

Abstract
Activated leukocyte cell adhesion molecule (ALCAM) is an immunoglobulin superfamily cell adhesion molecule that is aberrantly expressed in a wide variety of human tumors, including melanoma, prostate cancer, breast cancer, colorectal carcinoma, bladder cancer and pancreatic adenocarcinoma. This wide spectrum of human malignancies makes ALCAM a prospective pan-cancer immunoPET target to aid in detection and diagnosis in multiple malignancies. In this study, we assess site-specific versus non-site-specific conjugation strategies for 64Cu-DOTA (1,4,7,10-tetraazacyclododecane-1,4,7,10-tetraacetic acid) immunoPET imaging of a fully human ALCAM cys-diabody (cDb) with a reduced linker length that retains its bivalent binding ability. ALCAM constructs with linker lengths of eight, five and three amino acids were produced to make true non-covalent site-specifically modified cDbs. Characterization by gel electrophoresis, size exclusion chromatography, flow cytometry and mass spectrometry of the various constructs was performed. To demonstrate the increased utility of targeting multiple malignancies expressing ALCAM, we compare the targeting of the site-specific versus non-site-specific conjugated cDbs to the human colorectal cancer xenograft LS174T. Interestingly, the conjugation strategy not only affects tumor targeting but also hepatic and renal uptake/clearance.
Keywords: ALCAM, Cu-64, DOTA, immunoPET, site-specific conjugation
Introduction
Activated leukocyte cell adhesion molecule (ALCAM/CD166) is a 110 kDa cell surface glycoprotein that forms homotypic adhesion as well as heterotypic adhesion with CD6 expressed on activated leukocytes (Bowen et al., 1995; Patel et al., 1995). Altered ALCAM expression has been found to be important on a wide variety of malignancies, including melanoma (van Kempen et al., 2000; Swart et al., 2005), pancreatic cancer (Chen et al., 2005), prostate cancer (Kristiansen et al., 2005), breast cancer (King et al., 2004), ovarian cancer (Mezzanzanica et al., 2008), acute myeloid leukemia (Strassberger et al., 2014) and colorectal carcinoma (Weichert et al., 2004). The exact role of upregulation or downregulation of ALCAM expression in tumorigenesis and metastatic potential of these malignancies is not completely understood (Ofori-Acquah and King, 2008). However, ALCAM detection as an accessible target for antibody targeting (Schliemann et al., 2010) and the altered expression of ALCAM in multiple malignancies makes it a promising target for both detection (McCabe et al., 2012) and therapy (Wiiger et al., 2010). Recent findings demonstrating ALCAM expression on hematopoietic stem and progenitor cells might limit the use of ALCAM antibody-based therapy or require allogeneic stem cell transplantation post-therapy (Chitteti et al., 2014; Strassberger et al., 2014).
Immuno-positron emission tomography (immunoPET), whereby antibodies or their fragments are conjugated to PET radionuclides for subsequent PET acquisition, has generated considerable interest for the identification and quantification of tissue biomarkers in vivo (Wu and Senter, 200,5; Wu, 2009). It is the high sensitivity and intrinsic quantification of PET coupled with the specificity and affinity of antibodies that make immunoPET an attractive translatable imaging modality for the detection of disease-specific biomarkers in the clinic (Knowles and Wu, 2012).
To obtain high target-to-background immunoPET images, the pharmacokinetic properties of the imaging agent, i.e. target tissue uptake and blood clearance, need to be optimized for the specific radionuclide used. Intact antibodies interact with the neonatal Fc receptors via the Fc portion of the antibody in a pH-dependent manner allowing for antibody recycling and persistent circulation of ∼1–3 weeks. Copper-64 (64Cu) has many applications for the development of radiopharmaceuticals for PET imaging (Anderson and Ferdani, 2009). However, it has a half-life of 12.7 h, making it unsuitable for the conjugation to intact antibodies and imaging past 2 days post-injection. Therefore, engineered antibody fragments that lack the full Fc domain, like the diabody (Db) and minibody (Mb) that have terminal serum half-lives of 2–5 and 5–12 h, respectively, are more compatible for 64Cu-immunoPET (Wu, 2013).
Dbs (∼50 kDa) are non-covalent single-chain variable fragment (scFv ∼25 kDa) dimers that are constructed by reducing the linker length of an scFv between the VH and VL domains from 15–18 amino acids to 3–8 amino acids, forcing them to form interchain-VH and VL pairing resulting in bivalent Dbs and not intrachain-VH and VL pairing resulting in scFvs. Dbs exhibit increased tumor uptake due to their bivalent avidity and longer half-life compared with scFvs. Furthermore, cysteine residues can be engineered into the Db construct to produce covalent cysteine-modified diabodies, or cys-diabodies (cDbs) (Li et al., 2002; Olafsen et al., 2004). The engineered cysteine allows for the site-specific conjugation of thiol-reactive fluorophores (Sirk et al., 2008), quantum dots (Barat et al., 2009) or bifunctional chelators (Olafsen et al., 2004). As opposed to non-site-specific conjugation, site-specific conjugation allows for a single, uniform product instead of a distribution of conjugations to the protein of interest. Species with the greater number of conjugates using non-site-specific conjugations to radiometal chelators are likely less functional due to modification at the binding site of the antibody. Furthermore, these more highly-conjugated species are likely to carry more radioactivity, which when used for imaging purposes will result in most of the radioactive signal originating from inactive/non-functional radiolabeled protein. The importance of site specificity has been proven in many systems, including antibody–drug conjugation (Junutula et al., 2008), protein immobilization (Camarero, 2008) and molecular imaging (Tait et al., 2006; Tavaré et al., 2009), among others, due to defined, reproducible conjugations and directing the modification away from the active or binding site of the protein undergoing conjugation. Furthermore, groups have examined many other ways, including unnatural amino acids and click chemistry, to incorporate bio-orthogonal site-specific conjugations for protein modification (Sletten and Bertozzi, 2009; Liu and Schultz, 2010).
Recently, a cross-reactive human and mouse anti-ALCAM scFv was derived from an internalizing human scFv phage library (Liu et al., 2004; Liu et al., 2007). After reformatting into a diabody by reducing the linker length from 15 to 8 amino acids, it was conjugated to the chelator 1,4,7,10-tetraazacyclododecane-1,4,7,10-tetraacetic acid (DOTA) using a non-site-specific approach for subsequent 64Cu radiolabeling and immunoPET of human pancreatic adenocarcinoma xenografts (HPAF-II and BxPC-3) (McCabe et al., 2012). In this study, we initially optimized the linker length of the pre-existing ALCAM cDb to maximize diabody formation by reducing the linker length from eight amino acids to five or three amino acids to generate ALCAM-8L, -5L and -3L cDbs, respectively (Fig. 1A). With the validation of diabody formation, we analyzed the targeting capabilities of a site-specific versus non-site-specific DOTA-conjugated anti-ALCAM cDb to human LS174T colorectal carcinoma xenografts not only to monitor the effect of conjugation methodology but also to validate anti-ALCAM as a robust pan-cancer immunoPET agent.
Fig. 1.

(A) Anti-ALCAM gene assembly displaying linkers of eight, five or three amino acids and schematic representations of the resulting diabody and scFv dimer conformations. L, signal peptide for mammalian cell secretion; VH, variable heavy domain; VL, variable heavy domain. (B) SDS–PAGE of anti-ALCAM-8L, -5L and -3L cDbs under reducing (R) and non-reducing (NR) conditions. (C) Size exclusion chromatography using Superdex 75 column at 0.5 ml/min of anti-ALCAM-8L, -5L, and -3L cDbs using PBS as running buffer. Reference arrows indicate albumin (66 kDa) at 20.8 min, carbonic anhydrase (29 kDa) at 24.7 min, and cytochrome C (12.4 kDa) at 27.4 min.
Experimental procedures
Cell lines and media
The human colon carcinoma cell line LS174T (ATCC #CL-188) was maintained in minimum essential medium (Mediatech, Inc.) supplemented with 10% FetalPlex (Gemini Bio-Products), 1% l-glutamine, 1% non-essential amino acids and 1% sodium pyruvate. The rat glioma cell line C6 (ATCC #CCL-107) was maintained in deficient Dulbecco's modification of eagle's basal medium (DMEM) and high glucose (IrvineScientific) supplemented with 10% FetalPlex and 1% l-glutamine. NS0 mouse myeloma cell line (Sigma) (Galfre and Milstein, 1981) was maintained as described (Olafsen et al., 2007).
Cloning
The linker length of the previously described anti-ALCAM-8L cDb (McCabe et al., 2012) was reduced from eight amino acids (GGGGSGGG) to five (GGGGS) or three (GGS) amino acids by site-directed mutagenesis (Agilent Technologies) following manufacturer's instructions to create the anti-ALCAM-5L and -3L cDbs, respectively. The following primers were used:
ALCAM-3L-forward: 5′-GTCTCCTCAGGCGGTTCAAATTTTATGCTGACTCAG
ALCAM-3L-reverse: 5′-GCATAAAATTTGAACCGCCTGAGGAGACGGTGACCAG
ALCAM-5L-forward: 5′-GGAGGCGGTTCAAATTTTATGCTGACTCAGGAC
ALCAM-5L-reverse: 5′-CAGCATAAAATTTGAACCGCCTCCACCTGAGGAG
Protein production and purification
Anti-ALCAM-8L, -5L and -3L cDbs were produced and purified as described (McCabe et al., 2012). Briefly, NS0 cells were electroporated with SalI-linearized DNA. Using high dilution, clones were selected using selective glutamine-deficient DMEM (Mediatech, Inc.). Individual clones were screened for cDb production by harvesting supernatants and analyzed by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS–PAGE) and western blotting. Blots were probed with alkaline phosphatase-conjugated protein A (Sigma) followed by NBT/BCIP Color Development Substrate kit (Promega).
Alexa Fluor 488 conjugations
Anti-ALCAM-8L, -5L and -3L cDbs were initially conjugated to Alexa Fluor 488 C5-maleimide (mal488; Life Technologies) under reducing conditions. cDbs (∼100 μl at 1 mg/ml) were reduced with 10 mM tris[2-carboxyethyl]phosphine hydrochloride (TCEP; Pierce) for 2 h at room temperature. Subsequently, 200 mM of mal488 in DMSO was added directly to the reduced cDb for 2 h at room temperature. Excess dye was removed using a dye removal kit following manufacturer's instructions (Thermo Scientific). Conjugation efficiency was evaluated using the NanoDrop (Thermo Scientific) to calculate the ratio of moles Alexa488 to moles protein. Conjugation efficiency was also measured qualitatively by size exclusion chromatography using a Superdex 75 column (GE Healthcare) on an AKTA purifier (GE Healthcare) and SDS–PAGE-Coomassie stain analysis. Conjugation recovery is defined as the percentage of protein recovered post-conjugation.
DOTA conjugations
For DOTA conjugations, all solutions were made metal-free (MF) using Chelex 100 (1.2 g/l; BioRad). Anti-ALCAM-8L, -5L and -3L cDbs at 1 mg/ml were dialyzed overnight in MF-PBS using Slide-A-Lyzer MINI dialysis units (Thermo Scientific). Non-site-specific conjugation to anti-ALCAM-3L cDb was performed using 1,4,7,10-tetraazacyclododecane-1,4,7,10-tetraacetic acid mono (N-hydroxysuccinimide ester) (DOTA-NHS; Macrocyclics) to form anti-ALCAM-3L-amide-DOTA-cDb. Briefly, DOTA-NHS in MF-PBS was added at a 100-fold molar excess of ACLAM cDbs and the pH was adjusted to 8–8.5 immediately using 1 μl additions of 1 M MF-NaOH. The conjugation was allowed to proceed for 2 h before excess NHS-DOTA removal (see below). For site-specific conjugation to the ALCAM cDbs using 1,4,7,10-tetraazacyclododecane-1,4,7-tris-acetic acid-10-maleimidoethylacetamide (malDOTA; Macrocyclics), the ALCAM cDbs were reduced using a 10-fold molar excess of TCEP for 30 min at room temperature. Subsequently, a 20-fold molar excess of malDOTA was added directly to the reduced cDb for 2 h at room temperature. Excess NHS-DOTA or malDOTA was removed using PD-10 desalting columns (GE Healthcare) that were pre-equilibrated with MF-PBS. Eluted protein was concentrated with Amicon Ultra centrifugal filters (0.5 ml and 10 kDa MWCO; Millipore) that had been washed twice with MF-PBS. Conjugation efficiency was measured qualitatively by size exclusion chromatography using a Superdex 75 column on an AKTA purifier and SDS–PAGE-Coomassie stain analysis.
Mass spectrometry
The anti-ALCAM-3L cDb, anti-ALCAM-3L-amide-DOTA-cDb and anti-ALCAM-3L-thioether-DOTA-cDb liquid chromatography–mass spectrometry were acquired and analyzed at City of Hope. Briefly, mass measurements were determined by QToF mass spectrometry using an Agilent 6520, and Agilent nanoflow 1200 HPLC, and a Chip Cube interface with a C8 HPLC chip. Data analysis was performed using Agilent Mass Hunter Bioconfirm software.
MicroPET imaging and analysis
All animal studies were performed under an approved UCLA Chancellor's Animal Research Committee protocol. 3–4 × 106 cells (LS174T or C6) were harvested and resuspended in 50 μl of DMEM and mixed with 50 μl of Matrigel (BD Biosciences). Cells were implanted subcutaneously in the right (LS174T) and left (C6) shoulder areas of female nude mice (Charles River Laboratories International, Inc.) 9 days prior to microPET imaging. Both 64Cu-DOTA(amide)-cDb and 64Cu-DOTA(thioether)-cDb injected doses contained ∼98 μCi cDb (11.6 μg; 8.6 μCi/μg or 0.32 MBq/μg). Doses were prepared in a total volume of 200 μl and were injected via the tail vein. Mice were anesthetized at 4 h post-injection using 2% isoflurane, and 10-min static scans were acquired with an Inveon microPET (Siemens). Filtered back-projection was used for microPET image reconstruction and image analysis was performed using A Medical Imaging Data Examiner (AMIDE) (Loening and Gambhir, 2003).
Biodistribution
Mice were sacrificed after imaging, and tumors and organs were collected and weighed. Tissue activity was measured using a Wizard 3′ 1480 Automatic Gamma Counter (Perkin-Elmer), and decay-corrected values were used to calculate percent injected dose per gram (%ID/g). T-tests were performed to determine statistical significance.
Immunohistochemistry
LS174T and C6 tumors were harvested 2 weeks post-implantation for immunohistochemistry (IHC) analysis. Tumors were fixed in 4% formaldehyde overnight, paraffin-embedded and cut into 4 μm sections. Sections were stained for ALCAM as described previously (McCabe et al., 2012).
Results
The anti-ALCAM-8L cDb was initially produced, purified and conjugated to mal488 and malDOTA (Figs. 1–3). Upon SE analysis, however, it was apparent that the anti-ALCAM-8L cDb (Figs. 1C, 2B, 3B solid lines) was produced as a covalent dimer of scFvs and not a diabody. Due to this, site-specific mutagenesis was performed to reduce the linker length from eight to five or three amino acids, creating anti-ALCAM-5L and -3L cDbs respectively, to aid in inter-scFv pairing and diabody formation (Fig. 1A). All three constructs were produced and purified from NS0 supernatant using ProteinA affinity chromatography to >95% purity as detected by SDS–PAGE under reducing and non-reducing conditions (Fig. 1B). Under non-reducing conditions, the covalent cDbs run as 50 kDa dimers in the presence of denaturing SDS and as 25 kDa monomers upon reduction. SEC analysis showed the three anti-ALCAM constructs eluted at a correct size of ∼50 kDa and only anti-ALCAM-8L cDb shows a second peak in the SEC analysis of smaller molecular weight ∼25 kDa (Fig. 1C). Recoveries of the anti-ALCAM-8L, -5L and -3L cDbs ranged from 4 to 6.5 mg/l.
Fig. 3.

Analysis of malDOTA conjugation to anti-ALCAM-8L, -5L and -3L cDbs. (A) SDS–PAGE of anti-ALCAM-8L, -5L and -3L cDbs post-malDOTA conjugation. (B) Size exclusion chromatography analysis at 280 nm using the Superdex 75 column at 0.5 ml/min of malDOTA-conjugated anti-ALCAM-8L, -5L, and -3L cDbs using PBS as a running buffer. Reference arrows indicate albumin (66 kDa) at 20.8 min, carbonic anhydrase (29 kDa) at 24.7 min and cytochrome C (12.4 kDa) at 27.4 min.
Fig. 2.

Analysis of mal488 conjugation to anti-ALCAM-8L, -5L, and -3L cDbs. (A) SDS–PAGE of anti-ALCAM-8L, -5L, and -3L cDbs post-mal488 conjugation shown as Coomassie stain (left panel) and fluorescence (right panel). Size exclusion chromatography analysis at 280 nm (B) and 488 nm (C) using Superdex 75 column at 0.5 ml/min of mal488-conjugated anti-ALCAM-8L, -5L and -3L cDbs using PBS as running buffer. Reference arrows indicate albumin (66 kDa) at 20.8 min, carbonic anhydrase (29 kDa) at 24.7 min, and cytochrome C (12.4 kDa) at 27.4 min.
After conjugation to mal488, the anti-ALCAM-8L, -5L and -3L cDbs had conjugation recoveries of ∼40% and the dye-to-protein molar ratios were 0.9, 1.1 and 1.8, respectively. The anti-ALCAM-8L, -5L and -3L cDbs were analyzed by SDS–PAGE for the detection of protein by Coomassie staining and Alexa Fluor 488 by fluorescence detection (Fig. 2A). The anti-ALCAM-8L, -5L and -3L cDbs all showed protein and fluorescence signal at ∼25 kDa in the gel, as expected for a maleimide-conjugated cDb under denaturing SDS conditions (Fig. 2A). In SEC analysis using 280 nm for protein detection and 488 nm for fluorescence detection, it is apparent that both the 5L and 3L remained completely as ∼50 kDa diabodies (Fig. 2B) even though they run as ∼25 kDa in SDS–PAGE analysis. The 8L cDb, however, elutes as two peaks in both 280 and 488 nm detection at ∼50 and ∼25 kDa (Fig. 2C), indicating that some species behave as diabodies (51% by 280 nm and 26% by 488 nm absorbance) and some as scFv dimers (49% by 280 nm and 74% by 488 nm absorbance).
A similar analysis was performed for conjugating malDOTA to anti-ALCAM-8L, -5L and -3L cDbs (Fig. 3), showing again that the 8L cDb behaves as an scFv following reduction and site-specific conjugation to malDOTA. For this conjugation method, the 8L, 5L and 3L cDbs had conjugation recoveries of ∼80%. In contrast to the mal488 conjugation, the anti-ALCAM-8L cDb conjugation to malDOTA resulted in 100% conversion to the scFv as detected by SEC analysis. Elution profiles are summarized in Table I for the anti-ALCAM-8L, -5L and -3L cDbs under non-reducing conditions for unconjugated protein, mal488-conjugated protein and malDOTA-conjugation, as well as unconjugated protein under reducing conditions (SEC profiles not shown), demonstrating that the 8L cDb is produced as an scFv dimer.
Table I.
Elution times of unconjugated anti-ALCAM-8L, 5L, and 3L cDbs in non-reducing conditions and reducing conditions, and mal488- and malDOTA-conjugated anti-ALCAM-8L, 5L and 3L cDbs in non-reducing conditions using Superdex 75 size exclusion chromatography. Elution times are shown in minutes and numbers in parentheses indicate the percentage at each retention time if applicable. Note the reference elution times are as follows: albumin (66 kDa) at 20.8 min, carbonic anhydrase (29 kDa) at 24.7 min and cytochrome C (12.4 kDa) at 27.4 min
| Non-reducing |
Reducing |
Maleimide-488 |
Maleimide-DOTA |
|||||
|---|---|---|---|---|---|---|---|---|
| Construct | cDb | scFv | cDb | scFv | cDb | scFv | cDb | scFv |
| ALCAM-8L | 22.80 (82%) | 25.10 (18%) | n/a | 24.95 | 22.85 (51%) | 24.80 (49%) | n/a | 24.32 |
| ALCAM-5L | 22.98 | n/a | 23.31 | n/a | 22.91 | n/a | 22.86 | n/a |
| ALCAM-3L | 23.02 | n/a | 23.11 | n/a | 22.85 | n/a | 22.70 | n/a |
As the anti-ACLAM-3L cDb clearly retained its cross-paired diabody conformation following reduction and modification, it was used for all subsequent experiments. In order to compare non-site-specific with site-specific DOTA conjugation, the 3L cDb was conjugated to lysines using NHS-DOTA and to the reduced cysteines using maleimide-DOTA, resulting in ALCAM-3L-amide-DOTA-cDb and ALCAM-3L-thioether-DOTA-cDb, respectively. Mass spectrometry confirmed the addition of one to five DOTAs per diabody for the ALCAM-3L-amide-DOTA-cDb and exactly two DOTAs per diabody for the ALCAM-3L-thioether-DOTA-cDb (Table II).
Table II.
Mass spectrometry review of intact unconjugated anti-ALCAM-3L cDb, anti-ALCAM-3L-amide-DOTA-cDb and anti-ALCAM-3L-thioether-DOTA-cDb. Expected addition of one amide-DOTA conjugation is 386.2 kDa and one thioether-DOTA is 526.2 kDa
| Modification | Expected (kDa) | Found (kDa) | Difference (kDa) | |
|---|---|---|---|---|
| Unconjugated | n/a | 50344.2 | 50341.6 | −2.6 |
| NHS-DOTA | 0× DOTA | 50344.2 | 50342.3 | −1.9 |
| 1× DOTA | 50730.4 | 50729.3 | −1.1 | |
| 2× DOTA | 51116.8 | 51115.2 | −1.4 | |
| 3× DOTA | 51502.8 | 51501.3 | −1.5 | |
| 4× DOTA | 51889.0 | 51888.1 | −0.9 | |
| 5× DOTA | 52275.2 | 52275.5 | +0.3 | |
| malDOTA | 0× DOTA | 25173.1 | 25138.2 | −34.9 |
| 1× DOTA | 25699.3 | 25716.8 | +17.5 |
When radiolabeled with 64Cu, ALCAM-3L-amide-DOTA-cDb and ALCAM-3L-thioether-DOTA-cDb achieved specific activities of 8.6 μCi/μg (0.32 MBq/μg). Importantly, these conjugated ALCAM cDb's retained their ability to bind the ALCAM-expressing colon cancer cell line LS174T as the immunoreactive fraction of both 64Cu-radiolabeled ALCAM cDb's were 83%. Approximately 11.6 μg of cDb was injected into mice bearing ALCAM-positive LS174T tumors and antigen-negative C6 tumors. Four hours post-injection, immunoPET images were acquired (Fig. 4) and biodistributions were performed (Table III). Uptake in the LS174T tumor was 1.4 ± 0.15%ID/g and 2.6 ± 0.53%ID/g for ALCAM-3L-amide-DOTA-cDb and ALCAM-3L-thioether-DOTA-cDb, respectively, and 0.99 ± 0.06%ID/g and 1.4 ± 0.06%ID/g for the antigen-negative C6 tumor. The ALCAM-3L-thioether-DOTA-cDb also had higher blood activity (1.4 ± 0.15%ID/g) than the ALCAM-3L-amide-DOTA-cDb (0.97 ± 0.11%ID/g), indicating a slower blood clearance. The corresponding LS174T tumor-to-blood ratios of the ALCAM-3L-amide-DOTA-cDb and ALCAM-3L-thioether-DOTA-cDb were 1.4 and 1.9, respectively.
Fig. 4.

ImmunoPET images in athymic nude mice bearing ALCAM-positive LS174 T (right) and ALCAM-negative C6 (left) tumors acquired 4 h post-injection shown at different thresholds for comparing both tumor targeting and non-tumor biodistribution of the 64Cu-ALCAM-3L-amide-DOTA-cDb (A) and 64Cu-ALCAM-3L-thioether-DOTA-cDb (B). Coronal PET images are shown as 25 mm maximum intensity projections and transverse PET images are 2 mm maximum intensity projections.
Table III.
Biodistribution of 64Cu-ALCAM-3L-amide-DOTA-cDb and 64Cu-ALCAM-3L-thioether-DOTA-cDb at 4 h post-injection of female nude mice bearing ALCAM-positive LS174T and ALCAM-negative C6 xenografts
| % ID/g ± SD |
|||
|---|---|---|---|
| Organ | amide-DOTA (n = 4) | Thioether-DOTA (n = 4) | P |
| Blood | 0.97 ± 0.11 | 1.4 ± 0.15 | <0.01 |
| LS174T (+) | 1.4 ± 0.15 | 2.6 ± 0.53* | <0.01 |
| C6 (−) | 0.99 ± 0.06 | 1.4 ± 0.06 | <0.0001 |
| Spleen | 3.8 ± 0.38 | 2.6 ± 0.19 | <0.005 |
| Stomach | 1.5 ± 0.27 | 4.0 ± 0.29 | <0.0001 |
| Intestines | 2.0 ± 0.16 | 4.3 ± 0.04 | <0.0001 |
| Liver | 36 ± 2.9 | 20. ± 2.5 | <0.0005 |
| Kidneys | 51 ± 5.7 | 95 ± 9.5 | <0.0005 |
| Heart | 1.2 ± 0.11 | 1.4 ± 0.09 | <0.05 |
| Lungs | 2.2 ± 0.20 | 3.7 ± 0.35 | <0.0005 |
| Muscle | 0.19 ± 0.02 | 0.24 ± 0.03 | 0.054 |
| Carcass | 0.61 ± 0.04 | 0.78 ± 0.03 | <0.0005 |
*n = 3, P-values for LS174T versus C6 for amide-DOTA and thioether-DOTA are <0.005 and <0.01, respectively.
Interestingly, the different conjugation strategies altered both the liver and kidney uptakes; ALCAM-3L-amide-DOTA-cDb had kidney and liver uptakes of 51 ± 5.7%ID/g and 36 ± 2.9%ID/g, while the ALCAM-3L-thioether-DOTA-cDb had kidney and liver uptakes of 95 ± 9.5%ID/g and 20 ± 2.5%ID/g. Furthermore, the ALCAM-3L-thioether-DOTA-cDb shows enhanced uptake in the salivary glands and eyes when compared with the ALCAM-3L-amide-DOTA-cDb. PET images shown at different scales clearly represent the difference in uptake in the kidneys, liver, salivary glands, eyes and tumors of the site-specific versus non-site-specific DOTA-conjugation methods (Fig. 4). IHC was performed on both LS174T and C6 xenografts for ALCAM expression, indicating that the LS174T xenografts do express ALCAM in this model (Fig. 5). As shown here, conjugation strategy greatly affects the pharmacokinetics of cDb fragments and the site-specific conjugation strategy of the ALCAM cDb allows for increased tumor uptake and higher tumor-to-blood ratios than non-site-specifically conjugated cDb.
Fig. 5.
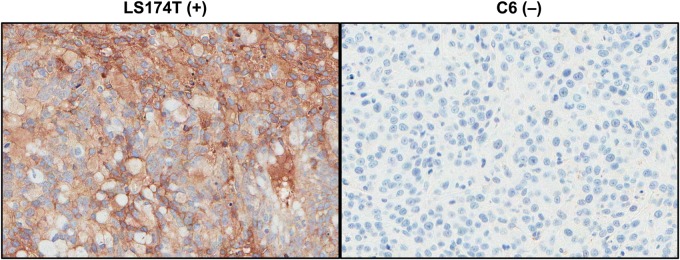
Ex vivo IHC of ALCAM-positive LS174T (left) and ALCAM-negative C6 (right) xenografts stained for ALCAM expression.
Discussion
Anti-ALCAM cDbs with linker lengths of eight, five and three amino acids were engineered, expressed at high levels in mammalian culture and efficiently purified by affinity chromatography. With extensive analysis using SDS–PAGE and SEC of native and site-specifically conjugated anti-ALCAM-8L, -5L and -3L cDbs to mal488 and malDOTA, it was determined that the 8L construct is a dimer of scFvs and not a true cDb. For the mal488 conjugations, the conjugation efficiencies varied from 0.9 to 1.8 fluorescent molecules per protein and the protein recoveries were very low. This conjugation method was not optimized further due to the low recovery but it did allow us to monitor the effect of conjugation to the engineered cysteine by the fluorescent Alexa Fluor 488. On the other hand, the malDOTA conjugations had recoveries of ∼80% and this could be due to the difference in hydrophobicity of the Alexa Fluor 488 and the DOTA chelator.
It has been shown that linker lengths of ≤12 aa cause an scFv to form Dbs (Whitlow et al., 1994; Kortt et al., 1997) and other examples of diabody construction using linker lengths as long or longer than the anti-ALCAM-8L cDb described in this work exist, for example anti-neuraminidase (10 aa (Kortt et al., 1997)), anti-CEA (8 aa (Wu et al., 1999)) and anti-CD20 (8 aa (Olafsen et al., 2010)). Furthermore, shorter linker lengths can cause multimerization into trimeric and/or tetrameric species (Kortt et al., 1997; Kelly et al., 2008). In this study, SEC showed a linker length of three amino acids did not induce multimerization. Individual pairs of VH and VL domains from different scFvs seem to behave differently in their ability to consistently form Dbs, and this could be due to V domain-interface stability, V domain stability and V domain orientation. Combined with reports that linkers of 18 aa can cause multimerization of certain scFv constructs (Whitlow et al., 1994; Le Gall et al., 1999), the work described here demonstrates the importance of rigorous characterization when constructing cDbs.
With the validation of true diabody conformation, we conjugated the ALCAM-3L cDb to the 64Cu chelator DOTA either site specifically via the engineered cysteines or non-site specifically via solvent-exposed lysines using maleimide-DOTA or NHS-DOTA, respectively. Mass spectrometry confirmed the conjugation strategy of exactly two DOTA conjugations per ALCAM-3L-thioether-DOTA-cDb and a distribution of one to five DOTA conjugations per ALCAM-3L-amide-DOTA-cDb. The ALCAM-3L-thioether-DOTA-cDb mass spectrometry showed a single peak consistent with the expected mass change from conjugation. The site-specific conjugation has allowed for the production of a single, chemically defined product for imaging that is still able to be radiolabeled with 64Cu and retains the ability to bind to ALCAM.
Upon 64Cu radiolabeling, the immuno-reactive fraction of both ALCAM-3L-thioether-DOTA-cDb and ALCAM-3L-amide-DOTA-cDb was very similar (∼83%), indicating that one to five DOTA conjugation per ALCAM-3L-amide-DOTA-cDb does not disrupt antigen binding. In xenograft models of C6 and LS174T tumors in nude mice, targeting of the ALCAM-positive LS174T xenograft was enhanced for the site-specific ALCAM-3L-thioether-DOTA-cDb even though both probes had similar immuno-reactivities. Enhanced tumor targeting could be due to the increased blood activity of the 64Cu-ALCAM-3L-thioether-DOTA-cDb compared with the 64Cu-ALCAM-3L-amide-DOTA-cDb, as indicated by their blood %ID/g at 4 h post-injection (1.4 ± 0.15 and 0.97 ± 0.11%ID/g, respectively). Increased blood activity enables greater tumor exposure, resulting in enhanced ALCAM-positive LS174T targeting of the 64Cu-ALCAM-3L-thioether-DOTA-cDb. Importantly, the 64Cu-ALCAM-3L-thioether-DOTA-cDb has higher LS174T tumor-to-blood ratios when compared with the 64Cu-ALCAM-3L-amide-DOTA-cDb even though the blood activity is higher. Altered serum half-life could be due to a variety of factors, including change in the overall surface charge due to conjugation strategy, increased flexibility of the ALCAM-3L-thioether-DOTA-cDb compared with the ALCAM-3L-amide-DOTA-cDb due to conjugation of the engineered thiol, and the effects of the proximity of two hexahistidine tags of the ALCAM-3L-amide-DOTA-cDb when the engineered thiols form a disulfide bond.
The internalization of this anti-ALCAM cDb construct (Liu et al., 2004) requires the use of residualizing PET radionuclides for retention of activity in target tissues. An offshoot is that organs of clearance (i.e. liver and kidney) also retain residualizing activity. High uptake in the kidneys, as seen here, is expected because the cDbs are below the renal filtration cutoff (Vegt et al., 2010). However, liver uptake is high when compared with other residualizing radioisotope-labeled Dbs (Yazaki et al., 2001; Schneider et al., 2009). This is most likely due to transchelation of 64Cu from DOTA to enzymes in the liver (Anderson and Ferdani, 2009). Interestingly, the resulting pharmacokinetic profiles of the two 64Cu-radiolabeled probes showed altered kidney and liver uptakes, as well as altered blood activity as mentioned above. The 64Cu-ALCAM-3L-amide-DOTA-cDb exhibited higher liver and lower kidney uptake when compared with the 64Cu-ALCAM-3L-thioether-DOTA-cDb. This could be due to the same reasons for altered blood clearance: surface charge, flexibility and histidine tags. As kidneys are radiosensitive, methods exist to reduce the renal radioactivity levels of the 64Cu-ALCAM-3L-amide-DOTA-cDb (Arano, 1998). Not only do the kidney and liver uptakes vary, but immunoPET clearly shows enhanced uptake of 64Cu-ALCAM-3L-thioether-DOTA-cDb to the salivary glands and eyes. This anti-ALCAM construct is cross-reactive with mouse and human ALCAM (McCabe et al., 2012), and salivary glands and retinal endothelium are known to express ALCAM (Abidi et al., 2006; Smith et al., 2012). The utilization of ALCAM knockout mice could validate whether salivary gland and retinal targeting of the 64Cu-ALCAM-3L-thioether-DOTA-cDb is target specific.
The concept of altered pharmacokinetics for differing radiolabeling methods based on radioisotope, chelator and conjugation strategy has not been analyzed rigorously for antibody fragments. Previously, peptide linkers have been shown to alter the kidney uptake of DOTA-conjugated anti-CEA cDb (Li et al., 2002). However, many studies have been performed with affibodies that highlight the importance of His-tag sequence, location of His-tag, chelator, radiometal and method of conjugation (Tolmachev and Orlova, 2010; Hofstrom et al., 2013; Strand et al., 2013). These studies highlight the potential effects that radiolabeling strategy can have on diabody tumor targeting and pharmacokinetics.
In conclusion, both conjugation strategies allowed for the targeting and imaging of ALCAM-positive LS174T xenografts in vivo. Interestingly, even though the non-site-specific conjugation did not affect the immunoreactivity of the radiolabeled cDb when compared with the site-specific conjugation, the site-specific ALCAM-3L-thioether-DOTA-cDb variant exhibited enhanced tumor-to-blood and positive-to-negative tumor ratios when compared with the ALCAM-3L-amide-DOTA-cDb. Furthermore, the nature of the conjugation had a drastic impact on renal and hepatic uptake and needs to be considered when developing immunoPET agents. The altered tumor targeting and pharmacokinetic profile could be due to the change in surface charge of the diabody post-DOTA-conjugation and the change in physical structure that results from the reduction and subsequent conjugation to the engineered cysteine residue of the site-specific ALCAM-3L-thioether-DOTA-cDb conjugate. Successful immunoPET imaging of ALCAM expression in both pancreatic (McCabe et al., 2012) and colon cancers suggests further investigation of ALCAM as a pan-cancer imaging biomarker.
Funding
This work was supported by the National Institutes of Health Scholars in Oncological Medical Imaging training program at UCLA [R25T CA098010]. Imaging studies were funded in part by the Cancer Center Support Grant [NIH P30 CA016042].
Acknowledgements
The authors would like to thank Waldemar Ladno, Darin Williams and Dr. David Stout at the Crump Institute for Molecular Imaging at UCLA for their help with the PET scans and both Roger Moore and Bogdan Gugiu at City of Hope for Mass Spectrometry analysis. Furthermore, the authors thank the Genoseq Core Facility at UCLA and the Jonsson Comprehensive Cancer Center.
Conflict of interest: A.M.W. is a founder and consultant to ImaginAb, Inc. and J.D.M. is a consultant to ImaginAb, Inc.
References
- Abidi S.M., Saifullah M.K., Zafiropulos M.D., Kaput C., Bowen M.A., Cotton C., Singer N.G. J. Clin. Immunol. 2006;26:12–21. doi: 10.1007/s10875-006-7119-6. [DOI] [PubMed] [Google Scholar]
- Anderson C.J., Ferdani R. Cancer Biother. Radiopharm. 2009;24:379–393. doi: 10.1089/cbr.2009.0674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arano Y. Q. J. Nucl. Med. 1998;42:262–270. [PubMed] [Google Scholar]
- Barat B., Sirk S.J., McCabe K.E., et al. Bioconjug. Chem. 2009;20:1474–1481. doi: 10.1021/bc800421f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowen M.A., Patel D.D., Li X., et al. J. Exp. Med. 1995;181:2213–2220. doi: 10.1084/jem.181.6.2213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camarero J.A. Biopolymers. 2008;90:450–458. doi: 10.1002/bip.20803. [DOI] [PubMed] [Google Scholar]
- Chen R., Yi E.C., Donohoe S., et al. Gastroenterology. 2005;129:1187–1197. doi: 10.1053/j.gastro.2005.08.001. [DOI] [PubMed] [Google Scholar]
- Chitteti B.R., Kobayashi M., Cheng Y., et al. Blood. 2014 doi: 10.1182/blood-2014-03-565721. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galfre G., Milstein C. Methods Enzymol. 1981;73:3–46. doi: 10.1016/0076-6879(81)73054-4. [DOI] [PubMed] [Google Scholar]
- Hofstrom C., Altai M., Honarvar H., Strand J., Malmberg J., Hosseinimehr S.J., Orlova A., Graslund T., Tolmachev V. J. Med. Chem. 2013;56:4966–4974. doi: 10.1021/jm400218y. [DOI] [PubMed] [Google Scholar]
- Junutula J.R., Raab H., Clark S., et al. Nat. Biotechnol. 2008;26:925–932. doi: 10.1038/nbt.1480. [DOI] [PubMed] [Google Scholar]
- Kelly M.P., Lee F.T., Tahtis K., Power B.E., Smyth F.E., Brechbiel M.W., Hudson P.J., Scott A.M. Cancer Biother. Radiopharm. 2008;23:411–423. doi: 10.1089/cbr.2007.0450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King J.A., Ofori-Acquah S.F., Stevens T., Al-Mehdi A.B., Fodstad O., Jiang W.G. Breast Cancer Res. 2004;6:R478–R487. doi: 10.1186/bcr815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knowles S.M., Wu A.M. J Clin Oncol. 2012;30:3884–3892. doi: 10.1200/JCO.2012.42.4887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kortt A.A., Lah M., Oddie G.W., et al. Protein Eng. 1997;10:423–433. doi: 10.1093/protein/10.4.423. [DOI] [PubMed] [Google Scholar]
- Kristiansen G., Pilarsky C., Wissmann C., et al. J. Pathol. 2005;205:359–376. doi: 10.1002/path.1676. [DOI] [PubMed] [Google Scholar]
- Le Gall F., Kipriyanov S.M., Moldenhauer G., Little M. FEBS Lett. 1999;453:164–168. doi: 10.1016/s0014-5793(99)00713-9. [DOI] [PubMed] [Google Scholar]
- Li L., Olafsen T., Anderson A.L., Wu A., Raubitschek A.A., Shively J.E. Bioconjug. Chem. 2002;13:985–995. doi: 10.1021/bc025565u. [DOI] [PubMed] [Google Scholar]
- Liu B., Conrad F., Cooperberg M.R., Kirpotin D.B., Marks J.D. Cancer Res. 2004;64:704–710. doi: 10.1158/0008-5472.can-03-2732. [DOI] [PubMed] [Google Scholar]
- Liu B., Conrad F., Roth A., Drummond D.C., Simko J.P., Marks J.D. J. Mol. Med. (Berl) 2007;85:1113–1123. doi: 10.1007/s00109-007-0208-z. [DOI] [PubMed] [Google Scholar]
- Liu C.C., Schultz P.G. Annu. Rev. Biochem. 2010;79:413–444. doi: 10.1146/annurev.biochem.052308.105824. [DOI] [PubMed] [Google Scholar]
- Loening A.M., Gambhir S.S. Mol. Imaging. 2003;2:131–137. doi: 10.1162/15353500200303133. [DOI] [PubMed] [Google Scholar]
- McCabe K.E., Liu B., Marks J.D., Tomlinson J.S., Wu H., Wu A.M. Mol. Imaging Biol. 2012;14:336–347. doi: 10.1007/s11307-011-0500-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mezzanzanica D., Fabbi M., Bagnoli M., et al. Clin. Cancer Res. 2008;14:1726–1733. doi: 10.1158/1078-0432.CCR-07-0428. [DOI] [PubMed] [Google Scholar]
- Ofori-Acquah S.F., King J.A. Transl. Res. 2008;151:122–128. doi: 10.1016/j.trsl.2007.09.006. [DOI] [PubMed] [Google Scholar]
- Olafsen T., Cheung C.W., Yazaki P.J., et al. Protein Eng. Des. Sel. 2004;17:21–27. doi: 10.1093/protein/gzh009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olafsen T., Gu Z., Sherman M.A., et al. J. Immunother. 2007;30:396–405. doi: 10.1097/CJI.0b013e318031b53b. [DOI] [PubMed] [Google Scholar]
- Olafsen T., Sirk S.J., Betting D.J., Kenanova V.E., Bauer K.B., Ladno W., Raubitschek A.A., Timmerman J.M., Wu A.M. Protein Eng. Des. Sel. 2010;23:243–249. doi: 10.1093/protein/gzp081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel D.D., Wee S.F., Whichard L.P., Bowen M.A., Pesando J.M., Aruffo A., Haynes B.F. J. Exp. Med. 1995;181:1563–1568. doi: 10.1084/jem.181.4.1563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schliemann C., Roesli C., Kamada H., Borgia B., Fugmann T., Klapper W., Neri D. Blood. 2010;115:736–744. doi: 10.1182/blood-2009-08-239004. [DOI] [PubMed] [Google Scholar]
- Schneider D.W., Heitner T., Alicke B., et al. J. Nucl. Med. 2009;50:435–443. doi: 10.2967/jnumed.108.055608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sirk S.J., Olafsen T., Barat B., Bauer K.B., Wu A.M. Bioconjug. Chem. 2008;19:2527–2534. doi: 10.1021/bc800113v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sletten E.M., Bertozzi C.R. Angew. Chem. Int. Ed. Engl. 2009;48:6974–6998. doi: 10.1002/anie.200900942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith J.R., Chipps T.J., Ilias H., Pan Y., Appukuttan B. Exp. Eye Res. 2012;104:89–93. doi: 10.1016/j.exer.2012.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strand J., Honarvar H., Perols A., Orlova A., Selvaraju R.K., Karlstrom A.E., Tolmachev V. PloS ONE. 2013;8:e70028. doi: 10.1371/journal.pone.0070028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strassberger V., Gutbrodt K.L., Krall N., Roesli C., Takizawa H., Manz M.G., Fugmann T., Neri D. J. Proteomic. 2014;99:138–151. doi: 10.1016/j.jprot.2014.01.022. [DOI] [PubMed] [Google Scholar]
- Swart G.W., Lunter P.C., Kilsdonk J.W., Kempen L.C. Cancer Metastasis Rev. 2005;24:223–236. doi: 10.1007/s10555-005-1573-0. [DOI] [PubMed] [Google Scholar]
- Tait J.F., Smith C., Levashova Z., Patel B., Blankenberg F.G., Vanderheyden J.L. J. Nucl. Med. 2006;47:1546–1553. [PubMed] [Google Scholar]
- Tavaré R., Torres Martin De Rosales R., Blower P.J., Mullen G.E. Bioconjug. Chem. 2009;20:2071–2081. doi: 10.1021/bc900160j. [DOI] [PubMed] [Google Scholar]
- Tolmachev V., Orlova A. Curr. Med. Chem. 2010;17:2636–2655. doi: 10.2174/092986710791859397. [DOI] [PubMed] [Google Scholar]
- van Kempen L.C., van den Oord J.J., van Muijen G.N., Weidle U.H., Bloemers H.P., Swart G.W. Am. J. Pathol. 2000;156:769–774. doi: 10.1016/S0002-9440(10)64943-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vegt E., de Jong M., Wetzels J.F.M., Masereeuw R., Melis M., Oyen W.J.G., Gotthardt M., Boerman O.C. J. Nucl. Med. 2010;51:1049–1058. doi: 10.2967/jnumed.110.075101. [DOI] [PubMed] [Google Scholar]
- Weichert W., Knosel T., Bellach J., Dietel M., Kristiansen G. J. Clin. Pathol. 2004;57:1160–1164. doi: 10.1136/jcp.2004.016238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitlow M., Filpula D., Rollence M.L., Feng S.L., Wood J.F. Protein Eng. 1994;7:1017–1026. doi: 10.1093/protein/7.8.1017. [DOI] [PubMed] [Google Scholar]
- Wiiger M.T., Gehrken H.B., Fodstad O., Maelandsmo G.M., Andersson Y. Cancer Immunol. Immunother. 2010;59:1665–1674. doi: 10.1007/s00262-010-0892-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu A.M. J. Nucl. Med. 2009;50:2–5. doi: 10.2967/jnumed.108.056887. [DOI] [PubMed] [Google Scholar]
- Wu A.M. Methods. 2014;65:139–147. doi: 10.1016/j.ymeth.2013.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu A.M., Senter P.D. Nat. Biotechnol. 2005;23:1137–1146. doi: 10.1038/nbt1141. [DOI] [PubMed] [Google Scholar]
- Wu A.M., Williams L.E., Zieran L., et al. Tumor Target. 1999;4:47–58. [Google Scholar]
- Yazaki P.J., Wu A.M., Tsai S.W., Williams L.E., Ikle D.N., Wong J.Y.C., Shively J.E., Raubitschek A.A. Bioconjug. Chem. 2001;12:220–228. doi: 10.1021/bc000092h. [DOI] [PubMed] [Google Scholar]