lee S, Stegemann JP. 2007. Influence of thrombin concentration on the mechanical and morphological properties of cell-seeded fibrin hydrogels. Acta Biomater. 3, 59–67. ( 10.1016/j.actbio.2006.08.006) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Catelas I, Sese N, Wu BM, Dunn JCY, Helgerson SAM, Tawil B. 2006. Human mesenchymal stem cell proliferation and osteogenic differentiation in fibrin gels in vitro. Tissue Eng. 12, 2385–2396. ( 10.1089/ten.2006.12.2385) [DOI] [PubMed] [Google Scholar]
- 186.Ye Q, Zünd G, Benedikt P, Jockenhoevel S, Hoerstrup SP, Sakyama S, Hubbell JA, Turina M. 2000. Fibrin gel as a three dimensional matrix in cardiovascular tissue engineering. Eur. J. Cardio-Thoracic Surg. 17, 587–591. ( 10.1016/S1010-7940(00)00373-0) [DOI] [PubMed] [Google Scholar]
- 187.Lorentz KM, Kontos S, Frey P, Hubbell JA. 2011. Engineered aprotinin for improved stability of fibrin biomaterials. Biomaterials 32, 430–438. ( 10.1016/j.biomaterials.2010.08.109) [DOI] [PubMed] [Google Scholar]
- 188.Christman KL, Vardanian AJ, Fang Q, Sievers RE, Fok HH, Lee RJ. 2004. Injectable fibrin scaffold improves cell transplant survival, reduces infarct expansion, and induces neovasculature formation in ischemic myocardium. J. Am. College Cardiol. 44, 654–660. ( 10.1016/j.jacc.2004.04.040) [DOI] [PubMed] [Google Scholar]
- 189.Christman KL, Fok HH, Sievers RE, Fang Q, Lee RJ. 2004. Fibrin glue alone and skeletal myoblasts in a fibrin scaffold preserve cardiac function after myocardial infarction. Tissue Eng. 10, 403–409. ( 10.1089/107632704323061762) [DOI] [PubMed] [Google Scholar]
- 190.Zhang X, Wang H, Ma X, Adila A, Wang B, Liu F, Chen B, Wang C, Ma Y. 2010. Preservation of the cardiac function in infarcted rat hearts by the transplantation of adipose-derived stem cells with injectable fibrin scaffolds. Exp. Biol. Med. 235, 1505–1515. ( 10.1258/ebm.2010.010175) [DOI] [PubMed] [Google Scholar]
- 191.Yamada Y, Seong Boo J, Ozawa R, Nagasaka T, Okazaki Y, Hata K-I, Ueda M. 2003. Bone regeneration following injection of mesenchymal stem cells and fibrin glue with a biodegradable scaffold. J. Cranio-Maxillofac. Surg. 31, 27–33. ( 10.1016/S1010-5182(02)00143-9) [DOI] [PubMed] [Google Scholar]
- 192.Costa R, Martín L, Mano J, Rodríguez-Cabello J. 2012. Elastin-like macromolecules. In Biomimetic approaches for biomaterials development, ch. 5 (ed. Mano JAF.), pp. 93–116. Weinheim, Germany: Wiley. [Google Scholar]
- 193.Wise SG, Weiss AS. 2009. Tropoelastin. Int. J. Biochem. Cell Biol. 41, 494–497. ( 10.1016/j.biocel.2008.03.017) [DOI] [PubMed] [Google Scholar]
- 194.Mithieux SM, Weiss AS. 2005. Elastin. Adv. Protein Chem. 70, 437–461. ( 10.1016/S0065-3233(05)70013-9) [DOI] [PubMed] [Google Scholar]
- 195.Sato F, Wachi H, Ishida M, Nonaka R, Onoue S, Urban Z, Starcher BC, Seyama Y. 2007. Distinct steps of cross-linking, self-association, and maturation of tropoelastin are necessary for elastic fiber formation. J. Mol. Biol. 369, 841–851. ( 10.1016/j.jmb.2007.03.060) [DOI] [PubMed] [Google Scholar]
- 196.Betre H, Setton LA, Meyer DE, Chilkoti A. 2002. Characterization of a genetically engineered elastin-like polypeptide for cartilaginous tissue repair. Biomacromolecules 3, 910–916. ( 10.1021/bm0255037) [DOI] [PubMed] [Google Scholar]
- 197.Nettles DL, Chilkoti A, Setton LA. 2010. Applications of elastin-like polypeptides in tissue engineering. Adv. Drug Delivery Rev. 62, 1479–1485. ( 10.1016/j.addr.2010.04.002) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Rodríguez-Cabello JC, Martín L, Alonso M, Arias FJ, Testera AM. 2009. ‘Recombinamers’ as advanced materials for the post-oil age. Polymer 50, 5159–5169. ( 10.1016/j.polymer.2009.08.032) [DOI] [Google Scholar]
- 199.Garcia Y, Hemantkumar N, Collighan R, Griffin M, Rodriguez-Cabello JC, Pandit A. 2009. In vitro characterization of a collagen scaffold enzymatically cross-linked with a tailored elastin-like polymer. Tissue Eng. Part A 15, 887–899. ( 10.1089/ten.tea.2008.0104) [DOI] [PubMed] [Google Scholar]
- 200.Shah M, Hsueh P-Y, Sun G, Chang HY, Janib SM, MacKay JA. 2012. Biodegradation of elastin-like polypeptide nanoparticles. Protein Sci. 21, 743–750. ( 10.1002/pro.2063) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Costa RR, Custódio CA, Testera AM, Arias FJ, Rodríguez-Cabello JC, Alves NM, Mano JF. 2009. Stimuli-responsive thin coatings using elastin-like polymers for biomedical applications. Adv. Funct. Mater. 19, 3210–3218. ( 10.1002/adfm.200900568) [DOI] [Google Scholar]
- 202.Costa RR, Custódio CA, Arias FJ, Rodríguez-Cabello JC, Mano JF. 2011. Layer-by-layer assembly of chitosan and recombinant biopolymers into biomimetic coatings with multiple stimuli-responsive properties. Small 7, 2640–2649. ( 10.1002/smll.201100875) [DOI] [PubMed] [Google Scholar]
- 203.Costa RR, Castro E, Arias FJ, Rodríguez-Cabello JC, Mano JF. 2013. Multifunctional compartmentalized capsules with a hierarchical organization from the nano to the macro scales. Biomacromolecules 14, 2403–2410. ( 10.1021/bm400527y) [DOI] [PubMed] [Google Scholar]
- 204.Servoli E, Maniglio D, Motta A, Migliaresi C. 2008. Folding and assembly of fibroin driven by an AC electric field: effects on film properties. Macromol. Biosci. 8, 827–835. ( 10.1002/mabi.200800057) [DOI] [PubMed] [Google Scholar]
- 205.Michelle G, Agostini M, Moraes D, Cecília A, Rodas D, Zazuco O, Masumi M. 2011. Hydrogels from silk fibroin metastable solution: formation and characterization from a biomaterial perspective. Mater. Sci. Eng. C 31, 997–1001. ( 10.1016/j.msec.2011.02.019) [DOI] [Google Scholar]
- 206.Motta A, Fambri L, Migliaresi C. 2002. Regenerated silk fibroin films: thermal and dynamic mechanical analysis. Macromol. Chem. Phys. 203, 1658–1665. ( 10.1002/1521-3935(200207)203:10/11<1658::AID-MACP1658>3.0.CO;2-3) [DOI] [Google Scholar]
- 207.Kim U-J, Park J, Li C, Jin H-J, Valluzzi R, Kaplan DL. 2004. Structure and properties of silk hydrogels. Biomacromolecules 5, 786–792. ( 10.1021/bm0345460) [DOI] [PubMed] [Google Scholar]
- 208.Matsumoto A, Chen J, Collette AL, Kim U-J, Altman GH, Cebe P, Kaplan DL. 2006. Mechanisms of silk fibroin sol-gel transitions. J. Phys. Chem. B 110, 21 630–21 638. ( 10.1021/jp056350v) [DOI] [PubMed] [Google Scholar]
- 209.Sun W, Incitti T. In press. Genipin-crosslinked gelatin-silk fibroin hydrogels for modulating the behaviour of pluripotent cells. J. Tissue Eng. Regener. Med. ( 10.1002/term.1868) [DOI] [PubMed] [Google Scholar]
- 210.Li M, Ogiso M, Minoura N. 2003. Enzymatic degradation behavior of porous silk fibroin sheets. Biomaterials 24, 357–365. ( 10.1016/S0142-9612(02)00326-5) [DOI] [PubMed] [Google Scholar]
- 211.Lu Q, Zhang B, Li M, Zuo B, Kaplan DL, Huang Y, Zhu H. 2011. Degradation mechanism and control of silk fibroin. Biomacromolecules 12, 1080–1086. ( 10.1021/bm101422j) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Mazzitelli S, Tosi A, Balestra C, Nastruzzi C, Luca G, Mancuso F, Calafiore R, Calvitti M. 2008. Production and characterization of alginate microcapsules produced by a vibrational encapsulation device. J. Biomater. Appl. 23, 123–145. ( 10.1177/0885328207084958) [DOI] [PubMed] [Google Scholar]
- 213.Tian YF, Devgun JM, Collier JH. 2011. Fibrillized peptide microgels for cell encapsulation and 3D cell culture. Soft Matter 7, 6005–6011. ( 10.1039/C1SM05504F) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Xu JH, Li SW, Tan J, Wang YJ, Luo GS. 2006. Preparation of highly monodisperse droplet in a T-junction microfluidic device. AIChE J. 52, 3005–3010. ( 10.1002/aic.10924) [DOI] [Google Scholar]
- 215.Song W, Lima AC, Mano JF. 2010. Bioinspired methodology to fabricate hydrogel spheres for multi-applications using superhydrophobic substrates. Soft Matter 6, 5868–5871. ( 10.1039/c0sm00901f) [DOI] [Google Scholar]
- 216.Oliveira MB, Mano JF. 2011. Polymer-based microparticles in tissue engineering and regenerative medicine. Biotechnol. Progress 27, 897–912. ( 10.1002/btpr.618) [DOI] [PubMed] [Google Scholar]
- 217.Poncelet D, Neufeld RJ, Goosen MFA, Burgarski B, Babak V. 1999. Formation of microgel beads by electric dispersion of polymer solutions. AIChE J. 45, 2018–2023. ( 10.1002/aic.690450918) [DOI] [Google Scholar]
- 218.Popa EG, Gomes ME, Reis RL. 2011. Cell delivery systems using alginate–carrageenan hydrogel beads and fibers for regenerative medicine applications. Biomacromolecules 12, 3952–3961. ( 10.1021/bm200965x) [DOI] [PubMed] [Google Scholar]
- 219.Lasheras JC, Hopfinger EJ. 2000. Liquid jet instability and atomization in a coaxial gas stream. Annu. Rev. Fluid Mech. 32, 275–308. ( 10.1146/annurev.fluid.32.1.275) [DOI] [Google Scholar]
- 220.Zhou H, Xu HHK. 2011. The fast release of stem cells from alginate-fibrin microbeads in injectable scaffolds for bone tissue engineering. Biomaterials 32, 7503–7513. ( 10.1016/j.biomaterials.2011.06.045) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Mazzitelli S, Borgatti M, Breveglieri G, Gambari R, Nastruzzi C. 2011. Encapsulation of eukaryotic cells in alginate microparticles: cell signaling by TNF-alpha through capsular structure of cystic fibrosis cells. J. Cell Commun. Signal. 5, 157–165. ( 10.1007/s12079-010-0105-z) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Sakai S, Ito S, Ogushi Y, Hashimoto I, Hosoda N, Sawae Y, Kawakami K. 2009. Enzymatically fabricated and degradable microcapsules for production of multicellular spheroids with well-defined diameters of less than 150 μm. Biomaterials 30, 5937–5942. ( 10.1016/j.biomaterials.2009.07.031) [DOI] [PubMed] [Google Scholar]
- 223.Prüsse BU, Dalluhn J, Breford J, Vorlop K-D. 2000. Production of spherical beads by JetCutting. Chem. Eng. Technol. 23, 1105–1110. ( 10.1002/1521-4125(200012)23:12<1105::AID-CEAT1105>3.0.CO;2-V) [DOI] [Google Scholar]
- 224.Xie J, Wang C-H. 2007. Electrospray in the dripping mode for cell microencapsulation. J. Colloid Interface Sci. 312, 247–255. ( 10.1016/j.jcis.2007.04.023) [DOI] [PubMed] [Google Scholar]
- 225.Gupta A, Seifalian AM, Ahmad Z, Edirisinghe MJ, Winslet MC. 2007. Novel electrohydrodynamic printing of nanocomposite biopolymer scaffolds. J. Bioactive Compatible Polym. 22, 265–280. ( 10.1177/0883911507078268) [DOI] [Google Scholar]
- 226.Taylor G. 1964. Disintegration of water drops in an electric field. Proc. R. Soc. Lond. A 280, 383–397. ( 10.1098/rspa.1964.0151) [DOI] [Google Scholar]
- 227.Park J-U, et al. 2007. High-resolution electrohydrodynamic jet printing. Nat. Mater. 6, 782–789. ( 10.1038/nmat1974) [DOI] [PubMed] [Google Scholar]
- 228.Selimović V, Oh J, Bae H, Dokmeci M, Khademhosseini A. 2012. Microscale strategies for generating cell-encapsulating hydrogels. Polymers 4, 1554–1579. ( 10.3390/polym4031554) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Xia Y, Whitesides GM. 1998. Soft lithography. Annu. Rev. Mater. Sci. 28, 153–184. ( 10.1146/annurev.matsci.28.1.153) [DOI] [Google Scholar]
- 230.Liu VA, Bhatia SN. 2002. Three-dimensional photopatterning of hydrogels containing living cells. Biomed. Microdevices 4, 257–266. ( 10.1023/A:1020932105236) [DOI] [Google Scholar]
- 231.Shin SR, et al. 2013. Cell-laden microengineered and mechanically tunable hybrid hydrogels of gelatin and graphene oxide. Adv. Mater. 25, 6385–6391. ( 10.1002/adma.201301082) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Lima AC, Sher P, Mano JF. 2012. Production methodologies of polymeric and hydrogel particles for drug delivery applications. Expert Opin. Drug Deliv. 9, 231–248. ( 10.1517/17425247.2012.652614) [DOI] [PubMed] [Google Scholar]
- 233.Elbert DL. 2011. Liquid-liquid two phase systems for the production of porous hydrogels and hydrogel microspheres for biomedical applications: a tutorial review. Acta Biomater. 7, 31–56. ( 10.1016/j.actbio.2010.07.028) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Batorsky A, Liao J, Lund AW, Plopper GE, Stegemann JP. 2005. Encapsulation of adult human mesenchymal stem cells within collagen-agarose microenvironments. Biotechnol. Bioeng. 92, 492–500. ( 10.1002/bit.20614) [DOI] [PubMed] [Google Scholar]
- 235.Suh YK, Kang S. 2010. A review on mixing in microfluidics. Micromachines 1, 82–111. ( 10.3390/mi1030082) [DOI] [Google Scholar]
- 236.Zhang H, Tumarkin E, Sullan RMA, Walker GC, Kumacheva E. 2007. Exploring microfluidic routes to microgels of biological polymers. Macromol. Rapid Commun. 28, 527–538. ( 10.1002/marc.200600776) [DOI] [Google Scholar]
- 237.Garstecki P, Fuerstman MJ, Stone HA, Whitesides GM. 2006. Formation of droplets and bubbles in a microfluidic T-junction-scaling and mechanism of break-up. Lab Chip 6, 437–446. ( 10.1039/b510841a) [DOI] [PubMed] [Google Scholar]
- 238.Garstecki P, Gitlin I, DiLuzio W, Whitesides GM, Kumacheva E, Stone HA. 2004. Formation of monodisperse bubbles in a microfluidic flow-focusing device. Appl. Phys. Lett. 85, 2649 ( 10.1063/1.1796526) [DOI] [Google Scholar]
- 239.Anna SL, Bontoux N, Stone HA. 2003. Formation of dispersions using flow focusing in microchannels. Appl. Phys. Lett. 82, 364 ( 10.1063/1.1537519) [DOI] [Google Scholar]
- 240.Fu T, Ma Y, Funfschilling D, Zhu C, Li HZ. 2010. Squeezing-to-dripping transition for bubble formation in a microfluidic T-junction. Chem. Eng. Sci. 65, 3739–3748. ( 10.1016/j.ces.2010.03.012) [DOI] [Google Scholar]
- 241.Jun Y, Kim MJ, Hwang YH, Jeon EA, Kang AR, Lee S-H, Lee DY. 2013. Microfluidics-generated pancreatic islet microfibers for enhanced immunoprotection. Biomaterials 34, 8122–8130. ( 10.1016/j.biomaterials.2013.07.079) [DOI] [PubMed] [Google Scholar]
- 242.Mironov V, Prestwich G, Forgacs G. 2007. Bioprinting living structures. J. Mater. Chem. 17, 2054–2060. ( 10.1039/b617903g) [DOI] [Google Scholar]
- 243.Guillemot F, Mironov V, Nakamura M. 2010. Bioprinting is coming of age: report from the International Conference on Bioprinting and Biofabrication in Bordeaux (3B'09). Biofabrication 2, 010201 ( 10.1088/1758-5082/2/1/010201) [DOI] [PubMed] [Google Scholar]
- 244.Gasperini L, Maniglio D, Motta A, Migliaresi C. In press. An electrohydrodynamic bioprinter for alginate hydrogels containing living cells. Tissue Eng. C Methods. ( 10.1089/ten.TEC.2014.0149) [DOI] [PubMed] [Google Scholar]
- 245.Billiet T, Gevaert E, De Schryver T, Cornelissen M, Dubruel P. 2014. The 3D printing of gelatin methacrylamide cell-laden tissue-engineered constructs with high cell viability. Biomaterials 35, 49–62. ( 10.1016/j.biomaterials.2013.09.078) [DOI] [PubMed] [Google Scholar]
- 246.Lima AC, Batista P, Valente TAM, Silva AS, Correia IJ, Mano JF. 2013. Novel methodology based on biomimetic superhydrophobic substrates to immobilize cells and proteins in hydrogel spheres for applications in bone regeneration. Tissue Eng. A 19, 1175–1187. ( 10.1089/ten.TEA.2012.0249) [DOI] [PubMed] [Google Scholar]
- 247.Lima AC, Custódio CA, Alvarez-Lorenzo C, Mano JF. 2013. Biomimetic methodology to produce polymeric multilayered particles for biotechnological and biomedical applications. Small 9, 2487–2492. ( 10.1002/smll.201202147) [DOI] [PubMed] [Google Scholar]