

Abstract
Parallel analysis of RNA ends (PARE) is a technique utilizing high-throughput sequencing to profile uncapped, mRNA cleavage or decay products on a genome-wide basis. Tools currently available to validate miRNA targets using PARE data employ only annotated genes, whereas important targets may be found in unannotated genomic regions. To handle such cases and to scale to the growing availability of PARE data and genomes, we developed a new tool, ‘sPARTA’ (small RNA-PARE target analyzer) that utilizes a built-in, plant-focused target prediction module (aka ‘miRferno’). sPARTA not only exhibits an unprecedented gain in speed but also it shows greater predictive power by validating more targets, compared to a popular alternative. In addition, the novel ‘seed-free’ mode, optimized to find targets irrespective of complementarity in the seed-region, identifies novel intergenic targets. To fully capitalize on the novelty and strengths of sPARTA, we developed a web resource, ‘comPARE’, for plant miRNA target analysis; this facilitates the systematic identification and analysis of miRNA-target interactions across multiple species, integrated with visualization tools. This collation of high-throughput small RNA and PARE datasets from different genomes further facilitates re-evaluation of existing miRNA annotations, resulting in a ‘cleaner’ set of microRNAs.
INTRODUCTION
Plant small RNAs (20–24 nt) play essential regulatory roles in growth, development as well as defense processes. These sRNAs are typically classified into two classes: miRNA (microRNA) and siRNA (short interfering RNA), both capable of post-transcriptional regulation via homology-dependent cleavage of their targets, with siRNA biogenesis dependent on RNA-dependent RNA polymerase activity. Since the first reports of miRNAs in plants (1,2), there has been a steep escalation in the number of known miRNAs, fuelled primarily by concurrent advances in sequencing technologies and computational methodologies. At the time of writing, there are over 7,385 mature miRNAs reported from 72 plant species in miRBASE (version 20). However, identification of a miRNA does not provide insights into its function or regulatory targets, nor is an understanding of targets part of the process of miRNA identification (3). Nonetheless, a key to understanding the biological relevance of a miRNA lies in discovering and validating its targets.
Parallel analysis of RNA ends (PARE) is a high-throughput sequencing technique which profiles uncapped mRNAs, products of cleavage or decay, facilitating studies of miRNA targets (4). Nearly identical techniques have been termed ‘degradome analysis’ or ‘GMUCT’ and they generate equivalent data (5,6). Because of our role in development of the technique called PARE, we are partial to this terminology and will use it hereafter. Computational tools to predict miRNA targets and validate those targets using PARE data are limited in both number and functionality. In addition, among these tools, there is divergence in the methodology used to predict targets and assign significance scores. CleaveLand, the most-cited and perhaps most commonly-used tool for computational validation of miRNA targets using PARE datasets, presumes that there exists a positive correlation between complementarity at a canonical seed region (2 to 13 nt from the 5′ end of the miRNA) of a miRNA::target duplex and probability of actual cleavage (7,8). Therefore, CleaveLand implements a ‘seed region’-based target scoring schema along with a penalty score cutoff of ≥4, to model the P-values for validated interactions. However, cleavage of potential targets can occur even with poor complementarity in the seed region or mismatches at canonical positions (9,10) (Figure 1).
Figure 1.
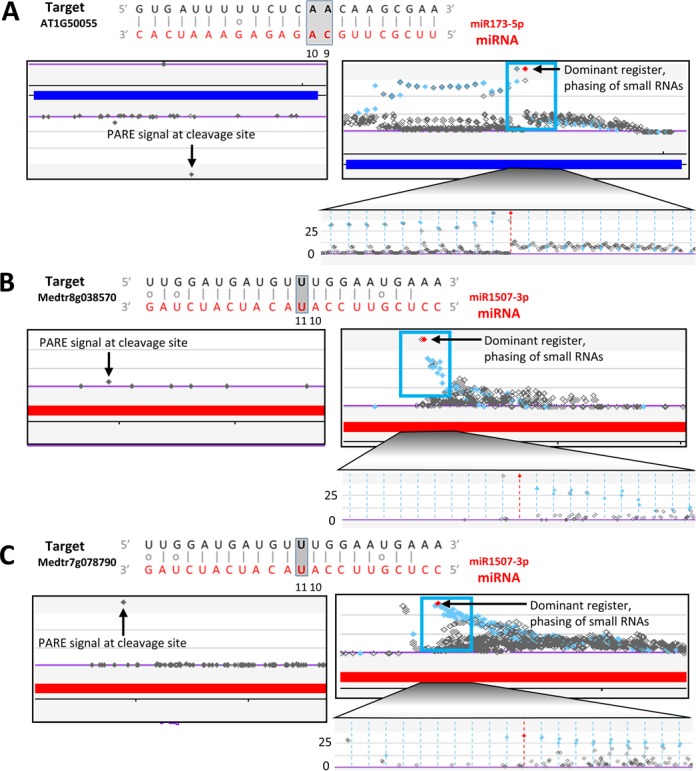
MicroRNA (miRNA) targets with weak or non-canonical interactions are missed by existing PARE-based validation tools. Each example shows the target-miRNA alignment at the top, with screenshots below from our website (http://mpss.udel.edu/); the upper panel shows the PARE data, the middle panel in each case, phased small RNA production from cleavage sites further substantiates the cleavage events. (A) at-miR173–5p cleaves the Arabidopsis thaliana TAS1B gene with a penalty score = 4.5, and with mismatches at both the 10th and 11th positions. (B) mtr-miR1507 cleaves the Medicago truncatula NBS-LRR type disease resistance gene (Medtr8g038570) with a penalty score = 7, and with a mismatch at the 11th position. (C) mtr-miR1507 cleaves the M. truncatula NBS-LRR type disease resistance gene (Medtr7g078790) with a penalty score = 7, and with a mismatch at the 11th position.
PAREsnip is an accelerated approach to extend PARE validation of targets from a small set of miRNAs up to a more extensive library of small RNAs (11). Yet PAREsnip suffers from the same inductive bias as CleaveLand, which is the assumption that there exists a positive correlation between complementarity in the canonical seed region and probability of actual cleavage. These assumptions about miRNA-target interactions are not easily modified or refined. Furthermore, the target search algorithm implemented in PAREsnip expects a perfect match at canonical 10th–11th positions and is dependent on ‘seed region’-based rules for its speed. Therefore, both PAREsnip and CleaveLand tend to bias the results by assigning significant P-values to only those interactions that either have a fairly good complementarity in seed regions or to those miRNAs that have limited number of interactions. Another existing tool for PARE-based validation of miRNA targets, SeqTar (9), broadens the complementarity-based prediction rules but it is currently moderately slow and therefore best employed for a pre-selected set of miRNA (or sRNAs) rather than a complex sRNA population.
These three tools for working with PARE data, Cleaveland (7), PAREsnip (11) and SeqTar (9) focus exclusively on the annotated portion of the genome, utilizing cDNA sets as their input. But many new genomes are poorly annotated, at least in their initial release, and recent studies indicate that even in well-annotated genomes, target mRNA still remain to be found in un-annotated, intergenic regions (IGRs), evidenced in reports of large numbers of miRNA-targeted long, noncoding RNAs in the grasses (12–14). For example, one recent report describes these loci (and their miRNA triggers) in un-annotated regions of the Brachypodium genome (15). Such analyses depend on approaches for target identification at a full-genome level, not just using annotated genes. As mentioned above, all existing algorithms to validate miRNA targets from PARE data are built on the assumption that the relevant interactions are within annotated transcripts. Since this is inaccurate, we sought a new approach.
In the past decade, the increase in yield-per-dollar cost of sequencing has democratized the use of high-throughput sequencing technologies. This has initiated a shift in plant genomics, from the study of model plants with modest genome sizes, to diverse crops, and now even including species with genomes many times larger than most model genomes. For example, the recently sequenced Picea abies and Triticum aestivum genomes are both >100× larger than Arabidopsis thaliana. Furthermore, an increased DNA sequencing throughput has commoditized the sequencing of RNA samples. A single small RNA or PARE library now includes tens millions of reads that can be analyzed and integrated to predict new miRNAs and their targets. These advances and cost reductions in genome and RNA sequencing warrant the development of a PARE validation approach capable of high efficiency to (i) handle enormous non-model genomes and (ii) quickly analyze all possible sRNA::PARE interactions from multiple libraries. Fortunately, there exist numerous technologies that could be deployed to meet these needs, via the development of algorithms capable of efficiently exploiting available computing power.
With increased studies of small RNAs, many groups have developed approaches to identify from sequence data novel miRNAs and their targets. Databases that computationally predict, curate and collect experimentally verified miRNA-target interactions include TarBase (16), StarBase (17), miRTarBase (18) and MiRecords (19). For biologists, the most effective use is to combine the best aspects of different databases and interpret the data using graphical interfaces. However, the database mentioned here lack integrated genome viewers, with the exception of StarBase (17) which uses the purpose-built deepView for visualization of mapped reads, target peaks and target plots. A limitation for plant biologists is that most of these databases focus on animals with only limited plant data. The extensive availability of data in plants is an opportunity for greater integration of small RNA, PARE and RNA-seq data to advance data-driven small RNA analyses.
Motivated by the prospect of discovering novel regulatory modules, the shortcomings of existing algorithms, explosive growth in the number of miRNAs, the number of sequenced plant genomes, and the amount of available PARE data, we developed a novel method for computational characterization of sRNAs. The package that we describe here is capable of predicting and validating targets at a whole genome level, and for all reported miRNAs or a given library of small RNAs. Unlike earlier tools, sPARTA employs true parallel computing to gain significant advances in speed, and it implements a data-partitioning scheme for both scalability and to maintain a low memory footprint. Thus, sPARTA is efficient in handling large genomes as well as large input sets of RNA data.
MATERIALS AND METHODS
The sPARTA algorithm has four main steps that are implemented in series. With the exception of the first step in which user-defined features (gene or intergenic) are extracted and fragmented, the three subsequent steps use single-instruction multiple-data (SIMD) parallel processing via Python (v3.3) multiprocessing module. The two most data intensive steps (i) mapping reads from multiple PARE libraries and (ii) the prediction of sRNA or miRNA targets (by miRferno), both benefit from two-way SIMD parallelism (Figure 2).
Figure 2.
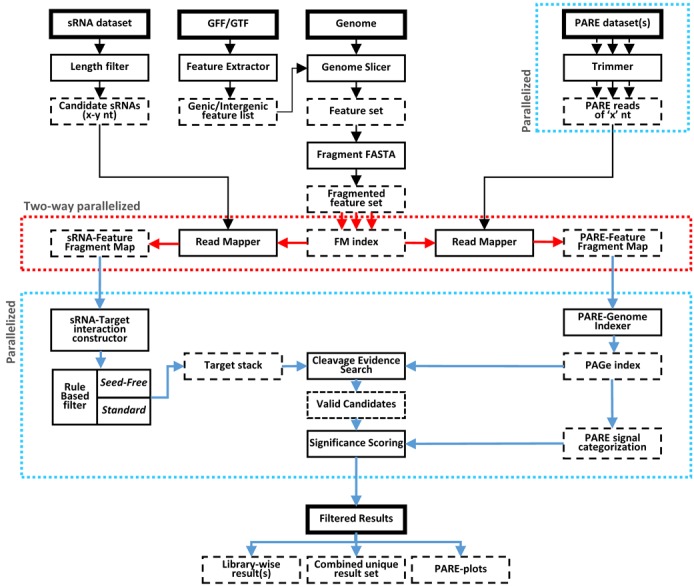
sPARTA schematic, showing order of steps in workflow. Solid boxes represent sPARTA functions, dashed boxes represent the product of an applied function. Multiple arrows indicates multiple output files from the preceding function. Steps executed in parallelized environment are enclosed within colored dotted lines.
Feature extraction and input file partitioning
To build a ‘feature set’ or input library of sequences in which targets will be identified for a species of interest, sPARTA starts with a GFF file (Generic Feature Format, version 3) containing gene annotations along with the corresponding genome sequence. In many cases, this is downloaded from Phytozome (20). These GFF and genome sequence files are used by the built-in Genome Slicer function to extract first the coordinates of selected features (i.e. genic or IGRs) from the GFF files and next to extract the sequences from the genome. These intergenic and genic sequence sets comprise the main feature set, which is further partitioned into different data elements (features) so as to implement data parallelism.
PARE data processing and read mapping
The next step in sPARTA is to map PARE reads to the feature set. An FM index (21) for each component of the feature-set is created using Bowtie (version 2, in the current sPARTA implementation) (22) with the default off-rate parameter. PARE reads in tag-count format (a tab-separated file of read sequences and normalized frequencies) from each dataset were then aligned to the partitioned FM indexes using Bowtie, with default end-to-end settings and no mismatch allowed, to generate PARE-fragment maps. PARE datasets in format other than tag-count could be easily converted to tag-count using publicly available Tally (23). The PARE mapping step implements SIMD parallel processing on both the involved datasets, i.e. the feature set and the PARE reads. The feature set file size for different species could range up to tens of gigabytes, while the number of reads in a PARE dataset range from millions to hundreds of millions. So, two-way parallelization further enhances both scalability and load balancing, improving the parallel processing efficiency of the sPARTA algorithm. The parallelization on the number of reads is achieved by Bowtie's built-in parallel processing function that makes use of the pthreads library to distribute reads across concurrent search threads (24).
Prediction of targets using novel miRferno algorithm
In the third major step, targets of small RNAs are identified in the sequences of the feature set. sPARTA has a newly developed, built-in target prediction module—miRferno—which has two prediction modes, greedy and exhaustive, described below. In both modes, the miRNA or sRNA sequences used as an input to find targets are mapped to the fragmented features using Bowtie. The advantage of using version 2 of Bowtie is that it allows gapped alignments, and therefore it can find miRNA-target interaction which include gaps and bulges. The inclusion of these mismatches substantially increases the sensitivity of target prediction, but gaps also greatly inflate the size of the search space and slow down the process of finding targets. Prior decomposition of the feature set into smaller partitions (i.e. features) by sPARTA reduces the index size and associated search space for gapped alignments. This increases the efficiency of alignments, and in combination with parallelization on the number of PARE reads and genomic partitions (i.e. two-way parallelization), comprises an effective combination of speed, sensitivity and scalability.
The two prediction modes of miRferno allow the user to optimize for time versus sensitivity. The greedy mode is designed to be fast but less sensitive. In this mode, multiple seeds are extracted from the miRNA or sRNA sequence. These seeds are 6 nt in length and extracted in 4 nt intervals, and they are aligned to the FM indexes from the partitioned feature set with a maximum allowed mismatch of 1 nt. Matched instances of these seeds are further extended to complete the alignment of the small RNA, unless three consecutive seed extension attempts fail, resulting in the termination of the extension. On other hand, the exhaustive mode is designed for improved sensitivity; it extracts a smaller seed of 4 nt spaced in a 3 nt interval from miRNA or sRNA sequence. The use of multiple 4 nt seeds from a single miRNA or sRNA along with one allowed mismatch improves the efficiency of finding targets, as the probability is high of at least one seed (out of seven in total, for a 21 nt small RNA) being extracted from the region of the miRNA which binds its target at a region with 3 nt matches. In addition to sensitive mapping parameters, if none of the extracted seeds reports a valid alignment, then a second, ‘re-seeding’ pass is allowed. In second pass, a new set of seeds is generated, slightly offset, and used to search for targets.
miRferno also offers the user two different systems for target scoring, standard and seed-free. Standard scoring provides backward compatibility for earlier miRNA-target prediction or validation experiments; in other words, it is based on previously described, complementarity rules based on a seed region (8). However, we added the seed-free scoring because several recent studies have shown that there exist miRNA-target interactions which deviate from the standard or canonical complementarity rules that utilize a seed region (9,10). Seed-free scoring may have broader utility; several early (25,26) as well as recent studies (27–31) from animals also indicate that formation of a functional miRNA-target duplex does not require strict complementarity between a miRNA seed and its target. These non-canonical targets in both plants and animals have been validated and support an ‘expanded’ range of miRNA-target interactions. Moreover, the targets sites from IGRs are often left unanalyzed because target-prediction tools focus on annotated genes; poorly annotated non-coding RNAs may interact differently with miRNAs in ways that are not yet well defined. So, we wanted to avoid over-fitting of complementarity rules based on seed regions that might not only restrict our ability to find non-canonical targets but also introduce bias into the results. The seed-free scoring achieved this, based on the assumption that a target site could be functional even with weak seed-region complementarity. Therefore, unlike the standard scoring system, within this region, G:U wobbles, gaps and mismatches have the same penalty score as elsewhere in the miRNA-target pairing. Finally, in the seed-free scoring system, mismatches at the critical 10th and 11th positions are permissible (32,33). While the seed-free scoring system relaxes many of the conventional miRNA-target interaction constraints, by assigning strong mismatch penalties, it retains a requirement of a correlation between sequence complementarity and cleavage efficacy. Each miRNA-target alignment is scored using following position specific rules, starting from the 5’ end of the miRNA:
Mismatches at either the 10th or 11th positions carry a penalty of 2.5.
A wobble with a single flanking mismatch or mismatches on both sides carries a penalty of 1.5 or 2.0, respectively.
A single gap, mismatch and wobble at any position carries a penalty of 1.5, 1.0 or 0.5, respectively.
Finally, in sPARTA, the Bowtie scoring system was modified to reject miRNA-target alignments with more than one gap or six ‘edits’ (mismatches or G:U wobbles). These settings are user-configurable and can be relaxed using the depth parameter with input values ranging from 0 (default) to 3 (relaxed).
Indexing and prediction of validated interactions
In the final step of sPARTA, the PARE read abundances and positions are assessed relative to the predicted miRNA or sRNA targets, with the aim of validating ‘real’ cleavage events. First, for each PARE library, map files generated for all partitions of the feature set (from the second step of sPARTA) are combined and transformed into an index. This PARE-Genome (PAGe) index is specific to PARE libraries and consists of coordinates in which the 5′ end of the PARE reads is mapped to the genic or intergenic feature set, along with the read abundance. PAGe indexes are used to classify the mapped reads (the evidence of cleavage at a specific site) into separate classes on the basis of their abundance (the strength of this evidence of cleavage). For a genic feature set, sPARTA implements the same signal classification schema described in earlier studies (7,11). This schema uses five ‘classes’ to rank the evidence of cleavage based upon normalized or raw tag count input file; in other words, each PARE read in a gene is assigned to one of the five classes. Class 0 indicates a PARE signal with abundance greater than one read that is also the maximal signal on the transcript; this is ultimately the most promising site for miRNA-directed cleavage. Class 1 is similar to class 0 except there exists more than one maximal PARE signal on the transcript with the same abundance. Class 2 is a PARE read above the median for the gene, and with an abundance of more than one read. Class 3 is a PARE read below the median, but still with an abundance of more than one read. Lastly, class 4 are PARE reads with an abundance of one, essentially not discernable from ‘background’.
IGRs may be more challenging to analyze than genic regions, for the purposes of finding and validating sRNA targets, for several reasons. First and foremost, a single IGR might contain more than one transcript and these transcripts could be coordinately regulated, potentially by one or more miRNAs. In such a scenario, a transcript with the highest expression could impact the signal of a more weakly expressed transcript sharing the IGR, pushing it to a lower class (2 or 3) and thus diluting the score and detection of a genuine cleavage event in the weaker transcript. Second, an IGR may contain no transcripts cleaved by an sRNA, with the only mapped PARE signals resulting from decay or nonspecific effects; in absence of strong signal from a cleavage event, these low strength signals will be assigned to class 0 or 1 thereby inflating these top categories and confounding the calculation of the confidence score. Therefore, to fit the variable and difficult-to-assess nature of IGRs, sPARTA classifies PARE signals on the basis of the global abundance of PARE reads. For each PARE library, the signals in the bottom 20% (by abundance) are assigned to class 4 and excluded from further calculations. The remaining signals are then classified as follows:
Class 0: > 90th percentile (of PARE read abundances excluding class 4)
Class 1: 90th percentile ≥ PARE read abundance > 75th percentile
Class 2: 75th percentile ≥ PARE read abundance > median (50th percentile)
Class 3: median ≥ PARE read abundance
sPARTA calculates the confidence score (P-value) as defined in Cleaveland (v3, or 'CL3') but with slight modification so as to improve the P-value for cases where miRNA-target interactions have weak complementarity or when a single miRNA cleaves hundreds of targets, for example, the miR2118 or miR2275 targets described previously for rice and maize (13). This P-value is further corrected for the noise around the cleavage site. The calculation of the P-value is as follows:
P-value (at least one significant result) = 1 − pbinom (0, trials, probability of success)
Corrected P-value = P-value of an interaction/signal to noise ratio
Where,
trials = total number of miRferno predicted targets within a score bracket, i.e. the number of predicted targets with score ≥5 and <6, instead of cumulative number of predicted targets for a miRNA at specific score as in CL3.
probability of success = fraction of total (eligible) bases in the feature set occupied by a specific PARE signal class (7).
And,
P-value of an interaction = PARE-validated interaction with P-value <0.25 and signal-to-noise ratio >0.25
Signal to noise ratio = fraction of PARE abundance at cleavage site in a 10 nt window around the cleavage site (5 nt in each the 3′ and 5′ directions).
This relaxed P-value calculation gives more weight to the evidence from PARE data and it yields a greater number of validated targets as compared to CL3, but it could also have a higher proportion of false positives. We believe that this trade-off can be reasonably reduced by either (i) including replicates of PARE datasets (11) or (ii) by establishing the anti-correlation in expression levels between miRNA and their targets.
Finally, for the analyses that we described here, publically available PARE, sRNA and RNA-seq datasets for A. thaliana, Oryza sativa, Medicago truncatula and Brachypodium distachyon were downloaded from NCBI GEO (Table 1). sPARTA (in the seed-free mode) was used to generate species-specific sets of PARE-validated miRNA-target interactions. The back-end for the comPARE web resource, which stores the data and perform searches, consists of a relational database implemented with MySQL on CentOS release 6.4. The graphical user interface (GUI) was developed in PHP for seamless integration with our customized genome browser (34) for visualization as well as in-depth exploration of data from different sources such as PARE, small RNA, RNA-seq (when available) integrated with genomic annotations and features.
Table 1. Small RNA and PARE data used in these analyses.
| Species | miRNAs | Annotation version | PARE datasets | Small RNA datasets |
|---|---|---|---|---|
| A. thaliana | 337 | TAIR 10.0 | GSM280226 | None used. |
| GSM280227 | ||||
| B. distachyon | 882 | MIPS 1.0 | BDI25, BDI20 (15) | GSM506621,GSM506620 |
| M. truncatula | 599 | JCVI 3.5 | MEDFL3 (35), GSM643818, GSM643817 | GSM767273, GSM769274, GSM769275, GSM769276, GSM729277, GSM729279 |
| O. sativa | 713 | MSU 7.0 | GSM476257 | None used. |
| GSM434596 |
RESULTS
Data and Tools
To assess the performance of sPARTA (greedy mode), real datasets (PARE, small RNA, genomes and miRNAs) were used to determine metrics, as it would be in an actual miRNA target identification experiment. Publically-available PARE datasets generated using Illumina sequencing from four different species (A. thaliana, B. distachyon, M. truncatula, and O. sativa; Table 1) were downloaded from our Massively Parallel Signature Sequencing database (34), and the corresponding genome sequences and annotation information were fetched from their respective repositories (Table 1). miRNA sequences for all four species were downloaded from miRBASE (version 20). CleaveLand (version 3, CL3) and PARESnip (version 2.1) are currently the only publicly-available, command line tools for PARE-based miRNA-target validation. We used CL3 for comparative benchmarking primarily because it's is the most cited tool for these types of analyses and secondly due to unresolved technical problems with the execution of PAREsnip.
Evaluation of sPARTA speed
sPARTA was evaluated on a machine equipped with 4 × 64 bit 8-core 2.4 GHz Intel Xeon (32 cores total) running CentOS release version 6.4. Python 3.3 and R 3.0 (36) were installed ‘as is’ available from their respective sources. In the comparisons below, the added time to extract features, i.e. genic or intergenic transcripts, from the genome is not included as this feature is not present in any available tools. All the runtimes reflect an average of five independent trials.
We first evaluated the total time required by sPARTA to predict and validate targets at a whole-genome level for all four species. All available miRNAs for each species were used for target prediction, followed by validation using two separate PARE libraries (Table 1). Two different scenarios were tested: sequential and parallel. As the name suggests, the sequential run used just one core for the analysis, whereas the parallel run utilized ∼85% of the available cores (n = 28). For the total runtimes, we excluded the execution time for the step which maps the PARE dataset to the genome, as mapping step is performed by Bowtie (22), for which the settings can optimized to run using the same number of processors as the sPARTA parallel mode. The comparison demonstrated a minimum speed gain of 10.56× with the genic feature set of O. sativa, and a maximum speed gain of 22.31× with the intergenic feature set of A. thaliana. At a whole-genome level, a maximum speed gain of 16.7x and minimum speed gain of 11.2x was achieved by the parallel mode of sPARTA (Figure 3A).
Figure 3.
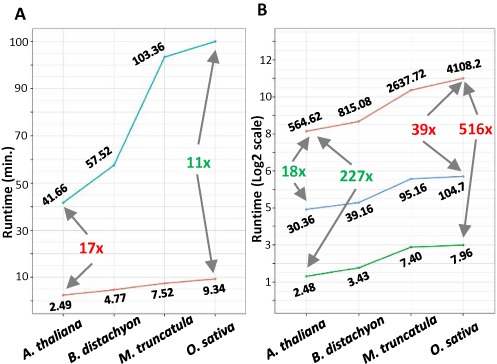
Comparative benchmarking of the sPARTA algorithm in parallelized mode and in comparison to CleaveLand version 3 (CL3). In both comparisons, four different plant genomes were used, as indicated on the X-axis. In each set of pairwise run comparisons, the minimum fold difference is indicated in green text and the maximum in red text. (A) Runtime comparisons between sPARTA in serial (blue line) and parallel (red line) modes exhibit a minimum speed gain of 11× and maximum speed gain of 16.8× for the parallel mode compared to the use of a 28-core single node. (B) sPARTA run in parallel mode (green line) is a minimum of 227× and maximum 516× faster than the comparable software package CL3 (red line). Using a single core (blue line), the sPARTA package is a minimum of 18× and a maximum 39× faster than CL3.
Next, we compared sPARTA performance to CleaveLand (CL3) which is the most-widely used tool for the evaluation of plant miRNA targets. CL3 consists of two sequentially executed scripts, requiring input from two third-party tools, TargetFinder (8) and Bowtie (22). To enable a comparison with sPARTA, we implemented the CL3-based pipeline using its bundled scripts and required tools, with no modification to those original scripts or settings. For the fairest comparison between algorithms, the PARE mapping step for CL3 was assigned the same number of cores as sPARTA. CL3 lacks the functionality to predict intergenic targets, therefore a comparison was made just for the genic feature set. Outperforming CL3, sPARTA exhibited a minimum speed gain of 227.39x (564.62 to 2.48 min) with A. thaliana and a maximum speed gain of 515.12x (4108 to 7.964 min) with O. sativa (Figure 3B). Even in the serial mode, sPARTA was found to be a minimum 18x (564.62 to 30.36 min) and maximum 39.5x (4108.2 to 104.7 min) faster than CL3 with A. thaliana and O. sativa respectively (Figure 3B).
Prediction performance of sPARTA
Strong experimental support is required to validate miRNA-target interactions identified by sPARTA. Such experimental data may be either modified 5′ RACE, applied to individual targets, or genome-level data sets from PARE, an extension of 5′ RACE to the genome level. For PARE data, there are a number of earlier miRNA-target validation studies (15,37–38). Yet, since these earlier studies were also computational (i.e. had their own set of parameters for PARE validation), their sensitivity is unknown and therefore cannot be used as a ‘gold standard’ to calculate the degree to which the sPARTA predictions generated false positives or false negatives. Moreover, since there is no earlier published approach or tool to cross-validate miRNA targets from IGRs, it is not possible to appraise the sensitivity of these intergenic predictions from sPARTA. In the context of these limitations, we performed an assessment of the predictive power of sPARTA by comparison to CL3.
A subset of plant miRNAs, including miR2118 (13), miR2275 (13), miR173 (39) and miR390 (32) induce the production of secondary siRNAs in a phased arrangement from their target RNA transcript, via the recruitment of RDR6 and DCL4 or DCL5. The start site of the register of this phasing is determined by the position of miRNA-guided cleavage. Since the presence of phased sRNAs (phasiRNAs) from a locus indicates a real miRNA-target interaction occurred, finding a PARE-validated trigger site that was responsible for phasiRNA production further supports the validity of some miRNA-target interaction. We used this cross-validation of a computationally predicted miRNA-target interaction with phasiRNA (‘PHAS’) loci as the basis to assess and compare the ability of different software tools to identify miRNA target sites.
Two recent studies have reported numerous 21-nt phased loci from genic regions of M. truncatula and both 21- and 24-nt phased loci from IGRs of B. distachyon (15,35).Using the small RNA data from these studies, we repeated those analyses to identify a total of 310 (24-nt phasing) and 755 (21-nt phasing) PHAS loci from IGRs of B. distachyon, and 129 (21-nt phasing) PHAS loci from genic regions of M. truncatula (Figure 4). For every phased locus, an index of potential miRNA target sites was generated. This index consisted of 11 coordinates (+/− 5 cycles) in correspondence to the phased (21- or 24-nt) periodicity from the initiation site of phased locus (Supplementary Figure S3). PARE datasets from both studies were used to generate a list of PARE-validated targets against all miRNAs for each species, using sPARTA and CL3 independently. Though the CL3 functionality is limited to transcriptome or genic regions, our aim was to compare an existing algorithm with sPARTA to assess its advantage or disadvantage in its prediction power. For this particular analysis, we rectified one of the main technical shortcoming of CL-the based pipeline by capacitating a parallelized prediction of targets; for this, we developed a parallelized version of TargetFinder (8). No changes were made to the target prediction scoring schema, so as to retain the original approach of CL3. Finally, triggers of phased loci were identified by searching for a match between the phase-index of an individual locus and validated cleavage sites from both CL3 and sPARTA.
Figure 4.
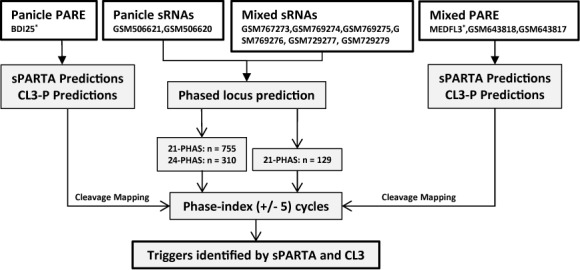
Our approach to assessing the comparative benchmark of the prediction power. Loci generating phased sRNAs were identified from published small RNA datasets of Brachypodium distachyon and Medicago truncatula, while genome-wide target prediction and validation was performed using their associated PARE datasets against all species-specific miRNAs. GEO accession numbers are indicated in the top row of boxes; asterisks indicate data either from http://mpss.udel.edu/brachy_pare2 or http://mpss.udel.edu/mt_pare/. Triggers of phased sRNA loci validated by sPARTA and CL3 were identified and used for a comparison of predictive power.
sPARTA demonstrated advantages over CL3 by identifying more triggers, as well as by exhibiting a high enrichment in P-value of correct predictions. In the case of phased loci from IGR of B. distachyon, 3-fold (total = 56) and 4.5-fold (total = 88) more triggers were validated by sPARTA (P-value ≤ 0.05) for 24- and 21-phased loci respectively (Figure 5A). Of all the miRNA triggers identified by sPARTA, 70% of 21-phased and 90% of 24-phased triggers were predicted under a P-value of 0.05. Interestingly, miR2118 was identified as trigger in 126 out of 127 validations of 21-phased loci whereas for 24-phased loci, miR2275 was identified as trigger in all the validations. This is consistent with earlier reports of the miR2118 and miR2275 families (14,35) as triggers of reproductive-specific 21- and 24-nt phased loci, respectively. This observation further supports the robustness of our approach used for comparative benchmark of predictive power, by showing that PARE-validated triggers of phased loci are not products of chance. For phased loci from genic regions of Medicago, sPARTA identified 2.5-fold more triggers under a P-value of 0.05 as compared to CL3 (Figure 5B). As in B. distachyon, sPARTA exhibited an enrichment of P-values by predicting 68% of 21-phased loci triggers under a P-value of 0.05.
Figure 5.
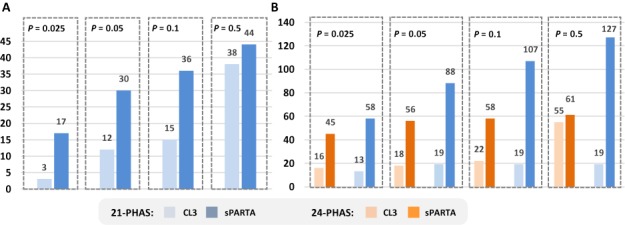
sPARTA validates more triggers and exhibit high P-value enrichment as compared to CL3. We performed comparative benchmarking of the predictive power of sPARTA, as outlined in Figure 4. (A) In an analysis of only 21-PHAS loci from genic regions from Medicago truncatula, sPARTA identified 2.5 times more miRNA triggers than CL3, with 68% of correct validations under a P-value of 0.05. (B) For 21- and 24-PHAS loci from intergenic regions of Brachypodium distachyon, sPARTA identified 3 and 4.5× more miRNA triggers with 70 and 90% of correct predictions under a P-value of 0.05, respectively.
Targets identified from intergenic regions
A total of 506 credible targets (with a corrected P-value ≤ 0.05, PARE signal class ≤3) for 70 different miRNA families (Figure 6) were identified from the IGRs of B. distachyon, using the published PARE datasets from root, leaf, stem and panicle tissues (15). These targets would certainly have been missed by existing PARE validations tools as those tools are limited to analysis of just the annotated genic regions. We also found multiple targets from a single IGR, each with different expression dynamics; our approach for the classification of PARE reads mapped to IGRs was developed with this scenario in mind.
Figure 6.
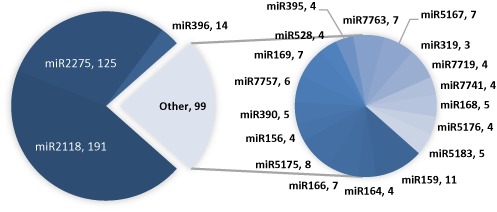
Intergenic targets in Brachypodium distachyon for 80 different miRNAs. A total of 506 credible intergenic targets were validated in B. distachyon from root, leaf, stem and panicle tissue. miRNAs bdi-miR2118 and bdi-miR2275 accounted for half of the intergenic targets. The pie charts show miRNA families with more than three targets, with the number of targets following the miRNA name.
From the total set of intergenic targets, the panicle data alone accounted for most validated interactions (n = 344) with 157 and 114 unique cleavages triggered by just two miRNA families, miR2118 and miR2275, respectively. Both miRNA families are known to trigger phasiRNA biogenesis (13). In 48% of cleavage site identified, we found an overlap of the cleavage site within +/− 5 phased positions or ‘indexes’ from the dominant register of phasing, i.e. the position with the highest phasing score. For those cleavage sites which did not match with the phased index, upon inspection, we found presence of a phased locus in close vicinity (∼250 nt). The reason for this disagreement between the cleavage site and phase index could be the depletion of some sRNAs from a few phasiRNA cycles, consistent with the non-stoichiometric abundances of tasiRNAs from Arabidopsis TAS loci, leading to a shift in the predicted position of the predominant register for the phasiRNAs. We also observed PARE validation of cleavage by miR2118 and miR2275 in leaf with the same cleavage co-ordinates as panicles (Figure 7). For these interactions shared with those that lead to phased sRNA generation in panicles, no associated phased sRNAs were found near the cleavage site in leaf, yet the abundance of PARE reads at the cleavage sites indicates strong expression of the precursors in both panicle and leaf tissues. These observations suggests that there are other factors influencing the production of phased sRNAs.
Figure 7.
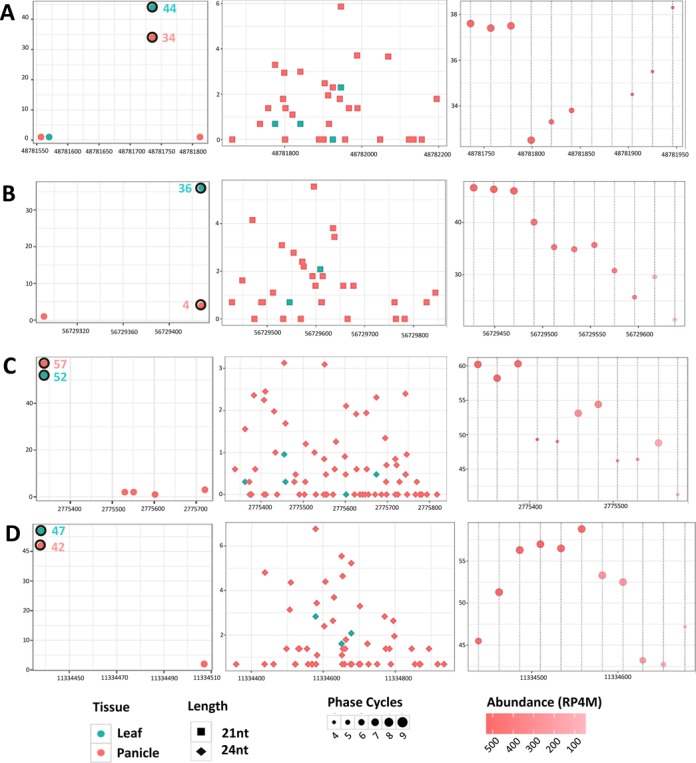
Shared but unphased, PARE-validated targets of miR2118 and miR2275 in leaf suggests additional factors in phased small RNA production. Examples of two targets each of miR2118 (panels A and B) and miR2275 (panels C and D) from Brachypodium distachyon, validated in leaf data and with cleavage sites identical to those in panicles. These miRNA-target interactions trigger phased sRNA generation in panicles but not leaves. Each panel shows three plots that correspond to the abundance (TP15M) of PARE reads at cleavage site, small RNA abundances (TP4M), and the phasing score profile for the region. (A) miR2118 target site on chromosome 1, with the cleavage site at 48781736. (B) miR2118 target site on chromosome 4, with the cleavage site at 56729428. (C) miR2275 target site on chromosome 3, with the cleavage site at 2775383. (D) miR2275 target site on chromosome 4, with the cleavage site at 11334438.
Unlike miR2118 and miR2275 families, whose activity was found to be conserved to leaf and panicle, a few miRNAs like miR396 shared targets across different combination of tissues. miR396 has been previously demonstrated to coordinate cell proliferation in leaf meristem by regulating transcription factors belonging to the family of growth-regulating factor (GRF) (40). Another transcription factor, bHLH74, crucial to margin and vein pattern formation of Arabidopsis leaves has been found to be a target of miR396 (41). Recently, it was reported that the miR396 regulatory network and tasiRNA biogenesis pathway synergistically interact to regulate leaf development (42). We found miR396 to be highly expressed not only in leaf but also seedling, stem and panicle of B. distachyon (15); it is also found in roots but at a comparatively low level. In panicle, a total of nine validated targets of miR396 were identified from genic (n = 4) and IGRs (n = 5). All four genic targets from panicle were found to be member of GRF family (Supplementary Figure S1) like earlier published studies on leaf development. Moreover, the PARE signal at the cleavage site of all four of these targets belonged to class 0, i.e. PARE read abundance ≥ 90th percentile of all PARE reads mapped to the genic regions (Supplementary Figure S1 A, B and C), suggesting moderate expression of cleaved GRF transcripts. There are also several IGRs (Supplementary Figure S1 D, E and F) with strong signals of miR396 activity, highly enriched in panicle. These observations indicate that in addition to leaf development, miR396 might also play a role in panicle development.
In the process of these analyses, we noted that reliance on annotated miRNAs without their critical assessment can lead to spurious conclusions. As an example, three annotated miRNA families, miR5174, miR5181 and miR5180 (43), accounted for the greatest number of validated targets (n = 132), after the miR2118 and mi2275 families. Further inspection of these miRNAs revealed that they originate from repetitive regions, rich in heterochromatic small RNAs (24 nt) and their abundance is quite low in the libraries used for prediction (Supplementary Figure S2). The approach (43) implemented to annotate these three miRNA families used default Bowtie parameters therefore only first valid sRNA alignment to genome was reported instead of all the mappings of sRNA to the genome which lead to ‘clustering’ with incomplete set of small RNA mappings, also no hit- or abundance-based filter was applied to remove lowly expressed or reads with large number of hits to genome. Moreover, through sPARTA-based analysis all of the targets of these three families were found to be in highly repetitive regions. These data strongly suggested that these are incorrectly annotated miRNAs; as miRNAs are largely predicted computationally using different pipelines and parameters, mostly on basis of small RNA sequencing datasets and submitted to miRBASE without experimental validation, researchers need to be wary of such false predictions. Presence of such spurious miRNAs in public repositories suggested the need for a resources which allows visualization of miRNAs and their targets in their genomic contexts, to allow manual inspection.
The comPARE web interface
We developed a web-based tool, which we call ‘comPARE’, for two purposes: (i) to serve as a single point of access for plant miRNA-target interactions that we have validated with PARE data, (ii) to facilitate connections of those data to our custom-built genome browser, specialized for small RNA (34). This interface is designed to be easy to use, yet incorporate advanced functionality such as modifiable search parameters, combined searches of sRNA or PARE datasets, and analysis of library-based data. The comPARE site is accessible at: https://mpss.udel.edu/tools/mirna_apps/comPARE.php. To use this site, shown in Figure 8, first, a user chooses the PARE database for species of interest from the main query page, additional information about the available databases and included libraries are found at our lab's main page http://mpss.udel.edu. For specific miRNAs and/or targets of interest, their identifiers are entered into the respective text boxes on the main query page, generating results by clicking on ‘Search with default values’. This then will display all the interactions that pass the criteria, set at a default for convenience. A more advanced search could be performed by modifying the values of search parameters, including the miRNA-target complementarity score (Target score), P-value cutoff, normalized abundance of the PARE signal at cleavage site (small window) and signal class (see ‘Materials and Methods’ section); the search is executed by clicking on ‘Search with selected values’. The results for both a simple or modified query are presented in a simplified table format consisting of the miRNA name, miRNA sequence and the list of targets. However, a detailed view can be opened by clicking on ‘Show extra columns’ located in the header of the results table, which displays additional information including the target score, P-value, small window (a 1 nt region flanking the cleavage site), large window (a 5 nt region flanking the cleavage site), signal class, the cleavage site coordinates, and the annotated function of the target (if available) for each interaction that passed the selected or default search parameters. A user can also search for all the interactions from the selected species with either modified or default search parameters. The results from the user query in comPARE then integrate small RNA and PARE data, layered on an annotated genome. This provides a comprehensive view of cleavage sites, facilitating an in depth exploration of miRNA-target interactions. For additional functionalities of comPARE, please refer to the Supplementary Text.
Figure 8.
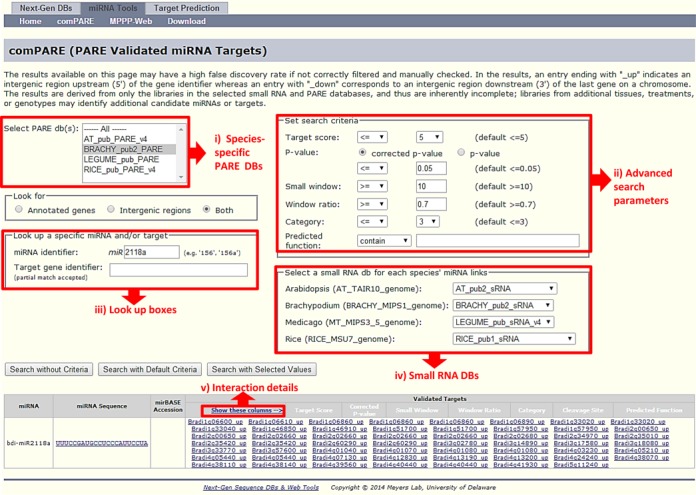
The interface to comPARE, web-based access to PARE-validated sets of miRNAs targets. A screenshot of the comPARE web interface. The red boxes highlight different types of user options. For example, in the upper left (i), the user can choose single or multiple species specific PARE databases to search for miRNA-target interactions. In the upper right (ii), in advanced search could be performed by setting the search parameters as per the required confidence level. In the lower left (iii), for a miRNA or target of interest, a search could be executed using a miRNA name and/or genome-specific target identifier as a query. Lower right (iv), if these options are listed, multiple sRNA databases for a species of interest other than the initial selection could be made. Finally (v), at the very bottom, the links, if clicked, display additional information about each interaction.
In addition to searches, visualization and exploration of miRNA-target interactions, one of the main strengths of comPARE is that it enables the discovery of conserved miRNA targets across different species. This functionality is of high value for revealing not only the evolutionarily conserved targets of specific miRNAs but, most interestingly, the non-conserved targets of different species or libraries. A quick search with ‘miR2118’ from the main query page shows that its targets are unrelated, genic and intergenic in M. truncatula and B. distachyon respectively, in consensus with earlier studies (35,44). Such cross-species contrasting patterns of miRNA targets are of high biological significance, and comPARE could aid in discovering this patterns as it allows identification of genome wide targets for miRNAs from different species.
DISCUSSION
sPARTA is a powerful tool for plant miRNA target prediction and PARE validation. It can search for targets in unannotated genomic regions, which is useful to discover novel regulatory modules, independent of genome annotations that may be incomplete. Earlier tools like PAREsnip use seed-region complementarity rules to accelerate the analysis, whereas sPARTA implements true parallelization to reduce the run-time for miRNA-target validation experiments from hours or days to minutes. This speed enables target analysis of hundreds, thousands or even millions of small RNAs at once. The novel ‘seed-free’ mode is based on recent empirical observations regarding miRNA-target interactions, and it identifies targets with weak seed-region complementarities or mismatches at canonical positions. sPARTA forms the core of comPARE, a web resource that allows the discovery, visualization and in-depth exploration of genome wide miRNA-target interactions in heterogeneous yet highly integrative environment. comPARE was developed to serve as repository of our validated miRNA interactions, collating small RNA and PARE datasets along with their genomic context. In conclusion, this study presents three novel tools: miRferno for target prediction, sPARTA for PARE based target validation and comPARE for visualization, exploration and comparative analysis of miRNAs targets. We believe that these three tools will allow us to effectively exploit advanced computing power, discover novel regulatory modules and dispense high quality miRNA-target interactions.
AVAILABILITY
sPARTA is released under GPL, and both the source code and a reference manual are available for download at: http://mpss.udel.edu/tools/mirna_apps/download.php.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
NSF IOS award [1257869]; project funding from the United Soybean Board and North Central Soybean Research Program.
Conflict of interest statement. None declared.
REFERENCES
- 1.Llave C., Kasschau K.D., Rector M.A., Carrington J.C. Endogenous and silencing-associated small RNAs in plants. Plant Cell. 2002;14:1605–1619. doi: 10.1105/tpc.003210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Reinhart B.J., Weinstein E.G., Rhoades M.W., Bartel B., Bartel D.P. MicroRNAs in plants. Genes Dev. 2002;16:1616–1626. doi: 10.1101/gad.1004402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Meyers B.C., Axtell M.J., Bartel B., Bartel D.P., Baulcombe D., Bowman J.L., Cao X., Carrington J.C., Chen X., Green P.J., et al. Criteria for annotation of plant MicroRNAs. Plant Cell. 2008;20:3186–3190. doi: 10.1105/tpc.108.064311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.German M.A., Luo S., Schroth G., Meyers B.C., Green P.J. Construction of parallel analysis of RNA Ends (PARE) libraries for the study of cleaved miRNA targets and the RNA degradome. Nat. Protoc. 2009;4:356–362. doi: 10.1038/nprot.2009.8. [DOI] [PubMed] [Google Scholar]
- 5.Gregory B.D., O'Malley R.C., Lister R., Urich M.A., Tonti-Filippini J., Chen H., Millar A.H., Ecker J.R. A link between RNA metabolism and silencing affecting Arabidopsis development. Dev. Cell. 2008;14:854–866. doi: 10.1016/j.devcel.2008.04.005. [DOI] [PubMed] [Google Scholar]
- 6.Addo-Quaye C., Eshoo T.W., Bartel D.P., Axtell M.J. Endogenous siRNA and miRNA targets identified by sequencing of the Arabidopsis degradome. Curr. Biol. 2008;18:758–762. doi: 10.1016/j.cub.2008.04.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Addo-Quaye C., Miller W., Axtell M.J. CleaveLand: a pipeline for using degradome data to find cleaved small RNA targets. Bioinformatics. 2009;25:130–131. doi: 10.1093/bioinformatics/btn604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fahlgren N., Carrington J.C. miRNA target prediction in plants. Methods Mol. Biol. 2010;592:51–57. doi: 10.1007/978-1-60327-005-2_4. [DOI] [PubMed] [Google Scholar]
- 9.Zheng Y., Li Y.-F., Sunkar R., Zhang W. SeqTar: an effective method for identifying microRNA guided cleavage sites from degradome of polyadenylated transcripts in plants. Nucleic Acids Res. 2012;40:e28. doi: 10.1093/nar/gkr1092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Brousse C., Liu Q., Beauclair L., Deremetz A., Axtell M.J., Bouché N. A non-canonical plant microRNA target site. Nucleic Acids Res. 2014;42:5270–5279. doi: 10.1093/nar/gku157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Folkes L., Moxon S., Woolfenden H.C., Stocks M.B., Szittya G., Dalmay T., Moulton V. PAREsnip: a tool for rapid genome-wide discovery of small RNA/target interactions evidenced through degradome sequencing. Nucleic Acids Res. 2012;40:e103. doi: 10.1093/nar/gks277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Arikit S., Zhai J., Meyers B.C. Biogenesis and function of rice small RNAs from non-coding RNA precursors. Curr. Opin. Plant Biol. 2013;16:170–179. doi: 10.1016/j.pbi.2013.01.006. [DOI] [PubMed] [Google Scholar]
- 13.Johnson C., Kasprzewska A., Tennessen K., Fernandes J., Nan G.-L., Walbot V., Sundaresan V., Vance V., Bowman L.H. Clusters and superclusters of phased small RNAs in the developing inflorescence of rice. Genome Res. 2009;19:1429–1440. doi: 10.1101/gr.089854.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Song X., Li P., Zhai J., Zhou M., Ma L., Liu B., Jeong D.H., Nakano M., Cao S., Liu C. Roles of DCL4 and DCL3b in rice phased small RNA biogenesis. Plant J. 2012;69:462–474. doi: 10.1111/j.1365-313X.2011.04805.x. [DOI] [PubMed] [Google Scholar]
- 15.Jeong D.H., Schmidt S.A., Rymarquis L.A., Park S., Ganssmann M., German M.A., Accerbi M., Zhai J., Fahlgren N., Fox S.E. Parallel analysis of RNA ends enhances global investigation of microRNAs and target RNAs of Brachypodium distachyon. Genome Biol. 2013;14:R145. doi: 10.1186/gb-2013-14-12-r145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Vergoulis T., Vlachos I.S., Alexiou P., Georgakilas G., Maragkakis M., Reczko M., Gerangelos S., Koziris N., Dalamagas T., Hatzigeorgiou A.G. TarBase 6.0: capturing the exponential growth of miRNA targets with experimental support. Nucleic Acids Res. 2012;40:D222–D229. doi: 10.1093/nar/gkr1161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Li J.-H., Liu S., Zhou H., Qu L.-H., Yang J.-H. starBase v2.0: decoding miRNA-ceRNA, miRNA-ncRNA and protein-RNA interaction networks from large-scale CLIP-Seq data. Nucleic Acids Res. 2014;42:D92–D97. doi: 10.1093/nar/gkt1248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hsu S.-D., Tseng Y.-T., Shrestha S., Lin Y.-L., Khaleel A., Chou C.-H., Chu C.-F., Huang H.-Y., Lin C.-M., Ho S.-Y., et al. miRTarBase update 2014: an information resource for experimentally validated miRNA-target interactions. Nucleic Acids Res. 2014;42:D78–D85. doi: 10.1093/nar/gkt1266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Xiao F., Zuo Z., Cai G., Kang S., Gao X., Li T. miRecords: an integrated resource for microRNA-target interactions. Nucleic Acids Res. 2009;37:D105–D110. doi: 10.1093/nar/gkn851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Goodstein D.M., Shu S., Howson R., Neupane R., Hayes R.D., Fazo J., Mitros T., Dirks W., Hellsten U., Putnam N., et al. Phytozome: a comparative platform for green plant genomics. Nucleic Acids Res. 2012;40:1178–1186. doi: 10.1093/nar/gkr944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Simpson J.T., Durbin R. Efficient construction of an assembly string graph using the FM-index. Bioinformatics. 2010;26:i367–i373. doi: 10.1093/bioinformatics/btq217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Langmead B., Salzberg S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods. 2012;9:357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Davis M.P.A., van Dongen S., Abreu-Goodger C., Bartonicek N., Enright A.J. Kraken: a set of tools for quality control and analysis of high-throughput sequence data. Methods. 2013;63:41–49. doi: 10.1016/j.ymeth.2013.06.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Langmead B., Trapnell C., Pop M., Salzberg S.L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009;10:R25. doi: 10.1186/gb-2009-10-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ha I., Wightman B., Ruvkun G. A bulged lin-4/lin-14 RNA duplex is sufficient for Caenorhabditis elegans lin-14 temporal gradient formation. Genes Dev. 1996;10:3041–3050. doi: 10.1101/gad.10.23.3041. [DOI] [PubMed] [Google Scholar]
- 26.Wightman B., Ha I., Ruvkun G. Posttranscriptional regulation of the heterochronic gene lin-14 by lin-4 mediates temporal pattern formation in C. elegans. Cell. 1993;75:855–862. doi: 10.1016/0092-8674(93)90530-4. [DOI] [PubMed] [Google Scholar]
- 27.Didiano D., Hobert O. Perfect seed pairing is not a generally reliable predictor for miRNA-target interactions. Nat. Struct. Mol. Biol. 2006;13:849–851. doi: 10.1038/nsmb1138. [DOI] [PubMed] [Google Scholar]
- 28.Chi S.W., Zang J.B., Mele A., Darnell R.B. Argonaute HITS-CLIP decodes microRNA-mRNA interaction maps. Nature. 2009;460:479–486. doi: 10.1038/nature08170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chi S.W., Hannon G.J., Darnell R.B. An alternative mode of microRNA target recognition. Nat. Struct. Mol. Biol. 2012;19:321–327. doi: 10.1038/nsmb.2230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Xia Z., Clark P., Huynh T., Loher P., Zhao Y., Chen H.-W., Rigoutsos I., Zhou R. Molecular dynamics simulations of Ago silencing complexes reveal a large repertoire of admissible “seed-less” targets. Sci. Rep. 2012;2:569. doi: 10.1038/srep00569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Khorshid M., Hausser J., Zavolan M., van Nimwegen E. A biophysical miRNA-mRNA interaction model infers canonical and noncanonical targets. Nat. Methods. 2013;10:253–255. doi: 10.1038/nmeth.2341. [DOI] [PubMed] [Google Scholar]
- 32.Allen E., Xie Z., Gustafson A.M., Carrington J.C. microRNA-directed phasing during trans-acting siRNA biogenesis in plants. Cell. 2005;121:207–221. doi: 10.1016/j.cell.2005.04.004. [DOI] [PubMed] [Google Scholar]
- 33.Li Y.-F., Zheng Y., Addo-Quaye C., Zhang L., Saini A., Jagadeeswaran G., Axtell M.J., Zhang W., Sunkar R. Transcriptome-wide identification of microRNA targets in rice. Plant J. 2010;62:742–759. doi: 10.1111/j.1365-313X.2010.04187.x. [DOI] [PubMed] [Google Scholar]
- 34.Nakano M., Nobuta K., Vemaraju K., Tej S.S., Skogen J.W., Meyers B.C. Plant MPSS databases: signature-based transcriptional resources for analyses of mRNA and small RNA. Nucleic Acids Res. 2006;34:D731–D735. doi: 10.1093/nar/gkj077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhai J., Jeong D.H., De Paoli E., Park S., Rosen B.D., Li Y., González A.J., Yan Z., Kitto S.L., Grusak M.a., et al. MicroRNAs as master regulators of the plant NB-LRR defense gene family via the production of phased, trans-acting siRNAs. Genes Dev. 2011;25:2540–2553. doi: 10.1101/gad.177527.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.R Development Core Team, R. R: a Language and Environment for Statistical Computing. R Found. Stat. Comput. 2011;1:409. [Google Scholar]
- 37.Gong L., Kakrana A., Arikit S., Meyers B.C., Wendel J.F. Composition and expression of conserved microRNA genes in diploid cotton (Gossypium) species. Genome Biol. Evol. 2013;5:2449–2459. doi: 10.1093/gbe/evt196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rymarquis L.A., Souret F.F., Green P.J. Evidence that XRN4, an Arabidopsis homolog of exoribonuclease XRN1, preferentially impacts transcripts with certain sequences or in particular functional categories. RNA. 2011;17:501–511. doi: 10.1261/rna.2467911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Axtell M.J., Jan C., Rajagopalan R., Bartel D.P. A two-hit trigger for siRNA biogenesis in plants. Cell. 2006;127:565–577. doi: 10.1016/j.cell.2006.09.032. [DOI] [PubMed] [Google Scholar]
- 40.Rodriguez R.E., Mecchia M.A., Debernardi J.M., Schommer C., Weigel D., Palatnik J.F. Control of cell proliferation in Arabidopsis thaliana by microRNA miR396. Development. 2010;137:103–112. doi: 10.1242/dev.043067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Debernardi J.M., Rodriguez R.E., Mecchia M.A., Palatnik J.F. Functional specialization of the plant miR396 regulatory network through distinct microRNA-target interactions. PLoS Genet. 2012;8 doi: 10.1371/journal.pgen.1002419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mecchia M.A., Debernardi J.M., Rodriguez R.E., Schommer C., Palatnik J.F. MicroRNA miR396 and RDR6 synergistically regulate leaf development. Mech. Dev. 2013;130:2–13. doi: 10.1016/j.mod.2012.07.005. [DOI] [PubMed] [Google Scholar]
- 43.Baev V., Milev I., Naydenov M., Apostolova E., Minkov G., Minkov I., Yahubyan G. Implementation of a de novo genome-wide computational approach for updating Brachypodium miRNAs. Genomics. 2011;97:282–293. doi: 10.1016/j.ygeno.2011.02.008. [DOI] [PubMed] [Google Scholar]
- 44.Jeong D.H., Thatcher S.R., Brown R.S.H., Zhai J., Park S., Rymarquis L.A., Meyers B.C., Green P.J. Comprehensive investigation of microRNAs enhanced by analysis of sequence variants, expression patterns, ARGONAUTE loading, and target cleavage. Plant Physiol. 2013;162:1225–1245. doi: 10.1104/pp.113.219873. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.