

Abstract
The heart is a self-renewing biological pump that converts chemical energy into mechanical energy. The entire process of energy conversion is subject to complex regulation at the transcriptional, translational and post-translational levels. Within this system, energy transfer occurs with high efficiency, facilitated by a series of compound-conserved cycles. At the same time, the constituent myocardial proteins themselves are continuously made and degraded in order to adjust to changes in energy demand and changes in the extracellular environment. We recently have identified signals arising from intermediary metabolism that regulate the cycle of myocardial protein turnover. Using a new conceptual framework, we discuss the principle of metabolic cycles and their importance for refueling and for rebuilding the failing heart.
Introduction
As a self-renewing biological pump, the heart converts chemical to mechanical energy. Although the sources of energy-providing substrates are diverse, for a given physiologic environment, the heart utilizes and/or oxidizes the most efficient fuel to produce adenosine triphosphate (ATP) for contraction. Substrate selection occurs at a transcriptional, translational or post-translational level through the regulation of specific enzymes in specific metabolic pathways. Other factors include substrate availability in general [1], and substrate availability in the specific metabolic environments created by feeding, fasting, exercise, or neurohumoral factors, epitomized by Randle’s “glucose-fatty acid cycle” [2,3]. In other words, the mammalian heart is a metabolic omnivore [4].
Energy substrate metabolism has been a field of active research for more than a century [5]. The new tools of transcript analysis, proteomics and metabolomics have added much to conventional biochemical methods and resulted in the discovery of new metabolic “signatures” and gene regulatory factors [6]. Powerful non-invasive tools, such as magnetic resonance spectroscopy (MRS) and positron emission tomography (PET), have added to the armamentarium [7]. Yet there are still many unanswered questions on the optimal fuel supply and utilization by the heart. Examples of these issues are the concepts of metabolic adaptation and maladaptation, the ongoing debates on glucose protection versus glucotoxicity [8,9], and on lipoprotection versus lipotoxicity [10].
In this article, we are proposing a new approach to metabolism with the aim of providing a conceptual framework for the treatment of heart failure. We consider evidence in support of the hypothesis that the cardiomyocyte is a dynamic structure in which energy transfer is linked to a series of compound-conserved cycles. At the same time, we propose that the heart muscle itself is subject to continuous self-renewal through breakdown and resynthesis of its constituent proteins, and that this cycle of intramyocellular self-renewal may be closely linked to the intermediary metabolism of energy-providing substrates. Alternatively stated, changes in the concentrations of intermediary metabolites such as glucose 6-phospate (G6P), ATP, or adenosine monophosphate (AMP) may regulate rates of protein synthesis or degradation in the heart.
A definition of heart failure
In heart muscle, like in any muscle, contraction and the metabolism of energy-providing substrates are inextricably linked through hydrolysis and rephosphorylation of ATP (Figure 1). Within this framework, it has been proposed that defective metabolism of energy-providing substrates is a cause for contractile dysfunction of the heart [11,12]. While this is undoubtedly the case in the setting of myocardial ischemia, where there is insufficient supply of oxygen for the oxidative phosphorylation of adenosine diphosphate (ADP) to ATP [13,14], and while this is also the case in metabolic derangements such as thiamine deficiency [15], and in mitochondrial disorders [16,17], support for this hypothesis is not as self-evident in other settings of heart failure. It still remains a circular argument as to whether changes in the metabolism of energy-providing substrates are the cause or consequence of contractile dysfunction. Stefan Neubauer has proposed that the failing heart is “an engine out of fuel” [14], meaning that the heart is running out of sources for ATP, the molecular unit of currency for energy transfer. In the non-ischemic failing heart, the supply of energy-providing fuels should never be a rate limiting factor in itself (unless the microcirculation is impaired) [18]. In other words, the heart is supplied with sufficient fuel as long as substrates are delivered in the blood stream and the pathways of fuel metabolism are unimpeded. Not surprisingly, many attempts to treat heart failure with metabolic interventions, by manipulating substrate supply or enzymatic activities, have either not been successful or are inconsistent in their results [19–22]. Why should this be the case?
Figure 1. Energy substrate metabolism and contraction are tightly linked.
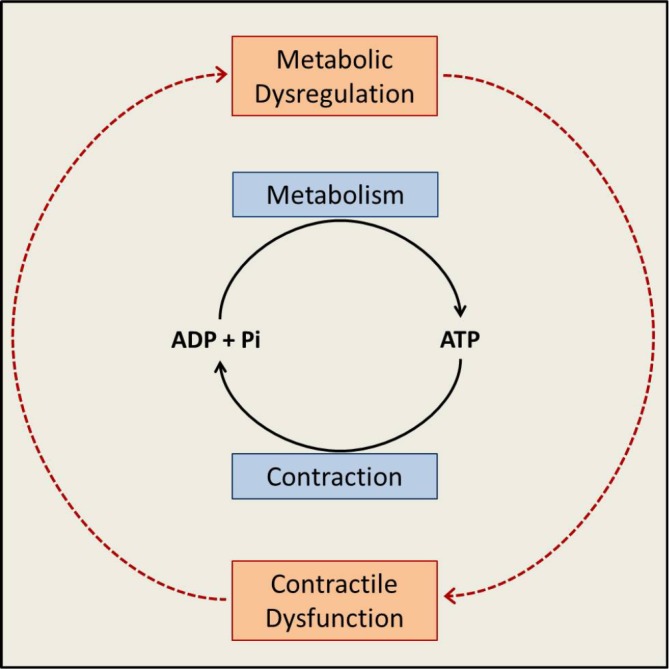
Intermediary metabolism of energy-providing substrates provides the energy needed for rephosphorylation of ADP to ATP. ATP hydrolysis provides the energy for contraction. In the schematic presented here, it is apparent that metabolic dysregulation begets contractile dysfunction and, vice versa, contractile dysfunction begets metabolic dysregulation. The system also reveals that metabolic dysregulation can either be the cause or the consequence of contractile dysfunction. See text for further discussion. Abbreviations: ADP, adenosine diphosphate; ATP, adenosine triphosphate; Pi, inorganic phosphate.
Heart failure and impaired energy transfer go hand in hand. If one defines heart failure as a systemic disorder that begins and ends with the heart, the unimpeded flow of energy is a prerequisite for the normal pump action of the heart. The traditional view is that systemic and organ-specific energy substrate metabolism is impaired when the heart fails. However, this is not the complete picture, as the following example shows.
When Schoenheimer proposed in 1942 that “any biological system represents one great cycle of closely linked chemical reactions”, it was the beginning of a new era of metabolism and metabolic regulation [23]. Using stable isotopes (chiefly 15N and 2H) to assess the fate of nitrogen in the mammalian body, Schoenheimer came to the conclusion that “not only the fuel, but also the structural materials are in a steady state of flux. The classical picture must thus be replaced by one which takes account of the dynamic state of body structure”. In other words, there is nothing static about the cardiomyocyte even though the cell is in the post-mitotic state. However, the structure of a cardiomyocyte is subject to continuous turnover. It is the result of balanced protein synthesis and protein degradation, the dynamics of which will be discussed in more detail below. Here, it suffices to state that the concept of protein turnover is valid for all proteins that make up the cell, although the turnover rate of individual proteins may vary considerably, ranging from minutes to days [24]. Functional and metabolic adaptation of the heart, which requires the adjustment of many enzymes, would not be possible unless enzymes—being proteins—were not continuously turned over. Although rates of synthesis and degradation are apparently independently regulated (see below), they are also linked in some way so that in the normal steady state, rates are maintained equally [24]. For the mammalian heart, we have estimated that the entire organ turns over all of its constituent proteins at least once every 30 days [25].
In this context, we now propose a new metabolic definition of heart failure, which encompasses both the impaired transfer of energy in the heart from energy-providing substrates and the impaired synthesis and degradation of structural and functional myocardial proteins. Although we strongly consider a link between metabolism and protein turnover, it remains still largely unknown which cardio-metabolic changes are causes and which are consequences of impaired contractile function. We propose that a new conceptual framework may help here, and this is described below.
Metabolic cycles in heart failure
Our recent work on protein turnover in the heart has led us to appreciate a full spectrum of metabolic cycles and a prevailing principle in biology: from the Krebs cycle to the cross-bridges, there is no life without cycles. The cycles involved in the transfer of energy from substrates to ATP are well described in textbooks of biochemistry [26], and they also apply to energy transfer in the heart [27]. In the year he died, Krebs wrote on the evolution of metabolic cycles that “the cycle must have evolved because in a competitive environment the chances for survival are greatest if resources are optimal” [28]. In other words, cycles improve efficiency [29]. This principle can be applied to energy metabolism in as much as it follows the laws of physics.
From the above, it is reasonable to conclude that energy metabolism does not consist of unidirectional pathways but rather consists of a series of compound-conserved cycles, some of which are depicted in Figure 2. The principle is derived from a review by Guy Brown [30], but includes the circulation as another compound-conserved cycle and applies to the physiology and pathophysiology of the heart. According to the schematic presented here (Figure 2A), energy transfer in a heart muscle cell begins with the delivery of substrates and oxygen through the circulation. Inside the cell, energy transfer continues with the metabolism of energy-providing substrates that fuel the citric acid cycle. The reactions of the citric acid cycle provide reducing equivalents in the form of reduced nicotinamide dinucleotide (NADH) and reduced flavine adenine dinucleotide (FADH), which, in turn, provide the electrons and the protons for the generation of the proton gradient. The collapse of the proton gradient is coupled to the phosphorylation of ADP and to the generation of H2O. ATP, in turn, provides the energy for contraction. The response of the heart to hemodynamic or neurohumoral stress is accompanied by an increase in flux through all cycles (Figure 2B). In the failing heart, contractile dysfunction of the cardiomyocyte results in a decrease in the circulation of blood, hence, a decrease in the delivery of energy-providing substrates and oxygen to the body, including the heart (Figure 2C). As a consequence, the activity of certain mitochondrial enzymes (but not the respiratory chain) is decreased. Processes that exchange metabolic intermediates (and cations) between the cytosol and mitochondria are part of the flux of energy [31]. A disruption is implicated in the pathogenesis of heart failure, which also applies to a disruption of Krebs cycle flux, and sets up a vicious cycle, which may be interrupted by support of the circulation with a left ventricular assist device (LVAD). We and others have recently reported that mechanical unloading promotes energy recovery in the human heart [32–34]. The improved circulatory support is accompanied by restoration of citric acid flux, most likely through anaplerotic mechanisms [33]. Because, in the same failing heart muscle samples, the same investigators using the same hearts had already shown that respiratory chain activity is preserved [35], it would then follow that the restoration of both substrate and oxygen supply, as well as the citric acid cycle activity, results in an increase in ATP production and increased availability of ATP for contractile function. This line of argument assumes that the contractile apparatus, or the Ca2+ handling proteins, also have not undergone irreversible changes. Indeed, functional and structural remission of heart failure has been reported with the provision of anaplerotic substrates in patients in which heart failure is due to impaired long chain fatty acid oxidation [16] and, more importantly, after the insertion of an LVAD [32,36]. Consequently, great strides are currently being made to unveil the mechanisms regulating “reverse remodeling” of the failing heart. However, many fundamental questions still remain unanswered in this context. For now, we propose that the boost to the circulation provided by the LVAD also restores anaplerotic flux in the mitochondria, while the mechanism for the phenomenon is unknown.
Figure 2. Cycles improve efficiency of energy transfer.
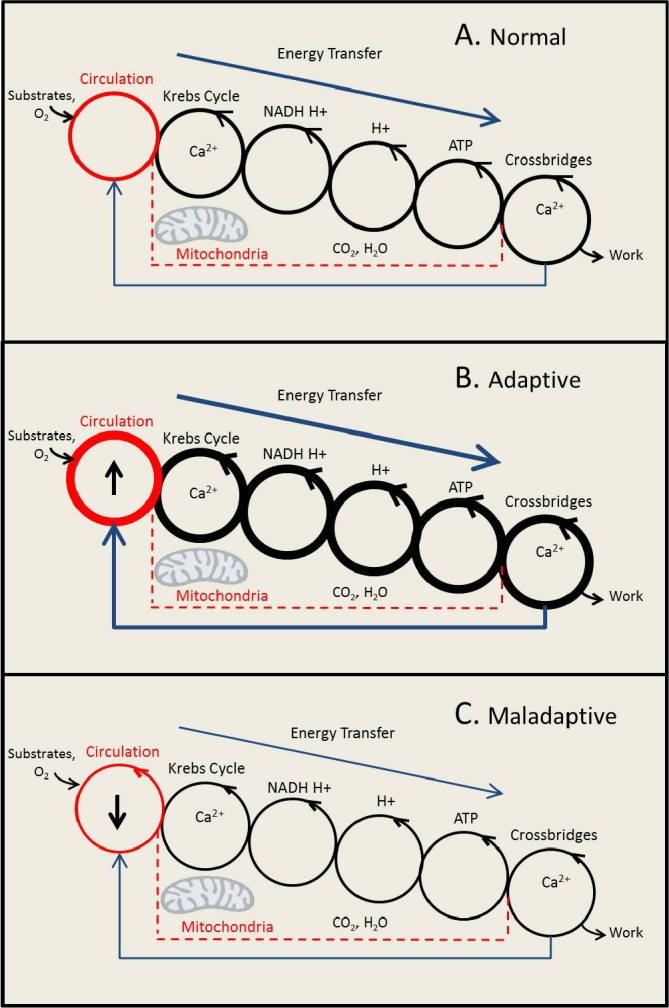
The three panels show six compound-conserved cycles, beginning with the circulation (first cycle on the left) and ending with the cross-bridges in the sarcomeres (last cycle on the right). There are four interlinked cycles in the mitochondria: the Krebs cycle, the NAD+/NADH-H+ and FAD+/FADH-H+ cycle, the build-up and collapse of the proton (H+) gradient across the inner mitochondrial membrane, and the ADP/ATP cycle. Release and reuptake of Ca2+ by the sarcoendoplasmic reticulum regulates cross-bridge formation in the sarcomeres and also regulates dehydrogenase activities of the Krebs cycle in the mitochondria. Panel A depicts a model of normal energy transfer (arrow on top of each panel). Panel B depicts a model of increased energy transfer in the adaptive response to an increase in workload of the heart. Panel C depicts a model of decreased energy transfer in the maladaptive state of heart failure. Note the feedback loop from crossbridges to the circulation (arrow on the bottom of each panel). In the text we propose that boosting the circulation (↑) with mechanical support may restore the flow of energy through the series of interconnected moiety-conserved cycles (lower panel).
Abbreviations: ADP, adenosine diphosphate; ATP, adenosine triphosphate; FAD, flavine adenine dinucleotide; FADH, reduced flavine adenine dinucleotide; NAD, nicotinamide adenine dinucleotide; NADH, reduced nicotinamide dinucleotide.
The cycle of intramyocardial protein turnover
There are many other dimensions to the concept of biological cycles in the heart. Of immediate relevance is that intracellular protein synthesis and degradation also represents a cycle (Figure 3). Even though the rate of turnover for this cycle cannot be compared to the rapid turnover rate of metabolic cycles, the principle is the same. It seems reasonable to propose that varying the rate of turnover for specific myocardial proteins allows the heart to respond and adapt to environmental changes. We propose that altered rates of myocardial protein turnover underlie the concept of cardiac plasticity [37], and are mediated by specific metabolic signals. Increasing evidence suggests that defective myocardial protein turnover results in proteotoxicity, which is an underlying feature of heart failure [25,38–40]. Another, better known, aspect of regenerative biology merits consideration in this context. At present, cellular regeneration of the heart occupies center stage in cardiovascular medicine [41–43]. The principle begins with programmed cell death (or apoptosis) as cause for replacement fibrosis, cardiac dilatation and contractile dysfunction [44] and finds its culmination in various forms of cell-based regenerative therapies (stem cells) leading to improved contractile function [45]. In contrast, the concepts of intracellular self-renewal of the cardiomyocyte, the dynamic processes of intracellular protein turnover, and heart failure as a consequence of protein quality control, are only considered by a minority [38,40]. Stem cells contribute little to the renewal of cardiomyocytes in the normal aging process [40,46]. The idea that the cardiomyocyte is a dynamic structure that continuously renews itself from within is still fairly new, as is the idea of proteotoxicity causing cardiac dysfunction [40]. By “self-renewal” we stress intracellular, rather than cellular, self-renewal. Inside the cardiomyocyte, damaged or useless proteins are degraded and replaced by new and functional proteins. Below, we discuss how metabolic stress may affect protein homeostasis in the cardiomyocyte. However, in order to do this, we first wish to consider the broader concept of metabolic homeostasis.
Figure 3. Structure and function of the cardiomyoctye is determined by the balance of protein synthesis and protein degradation.
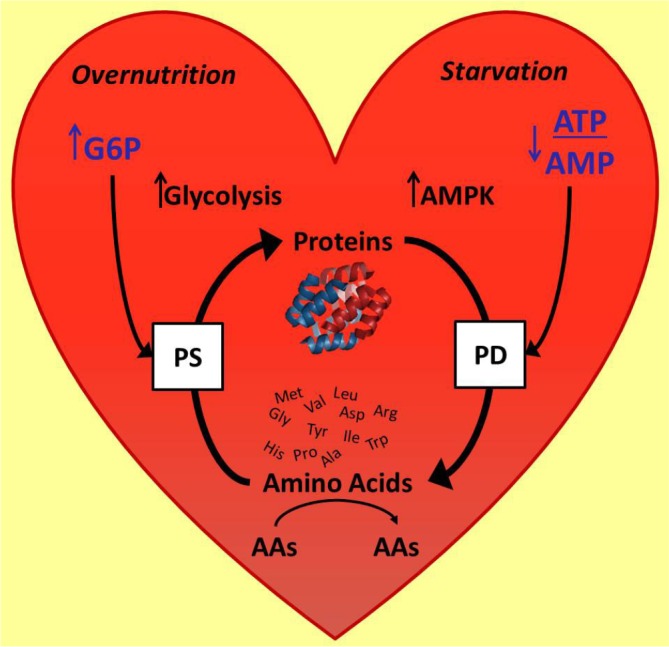
Activation of the enzyme AMPK by a decrease in the ATP/AMP ratio increases protein degradation, while the glycolytic intermediate, G6P, regulates the nutrient sensor mTOR and protein synthesis. Of note, amino acids themselves are also metabolized and some may serve as regulators of protein synthesis.
Abbreviations: AAs, Amino Acids; AMP, adenosine monophosphate; AMPK, 5’AMP activated kinase; ATP, adenosine triphosphate; G6P, glucose 6-phosphate; mTOR, mammalian target of rapamycin; PD, protein degredation; PS, protein systhesis.
Homeostasis and metabolic stress
The concept of cellular homeostasis means that the cell maintains a delicate balance between fuel uptake and fuel utilization as well as synthesis and degradation of its constituent proteins. In other words, the cell tends to maintain a stable condition by regulating its internal environment in the face of a highly variable external environment [47]. The concept of homeostasis was preceded by Claude Bernard’s “milieu intérieur” [48] and was subsequently introduced by Walter Cannon in 1932 as the self-regulating process that all biological systems obey [47].
Cannon’s Wisdom of the Body [47] also applies to the wisdom of the cell [49]. Cellular metabolism obeys the concept of homeostasis through pathways that seek to maintain a delicate balance between energy production and energy utilization despite changes in the external environment. In an effort to maintain homeostasis, the heart’s initial response to stress, irrespective of the stressor, is to adapt its metabolic machinery to meet the energy needs of contraction [5]. A disruption in myocardial energy homeostasis is fundamental to the development of many disease states of the heart. A key feature of heart failure is the dysregulated metabolism of energy-providing substrates and of its constituent proteins. This change in the metabolic milieu imposes a metabolic stress on the heart akin to, but different from, hemodynamic or neurohumoral stress. The key point is the accumulation of intermediary metabolites inside the cardiomyocyte that are potential regulators of myocardial protein synthesis and degradation.
Aside from ischemia, metabolic stress is the consequence of either nutrient deprivation [50] or nutrient oversupply [51–53]. On the one hand, in the fully oxygenated heart, a decrease in fuel supply results in cardiac atrophy [50,54]. On the other hand, an oversupply of glucose or amino acids results in the activation of the mammalian target of rapamycin (mTOR) pathway [51,55]. One of the consequences of systemic overnutrition is substrate activation of insulin secretion and of the insulin signaling pathway and, consequently, activation of the insulin signaling pathway in peripheral tissues [56]. The metabolic signals arising from either substrate deprivation or substrate overload and their effects on the cycle of myocardial protein turnover are shown in Figure 3.
Undernutrition: enhanced myocardial protein degradation as a consequence of nutrient deprivation
In 1983, the death of an iconic American singer, Karen Carpenter, brought world-wide attention to the dangers of eating disorders. Her death was secondary to heart failure, a complication of her long standing battle with anorexia nervosa [57]. Thirty years later, the mechanism by which nutrient deprivation results in heart failure is still a matter of speculation.
Recent work on systemic ketone body metabolism and its relationship to cardiac metabolism provides new insight into the potential role that ketone body metabolism plays on the structure and function of the heart, and elegantly displays the dynamic relationships within interwoven metabolic networks. The three ketone bodies, acetoacetate, beta-hydroxybutyrate, and acetone, are all predigested fatty acids that are produced by the liver during starvation [57] when the capacity of the citric acid cycle is limited by the availability of oxaloacetate, which is rerouted to gluconeogenesis. In the heart, ketone bodies affect protein homeostasis due to their ability to serve as energy-providing substrates and to be used as such in the place of glucose [58–60]. Kochel et al. [54], and earlier observations by Owen et al. [61], had shown that with prolonged fasting, ketone bodies, specifically beta-hydroxybutyrate and acetoacetate, displace glucose oxidation in the brain, thereby sparing gluconeogenesis and, in turn, sparing body protein. In the isolated working rat heart, however, when ketone bodies are present as the only substrate, ketone bodies induce contractile dysfunction due to the reversible impairment of citric acid cycle flux, which is reversed by glucose and/or pyruvate [1,62,63]. Both are anaplerotic substrates. A mixture of glucose and non-carbohydrate substrates not only restores citric acid cycle flux but also elevates G6P levels, which activate mTOR and protein synthesis (see below) [51,53].
Extreme nutrient deprivation ex vivo or starvation in vivo turns on cycles of protein degradation. In rabbit heart, nutrient deprivation for 7 days decreases protein synthesis and increases protein degradation in the heart in vivo [50]. Starvation decreases the intracellular concentration of ATP and subsequently, the enzyme 5’ activated AMP kinase (AMPK) is activated to provide energy to maintain normal cellular function by fueling the Krebs Cycle with amino acids [64]. The line of reasoning is as follows: with substrate deprivation, ATP levels fall while AMP levels rise, resulting in the activation of AMPK. AMPK regulates not only energy substrate metabolism [65] but also inhibits protein synthesis [66] and regulates transcription of a number of metabolic genes [67,68]. Starvation also induces autophagy and proteasome-mediated protein degradation in cardiomyocytes through AMPK [69,70]. AMPK is a regulator of the ubiquitin ligases Atrogin-1 and MuRF1, mediators of cardiac protein degradation and cardiac atrophy, which are both increased with starvation [71]. The transcription and regulation of AMPK by ubiquitin ligases allows for the liberation of nutrients at the expense of cardiac mass and contractile function [72]. In other words, the cell survives intact but has shrunk to essential constituents. We propose that restoring fuel homeostasis will also restore the cycle of protein turnover.
Overnutrition: modification of myocardial proteins by the oversupply of nutrients
Of four classes of energy-providing substrates for the heart—fat, carbohydrates (glucose and lactate), ketone bodies, and amino acids [5]—we wish to consider only dysregulated glucose metabolism in more detail.
We have proposed that metabolic stress and the ensuing metabolic signals contribute to structural remodeling of the cardiomyocyte, an adaptive response to an altered metabolic milieu. The supporting evidence is that metabolic signals directly activate pathways of myocardial protein turnover [53]. Now, we propose that impaired myocardial protein turnover is a consequence of chronic derangements in fuel supply, and a potential key player in the development of non-ischemic heart failure (i.e. diabetic or lipotoxic cardiomyopathy). Dysregulated glucose metabolism adversely affects myocardial protein turnover and overall cardiac function [53].
We have new evidence supporting the idea that metabolic remodeling regulates structural and functional remodeling of the heart. Insulin is a regulator of cardiac mass via the mTOR pathway. Firstly, we have found that the activation of the insulin signaling pathway is tied to the metabolism of glucose in the glycolytic pathway [51]. Secondly, we found that rat hearts subjected to increased workload had enhanced glucose uptake and increased activation of mTOR, a known regulator of protein synthesis and cardiac growth. This led us to postulate that intermediates of glucose metabolism act as metabolic signals to induce protein synthesis in the heart. Indeed, “load-induced” hypertrophic signaling through mTOR complex 1 (mTORC1) is glucose dependent and mediated by G6P. A similar phenomenon exists in hearts deficient in fatty acyl-CoA synthetase, which are dependent on glucose metabolism [73].
The new data also suggest that G6P-mediated mTOR activation leads to endoplasmic reticulum (ER) stress and impairs contractile function in hearts subject to high workload and supplied with glucose. When subjected to hemodynamic stress, the heart increases its reliance on glucose by returning to the fetal gene program, which favors uptake and oxidation of glucose. However, rates of glucose uptake now exceed rates of glucose oxidation and result in the accumulation of G6P [53]. We have reasoned that the observed increase in unfolded protein response results from the sustained activation of mTOR [53]. This causes increased rates of protein synthesis, which overwhelm the ER with unfolded and misfolded proteins beyond its capacity to cope with the load, leading to the ER stress response [53,74]. The functional consequences of ER stress and the unfolded protein response in the heart (in terms of protein synthesis and degradation, cell growth and metabolism regulation) are manifold, and can be both adaptive and maladaptive [75]. In 2012, a thrombospondrin-dependent pathway for a protective ER stress response was discovered [76]. In our hands, rates of glucose uptake in excess of rates of glucose oxidation by the heart promote dysregulated protein synthesis and ER stress, which contributes to an impairment of contractile function [53]. Irrespective of the adaptive or maladaptive role of the ER stress response, our observations prompted us to propose that insulin resistance is a protective mechanism for the heart ameliorating the consequences of myocardial fuel overload [77]. In short, the heart develops mechanisms to protect itself from the consequences of fuel toxicity and the consequent adverse effects on intracellular homeostasis.
Conclusions and outlook
We have described the heart as a biological pump that maintains intracellular homeostasis through a variety of cycles. We have described the flux of energy from energy-providing substrates to the crossbridges of actin and myosin, which is facilitated by a series of content-conserved cycles as the most efficient form of energy transfer. At the same time, the cycle of synthesis and degradation of myocardial proteins offers the postmitotic cardiomyocyte a means to respond to a wide range of environmental changes, by destroying unneeded or defective proteins and replacing them with new, functional proteins. In short, the biological system of the cardiomyocyte represents one big cycle of closely linked chemical reactions [23]. Targeting the biological cycles described in this review may offer new opportunities to rebuild the failing heart. To quote Steven McKnight, “The one field etiolated by the cloud of molecular biology has been metabolism” [78]. In the biology of the heart, it seems that interest in all forms of metabolism begins to re-emerge because metabolism is indeed the missing link between the structure and function of the heart. Rethinking cardiac metabolism and resting metabolic cycles to refuel and rebuild the failing heart may be a reasonable way to exploit the power of metabolism.
Acknowledgments
We thank Mrs. Roxy A. Tate for her expert editorial assistance, the reviewers for excellent suggestions and the NIH for grant support (5R01HL061483).
Abbreviations
- ADP
adenosine diphosphate
- AMP
adenosine monophosphate
- AMPK
5’AMP activated kinase
- ATP
adenosine triphosphate
- ER
endoplasmic reticulum
- FADH
reduced flavine adenine dinucleotide
- G6P
glucose 6-phosphate
- LVAD
left ventricular assist device
- MRS
magnetic resonance spectroscopy
- mTOR
mammalian target of rapamycin
- mTORC1
mTOR complex 1
- NADH
reduced nicotinamide dinucleotide
- PET
positron emission tomography
- Pi
inorganic phosphate
Disclosures
The authors declare that they have no disclosures.
The electronic version of this article is the complete one and can be found at: http://f1000.com/prime/reports/m/6/90
References
- 1.Taegtmeyer H, Hems R, Krebs HA. Utilization of energy-providing substrates in the isolated working rat heart. Biochem J. 1980;186:701–11. doi: 10.1042/bj1860701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Randle PJ, Garland PB, Hales CN, Newsholme EA. The glucose fatty-acid cycle. Its role in insulin sensitivity and the metabolic disturbances of diabetes mellitus. Lancet. 1963;1:785–9. doi: 10.1016/S0140-6736(63)91500-9. [DOI] [PubMed] [Google Scholar]
- 3.Hue L, Taegtmeyer H. The Randle cycle revisited: a new head for an old hat. Am J Physiol Endocrinol Metab. 2009;297:E578–91. doi: 10.1152/ajpendo.00093.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Taegtmeyer H. Carbohydrate interconversions and energy production. Circulation. 1985;72:IV1–8. [PubMed] [Google Scholar]
- 5.Taegtmeyer H. Energy metabolism of the heart: from basic concepts to clinical applications. Curr Probl Cardiol. 1994;19:59–113. doi: 10.1016/0146-2806(94)90008-6. [DOI] [PubMed] [Google Scholar]
- 6.Scarpulla RC, Vega RB, Kelly DP. Transcriptional integration of mitochondrial biogenesis. Trends Endocrinol Metab. 2012;23:459–66. doi: 10.1016/j.tem.2012.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gropler RJ. Recent advances in metabolic imaging. J Nucl Cardiol. 2013;20:1147–72. doi: 10.1007/s12350-013-9786-z. [DOI] [PubMed] [Google Scholar]
- 8.Depre C, Taegtmeyer H. Metabolic aspects of programmed cell survival and cell death in the heart. Cardiovasc Res. 2000;45:538–48. doi: 10.1016/S0008-6363(99)00266-7. [DOI] [PubMed] [Google Scholar]
- 9.Young ME, McNulty P, Taegtmeyer H. Adaptation and maladaptation of the heart in diabetes: Part II: potential mechanisms. Circulation. 2002;105:1861–70. doi: 10.1161/01.CIR.0000012467.61045.87. [DOI] [PubMed] [Google Scholar]
- 10.Taegtmeyer H, Stanley WC. Too much or not enough of a good thing? Cardiac glucolipotoxicity versus lipoprotection. J Mol Cell Cardiol. 2011;50:2–5. doi: 10.1016/j.yjmcc.2010.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Taegtmeyer H. Cardiac metabolism as a target for the treatment of heart failure. Circulation. 2004;110:894–6. doi: 10.1161/01.CIR.0000139340.88769.D5. [DOI] [PubMed] [Google Scholar]
- 12.Beadle RM, Frenneaux M. Modification of myocardial substrate utilisation: a new therapeutic paradigm in cardiovascular disease. Heart. 2010;96:824–30. doi: 10.1136/hrt.2009.190256. [DOI] [PubMed] [Google Scholar]
- 13.Gorlin R. Coronary artery disease. Philadelphia: W.B. Saunders Co; 1976. [Google Scholar]
- 14.Neubauer S. The failing heart--an engine out of fuel. N Engl J Med. 2007;356:1140–51. doi: 10.1056/NEJMra063052. [DOI] [PubMed] [Google Scholar]; http://f1000.com/prime/718541548
- 15.Dinicolantonio JJ, Lavie CJ, Niazi AK, O'Keefe JH, Hu T. Effects of thiamine on cardiac function in patients with systolic heart failure: systematic review and metaanalysis of randomized, double-blind, placebo-controlled trials. Ochsner J. 2013;13:495–9. [PMC free article] [PubMed] [Google Scholar]; http://f1000.com/prime/718541551
- 16.Roe CR, Sweetman L, Roe DS, David F, Brunengraber H. Treatment of cardiomyopathy and rhabdomyolysis in long-chain fat oxidation disorders using an anaplerotic odd-chain triglyceride. J Clin Invest. 2002;110:259–69. doi: 10.1172/JCI0215311. [DOI] [PMC free article] [PubMed] [Google Scholar]; http://f1000.com/prime/718541552
- 17.Bohles H, Sewell AC. Metabolic cardiomyopathy. Stuttgart: Medpharm Scientific Publishers; 2004. [Google Scholar]
- 18.Taegtmeyer H. The failing heart. N Engl J Med. 2007;356:2545–6. author reply 2546. [PubMed] [Google Scholar]
- 19.Lincoff AM, Wolski K, Nicholls SJ, Nissen SE. Pioglitazone and risk of cardiovascular events in patients with type 2 diabetes mellitus: a meta-analysis of randomized trials. JAMA. 2007;298:1180–8. doi: 10.1001/jama.298.10.1180. [DOI] [PubMed] [Google Scholar]
- 20.Singh S, Loke YK, Furberg CD. Long-term risk of cardiovascular events with rosiglitazone: a meta-analysis. JAMA. 2007;298:1189–95. doi: 10.1001/jama.298.10.1189. [DOI] [PubMed] [Google Scholar]
- 21.Graham DJ, Ouellet-Hellstrom R, MaCurdy TE, Ali F, Sholley C, Worrall C, Kelman JA. Risk of acute myocardial infarction, stroke, heart failure, and death in elderly Medicare patients treated with rosiglitazone or pioglitazone. JAMA. 2010;304:411–8. doi: 10.1001/jama.2010.920. [DOI] [PubMed] [Google Scholar]; http://f1000.com/prime/5325956
- 22.Selker HP, Beshansky JR, Sheehan PR, Massaro JM, Griffith JL, D’Agostino RB, Ruthazer R, Atkins JM, Sayah AJ, Levy MK, Richards ME, Aufderheide TP, Braude DA, Pirrallo RG, Doyle DD, Frascone RJ, Kosiak DJ, Leaming JM, Van Gelder, Carin M, Walter G, Wayne MA, Woolard RH, Opie LH, Rackley CE, Apstein CS, Udelson JE. Out-of-hospital administration of intravenous glucose-insulin-potassium in patients with suspected acute coronary syndromes: the IMMEDIATE randomized controlled trial. JAMA. 2012;307:1925–33. doi: 10.1001/jama.2012.426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schoenheimer R. The dynamic state of body constituents. Cambridge, MA: Harvard University Press; 1942. [Google Scholar]
- 24.Waterlow JC, Garlick PJ, Millward DJ. Protein turnover in mammalian tissues and in the whole body. Amsterdam: North-Holland Publishing Company; 1978. [Google Scholar]
- 25.Razeghi P, Taegtmeyer H. Cardiac remodeling: UPS lost in transit. Circ Res. 2005;97:964–6. doi: 10.1161/01.RES.0000193563.53859.3e. [DOI] [PubMed] [Google Scholar]
- 26.Lehninger AL. Biochemistry. New York: Worth Publishers; 1970. [Google Scholar]
- 27.Taegtmeyer H. Tracing cardiac metabolism in vivo: one substrate at a time. J Nucl Med. 2010;51(Suppl 1):80S–87S. doi: 10.2967/jnumed.109.068205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Baldwin JE, Krebs H. The evolution of metabolic cycles. Nature. 1981;291:381–2. doi: 10.1038/291381a0. [DOI] [PubMed] [Google Scholar]; http://f1000.com/prime/718541553
- 29.Racker E. Energy cycles in health and disease. Curr Top Cell Regul. 1981;18:361–76. doi: 10.1016/B978-0-12-152818-8.50027-X. [DOI] [PubMed] [Google Scholar]
- 30.Brown GC. Control of respiration and ATP synthesis in mammalian mitochondria and cells. Biochem J. 1992;284(Pt 1):1–13. doi: 10.1042/bj2840001. [DOI] [PMC free article] [PubMed] [Google Scholar]; http://f1000.com/prime/718541554
- 31.Carley AN, Taegtmeyer H, Lewandowski ED. Matrix revisited: mechanisms linking energy substrate metabolism to the function of the heart. Circ Res. 2014;114:717–29. doi: 10.1161/CIRCRESAHA.114.301863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Razeghi P, Myers TJ, Frazier OH, Taegtmeyer H. Reverse remodeling of the failing human heart with mechanical unloading. Emerging concepts and unanswered questions. Cardiology. 2002;98:167–74. doi: 10.1159/000067313. [DOI] [PubMed] [Google Scholar]
- 33.Gupte AA, Hamilton DJ, Cordero-Reyes AM, Youker KA, Yin Z, Estep JD, Stevens RD, Wenner B, Ilkayeva O, Loebe M, Peterson LE, Lyon CJ, Wong Stephen TC, Newgard CB, Torre-Amione G, Taegtmeyer H, Hsueh WA. Mechanical unloading promotes myocardial energy recovery in human heart failure. Circ Cardiovasc Genet. 2014;7:266–76. doi: 10.1161/CIRCGENETICS.113.000404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chokshi A, Drosatos K, Cheema FH, Ji R, Khawaja T, Yu S, Kato T, Khan R, Takayama H, Knöll R, Milting H, Chung CS, Jorde U, Naka Y, Mancini DM, Goldberg IJ, Schulze PC. Ventricular assist device implantation corrects myocardial lipotoxicity, reverses insulin resistance, and normalizes cardiac metabolism in patients with advanced heart failure. Circulation. 2012;125:2844–53. doi: 10.1161/CIRCULATIONAHA.111.060889. [DOI] [PMC free article] [PubMed] [Google Scholar]; http://f1000.com/prime/718541555
- 35.Cordero-Reyes AM, Gupte AA, Youker KA, Loebe M, Hsueh WA, Torre-Amione G, Taegtmeyer H, Hamilton DJ. Freshly isolated mitochondria from failing human hearts exhibit preserved respiratory function. J Mol Cell Cardiol. 2014;68:98–105. doi: 10.1016/j.yjmcc.2013.12.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Müller J, Wallukat G, Weng YG, Dandel M, Spiegelsberger S, Semrau S, Brandes K, Theodoridis V, Loebe M, Meyer R, Hetzer R. Weaning from mechanical cardiac support in patients with idiopathic dilated cardiomyopathy. Circulation. 1997;96:542–9. doi: 10.1161/01.CIR.96.2.542. [DOI] [PubMed] [Google Scholar]; http://f1000.com/prime/718541556
- 37.Hill JA, Olson EN. Cardiac plasticity. N Engl J Med. 2008;358:1370–80. doi: 10.1056/NEJMra072139. [DOI] [PubMed] [Google Scholar]; http://f1000.com/prime/718541557
- 38.Wang X, Robbins J. Heart failure and protein quality control. Circ Res. 2006;99:1315–28. doi: 10.1161/01.RES.0000252342.61447.a2. [DOI] [PubMed] [Google Scholar]; http://f1000.com/prime/718541558
- 39.Del Monte F, Hajjar RJ. Intracellular devastation in heart failure. Heart Fail Rev. 2008;13:151–62. doi: 10.1007/s10741-007-9071-9. [DOI] [PubMed] [Google Scholar]
- 40.Willis MS, Patterson C. Proteotoxicity and cardiac dysfunction--Alzheimer's disease of the heart? N Engl J Med. 2013;368:455–64. doi: 10.1056/NEJMra1106180. [DOI] [PubMed] [Google Scholar]; http://f1000.com/prime/718541560
- 41.Chien KR. Regenerative medicine and human models of human disease. Nature. 2008;453:302–5. doi: 10.1038/nature07037. [DOI] [PubMed] [Google Scholar]
- 42.Mazhari R, Hare JM. Translational findings from cardiovascular stem cell research. Trends Cardiovasc Med. 2012;22:1–6. doi: 10.1016/j.tcm.2012.05.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Anversa P, Kajstura J, Rota M, Leri A. Regenerating new heart with stem cells. J Clin Invest. 2013;123:62–70. doi: 10.1172/JCI63068. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 44.Sabbah HN, Sharov VG, Goldstein S. Cell death, tissue hypoxia and the progression of heart failure. Heart Fail Rev. 2000;5:131–8. doi: 10.1023/A:1009880720032. [DOI] [PubMed] [Google Scholar]
- 45.Sanganalmath SK, Bolli R. Cell therapy for heart failure: a comprehensive overview of experimental and clinical studies, current challenges, and future directions. Circ Res. 2013;113:810–34. doi: 10.1161/CIRCRESAHA.113.300219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hsieh Patrick CH, Segers Vincent FM, Davis ME, MacGillivray C, Gannon J, Molkentin JD, Robbins J, Lee RT. Evidence from a genetic fate-mapping study that stem cells refresh adult mammalian cardiomyocytes after injury. Nat Med. 2007;13:970–4. doi: 10.1038/nm1618. [DOI] [PMC free article] [PubMed] [Google Scholar]; http://f1000.com/prime/718541561
- 47.Cannon WB. The wisdom of the body. New York: W.W. Norton and Company; 1932. [Google Scholar]
- 48.Gross CG. Claude bernard and the constancy of the internal environment. Neuroscientist. 1998;4:380–5. doi: 10.1177/107385849800400520. [DOI] [Google Scholar]
- 49.Thomas L. The lives of a cell: Notes of a biology watcher. New York: Viking Press; 1974. [Google Scholar]
- 50.Samarel AM, Parmacek MS, Magid NM, Decker RS, Lesch M. Protein synthesis and degradation during starvation-induced cardiac atrophy in rabbits. Circ Res. 1987;60:933–41. doi: 10.1161/01.RES.60.6.933. [DOI] [PubMed] [Google Scholar]
- 51.Sharma S, Guthrie PH, Chan SS, Haq S, Taegtmeyer H. Glucose phosphorylation is required for insulin-dependent mTOR signalling in the heart. Cardiovasc Res. 2007;76:71–80. doi: 10.1016/j.cardiores.2007.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Proud CG. mTORC1 regulates the efficiency and cellular capacity for protein synthesis. Biochem Soc Trans. 2013;41:923–6. doi: 10.1042/BST20130036. [DOI] [PubMed] [Google Scholar]; http://f1000.com/prime/718067234
- 53.Sen S, Kundu BK, Wu HC, Hashmi SS, Guthrie P, Locke LW, Roy RJ, Matherne GP, Berr SS, Terwelp M, Scott B, Carranza S, Frazier OH, Glover DK, Dillmann WH, Gambello MJ, Entman ML, Taegtmeyer H. Glucose regulation of load-induced mTOR signaling and ER stress in mammalian heart. J Am Heart Assoc. 2013;2:e004796. doi: 10.1161/JAHA.113.004796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kochel PJ, Kira Y, Gordon EE, Morgan HE. Effects of noncarbohydrate substrates on protein synthesis in hearts from fed and fasted rats. J Mol Cell Cardiol. 1984;16:371–83. doi: 10.1016/S0022-2828(84)80608-2. [DOI] [PubMed] [Google Scholar]
- 55.Harmancey R, Wilson CR, Taegtmeyer H. Adaptation and maladaptation of the heart in obesity. Hypertension. 2008;52:181–7. doi: 10.1161/HYPERTENSIONAHA.108.110031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Morgan HE, Earl DC, Broadus A, Wolpert EB, Giger KE, Jefferson LS. Regulation of protein synthesis in heart muscle. I. Effect of amino acid levels on protein synthesis. J Biol Chem. 1971;246:2152–62. [PubMed] [Google Scholar]
- 57.Cahill GF. Starvation in man. N Engl J Med. 1970;282:668–75. doi: 10.1056/NEJM197003192821209. [DOI] [PubMed] [Google Scholar]
- 58.Crawford PA, Crowley JR, Sambandam N, Muegge BD, Costello EK, Hamady M, Knight R, Gordon JI. Regulation of myocardial ketone body metabolism by the gut microbiota during nutrient deprivation. Proc Natl Acad Sci USA. 2009;106:11276–81. doi: 10.1073/pnas.0902366106. [DOI] [PMC free article] [PubMed] [Google Scholar]; http://f1000.com/prime/718541562
- 59.Wentz AE, d'Avignon DA, Weber ML, Cotter DG, Doherty JM, Kerns R, Nagarajan R, Reddy N, Sambandam N, Crawford PA. Adaptation of myocardial substrate metabolism to a ketogenic nutrient environment. J Biol Chem. 2010;285:24447–56. doi: 10.1074/jbc.M110.100651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Cotter DG, Schugar RC, Crawford PA. Ketone body metabolism and cardiovascular disease. Am J Physiol Heart Circ Physiol. 2013;304:H1060–76. doi: 10.1152/ajpheart.00646.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]; http://f1000.com/prime/718541563
- 61.Owen OE, Morgan AP, Kemp HG, Sullivan JM, Herrera MG, Cahill GF. Brain metabolism during fasting. J Clin Invest. 1967;46:1589–95. doi: 10.1172/JCI105650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Taegtmeyer H. On the inability of ketone bodies to serve as the only energy providing substrate for rat heart at physiological work load. Basic Res Cardiol. 1983;78:435–50. doi: 10.1007/BF02070167. [DOI] [PubMed] [Google Scholar]
- 63.Russell RR, Mrus JM, Mommessin JI, Taegtmeyer H. Compartmentation of hexokinase in rat heart. A critical factor for tracer kinetic analysis of myocardial glucose metabolism. J Clin Invest. 1992;90:1972–7. doi: 10.1172/JCI116076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kim AS, Miller EJ, Young LH. AMP-activated protein kinase: a core signalling pathway in the heart. Acta Physiol (Oxf) 2009;196:37–53. doi: 10.1111/j.1748-1716.2009.01978.x. [DOI] [PubMed] [Google Scholar]; http://f1000.com/prime/718541564
- 65.Hardie DG, Carling D. The AMP-activated protein kinase--fuel gauge of the mammalian cell? Eur J Biochem. 1997;246:259–73. doi: 10.1111/j.1432-1033.1997.00259.x. [DOI] [PubMed] [Google Scholar]
- 66.Bolster DR, Crozier SJ, Kimball SR, Jefferson LS. AMP-activated protein kinase suppresses protein synthesis in rat skeletal muscle through down-regulated mammalian target of rapamycin (mTOR) signaling. J Biol Chem. 2002;277:23977–80. doi: 10.1074/jbc.C200171200. [DOI] [PubMed] [Google Scholar]; http://f1000.com/prime/718541565
- 67.Bungard D, Fuerth BJ, Zeng P, Faubert B, Maas NL, Viollet B, Carling D, Thompson CB, Jones RG, Berger SL. Signaling kinase AMPK activates stress-promoted transcription via histone H2B phosphorylation. Science. 2010;329:1201–5. doi: 10.1126/science.1191241. [DOI] [PMC free article] [PubMed] [Google Scholar]; http://f1000.com/prime/5141956
- 68.McGee SL, van Denderen, Bryce J W, Howlett KF, Mollica J, Schertzer JD, Kemp BE, Hargreaves M. AMP-activated protein kinase regulates GLUT4 transcription by phosphorylating histone deacetylase 5. Diabetes. 2008;57:860–7. doi: 10.2337/db07-0843. [DOI] [PubMed] [Google Scholar]; http://f1000.com/prime/718541566
- 69.Matsui Y, Takagi H, Qu X, Abdellatif M, Sakoda H, Asano T, Levine B, Sadoshima J. Distinct roles of autophagy in the heart during ischemia and reperfusion: roles of AMP-activated protein kinase and Beclin 1 in mediating autophagy. Circ Res. 2007;100:914–22. doi: 10.1161/01.RES.0000261924.76669.36. [DOI] [PubMed] [Google Scholar]; http://f1000.com/prime/718541567
- 70.Baskin KK, Taegtmeyer H. AMP-activated protein kinase regulates E3 ligases in rodent heart. Circ Res. 2011;109:1153–61. doi: 10.1161/CIRCRESAHA.111.252742. [DOI] [PMC free article] [PubMed] [Google Scholar]; http://f1000.com/prime/13480982
- 71.Baskin KK, Taegtmeyer H. An expanded role for AMP-activated protein kinase: regulator of myocardial protein degradation. Trends Cardiovasc Med. 2011;21:124–7. doi: 10.1016/j.tcm.2012.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Baskin KK, Rodriguez MR, Kansara S, Chen W, Carranza S, Frazier OH, Glass DJ, Taegtmeyer H. MAFbx/Atrogin-1 is required for atrophic remodeling of the unloaded heart. J Mol Cell Cardiol. 2014;72:168–76. doi: 10.1016/j.yjmcc.2014.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Ellis JM, Mentock SM, Depetrillo MA, Koves TR, Sen S, Watkins SM, Muoio DM, Cline GW, Taegtmeyer H, Shulman GI, Willis MS, Coleman RA. Mouse cardiac acyl coenzyme a synthetase 1 deficiency impairs Fatty Acid oxidation and induces cardiac hypertrophy. Mol Cell Biol. 2011;31:1252–62. doi: 10.1128/MCB.01085-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Glembotski CC. The role of the unfolded protein response in the heart. J Mol Cell Cardiol. 2008;44:453–9. doi: 10.1016/j.yjmcc.2007.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Doroudgar S, Glembotski CC. New concepts of endoplasmic reticulum function in the heart: programmed to conserve. J Mol Cell Cardiol. 2013;55:85–91. doi: 10.1016/j.yjmcc.2012.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Lynch JM, Maillet M, Vanhoutte D, Schloemer A, Sargent MA, Blair NS, Lynch KA, Okada T, Aronow BJ, Osinska H, Prywes R, Lorenz JN, Mori K, Lawler J, Robbins J, Molkentin JD. A thrombospondin-dependent pathway for a protective ER stress response. Cell. 2012;149:1257–68. doi: 10.1016/j.cell.2012.03.050. [DOI] [PMC free article] [PubMed] [Google Scholar]; http://f1000.com/prime/718541568
- 77.Taegtmeyer H, Beauloye C, Harmancey R, Hue L. Insulin resistance protects the heart from fuel overload in dysregulated metabolic states. Am J Physiol Heart Circ Physiol. 2013;305:H1693–7. doi: 10.1152/ajpheart.00854.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.McKnight SL. On getting there from here. Science. 2010;330:1338–9. doi: 10.1126/science.1199908. [DOI] [PubMed] [Google Scholar]; http://f1000.com/prime/7966956