

Abstract
Toll-like receptor (TLR) 7 and 8 agonists are potential vaccine adjuvants, since they directly activate APCs and enhance Th1-driven immune responses. Previous SAR investigations in several scaffolds of small molecule TLR7/8 activators pointed to the strict dependence of the selectivity for TLR7 vis-à-vis TLR8 on the electronic configurations of the heterocyclic systems, which we sought to examine quantitatively with the goal of developing “heuristics” to define structural requisites governing activity at TLR7 and/or TLR8. We undertook a scaffold-hopping approach, entailing the syntheses and biological evaluations of 13 different chemotypes. Crystal structures of TLR8 in complex with the two most active compounds confirmed important binding interactions playing a key role in ligand occupancy and biological activity. Density functional theory based quantum chemical calculations on these compounds followed by linear discriminant analyses permitted the classification of inactive, TLR8-active, and TLR7/8 dual-active compounds, confirming the critical role of partial charges in determining biological activity.
Introduction
The human immune system can be conceived of as comprising two mutually nonexclusive subsystems: the innate and adaptive immune limbs work cooperatively to afford protection from numerous pathogenic microorganisms and toxins. Toll-like receptors (TLRs) are important pattern recognition receptors (PRRs) of the innate immune system which are expressed on leukocytes, as well as epithelial cells of mucosal surfaces.1,2 They are activated upon specific recognition of pathogen-associated molecular patterns (PAMPs) that are distinct to the pathogen,3 providing immediate defense mediated by a variety of effector mechanisms including the production of proinflammatory cytokines, up-regulation of major histocompatibility complex (MHC) molecules, and costimulatory signals in antigen-presenting cells (APCs), as well as the activation of natural killer (NK) cells. These events lead to priming and amplification of adaptive immune responses mediated via both T- and B-lymphocyte effector functions.4
Ten functional TLRs have been identified in humans so far;5 TLRs present in the intracellular compartments (TLR3, -7, -8, and -9) sense viral and bacterial nucleic acids, and those expressed on the cell surface (TLR1, -2, -4, -5, -6, and -11) sense outer membrane components of bacteria, fungi, and protozoan organisms.5,6 Aside from binding to exogenous PAMPs, TLRs are also known to interact with endogenous molecules released from damaged tissues or dead cells. This endogenous ligand-mediated TLR activation appears to play a central role in pathological conditions such as tissue injury, repair and regeneration, autoimmune diseases, and tumorigenesis.7
Several synthetic compounds have been identified as modulators of TLR signaling.8 Detailed mechanistic processes underlying TLR signaling that have emerged during the past 15 years not only have provided insights in the interfaces between innate and adaptive immune responses9 but also are paving the way for clinical applications.10 Given that TLRs are predominantly expressed on or in immune cells, appropriate stimulation of the immune cells via effective targeting of TLRs is increasingly recognized as a key factor in vaccinology. From this perspective, we have explored in detail structure–activity relationships (SARs) of several immunostimulatory chemotypes.11−18
In particular, TLR7/8 agonists have demonstrated potential as vaccine adjuvants, since they directly activate APCs and can enhance both humoral and cellular immune responses, especially Th1-biased responses. TLR7 is expressed in plasmacytoid dendritic cells (pDC) and B cells, whereas TLR8 is mainly expressed in conventional/myeloid dendritic cells (cDCs), monocytes, macrophages, and neutrophils.19 The natural ligands for TLR7 and TLR8 are single-stranded RNA (ssRNA); these endosomal TLRs can also be activated by synthetic small molecule TLR7/8 agonists.20 Small molecule TLR7/8 activators constitute a small set of compounds occupying a limited chemical space. Extensive SARs on several TLR7/8-agonistic scaffolds such as imidazo[4,5-c]quinolines,21,22 imidazo[4,5-c]pyridines,23 thiazolo[4,5-c]quinolines,24 furo[2,3-c]pyridines,25 and furo[2,3-c]quinolines26 have been reported from our laboratory (Figure 1). Recently, a C2-butyl furo[2,3-c]quinoline (5) having pure TLR8 agonistic activity was cocrystallized with the human TLR8 ectodomain.27 This served as the point of departure toward a focused structure-based ligand design study, leading to the identification of 3-pentylquinoline-2-amine (6, Figure 1) as a novel, structurally simple, and highly potent human TLR8-specific agonist.28 Our SAR investigations in several of these scaffolds, while continuing to incrementally improve our understanding of the structural features required for the TLR7/8 activity, pointed strongly also to the strict dependence of the selectivity for TLR7 vis-à-vis TLR8 on the electronic configurations of the heterocyclic systems, the nuances of which we desired to examine quantitatively with the goal of developing “heuristics” to clearly define structural requisites governing activity at TLR7 and/or TLR8. In order to systematically examine the effect of electronic properties on the activity profiles, we undertook a scaffold-hopping approach,29−31 entailing the syntheses and biological evaluations of 13 different chemotypes including oxazolo[4,5-c]quinoline, thiazolo/oxazolo[4,5-c]quinolin-2-amines, thiazolo/oxazolo[5,4-c]quinolines, imidazo[1,2-c]quinazoline, [1,2,4]triazolo[1,5-c]quinazoline, imidazo[1,2-a]quinoxaline, [1,2,4]triazolo[1,5-a]quinoxaline, [1,2,4]-triazolo[4,3-a]quinoxaline, and pyrazolo[3,4-c]quinoline.
Figure 1.

Structures of representative TLR7 and TLR8 agonists.
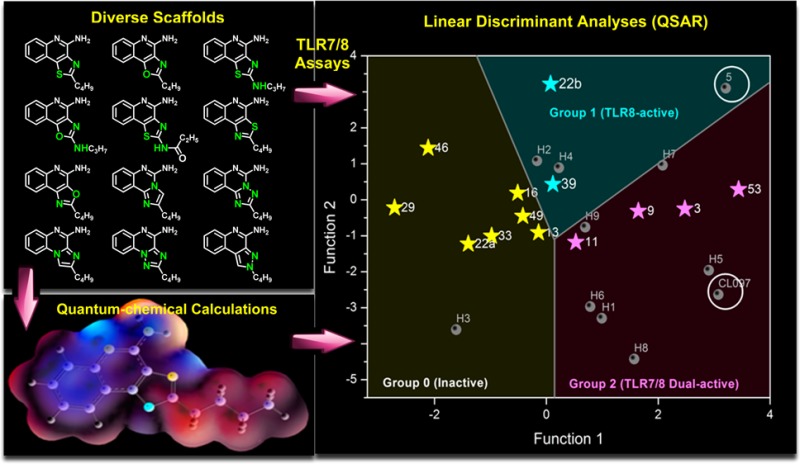
Crystal structures of TLR8 in complex with two most active compounds confirmed important binding interactions playing a key role in ligand occupancy and biological activity. Reasoning that stereoelectronic effects of heterocyclic ring systems could have a profound effect on the biological activity of TLR7/8 modulators, we undertook studies of three-dimensional molecular electrostatic potential (MESP) in an effort to obtain complementary and/or mechanistic information in characterizing active molecules.32 Density functional theory (DFT) based quantum chemical calculations and linear discriminant analyses were therefore performed. These studies allowed, for the first time, a clear delineation of inactive, TLR8-active, and TLR7/8 dual-active compounds, confirming the critical role of partial charges in determining biological activity.
Results and Discussion
As mentioned earlier, a number of leads including pure TLR7 agonists (1 and 2), dual TLR7/8 agonist (e.g., 3), and pure TLR8 agonists (4, 5, and 6) are undergoing preclinical evaluation as vaccine adjuvants in our laboratory. Our earlier structure–activity relationship studies on the imidazo[4,5-c]quinolines,21,22 thiazolo[4,5-c]quinolines,24 furo[2,3-c]pyridines,25 and furo[2,3-c]quinolines26 had all converged on the optimal chain length for the C2 alkyl substituent being butyl. Our goal was therefore to examine the electronic effects of heterocyclic modifications while holding the substituent at the C2 position invariant at four atoms. We envisioned that a reagent-based diversification approach33 could allow us to access several different heterocyclic scaffolds (including the thiazolo[4,5-c]quinolines) with substantial variations in the electronic configurations (Scheme 1). By employment of this diversification strategy, the previously described 2-butylthiazolo[4,5-c]quinoline was synthesized from aminoquinolin-4-ol and valeroyl chloride via a one-pot, sequential reaction involving acylation and subsequent microwave-accelerated (120 °C, 600 W) cyclization using P2S5 (7, Scheme 1), while replacement of P2S5 with P2O5 in this reaction resulted in its congener 8 (2-butyloxazolo[4,5-c]quinoline) in moderate yield. Microwave-assisted cyclization also yielded N-propylthiazolo[4,5-c]quinolin-2-amine 10 using propyl isothiocyanate, whereas conventional heating was unsuccessful. The synthesis of N-propyloxazolo[4,5-c]quinolin-2-amine 12 using P2O5 led to the formation of a mixture of compounds with very poor yields; substituting N-(3-dimethylaminopropyl)-N′-ethylcarbodiimide (EDC) for P2O5 in this reaction not only worked as a sulfur scavenger but greatly enhanced yields of the desired oxazolo analog 12 (Scheme 1). The C4 amine functionality was then installed using conventional methods22,24 to furnish the 2-butyloxazolo[4,5-c]quinolin-4-amine 9, the N-propylthiazolo[4,5-c]quinoline-2,4-diamine 11, and the N-propyloxazolo[4,5-c]quinoline-2,4-diamine 13.
Scheme 1.
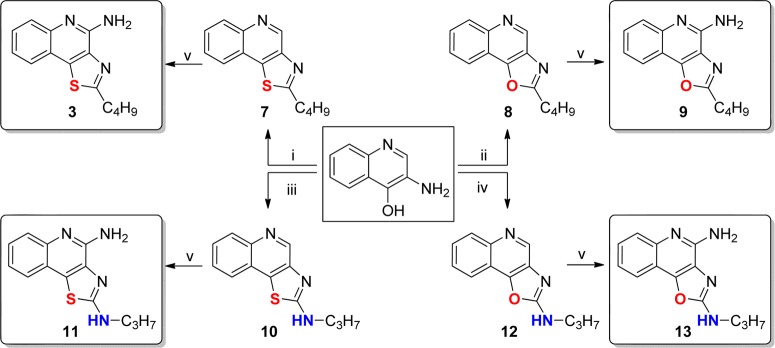
The 2-butyloxazolo[4,5-c]quinolin-4-amine 9 displayed more potent dual TLR7/8-agonistic activity compared to the thiazolo[4,5-c]quinolin-4-amine 3, with EC50 values of 0.55 and 0.18 μM in TLR7 and TLR8 assays, respectively (Figure 2). The N-propylthiazolo[4,5-c]quinoline-2,4-diamine 11, however, exhibited comparable TLR7-agonistic activity (EC50 = 0.73 μM) but a 10-fold reduction in TLR8 potency (EC50 = 3.94 μM). Astonishingly, the N-propyloxazolo[4,5-c]quinoline-2,4-diamines 13 was entirely devoid of any detectable TLR7- or TLR8-agonistic activity. The C2 N-acyl derivative N-(4-aminothiazolo[4,5-c]quinolin-2-yl)propionamide 16 and its des-acyl analog 17 were synthesized from commercially available thiazolo[4,5-c]quinolin-2-amine (Scheme 2). These compounds were also found to be inactive in cell based assays (Figure 2).
Figure 2.

TLR7- and TLR8-agonistic potencies (EC50 values) of the compounds determined in TLR-specific reporter gene assays. Mean values and standard deviations on quadruplicate samples are depicted.
Scheme 2.

The dramatic (and rather unexpected) differences in activity profiles of the closely related congeners warranted a detailed investigation of analogues with variable electronic properties. We first synthesized and evaluated the regioisomeric 2-butylthiazolo[5,4-c]quinolin-4-amine 22a and 2-butyloxazolo[5,4-c]quinolin-4-amine 22b (Scheme 3). The thiazolo[5,4-c]quinoline derivative (22a) was completely inactive in both TLR7 and TLR8 agonism assays, and the oxazolo[5,4-c]quinoline derivative (22b) was found to possess negligibly low TLR8-agonistic activity. This result, too, was unexpected, given that we had observed prominent and selective TLR8 agonism in the 2-butylfuro[2,3-c]quinolin-4-amine 5,26 but further strengthened the case for a systematic exploration of the role of electron densities in the heterocyclic core in determining TLR7/8 activity.
Scheme 3.

Further scaffold modifications were therefore carried out based on the pure TLR7-agonistic lead molecule 1-benzyl-2-butyl-1H-imidazo[4,5-c]quinolin-4-amine 1 (Figure 1). “Repositioning” of the nitrogen atoms in the imidazole ring was done, and triazole analogues were designed and synthesized (Schemes 4–8).34 The novel analogues 2,3-dihydroimidazo[1,2-c]quinazoline 27 and imidazo[1,2-c]quinazoline 29, with an altered imidazole fused ring (Scheme 4), were entirely inactive (Figure 2). We sought to examine if activity could be restored by incorporating an additional nitrogen atom in ring system, but the triazole analogue 33 (2-butyl-[1,2,4]triazolo[1,5-c]quinazolin-5-amine, Scheme 5) was also inactive. On the other hand, the 1,2-dihydroimidazo[1,2-a]quinoxaline 37 and the imidazo[1,2-a]quinoxaline 39 shown in Scheme 6 were found to be selective TLR8 agonists with EC50 values of 3.05 and 7.99 μM, respectively (Figure 2). Transitioning from the imidazo[1,2-a]quinoxaline scaffold to two other triazolo analogues (46 in Scheme 7 and 49 in Scheme 8) also resulted in complete loss of activity.
Scheme 4.

Scheme 8.

Scheme 5.

Scheme 6.

Scheme 7.

Our scaffold-hopping approach also led us to synthesize 2-butyl-2H-pyrazolo[3,4-c]quinolin-4-amine 53 (Scheme 9). Compound 53 was found to be extraordinarily potent as a TLR7 agonist (EC50 = 0.19 μM), significantly greater than that of the thiazoloquinoline 3 (EC50 = 0.86 μM), the oxazoloquinoline 9 (EC50 = 0.55 μM), and the aminothiazoloquinoline 11 (EC50 = 0.73 μM) and approaching that of our best-in-class, pure TLR7 agonistic imidazoquinoline 1 (EC50 = 0.059 μM). Furthermore, the pyrazolo[3,4-c]quinoline 53 was also found to be the most potent in TLR8 agonism assays (EC50 = 0.056 μM) among all TLR8-active compounds that we had hitherto characterized (Figure 2).
Scheme 9.

We confirmed TLR7/8 selectivity and potency of the active compounds in secondary screens including cytokine-inducing properties in human peripheral blood mononuclear cells (hPBMCs), as well as cellular activation in ex vivo whole human blood. In IFN-α induction assays, as expected and in accordance with our previous SAR, compounds with TLR7 agonistic activity (9, 11, and 53) showed IFN-α inducing ability and the TLR8 selective compound 39 did not (Figure 3). We also found strong type II interferon (IFN-γ), cytokine (IL-1α, IL-1β, IL-6, IL-10, TNF-α), and chemokine (IP-10/CXCL-10, MCP-1, MIP-1β) induction by the active compounds consistent with their TLR7/8 selectivity profiles (Figure 4). The extraordinary potency of 53 was also manifested in CD69 expression in whole blood assays, showing dramatically enhanced expression in cytokine-induced killer-, natural killer-, T-, and B-lymphocytic subsets (Figure 5).
Figure 3.
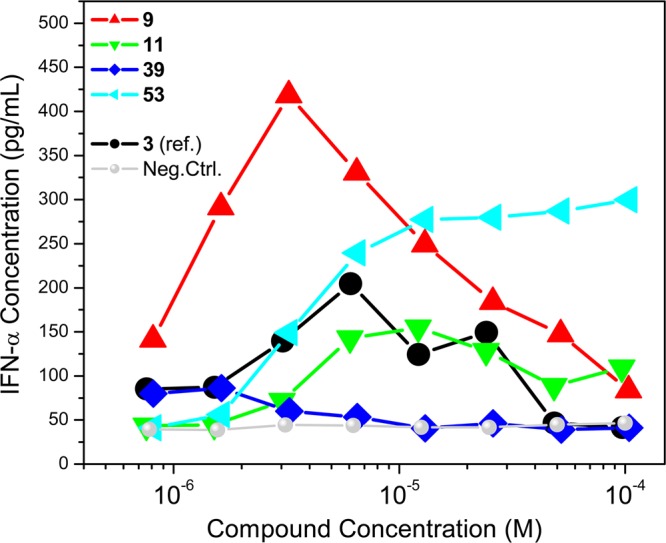
Dose–response profiles of IFN-α induction by the active compounds in human PBMCs. Mean values on triplicate samples of a representative experiment are shown.
Figure 4.

Cytokine and chemokine induction profiles in human PBMCs stimulated with 10 μM select compounds. Mean values on triplicate samples of a representative experiment are shown.
Figure 5.

CD69 up-regulation in human lymphocytic subsets by active analogues.
We were fortunate in also being able to obtain the crystal structures of TLR8 in complex with the two most active compounds: the oxazoloquinoline 9 and the pyrazoloquinoline 53. An examination of TLR8 liganded with 9 and 53 confirmed near-identical binding geometries of the two compounds (Figure 6). Major interactions include hydrogen bonding of the amidine group with Asp543 and the N atom of the oxazole/pyrazole ring with Thr574, π–π interactions of the quinoline ring with Phe405, and hydrophobic interactions of the alkyl chain in a pocket formed by Tyr348, Val378, and Phe405.
Figure 6.
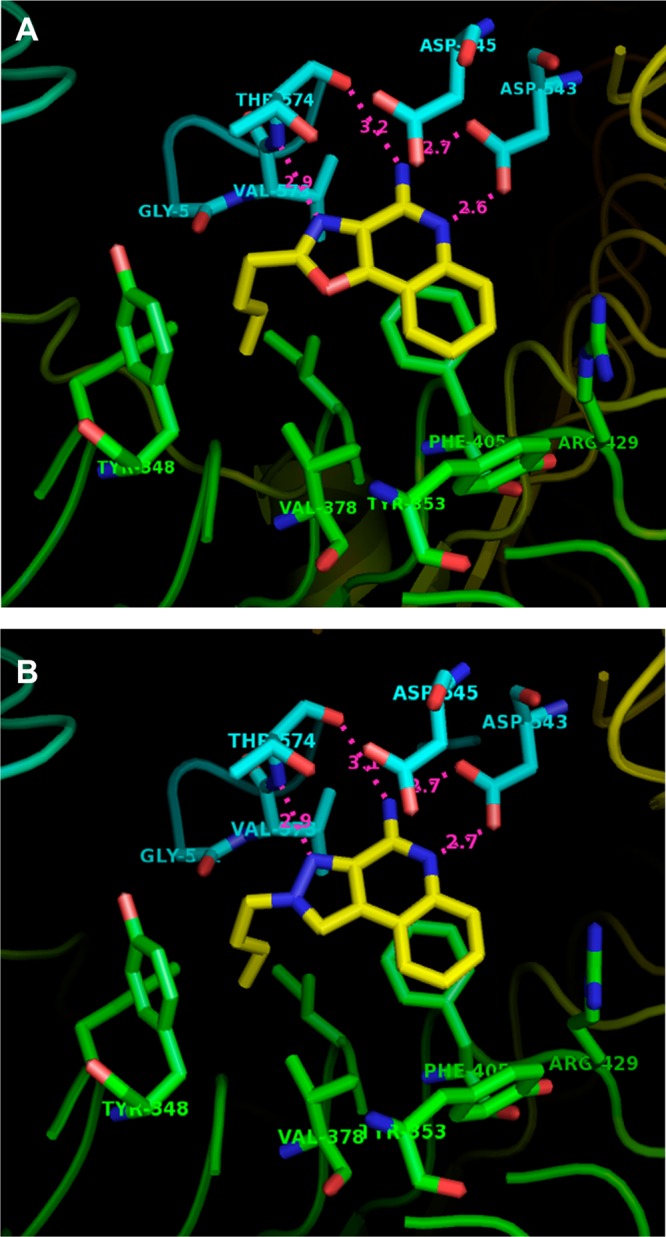
Crystal structures of human TLR8 ectodomain complexed with compound 9 (A) and compound 53 (B), showing key interactions in the binding pocket. PDB codes for compounds 9 and 53 are, respectively, 4QBZ and 4QC0.
Whereas the steric properties of most of these analogues are very similar, their activity profiles are considerably different and the data, taken together, strongly pointed to electronic densities of ring system(s) being dominant determinants of occupancy and activation of TLR7 and TLR8 by these analogues. We therefore undertook quantum chemical calculations of electron densities and of Mulliken atomic charges with the objective of obtaining insights into the properties of these molecules, which we hoped would lead to quantitative predictors of selectivity and potency at TLR7 and TLR8.
As described earlier, the crystal structures of TLR8 complexed with active analogues showed key H-bonds between the amidine group of the quinoline moiety with the side chain of Asp543, and the N atoms of the oxazole and pyrazole moieties of compounds 9 and 53, respectively, with Thr574, providing major contributions to overall binding interactions (Figure 6). Consistent with our expectation that the strength and geometry of the H-bonds are modulated by electron densities and Mulliken charges on appropriate heteroatoms, we observed clear differences in atoms known to be involved in H-bonding interactions. The active compounds 3, 9, 11, 39, and 53 (Figure 7 and Figure S1) display pronounced negative charges (−0.24, −0.24, −0.29, −0.27, and −0.23 electron units, respectively) at position M1 of the five-membered ring. Compounds that do not have the electronegative atom at position M1 (22a and 29; +0.15, 0.0) were found to be inactive. We also noticed a higher partial positive charge at the M2 position in the oxazolo[4,5-c]quinoline-2,4-diamine 13 (0.33 electron unit, Figure 7c) compared to other compounds, attributable to adjacent electronegative heteroatoms (N and O), possibly explaining the lack of activity. The quinazoline analogues 29 and 33 were unique in that the presence of an additional electronegative nitrogen atom at position M5 (−0.25 and −0.22, respectively) resulted in strong positive charge at position M6 (+0.26 and +0.29 electron units, respectively, Figure S1, and Figure 7e), again correlating with absence of agonistic activities.
Figure 7.

Molecular electrostatic potential surfaces of selected compounds plotted onto a surface of constant electron density (0.002 e/au3) showing the most positive potential (deepest blue color), the most negative potential (deepest red color), and the intermediate potential regions (intermediate shades). Also shown is the numbering scheme used to denote atoms (M1–M10) used in linear discriminant analyses (see Figure 8). Compounds 11 and 13 have electrostatic maps that could be only described at an isodensity value of 0.0002, different from the common value of 0.002 used in all other cases.
Given the importance of hydrogen bonding of the amidine group with Asp543, we examined electrostatic potentials at M7. Active compounds could be differentiated from inactive compounds (Figure 8A) with three exceptions: compounds 13, 16, and 22a. The absence of TLR7/8-agonistic activity in 16 and 22a could be explained readily; the presence of a polar amide side chain in 16 is expected to disfavor interactions in the hydrophobic pocket, while the regioisomeric thiazoloquinoline 22a possesses a bulky sulfur atom at M1. The misclassification of 13 as an active compound may, as mentioned earlier, be related to the higher partial positive charge at the M2 position.
Figure 8.

(A) Classification of inactive and active compounds based on electrostatic potentials at M7 calculated using the geometries fully optimized at M06-2X/cc-pVDZ level of theory. An arbitrary line of demarcation at −18.3825 classifies actives from inactives. Of the three exceptions (denoted in ellipses), the origin of the misclassification of 13 is unknown. (B) Demarcation of group 0 (inactive), group 1 (TLR8-specific), and group 2 (TLR7/8 dual-active) compounds obtained via linear discriminant analyses of Mulliken charges. The abscissa and ordinate axes denote discriminant functions, which are linear combinations of variables (calculated Mulliken charges on M1–M10), that best separate the groups of cases. The functions are expressed as dik = b0k + b1kxi1 + ... + bpkxip where dik is the value of the kth discriminant function for the ith case, p is the number of predictors, bjk is the value of the jth coefficient of the kth function, and xij is the value of the ith case of the jth predictor. The classification coefficients for M1–M6 for discriminant functions 1 and 2 are provided in Table S4. Also shown are the structures of test-set of compounds.
The availability of electron density and charge parameters for all atoms in all of the analogues prompted us to examine if a formal classification of active vis-à-vis inactive compounds could be arrived at, with our goal of being able to utilize such methodology in prospectively designing “bespoke” compounds with predefined selectivity. Stepwise linear discriminant analyses were performed with the 13 compounds shown in Figure 2 as a training set. A linear combination of the variables corresponding to M1–M6 explained 100% of the variance in two dimensions (discriminant functions 1 and 2; see Figure 8B), allowing a clear-cut classification of inactive (coded “0”), TLR8-active (coded “1”), and TLR7/8 dual-active (coded “2”) compounds (Figure 8B). The discriminant functions were utilized to examine a test set of compounds which included 5(26) (Figure 1), CL097,35 and nine “hypothetical” compounds (Figure 8B). Compound 5 and CL097 were correctly classified as being TLR8 (group 1) and TLR7/8 dual-active (group 2). All of the proposed analogues with the exception of H3 were predicted to be active. It is noteworthy that the isomeric compounds H8 and H9 (2-butyl-cyclopentaquinolin-4-amines) could be considered as conformationally constrained analogues of 3-alkyl-quinoline-2-amines28 (Figure 1), which we have recently designed and characterized as pure TLR8 agonists. The thienoquinolines H1 and H2 as well as the pyrroloquinolines H3 and H4 are of particular interest, and we are currently evaluating such analogues.
The question as to why the activity profiles of the oxazoloquinoline 9 and its 2-amino analogue 13 are completely divergent remains unclear, however, and crystallographic observations of the complex of 9 with TLR8 even in conjunction with electronic structure calculations only allow us to speculate at the present time as to the role of the water molecule by virtue of its permanent dipole moment and polarizability on stabilizing (or destabilizing interactions) depending on electron densities around the five-membered ring. We are gratified, nonetheless, that quantum chemical calculations in conjunction with rigorous multivariate analyses may afford an empirical but accessible means to evaluating analogues de novo.
Experimental Section
Chemistry
All of the solvents and reagents used were obtained commercially and used as such unless noted otherwise. Moisture- or air-sensitive reactions were conducted under nitrogen atmosphere in oven-dried (120 °C) glass apparatus. The solvents were removed under reduced pressure using standard rotary evaporators. Flash column chromatography was carried out using RediSep Rf “Gold” high performance silica columns on CombiFlash Rf instrument unless otherwise mentioned, while thin-layer chromatography was carried out on silica gel CCM precoated aluminum sheets. Purity for all final compounds was confirmed to be greater than 97% by LC–MS using a Zorbax Eclipse Plus 4.6 mm × 150 mm, 5 μm analytical reverse phase C18 column with H2O–CH3CN gradients and an Agilent 6520 ESI-QTOF Accurate Mass spectrometer (mass accuracy of 5 ppm) operating in the positive ion acquisition mode.
2-Butylthiazolo[4,5-c]quinoline (7)
To a solution of 3-aminoquinolin-4-ol (12 mg, 0.075 mmol) in pyridine (0.5 mL) was added valeroyl chloride (11 μL, 0.09 mmol), and the resulting mixture was heated in a sealed vial at 50 °C for 1 h. P2S5 (33 mg) was added, and the mixture was heated at 120 °C for 1 h under microwave irradiation. The solvents were removed and the crude residue was purified by flash chromatography (SiO2, MeOH in CH2Cl2: 0–5%) to give compound 7 (13.4 mg, 79%) as reddish brown solid. [1H NMR (500 MHz, CDCl3) δ 9.44 (s, 1H), 8.24 (d, J = 8.4 Hz, 1H), 7.96 (dd, J = 8.1, 0.9 Hz, 1H), 7.73 (ddd, J = 8.4, 5.4, 1.4 Hz, 1H), 7.63 (ddd, J = 8.1, 7.1, 1.1 Hz, 1H), 3.25–3.19 (m, 2H), 1.97–1.89 (m, 2H), 1.55–1.46 (m, 2H), 1.00 (t, J = 7.4 Hz, 3H). 13C NMR (126 MHz, CDCl3) δ 173.0, 147.9, 145.8, 144.2, 140.6, 130.6, 128.8, 128.8, 127.6, 125.0, 123.6, 34.2, 32.0, 22.4, 13.9. MS (ESI-TOF, m/z): calculated for C14H14N2S [M + H]+ 243.0950; found 243.0976 (data from Org. Biomol. Chem. 2013, 11, 1179.].
2-Butyloxazolo[4,5-c]quinoline (8)
To a solution of 3-aminoquinolin-4-ol (12 mg, 0.075 mmol) in pyridine (0.5 mL) was added valeroyl chloride (11 μL, 0.09 mmol), and the resulting mixture was heated in a sealed vial at 50 °C for 1 h. P2O5 (22 mg) was added, and the resulting mixture was heated at 120 °C for 1 h under microwave irradiation. The solvents were removed and the crude residue was purified by flash chromatography (SiO2, MeOH in CH2Cl2: 0–5%) to give compound 8 (11.5 mg, 63%) as a pale yellow solid. 1H NMR (500 MHz, CDCl3) δ 9.27 (s, 1H), 8.25 (d, J = 8.4 Hz, 1H), 8.20 (ddd, J = 8.1, 1.5, 0.6 Hz, 1H), 7.75 (ddd, J = 8.5, 7.0, 1.5 Hz, 1H), 7.68 (ddd, J = 8.1, 7.0, 1.1 Hz, 1H), 3.08 (dd, J = 9.0, 6.3 Hz, 2H), 1.96 (ddd, J = 15.2, 7.6, 6.0 Hz, 2H), 1.51 (dt, J = 14.8, 7.4 Hz, 2H), 1.01 (t, J = 7.4 Hz, 3H). 13C NMR (126 MHz, CDCl3) δ 167.5, 152.1, 145.9, 143.9, 134.9, 130.1, 128.7, 127.3, 120.1, 116.3, 28.9, 28.4, 22.3, 13.7. MS (ESI-TOF, m/z): calculated for C14H14N2O [M + H]+ 227.1179; found 227.1216.
2-Butyloxazolo[4,5-c]quinoline 5-Oxide
To a solution of compound 8 (68 mg, 0.30 mmol) in CHCl3 (5 mL) was added m-CPBA (≤77%, 100 mg, 0.45 mmol), and the reaction mixture was stirred at room temperature for 4 h. The solvent was removed under reduced pressure, and the crude residue was purified by flash chromatography (SiO2, MeOH in CH2Cl2: 0–5%) to give 2-butyloxazolo[4,5-c]quinoline 5-oxide. 1H NMR (500 MHz, CDCl3) δ 8.97 (s, 1H), 8.92–8.87 (m, 1H), 8.18 (ddd, J = 6.4, 2.2, 0.6 Hz, 1H), 7.83–7.77 (m, 2H), 3.06 (dd, J = 9.0, 6.3 Hz, 2H), 1.97–1.90 (m, 2H), 1.55–1.47 (m, 2H), 1.01 (t, J = 7.4 Hz, 3H). 13C NMR (126 MHz, CDCl3) δ 169.5, 144.3, 139.8, 134.6, 130.0, 129.7, 129.6, 121.5, 120.8, 116.6, 28.8, 28.4, 22.3, 13.7. MS (ESI-TOF, m/z): calculated for C14H14N2O2 [M + H]+ 243.1128; found 243.1146.
2-Butyloxazolo[4,5-c]quinolin-4-amine (9)
2-Butyloxazolo[4,5-c]quinoline 5-oxide (60 mg, 0.25 mmol) was dissolved in anhydrous CH2Cl2 (2 mL). Benzoyl isocyanate (74 mg, 0.50 mmol) was added, and the resulting mixture was refluxed for 1.5 h. The solvent was removed, and the residue was dissolved in anhydrous methanol (2 mL). Sodium methoxide (27 mg, 0.50 mmol) was added, and the resulting mixture was refluxed for 30 min. The solvent was removed under reduced pressure and the crude residue was purified by flash chromatography (SiO2, MeOH in CH2Cl2: 0–10%) to give compound 9 (37 mg, 61%) as white solid. 1H NMR (500 MHz, MeOD) δ 7.95 (ddd, J = 8.0, 1.5, 0.5 Hz, 1H), 7.70–7.65 (m, 1H), 7.56 (ddd, J = 8.5, 7.0, 1.5 Hz, 1H), 7.35 (ddd, J = 8.1, 7.0, 1.1 Hz, 1H), 3.09–3.03 (m, 2H), 1.92 (ddd, J = 13.7, 8.2, 6.9 Hz, 2H), 1.55–1.46 (m, 2H), 1.02 (t, J = 7.4 Hz, 3H). 13C NMR (126 MHz, MeOD) δ 168.0, 154.0, 152.9, 146.4, 130.2, 126.3, 125.6, 124.0, 121.1, 114.3, 30.0, 28.9, 23.3, 14.0. MS (ESI-TOF, m/z): calculated for C14H15N3O [M + H]+ 242.1288; found 242.1313.
N-Propylthiazolo[4,5-c]quinolin-2-amine (10)
To a solution of 3-aminoquinolin-4-ol (32 mg, 0.20 mmol) in pyridine (1 mL) was added propyl isothiocyanate (31 μL, 0.30 mmol), and the resulting mixture was heated in a sealed vial at 50 °C for 30 min. P2S5 (89 mg) was added, and the resulting mixture was heated at 120 °C for 1 h under microwave irradiation. The solvent was removed and the crude residue was purified by flash chromatography (SiO2, MeOH in CH2Cl2: 0–5%) to give compound 10 (34 mg, 70%) as a pale brown solid. 1H NMR (400 MHz, CDCl3) δ 9.12 (s, 1H), 8.15 (dd, J = 8.3, 0.8 Hz, 1H), 7.78–7.71 (m, 1H), 7.60 (ddd, J = 8.4, 7.0, 1.7 Hz, 1H), 7.55 (ddd, J = 8.2, 7.0, 1.4 Hz, 1H), 5.85 (s, 1H), 3.47 (dd, J = 11.7, 7.0 Hz, 2H), 1.78 (dd, J = 14.4, 7.3 Hz, 2H), 1.06 (t, J = 7.4 Hz, 3H). 13C NMR (126 MHz, CDCl3) δ 168.3, 147.0, 143.1, 143.1, 134.0, 130.3, 127.1, 127.0, 123.7, 123.6, 47.8, 22.7, 11.4. MS (ESI-TOF, m/z): calculated for C13H13N3S [M + H]+ 244.0903; found 244.0946.
N2-Propylthiazolo[4,5-c]quinoline-2,4-diamine (11)
1H NMR (500 MHz, MeOD) δ 7.57 (d, J = 8.4 Hz, 1H), 7.50 (d, J = 8.0 Hz, 1H), 7.42–7.36 (m, 1H), 7.26–7.19 (m, 1H), 3.42 (t, J = 7.0 Hz, 2H), 1.72 (h, J = 7.3 Hz, 2H), 1.02 (t, J = 7.4 Hz, 3H). 13C NMR (126 MHz, MeOD) δ 169.3, 152.0, 143.2, 137.6, 133.7, 128.3, 125.8, 124.4, 123.9, 121.0, 47.8, 23.5, 11.8. MS (ESI-TOF, m/z): calculated for C13H14N4S [M + H]+ 259.1012; found 259.1054.
N-Propyloxazolo[4,5-c]quinolin-2-amine (12)
To a solution of 3-aminoquinolin-4-ol (32 mg, 0.20 mmol) in pyridine (1 mL) was added propyl isothiocyanate (31 μL, 0.30 mmol), and the resulting mixture was heated in a sealed vial at 50 °C for 30 min. EDC (77 mg, 0.4 mmol) was added, and the resulting mixture was heated at 120 °C for 30 min under microwave irradiation. The solvent was removed and the crude residue was purified by flash chromatography (SiO2, MeOH in CH2Cl2: 0–5%) to give compound 12 (25 mg, 59%) as a brown solid. 1H NMR (500 MHz, MeOD) δ 8.85 (s, 1H), 8.08–8.03 (m, 1H), 8.04–8.00 (m, 1H), 3.51 (q, J = 7.3 Hz, 2H), 1.34 (t, J = 7.3 Hz, 3H). 13C NMR (126 MHz, MeOD) δ 164.6, 150.2, 145.1, 141.2, 137.8, 129.7, 128.7, 128.6, 120.2, 116.7, 38.9, 14.9. MS (ESI-TOF, m/z): calculated for C12H11N3O [M + H]+ 214.0957; found 214.1045. 1H NMR (500 MHz, MeOD) δ 8.83 (s, 1H), 8.06–8.02 (m, 1H), 8.01–7.97 (m, 1H), 7.67–7.58 (m, 2H), 3.42 (t, J = 7.1 Hz, 2H). 13C NMR (126 MHz, MeOD) δ 164.7, 150.1, 145.1, 141.2, 137.8, 129.7, 128.7, 128.6, 120.2, 116.7, 45.9, 23.6, 11.6. MS (ESI-TOF, m/z): calculated for C13H13N3O [M + H]+ 228.1131; found 228.1164.
2-(Propylamino)oxazolo[4,5-c]quinoline 5-Oxide
1H NMR (500 MHz, DMSO-d6) δ 8.83 (s, 1H), 8.58 (t, J = 5.8 Hz, 1H), 8.56 (d, J = 8.8 Hz, 1H), 7.97 (dd, J = 8.3, 0.6 Hz, 1H), 7.75 (ddd, J = 8.2, 6.9, 1.1 Hz, 1H), 7.69–7.64 (m, 1H), 3.36–3.30 (m, 10H), 1.69–1.59 (m, 2H), 0.94 (t, J = 7.4 Hz, 3H). 13C NMR (126 MHz, DMSO-d6) δ 163.8, 139.3, 137.2, 136.8, 129.4, 127.7, 127.3, 120.5, 119.6, 115.3, 44.4, 22.0, 11.3. MS (ESI-TOF, m/z): calculated for C13H13N3O2 [M + H]+ 244.1081; found 244.0974.
N2-Propyloxazolo[4,5-c]quinoline-2,4-diamine (13)
1H NMR (500 MHz, DMSO-d6) δ 11.96 (s, 1H), 8.16 (dd, J = 8.0, 1.2 Hz, 1H), 7.50–7.45 (m, 1H), 7.44 (dd, J = 8.2, 1.0 Hz, 1H), 7.16 (ddd, J = 8.1, 6.6, 1.5 Hz, 1H), 7.06 (t, J = 5.9 Hz, 1H), 3.24 (dd, J = 14.1, 6.3 Hz, 2H), 1.63–1.48 (m, 2H), 0.90 (t, J = 7.4 Hz, 3H). 13C NMR (126 MHz, DMSO-d6) δ 161.7, 155.7, 150.2, 137.4, 129.0, 124.9, 124.0, 120.2, 117.0, 112.8, 43.9, 22.6, 11.3. MS (ESI-TOF, m/z): calculated for C13H14N4O [M + H]+ 243.1240; found 243.1141.
2-Propionamidothiazolo[4,5-c]quinoline 5-Oxide (15)
To a solution of thiazolo[4,5-c]quinolin-2-amine (100 mg, 0.497 mmol) in pyridine (2 mL) was added propionyl chloride (54 μL, 0.62 mmol), and the resulting mixture was stirred at room temperature for 1 h. The solvent was removed and the crude residue was purified by flash chromatography (SiO2, MeOH in CH2Cl2: 0–5%) to give compound N-(thiazolo[4,5-c]quinolin-2-yl)propionamide 14 as an impure solid, which was dissolved in CHCl3 (4 mL). m-CPBA (≤77%, 224 mg, 1.0 mmol) was added, and the reaction mixture was stirred at room temperature for 4 h and then concentrated. The crude residue was purified by flash chromatography (SiO2, MeOH in CH2Cl2: 0–5%) to give compound 15 (98 mg, 72%) as white solid. 1H NMR (500 MHz, DMSO-d6) δ 12.75 (s, 1H), 9.11 (s, 1H), 8.68–8.62 (m, 1H), 8.24–8.17 (m, 1H), 7.86–7.76 (m, 2H), 2.56 (q, J = 7.5 Hz, 2H), 1.14 (t, J = 7.5 Hz, 3H). 13C NMR (126 MHz, DMSO-d6) δ 173.3, 160.0, 142.9, 137.8, 129.8, 129.7, 128.7, 125.1, 124.6, 123.6, 120.3, 28.4, 8.8. MS (ESI-TOF, m/z): calculated for C13H11N3O2S [M + H]+ 274.0645; found 274.0667.
N-(4-Aminothiazolo[4,5-c]quinolin-2-yl)propionamide (16)
Compound 15 (55 mg, 0.20 mmol) was dissolved in anhydrous CH2Cl2 (2 mL). Benzoyl isocyanate (59 mg, 0.40 mmol) was added, and the resulting mixture was refluxed for 1.5 h. The solvent was removed under reduced pressure, and the residue was dissolved in anhydrous MeOH (2 mL). Sodium methoxide (22 mg, 0.40 mmol) was added, and the reaction mixture was refluxed for 30 min. The solvent was removed and the crude residue was purified by flash chromatography (SiO2, MeOH in CH2Cl2: 0–10%) to give compound 16 (41 mg, 79%) as a white solid. 1H NMR (500 MHz, DMSO-d6) δ 12.53 (s, 1H), 7.80 (dd, J = 8.0, 1.0 Hz, 1H), 7.61 (dd, J = 8.3, 0.6 Hz, 1H), 7.48 (ddd, J = 8.4, 7.0, 1.4 Hz, 1H), 7.26 (ddd, J = 8.1, 7.0, 1.2 Hz, 1H), 2.55 (q, J = 7.5 Hz, 2H), 1.14 (t, J = 7.5 Hz, 3H). 13C NMR (126 MHz, DMSO-d6) δ 173.1, 157.0, 151.5, 144.2, 133.8, 133.1, 127.8, 125.9, 123.7, 122.2, 119.1, 28.3, 9.0. MS (ESI-TOF, m/z): calculated for C13H12N4OS [M + H]+ 273.0805; found 273.0832. The sodium methoxide solvolysis also resulted in a small amount of thiazolo[4,5-c]quinoline-2,4-diamine (17) (4 mg, 9%) which was isolated from the crude mixture as white solid. 1H NMR (500 MHz, MeOD) δ 7.58 (ddd, J = 8.4, 1.0, 0.5 Hz, 1H), 7.52 (ddd, J = 8.0, 1.4, 0.5 Hz, 1H), 7.41 (ddd, J = 8.4, 7.0, 1.5 Hz, 1H), 7.24 (ddd, J = 8.1, 7.1, 1.1 Hz, 1H). 13C NMR (126 MHz, MeOD) δ 169.6, 152.0, 143.6, 137.3, 134.7, 128.4, 126.0, 124.4, 123.9, 121.1. MS (ESI-TOF, m/z): calculated for C10H8N4S [M + H]+ 217.0542; found 217.0569.
N-(Quinolin-4-yl)valeramide (18)
To a solution of 4-aminoquinoline (302 mg, 2.1 mmol) in pyridine (6 mL) was added valeroyl chloride (0.28 mL, 2.4 mmol). The mixture was heated at 65 °C for 90 min and then concentrated. The crude residue was purified by flash chromatography (SiO2, MeOH in CH2Cl2: 0–10%) to give compound 18 (382 mg, 80%) as a dark brown solid. 1H NMR (500 MHz, DMSO-d6) δ 11.11 (s, 1H), 9.07 (d, J = 6.5 Hz, 1H), 8.95 (dd, J = 8.7, 1.1 Hz, 1H), 8.70 (d, J = 6.4 Hz, 1H), 8.32 (dd, J = 8.5, 1.1 Hz, 1H), 8.12 (ddd, J = 8.4, 6.9, 1.1 Hz, 1H), 7.93 (ddd, J = 8.4, 6.9, 1.1 Hz, 1H), 2.80 (t, J = 7.4 Hz, 2H), 1.66 (tt, J = 7.5 Hz, 2H), 1.39 (tq, J = 7.4 Hz, 2H), 0.94 (t, J = 7.4 Hz, 3H). 13C NMR (126 MHz, DMSO-d6) δ 174.2, 149.6, 145.0, 139.0, 133.8, 128.1, 123.8, 121.2, 119.0, 108.9, 36.4, 26.6, 21.6, 13.7. MS (ESI-TOF, m/z): calculated for C14H16N2O [M + H]+ 229.1341; found 229.1306.
N-(3-Bromoquinolin-4-yl)valeramide (19)
To a suspension of compound 19 (151 mg, 0.66 mmol) in benzene (anhydrous, 10 mL) were added N-bromosuccinimide (NBS, 143 mg, 0.81 mmol) and azobisisobutyronitrile (AIBN, 99 mg, 0.60 mmol). The mixture was refluxed for 5 h and then concentrated. The crude residue was purified by flash chromatography (SiO2, EtOAc in hexane: 0–30%) to give compound 19 (86 mg, 42%) as a light brown solid. 1H NMR (500 MHz, DMSO-d6) δ 10.31 (s, 1H), 9.05 (s, 1H), 8.08 (dd, J = 8.5, 1.2 Hz, 1H), 7.95 (dd, J = 8.4, 1.4 Hz, 1H), 7.84 (ddd, J = 8.4, 6.9, 1.5 Hz, 1H), 7.70 (ddd, J = 8.2, 6.8, 1.3 Hz, 1H), 2.54–2.46 (m, 2H), 1.68 (quin, J = 7.5 Hz, 2H), 1.43 (sex, J = 7.4 Hz, 2H), 0.95 (t, J = 7.4 Hz, 3H). 13C NMR (126 MHz, DMSO-d6) δ 171.4, 152.1, 147.1, 141.5, 130.2, 129.1, 127.8, 126.6, 123.8, 116.8, 35.2, 27.3, 21.9, 13.8. MS (ESI-TOF, m/z): calculated for C14H15BrN2O [M + H]+ 307.0446; found 307.0254.
2-Butylthiazolo[5,4-c]quinoline (20a)
To a solution of compound 19 (60.8 mg, 0.20 mmol) in pyridine (4 mL) was added Lawesson’s reagent (234 mg, 0.58 mmol). The resulting mixture was heated in a sealed vial under microwave irradiation (500 W, 140 °C) for 35 min and then concentrated. The crude residue was purified by flash chromatography (SiO2, EtOAc in hexanes: 0–10%) to give compound 20a (15 mg, 31%) as brown oil. 1H NMR (500 MHz, CDCl3) δ 9.33 (s, 1H), 8.71 (dd, J = 8.1, 1.5 Hz, 1H), 8.22 (brd, J = 8.3 Hz, 1H), 7.76 (ddd, J = 8.3, 7.0, 1.5 Hz, 1H), 7.70 (ddd, J = 8.1, 7.0, 1.2 Hz, 1H), 3.27 (t, J = 7.7 Hz, 2H), 1.95 (tt, J = 7.6, 7.7 Hz, 2H), 1.52 (qt, J = 7.3, 7.4 Hz, 2H), 1.01 (t, J = 7.4 Hz, 3H). 13C NMR (126 MHz, CDCl3) δ 177.3, 154.9, 145.9, 143.8, 129.4, 128.7, 127.8, 127.2, 123.6, 123.4, 34.3, 32.0, 22.3, 13.8. MS (ESI-TOF, m/z): calculated for C14H14N2S [M + H]+ 243.0956; found 243.1019.
2-Butylthiazolo[5,4-c]quinoline 5-Oxide (21a)
To a solution of compound 20a (14.5 mg, 0.060 mmol) in CH2Cl2 (1 mL) was added m-CPBA (≤77%, 35.2 mg). The reaction mixture was stirred at room temperature for 2.5 h and then concentrated. The crude residue was purified by flash chromatography (SiO2, MeOH in CH2Cl2: 0–5%) to give compound 21a (13.5 mg, 87%) as brown oil. 1H NMR (500 MHz, CDCl3) δ 9.04 (s, 1H), 8.90–8.76 (m, 1H), 8.78–8.58 (m, 1H), 7.95–7.72 (m, 2H), 3.25 (t, J = 7.7 Hz, 2H), 1.94 (tt, J = 7.6, 7.7 Hz, 2H), 1.63–1.44 (m, 2H), 1.02 (t, 3H, J = 7.3 Hz). 13C NMR (126 MHz, CDCl3) δ 176.4, 147.8, 140.1, 134.3, 132.6, 130.0, 129.5, 128.0, 124.4, 120.3, 34.1, 31.9, 22.3, 13.8. MS (ESI-TOF, m/z): calculated for C14H14N2OS [M + H]+ 259.0827; found 259.0755.
2-Butylthiazolo[5,4-c]quinolin-4-amine (22a)
To a solution of compound 21a (10.1 mg, 0.039 mmol) in CH2Cl2 (0.2 mL) was added benzoyl isocyanate (13.2 mg, 0.090 mmol). The mixture was heated to reflux for 3 h and then concentrated. The crude residue was purified by flash chromatography (SiO2, EtOAc in hexanes: 0–15%) to give intermediate (8.8 mg, 63%) as light brown oil. Benzamide (8.8 mg, 0.024 mmol) was dissolved in NaOMe solution (0.5 mL, 0.5 M in MeOH). The mixture was heated to reflux overnight and then concentrated. The crude residue was purified by flash chromatography (SiO2, MeOH in CH2Cl2: 0–5%) to give compound 22a (5.6 mg, 89%) as a yellow solid. 1H NMR (500 MHz, CDCl3) δ 8.50 (dd, J = 8.1, 1.2 Hz, 1H), 7.82 (brd, J = 8.4 Hz, 1H), 7.62 (ddd, J = 8.4, 7.0, 1.5 Hz, 1H), 7.44 (ddd, J = 8.0, 7.0, 1.1 Hz, 1H), 5.15 (brs, 2H), 3.25 (t, J = 7.7 Hz, 2H), 2.00–1.87 (m, 2H), 1.61–1.43 (m, 2H), 1.02 (t, J = 7.4 Hz, 3H). 13C NMR (126 MHz, CDCl3) δ 175.7, 157.0, 150.9, 145.8, 129.2, 125.7, 123.6, 123.5, 120.2, 116.7, 34.2, 32.0, 22.3, 13.8. MS (ESI-TOF, m/z): calculated for C14H15N3S [M + H]+ 258.1065; found 258.0964.
2-Butyloxazolo[5,4-c]quinoline (20b)
To a solution of compound 19 (50.4 mg, 0.16 mmol) in pyridine (2 mL) were added CuI (66 mg, 0.35 mmol) and K2CO3 (47 mg, 0.34 mmol). The resulting mixture was heated in a sealed vial under microwave irradiation (500 W, 140 °C) for 35 min and then concentrated. The crude residue was purified by flash chromatography (SiO2, EtOAc in hexanes: 0–10%) to give compound 20b (28 mg, 76%) as a brown solid. 1H NMR (500 MHz, CDCl3) δ 9.20 (bs, 1H), 8.46 (bs, 1H), 8.25 (bs, 1H), 7.76 (bt, J = 6.0 Hz, 1H), 7.72–7.64 (m, 1H), 3.09 (t, J = 7.6 Hz, 2H), 2.00–1.91 (m, 2H), 1.55–1.43 (m, 2H), 1.00 (t, J = 7.4 Hz, 3H). 13C NMR (126 MHz, CDCl3) δ 169.8, 145.5, 143.7, 135.1, 129.9, 128.3, 127.4, 122.2, 29.0, 28.7, 22.3, 13.7. MS (ESI-TOF, m/z): calculated for C14H14N2O [M + H]+ 227.1184; found 227.1051.
2-Butyloxazolo[5,4-c]quinoline 5-Oxide (21b)
To a solution of compound 20b (28 mg, 0.124 mmol) in CH2Cl2 (2 mL) was added m-CPBA (≤77%, 83.5 mg). The reaction mixture was stirred at room temperature for 4 h and then concentrated. The crude residue was purified by flash chromatography (SiO2, MeOH in CH2Cl2: 0–5%) to give compound 21b (22 mg, 73%) as a red solid. 1H NMR (500 MHz, CDCl3) δ 8.93 (s, 1H), 8.87–8.79 (m, 1H), 8.46–8.41 (m, 1H), 7.84–7.75 (m, 2H), 3.06 (t, J = 7.6 Hz, 2H), 1.93 (tt, J = 7.7, 7.6 Hz, 2H), 1.50 (qt, J = 7.4, 7.3 Hz, 2H), 1.01 (t, J = 7.4 Hz, 3H). 13C NMR (126 MHz, CDCl3) δ 169.9, 143.9, 140.1, 135.7, 129.6, 129.3, 123.6, 122.8, 121.8, 120.8, 28.9, 28.5, 22.3, 13.7. MS (ESI-TOF, m/z): calculated for C14H14N2O2 [M + H]+ 243.1134; found 243.1032.
2-Butyloxazolo[5,4-c]quinolin-4-amine (22b)
To a solution of compound 21b (17.8 mg, 0.073 mmol) in CH2Cl2 (0.5 mL) was added benzoyl isocyanate (38.4 mg, 0.26 mmol). The mixture was heated to reflux for 4 h and then concentrated. The crude residue was purified by flash chromatography (SiO2, EtOAc in hexanes: 0–20%) to give intermediate (8.7 mg, 34%) as colorless oil. Benzamide (6 mg, 0.017 mmol) was dissolved in NaOMe solution (0.5 mL, 0.5 M in MeOH). The mixture was heated to reflux for 8 h and then concentrated. The crude residue was purified by flash chromatography (SiO2, MeOH in CH2Cl2: 0–5%) to give compound 22b (3.3 mg, 79%) as a yellow solid. 1H NMR (500 MHz, CDCl3) δ 8.22 (dd, J = 8.2, 1.5 Hz, 1H), 7.80 (dd, J = 8.7, 1.5 Hz, 1H), 7.59 (ddd, J = 8.5, 7.0, 1.6 Hz, 1H), 7.42 (ddd, J = 8.2, 7.0, 1.1 Hz, 1H), 5.15 (bs, 2H), 3.05 (t, J = 7.7 Hz, 2H), 2.01–1.85 (m, 2H), 1.58–1.43 (m, 2H), 1.00 (t, J = 7.4 Hz, 3H). 13C NMR (126 MHz, CDCl3) δ 168.6, 144.9, 144.0, 135.2, 128.5, 127.3, 126.2, 123.5, 122.0, 119.0, 29.2, 28.6, 22.3, 13.7. MS (ESI-TOF, m/z): calculated for C14H15N3O [M + H]+ 242.1293; found 242.1240.
Quinazoline-2,4(1H,3H)-dione (23)
The mixture of anthranilic acid (500 mg, 3.65 mmol) and urea (2.2 g, mol) was heated at 150 °C for 6 h. The reaction mixture was cooled to room temperature and then water (50 mL) was added to quench the reaction. The crude product was obtained by filtration and then washed with water (20 mL × 3). The residue was dissolved in hot aqueous NaOH and cooled to 0 °C, and pH was adjusted to 5–6 using dilute HCl and stirred for 30 min. The crude mixture was filtered and washed with water and dried under vacuum to yield compound 23 as white solid (500 mg, 85%). 1H NMR (500 MHz, DMSO-d6) δ 11.28 (s, 1H), 11.14 (s, 1H), 7.88 (dd, J = 7.8, 1.1 Hz, 1H), 7.65–7.60 (m, 1H), 7.17 (ddd, J = 8.2, 6.1, 1.8 Hz, 2H). 13C NMR (126 MHz, DMSO-d6) δ 162.9, 150.3, 140.9, 135.0, 127.0, 122.4, 115.3, 114.4. MS (ESI-TOF, m/z): calculated for C8H6N2O2 [M + H]+ 163.0502; found 163.0491.
2-((2-Chloroquinazolin-4-yl)amino)hexan-1-ol (25)
POCl3 (5 mL) and DIPEA (430 μL, 2.47 mmol) were added to compound 23 (200 mg, 1.23 mmol), and the reaction mixture was heated to reflux for 4 h. The excess POCl3 was removed by evaporation. The residue was dissolved in ice water, and then the suspension was filtered and washed with water to afford compound 24 as white solid (220 mg, 90%). To a solution of compound 24 (200 mg, 1 mmol) in DMF (5 mL) was added dl-2-amino-1-hexanol (194 μL, 1.5 mmol). The resulting mixture was heated at 100 °C for 1 h, allowed to cool, and concentrated. The residue was purified by flash chromatography (SiO2, EtOAc in hexane: 0–50%) to give compound 25 as a yellow solid (95 mg, 34%). 1H NMR (500 MHz, DMSO-d6) δ 8.37 (dd, J = 8.4, 0.8 Hz, 1H), 8.24 (d, J = 8.4 Hz, 1H), 7.78 (ddd, J = 8.3, 7.0, 1.3 Hz, 1H), 7.60 (dd, J = 8.3, 0.8 Hz, 1H), 7.52 (ddd, J = 8.2, 7.0, 1.2 Hz, 1H), 4.80 (t, J = 5.7 Hz, 1H), 4.35 (qd, J = 5.7, 10.6 Hz, 1H), 3.56–3.46 (m, 2H), 1.74–1.65 (m, 1H), 1.62–1.52 (m, 1H), 1.29 (dddd, J = 14.8, 10.3, 8.2, 3.2 Hz, 4H), 0.85 (t, J = 6.7 Hz, 3H). 13C NMR (126 MHz, DMSO-d6) δ 161.4, 157.1, 150.4, 133.6, 126.6, 125.8, 123.5, 113.6, 62.8, 52.9, 30.1, 27.8, 22.1, 14.0. MS (ESI-TOF, m/z): calculated for C14H18ClN3O [M + H]+ 280.1211; found 280.1279.
2-Butyl-2,3-dihydroimidazo[1,2-c]quinazolin-5-amine (27)
To a solution of compound 25 (50 mg, 0.18 mmol) in CH2Cl2 (2 mL) was added triethylamine (38 μL, 0.27 mmol) and methanesulfonyl chloride (17 μL, 0.22 mmol), and the resulting mixture was stirred at room temperature overnight. CH2Cl2 (20 mL) was added, and the organic layer was washed with water (10 mL × 2), dried over anhydrous sodium sulfate, and evaporated to furnish the crude residue of compound 26 (45 mg, 96% crude yield). Ammonia in methanol (2M, 1 mL) was added to compound 26 (10 mg, 38 μmol), and the reaction mixture was heated at 80 °C for 2 h and concentrated. The residue was purified by flash chromatography (SiO2, MeOH in CH2Cl2: 0–10%) to give compound 27 (6 mg, 66%) as a yellow solid. 1H NMR (500 MHz, MeOD) δ 7.88 (dd, J = 8.0, 1.2 Hz, 1H), 7.53 (ddd, J = 8.5, 7.2, 1.5 Hz, 1H), 7.20 (d, J = 8.0 Hz, 1H), 7.12 (ddd, J = 8.1, 7.2, 1.0 Hz, 1H), 4.42–4.35 (m, 1H), 4.21 (t, J = 10.4 Hz, 1H), 3.77 (dd, J = 10.3, 7.6 Hz, 1H), 1.87–1.78 (m, 1H), 1.66 (ddd, J = 11.0, 6.6, 3.7 Hz, 1H), 1.50–1.37 (m, 4H), 0.97 (t, J = 7.1 Hz, 3H). 13C NMR (126 MHz, MeOD) δ 156.9, 151.8, 150.6, 135.4, 126.5, 124.6, 123.6, 113.2, 64.5, 51.9, 37.3, 28.6, 23.7, 14.4. MS (ESI-TOF, m/z): calculated for C14H18N4 [M + H]+ 243.1604; found 243.1589.
2-Butylimidazo[1,2-c]quinazolin-5-amine (29)
To a solution of compound 26 (28 mg, 0.11 mmol) in toluene (2 mL) was added MnO2 (47 mg, 0.54 mmol), and the mixture was heated at reflux for 20 h. Additional MnO2 was added, and the reaction mixture was refluxed for another 48 h. The mixture was allowed to cool, filtered, and purified by column chromatography (SiO2, EtOAc in hexane: 0–50%) to obtain compound 28 (12 mg, 41%) as a pale yellow solid. Compound 28 (10 mg) in ammonia solution (2 M in ammonia, 1 mL) was heated at 80 °C for 2 h, concentrated, and purified by column chromatography (SiO2, MeOH in CH2Cl2: 0–10%) to obtain compound 29 (4 mg, 36%) as a pale yellow solid. 1H NMR (500 MHz, MeOD) δ 8.28 (dd, J = 8.0, 0.9 Hz, 1H), 7.70 (s, 1H), 7.58–7.51 (m, 2H), 7.35 (ddd, J = 8.1, 6.8, 1.5 Hz, 1H), 2.84–2.78 (m, 2H), 1.79 (ddd, J = 13.1, 8.5, 6.6 Hz, 2H), 1.48 (dq, J = 14.8, 7.4 Hz, 2H), 1.01 (t, J = 7.4 Hz, 3H). 13C NMR (126 MHz, MeOD) δ 131.5, 125.5, 124.7, 123.5, 108.3, 32.4, 29.1, 23.5, 14.2. MS (ESI-TOF, m/z): calculated for C14H16N4 [M + H]+ 241.1448; found 241.1466.
Methyl (2-Cyanophenyl)carbamate (30)
2-Aminobenzonitrile (200 mg, 1.69 mmol) and sodium carbonate (359 mg, 3.39 mmol) was heated to reflux in methyl chloroformate (7 mL) for 4 h. The reaction mixture was concentrated and purified by column chromatography (SiO2, EtOAc in hexane: 0–20%) to obtain compound 30 (276 mg, 93%) as a white solid. 1H NMR (500 MHz, DMSO-d6) δ 9.77 (s, 1H), 7.79 (dd, J = 7.8, 1.3 Hz, 1H), 7.67 (ddd, J = 8.2, 7.6, 1.6 Hz, 1H), 7.52 (d, J = 8.1 Hz, 1H), 7.33 (td, J = 7.6, 1.1 Hz, 1H), 3.69 (s, 3H). 13C NMR (126 MHz, DMSO-d6) δ 154.5, 140.5, 133.9, 133.3, 125.5, 125.2, 116.8, 107.3, 52.2. MS (ESI-TOF, m/z): calculated for C9H8N2O2 [M + H]+ 177.0659; found 177.0694.
2-Butyl[1,2,4]triazolo[1,5-c]quinazolin-5(6H)-one (31)
To a solution of compound 30 (200 mg, 1.14 mmol) in N-methyl-2-pyrrolidone (NMP, 5 mL) was added valeric acid hydrazide (158 mg, 1.36 mmol), and the reaction mixture was heated at 180 °C for 3 h. Crushed ice was added to the reaction mixture and the solid obtained was filtered and purified by column chromatography (SiO2, EtOAc in hexanes: 0–50%) to obtain compound 31 (270 mg, 98%) as white solid. 1H NMR (500 MHz, DMSO-d6) δ 12.21 (s, 1H), 8.11 (dd, J = 7.9, 1.1 Hz, 1H), 7.69–7.64 (m, 1H), 7.41 (d, J = 8.2 Hz, 1H), 7.39–7.33 (m, 1H), 2.82 (t, J = 7.6 Hz, 2H), 1.79–1.72 (m, 2H), 1.43–1.33 (m, 2H), 0.92 (t, J = 7.4 Hz, 3H). 13C NMR (126 MHz, DMSO-d6) δ 166.5, 152.7, 143.8, 136.9, 132.6, 124.0, 123.5, 116.0, 110.3, 29.6, 27.7, 21.8, 13.7. MS (ESI-TOF, m/z): calculated for C13H14N4O [M + H]+ 243.1240; found 243.1350.
2-Butyl-5-chloro[1,2,4]triazolo[1,5-c]quinazoline (32)
To a suspension of compound 31 (18 mg, 74 μmol) in POCl3 (1 mL) was added DIPEA (29 μL, 0.15 mmol), and the resulting mixture was heated at 110 °C for 18 h. The mixture was cooled to room temperature and concentrated. The residue was purified by flash chromatography (SiO2, EtOAc in hexane: 0–30%) to give compound 32 as a pale yellow solid (15 mg, 79%). 1H NMR (500 MHz, DMSO-d6) δ 8.41 (d, J = 8.0 Hz, 1H), 7.99 (d, J = 8.3 Hz, 1H), 7.94 (t, J = 7.7 Hz, 1H), 7.82 (t, J = 7.5 Hz, 1H), 2.93 (t, J = 7.6 Hz, 2H), 1.84–1.76 (m, 2H), 1.47–1.37 (m, 2H), 0.94 (t, J = 7.4 Hz, 3H). 13C NMR (126 MHz, DMSO-d6) δ 167.0, 152.0, 142.3, 135.3, 132.7, 129.1, 127.6, 123.6, 116.8, 29.7, 27.8, 21.8, 13.7. MS (ESI-TOF, m/z): calculated for C13H13ClN4 [M + H]+ 261.0902; found 261.1021.
2-Butyl[1,2,4]triazolo[1,5-c]quinazolin-5-amine (33)
Compound 32 (10 mg, 38 μmol) was treated with 2 M ammonia in methanol (1 mL), heated at 80 °C for 20 h, and concentrated. The residue was purified by flash chromatography (SiO2, MeOH in CH2Cl2: 0–10%) to give compound 33 as a white solid (5 mg, 55%). 1H NMR (500 MHz, MeOD) δ 8.21 (ddd, J = 8.0, 1.5, 0.5 Hz, 1H), 7.65 (ddd, J = 8.5, 7.1, 1.5 Hz, 1H), 7.57 (dd, J = 8.4, 0.5 Hz, 1H), 7.37 (ddd, J = 8.1, 7.1, 1.1 Hz, 1H), 2.97–2.89 (m, 2H), 1.86 (dt, J = 15.3, 7.6 Hz, 2H), 1.50–1.38 (m, 2H), 0.98 (t, J = 7.4 Hz, 3H). 13C NMR (126 MHz, MeOD) δ 167.9, 153.1, 146.3, 146.1, 133.4, 126.0, 124.8, 124.5, 114.4, 31.4, 29.2, 23.4, 14.1. MS (ESI-TOF, m/z): calculated for C13H15N5 [M + H]+ 242.1400; found 242.1399.
2,3-Dichloroquinoxaline (34)
Quinoxaline-2,3-diol (500 mg, 3.08 mmol) and POCl3 (5 mL) in DMF (5 mL) were heated at 100 °C for 1.5 h, allowed to cool, and concentrated. The residue was treated with ice–water and filtered to give compound 34 (418 mg, 68%) as off white solid. 1H NMR (500 MHz, DMSO-d6) δ 8.10–8.06 (m, 2H), 7.96–7.91 (m, 2H). 13C NMR (126 MHz, DMSO-d6) δ 144.7, 140.1, 131.8, 128.0. MS (ESI-TOF, m/z): calculated for C8H4Cl2N2 [M + H]+ 198.9824; found 198.9853.
2-((3-Chloroquinoxalin-2-yl)amino)hexan-1-ol (35)
To a solution of compound 34 (200 mg, 1 mmol) in EtOH (5 mL), was added dl-2-amino-1-hexanol (194 μL, 1.5 mmol) in EtOH (3 mL). The mixture was heated at 90 °C for 18 h, allowed to cool, and concentrated. The residue was purified by flash chromatography (SiO2, EtOAc in hexane: 0–20%) to give compound 35 as a yellow solid (160 mg, 57%). 1H NMR (500 MHz, CDCl3) δ 7.80 (dd, J = 8.2, 1.0 Hz, 1H), 7.66 (dd, J = 8.3, 0.9 Hz, 1H), 7.58 (ddd, J = 8.4, 7.0, 1.4 Hz, 1H), 7.40 (ddd, J = 8.3, 7.0, 1.4 Hz, 1H), 5.65 (d, J = 6.6 Hz, 1H), 4.30–4.22 (m, 1H), 3.90 (dd, J = 11.1, 2.8 Hz, 1H), 3.76 (dd, J = 11.1, 6.3 Hz, 1H), 3.60 (s, 1H), 1.78–1.64 (m, 2H), 1.48–1.35 (m, 4H), 0.93 (t, J = 7.1 Hz, 3H). 13C NMR (126 MHz, CDCl3) δ 148.5, 140.6, 138.1, 136.6, 130.5, 128.1, 125.7, 125.4, 66.8, 54.6, 31.3, 28.5, 22.7, 14.1. MS (ESI-TOF, m/z): calculated for C14H18ClN3O [M + H]+ 280.1211; found 280.1296.
2-Butyl-4-chloro-1,2-dihydroimidazo[1,2-a]quinoxaline (36)
To a solution of compound 35 (100 mg, 0.36 mmol) in CHCl3 (1 mL) was added thionyl chloride (1 mL) at 0 °C, and the mixture was heated to reflux for 2 h. The mixture was cooled to room temperature and concentrated. The residue was purified by flash chromatography (SiO2, MeOH in CH2Cl2: 0–10%) to give compound 36 (75 mg, 80%) as a yellow solid. 1H NMR (500 MHz, DMSO-d6) δ 8.02 (dd, J = 8.1, 0.7 Hz, 1H), 7.94–7.89 (m, 1H), 7.67 (dd, J = 14.6, 7.6 Hz, 2H), 4.90 (m, 1H), 4.62–4.51 (m, 2H), 1.90–1.82 (m, 1H), 1.78–1.70 (m, 1H), 1.47–1.33 (m, 4H), 0.92 (t, J = 7.1 Hz, 3H). 13C NMR (126 MHz, DMSO-d6) δ 147.5, 136.5, 134.3, 133.3, 129.0, 128.8, 126.8, 115.5, 57.2, 53.4, 33.9, 26.3, 21.9, 13.9. MS (ESI-TOF, m/z): calculated for C14H16ClN3 [M + H]+ 262.1106; found 262.1173.
2-Butyl-1,2-dihydroimidazo[1,2-a]quinoxalin-4-amine (37)
Compound 36 (29 mg, 0.11 mmol) was dissolved in 2 M ammonia in methanol (1 mL) and heated in a sealed vial at 90 °C for 20 h and concentrated. The residue was purified by flash chromatography (SiO2, MeOH in CH2Cl2: 0–10%) to give compound 37 (18 mg, 67%) as a yellow solid. 1H NMR (500 MHz, MeOD) δ 7.25 (dd, J = 7.9, 1.2 Hz, 1H), 7.13 (td, J = 7.9, 1.4 Hz, 1H), 7.00 (td, J = 7.8, 1.3 Hz, 1H), 6.85 (dd, J = 8.0, 1.2 Hz, 1H), 4.38–4.30 (m, 1H), 4.26–4.20 (m, 1H), 3.76 (dd, J = 10.3, 8.6 Hz, 1H), 1.79–1.71 (m, 1H), 1.68–1.57 (m, 1H), 1.53–1.37 (m, 4H), 0.95 (t, J = 7.1 Hz, 3H). 13C NMR (126 MHz, MeOD) δ 149.4, 147.7, 134.7, 130.9, 125.9, 125.2, 123.0, 112.8, 65.8, 52.8, 37.8, 28.9, 23.8, 14.4. MS (ESI-TOF, m/z): calculated for C14H18N4 [M + H]+ 243.1604; found 243.1677.
2-Butyl-4-chloroimidazo[1,2-a]quinoxaline (38)
To a solution of compound 36 (357 mg, 1.36 mmol) in toluene (15 mL) was added MnO2 (593 mg, 6.82 mmol) and heated to reflux for 24 h. Additional MnO2 (593 mg, 6.82 mmol) was added, and after another 48 h, the mixture was allowed to cool and filtered. The solvent was evaporated and the crude product was purified by flash chromatography (SiO2, EtOAc in hexane: 0–50%) to give compound 38 (99 mg, 28%) as a yellow solid. 1H NMR (500 MHz, DMSO-d6) δ 8.73 (s, 1H), 8.33 (dd, J = 8.3, 0.9 Hz, 1H), 7.97 (dd, J = 8.2, 1.2 Hz, 1H), 7.78–7.73 (m, 1H), 7.67–7.62 (m, 1H), 2.80 (t, J = 7.6 Hz, 2H), 1.72 (dd, J = 15.2, 7.6 Hz, 2H), 1.44–1.34 (m, 2H), 0.94 (t, J = 7.4 Hz, 3H). 13C NMR (126 MHz, DMSO-d6) δ 158.1, 150.8, 144.0, 143.5, 138.8, 138.4, 136.3, 136.3, 125.5, 122.8, 40.3, 37.6, 31.4, 23.3. MS (ESI-TOF, m/z): calculated for C14H14ClN3 [M + H]+ 260.0949; found 260.0900.
2-Butylimidazo[1,2-a]quinoxalin-4-amine (39)
Compound 38 (70 mg, 0.27 mmol) was dissolved in 2 M ammonia in methanol (2 mL), heated in a sealed vial at 90 °C for 20 h, and concentrated. The residue was purified by flash chromatography (SiO2, EtOAc in hexane: 0–100%) to give compound 39 (21 mg, 33%) as a pale yellow solid. 1H NMR (500 MHz, MeOD) δ 8.18 (s, 1H), 7.94 (dd, J = 8.1, 1.2 Hz, 1H), 7.60 (dd, J = 8.1, 1.2 Hz, 1H), 7.46–7.41 (m, 1H), 7.36 (ddd, J = 8.5, 7.3, 1.4 Hz, 1H), 2.86–2.80 (m, 2H), 1.80 (ddd, J = 13.1, 8.4, 6.7 Hz, 2H), 1.47 (dd, J = 15.0, 7.5 Hz, 2H), 1.00 (t, J = 7.4 Hz, 3H). 13C NMR (126 MHz, MeOD) δ 149.5, 148.1, 137.3, 132.9, 127.7, 126.7, 125.8, 125.0, 116.2, 112.2, 32.8, 29.2, 23.5, 14.2. MS (ESI-TOF, m/z): calculated for C14H16N4 [M + H]+ 241.1448; found 241.1462.
N′-(2-Nitrophenyl)pentanehydrazide (40)
To a solution of (2-nitrophenyl)hydrazine (500 mg, 3.26 mmol) in CH2Cl2 (10 mL) was added N-methylmorpholine (298 μL, 4.24 mmol), followed by valeroyl chloride (415 μL, 3.43 mmol). The mixture was stirred at room temperature for 1 h and concentrated. The residue was purified by flash chromatography (SiO2, MeOH in CH2Cl2: 0–10%) to give compound 40 (564 mg, 73%) as a yellow solid. 1H NMR (500 MHz, DMSO-d6) δ 10.10 (s, 1H), 9.21 (s, 1H), 8.09 (dd, J = 8.5, 1.5 Hz, 1H), 7.61–7.56 (m, 1H), 7.05 (dd, J = 8.6, 1.1 Hz, 1H), 6.85 (ddd, J = 8.4, 7.0, 1.3 Hz, 1H), 2.23 (t, J = 7.5 Hz, 2H), 1.56 (dt, J = 15.0, 7.5 Hz, 2H), 1.33 (dq, J = 14.6, 7.4 Hz, 2H), 0.90 (t, J = 7.4 Hz, 3H). 13C NMR (126 MHz, DMSO-d6) δ 171.9, 145.5, 136.5, 131.6, 125.8, 117.7, 114.6, 33.0, 27.0, 21.8, 13.7. MS (ESI-TOF, m/z): calculated for C11H15N3O3 [M + H]+ 238.1186; found 238.1189.
N′-(2-Nitrophenyl)pentanehydrazonoyl Chloride (41)
Compound 40 (82 mg, 0.35 mmol) in POCl3 (3 mL) was heated at 110 °C for 2 h. The mixture was cooled to room temperature and concentrated. The residue was treated with ice–water and extracted with CH2Cl2. The organic layer was washed with brine, dried over Na2SO4, concentrated, and the crude product was purified by flash chromatography (SiO2, EtOAc in hexane: 0–10%) to give compound 41 (68 mg, 76%) as yellow oil. 1H NMR (500 MHz, DMSO-d6) δ 10.82 (s, 1H), 8.15 (dt, J = 8.6, 0.9 Hz, 1H), 7.71 (dd, J = 4.5, 1.0 Hz, 2H), 7.03–6.99 (m, 1H), 2.76–2.71 (m, 2H), 1.68 (dt, J = 20.6, 7.5 Hz, 2H), 1.42–1.32 (m, 2H), 0.92 (t, J = 7.4 Hz, 3H). 13C NMR (126 MHz, DMSO-d6) δ 140.1, 137.0, 134.1, 131.4, 125.9, 119.5, 115.8, 37.9, 28.1, 21.1, 13.6.
N′-(2-Nitrophenyl)pentanehydrazonamide (42)
Compound 41 (60 mg, 0.23 mmol) was treated with 2 M ammonia in methanol (1 mL) and stirred at room temperature for 20 h. The residue was concentrated to give compound 42 (38 mg, 70%) as a red solid. 1H NMR (500 MHz, DMSO-d6) δ 9.73 (s, 1H), 8.11 (dd, J = 8.5, 1.4 Hz, 1H), 7.61 (ddd, J = 8.5, 7.2, 1.4 Hz, 1H), 7.22 (d, J = 8.5 Hz, 1H), 6.89 (t, J = 7.6 Hz, 1H), 2.44 (t, J = 7.6 Hz, 2H), 1.71–1.62 (m, 2H), 1.38 (dt, J = 14.7, 7.4 Hz, 2H), 0.93 (t, J = 7.4 Hz, 3H). 13C NMR (126 MHz, DMSO-d6) δ 142.5, 136.4, 125.9, 118.1, 114.5, 30.8, 28.6, 21.6, 13.6. MS (ESI-TOF, m/z): calculated for C11H16N4O2 [M + H]+ 237.1346; found 237.1344.
Ethyl 3-Butyl-1-(2-nitrophenyl)-1H-1,2,4-triazole-5-carboxylate (43)
To a solution of compound 42 (78 mg, 0.33 mmol) in ether (3 mL) was added dropwise ethyl oxalyl chloride (110 μL, 0.99 mmol) in ether (2 mL). Toluene (5 mL) was added, and the mixture was heated at 110 °C for 3 h, and concentrated. The residue was purified by flash chromatography (SiO2, EtOAc in hexane: 0–50%) to give compound 43 (60 mg, 57%) as yellow oil. 1H NMR (500 MHz, DMSO-d6) δ 8.26 (dd, J = 8.5, 1.4 Hz, 1H), 7.94 (ddd, J = 8.0, 7.4, 1.5 Hz, 1H), 7.87–7.83 (m, 2H), 4.20 (q, J = 7.1 Hz, 2H), 2.73 (t, J = 7.4 Hz, 2H), 1.71–1.64 (m, 2H), 1.37–1.29 (m, 2H), 1.13 (t, J = 7.1 Hz, 3H), 0.90 (t, J = 7.4 Hz, 3H). 13C NMR (126 MHz, DMSO-d6) δ 164.6, 161.0, 156.5, 144.2, 134.6, 131.5, 131.2, 130.1, 125.3, 62.1, 29.6, 27.1, 21.5, 13.8, 13.6. MS (ESI-TOF, m/z): calculated for C15H18N4O4 [M + H]+ 319.1401; found 319.1410.
2-Butyl[1,2,4]triazolo[1,5-a]quinoxalin-4(5H)-one (44)
To a solution of compound 43 (58 mg, 0.18 mmol) in acetic acid (2 mL) was added iron powder (100 mg). The mixture was heated at 90 °C for 1 h, allowed to cool, filtered, and concentrated. The residue was purified by flash chromatography (SiO2, MeOH in CH2Cl2: 0–10%) to give compound 44 as a white solid (19 mg, 43%). 1H NMR (500 MHz, DMSO-d6) δ 12.31 (s, 1H), 8.04 (dd, J = 8.2, 1.0 Hz, 1H), 7.50–7.43 (m, 2H), 7.36 (ddd, J = 8.5, 7.1, 1.6 Hz, 1H), 2.90–2.83 (m, 2H), 1.76 (ddd, J = 13.3, 8.4, 6.7 Hz, 2H), 1.39 (dq, J = 14.7, 7.4 Hz, 2H), 0.93 (t, J = 7.4 Hz, 3H). 13C NMR (126 MHz, DMSO-d6) δ 166.0, 152.2, 144.3, 128.6, 127.8, 123.5, 122.4, 116.6, 115.4, 29.9, 27.6, 21.7, 13.7. MS (ESI-TOF, m/z): calculated for C13H14N4O [M + H]+ 243.1240; found 243.1227.
Syntheses of 5-chloro[1,2,4]triazolo[1,5-a]quinoxaline 45 and -[1,2,4]triazolo[1,5-a]quinoxalin-4-amine 46 were carried out as reported for 32 and 33, respectively.
2-Butyl-4-chloro[1,2,4]triazolo[1,5-a]quinoxaline (45)
1H NMR (500 MHz, DMSO-d6) δ 8.35 (dd, J = 8.3, 1.0 Hz, 1H), 8.13 (dd, J = 8.2, 1.0 Hz, 1H), 7.90 (ddd, J = 8.4, 7.3, 1.4 Hz, 1H), 7.79 (ddd, J = 8.5, 7.3, 1.4 Hz, 1H), 3.00–2.93 (m, 2H), 1.81 (ddd, J = 13.3, 8.4, 6.7 Hz, 2H), 1.47–1.36 (m, 2H), 0.94 (t, J = 7.4 Hz, 3H). 13C NMR (126 MHz, DMSO-d6) δ 167.0, 142.7, 140.3, 134.7, 130.9, 129.1, 128.0, 127.5, 115.3, 29.9, 27.8, 21.8, 13.7. MS (ESI-TOF, m/z): calculated for C13H13ClN4 [M + H]+ 261.0902; found 261.1890.
2-Butyl[1,2,4]triazolo[1,5-a]quinoxalin-4-amine (46)
1H NMR (500 MHz, MeOD) δ 8.17 (dd, J = 8.2, 1.0 Hz, 1H), 7.68 (dd, J = 8.2, 1.0 Hz, 1H), 7.54–7.50 (m, 1H), 7.44 (ddd, J = 8.5, 7.3, 1.3 Hz, 1H), 3.00–2.97 (m, 2H), 1.88 (dt, J = 15.2, 7.6 Hz, 2H), 1.48 (dq, J = 14.8, 7.4 Hz, 2H), 1.00 (t, J = 7.4 Hz, 3H). 13C NMR (126 MHz, MeOD) δ 167.8, 149.1, 140.2, 137.9, 128.6, 126.8, 126.5, 125.6, 116.0, 31.6, 29.2, 23.4, 14.1. MS (ESI-TOF, m/z): calculated for C13H15N5 [M + H]+ 242.1400; found 242.1391.
2-Chloro-3-hydrazinylquinoxaline (47)
To a solution of 2,3-dichloroquinoxaline 34 (160 mg, 0.81 mmol) in MeOH (4 mL) was added hydrazine hydrate (100 μL, 3.24 mmol), and the reaction mixture was stirred at room temperature overnight. The solvent was evaporated to obtain compound 47 as a pale yellow solid (148 mg, 95%). 1H NMR (500 MHz, MeOD) δ 7.78–7.71 (m, 2H), 7.61 (ddd, J = 8.4, 7.1, 1.5 Hz, 1H), 7.42 (ddd, J = 8.4, 7.1, 1.3 Hz, 1H). 13C NMR (126 MHz, MeOD) δ 150.9, 142.2, 138.3, 137.7, 131.4, 128.6, 126.7, 126.3. MS (ESI-TOF, m/z): calculated for C8H7ClN4 [M + H]+ 195.0432; found 195.0344.
1-Butyl-4-chloro[1,2,4]triazolo[4,3-a]quinoxaline (48)
Trimethyl orthovalerate (0.5 mL) was added to compound 47 (50 mg, 0.257 mmol), and the resulting mixture was heated at 100 °C for 1 h. The solvent was removed and the product obtained was purified by flash chromatography to obtain compound 48 as a white solid (38 mg, 57%). 1H NMR (500 MHz, CDCl3) δ 8.12 (dd, J = 8.4, 1.1 Hz, 1H), 8.09 (dd, J = 8.0, 1.4 Hz, 1H), 7.74 (ddd, J = 8.4, 7.4, 1.7 Hz, 1H), 7.68 (ddd, J = 7.9, 7.4, 1.3 Hz, 1H), 3.53–3.45 (m, 2H), 2.08–1.99 (m, 2H), 1.67–1.54 (m, 2H), 1.04 (t, J = 7.4 Hz, 3H). 13C NMR (126 MHz, CDCl3) δ 152.8, 143.3, 142.9, 135.8, 130.6, 130.0, 128.1, 126.4, 115.6, 28.8, 28.5, 22.6, 13.9. MS (ESI-TOF, m/z): calculated for C13H13ClN4 [M + H]+ 261.0902; found 261.1004.
1-Butyl[1,2,4]triazolo[4,3-a]quinoxalin-4-amine (49)
Compound 48 (25 mg, 0.096 mmol) was dissolved in ammonia solution (2 M in methanol, 1 mL) and heated in a sealed vial at 60 °C for 4 h. Concentration and purification by column chromatography (SiO2, MeOH in CH2Cl2: 0–5%) to give compound 49 (16 mg, 70%) as a white solid. 1H NMR (500 MHz, CDCl3) δ 7.96 (dd, J = 8.4, 1.1 Hz, 1H), 7.71 (dd, J = 8.1, 1.4 Hz, 1H), 7.53–7.47 (m, 1H), 7.38 (ddd, J = 8.7, 7.3, 1.5 Hz, 1H), 5.87 (bs, 2H), 3.48–3.39 (m, 2H), 2.07–1.98 (m, 2H), 1.60 (dq, J = 14.8, 7.4 Hz, 2H), 1.04 (t, J = 7.4 Hz, 3H). 13C NMR (126 MHz, CDCl3) δ 152.3, 147.1, 139.9, 137.6, 127.7, 127.4, 124.6, 124.5, 115.4, 28.8, 28.4, 22.6, 13.9. MS (ESI-TOF, m/z): calculated for C13H15N5 [M + H]+ 242.1400; found 242.1426.
Ethyl 2-(1H-Indol-3-yl)-2-oxoacetate (50)
To a solution of indole (1.01 g, 8.66 mmol) in anhydrous Et2O (17 mL) and pyridine (0.95 mL, 11.7 mmol) was added a solution of ethyl chlorooxoacetate (1.2 mL, 10.7 mmol) in anhydrous Et2O (3 mL) at 0 °C over a period of 15 min. The reaction mixture was stirred at 0 °C for 2 h and filtered. The resulting solid was then washed with cold Et2O and water, dried over high vacuum to give compound 50 (1.52 g, 81%) as a pale yellow powder. 1H NMR (500 MHz, DMSO-d6) δ 12.39 (bs, 1H), 8.42 (d, J = 3.3 Hz, 1H), 8.18–8.13 (m, 1H), 7.57–7.53 (m, 1H), 7.32–7.24 (m, 2H), 4.36 (q, J = 7.1 Hz, 2H), 1.34 (t, J = 7.1 Hz, 3H). 13C NMR (126 MHz, DMSO-d6) δ 179.1, 163.6, 138.3, 136.7, 125.5, 123.9, 122.9, 121.1, 112.8, 112.4, 61.6, 14.0. MS (ESI-TOF, m/z): calculated for C12H11NO3 [M + H]+ 218.0812; found 218.0796.
2-Butyl-2H-pyrazolo[3,4-c]quinolin-4(5H)-one (51)
To a solution of butylhydrazine hydrochloride salt (288 mg, 2.31 mmol) in absolute EtOH (20 mL) were added compound 50 (352 mg, 1.62 mmol) and acetic acid (0.4 mL). The reaction mixture was heated to reflux and stirred for 24 h, cooled to room temperature, and concentrated. The crude product was purified by flash chromatography (SiO2, MeOH in CH2Cl2: 0–3%) to give compound 51 (243 mg, 62%) as a gray powder. 1H NMR (500 MHz, DMSO-d6) δ 11.32 (s, 1H), 8.69 (s, 1H), 7.85 (d, J = 7.8 Hz, 1H), 7.36–7.29 (m, 2H), 7.17 (m, 1H), 4.37 (t, J = 7.0 Hz, 2H), 1.94–1.80 (m, 2H), 1.35–1.20 (m, 2H), 0.91 (t, J = 7.4 Hz, 3H). 13C NMR (126 MHz, DMSO-d6) δ 157.0, 139.9, 135.9, 127.2, 125.5, 123.5, 122.0, 121.2, 115.9, 115.3, 52.5, 31.8, 19.2, 13.4. MS (ESI-TOF, m/z): calculated for C14H15N3O [M + H]+ 242.1288; found 242.1272.
2-Butyl-4-chloro-2H-pyrazolo[3,4-c]quinoline (52)
To a mixture of compound 51 (163 mg, 0.675 mmol) and PCl5 (33.3 mg, 0.160 mmol) was added POCl3 (3 mL). The reaction mixture was heated to reflux, stirred for 2 h, and then concentrated. The residue was diluted with CH2Cl2 and washed with saturated NaHCO3, dried over Na2SO4, filtered, and concentrated. The crude product was purified by flash chromatography (EtOAc in hexanes: 0–20%) to give compound 52 (152 mg, 86%) as a yellow solid. 1H NMR (500 MHz, CDCl3) δ 8.31 (s, 1H), 8.08–8.04 (m, 1H), 8.02–7.98 (m, 1H), 7.63–7.55 (m, 2H), 4.53 (t, J = 7.3 Hz, 2H), 2.12–2.00 (m, 2H), 1.46–1.37 (m, 2H), 0.99 (t, J = 7.4 Hz, 3H). 13C NMR (126 MHz, CDCl3) δ 143.4, 142.1, 141.3, 129.4, 127.4, 127.4, 123.5, 123.1, 122.5, 121.9, 54.3, 32.7, 19.9, 13.6. MS (ESI-TOF, m/z): calculated for C14H14ClN3 [M + H]+ 260.0949; found 260.0989.
2-Butyl-2H-pyrazolo[3,4-c]quinolin-4-amine (53)
Ammonia solution (2 M in MeOH, 1 mL) was added to compound 52 (60 mg, 0.23 mmol), and the reaction mixture was heated in a sealed vial at 100 °C overnight and then concentrated. The crude product was purified by flash chromatography (MeOH in CH2Cl2: 0–10%) to give compound 53 (27 mg, 48%) as a pale yellow powder. 1H NMR (500 MHz, DMSO-d6) δ 8.82 (s, 1H), 7.95 (dd, J = 7.8 Hz, J = 1.5 Hz, 1H), 7.53 (dd, J = 8.2, 1.2 Hz, 1H), 7.39 (ddd, J = 8.3, 7.1, 1.5 Hz, 1H), 7.29–7.22 (m, 1H), 4.46 (t, J = 7.0 Hz, 2H), 3.42 (s, 2H), 2.00–1.84 (m, 2H), 1.37–1.24 (m, 2H), 0.92 (t, J = 7.4 Hz, 3H). 13C NMR (126 MHz, DMSO-d6) δ 150.1, 140.3, 135.5, 126.9, 125.3, 123.4, 123.3, 122.5, 120.6, 118.1, 52.8, 32.0, 19.2, 13.5. MS (ESI-TOF, m/z): calculated for C14H16N4 [M + H]+ 241.1448; found 241.1470.
Human TLR7/8 Reporter Gene Assays (NF-κB Induction)
The induction of NF-κB was quantified using HEK-Blue-7 (hTLR7-specific) and HEK-Blue-8 (hTLR8-specific) cells as previously described by us.12,22 HEK293 cells stably co-transfected with human TLR7 or human TLR8, MD2, and secreted alkaline phosphatase (sAP) were maintained in HEK-Blue Selection medium containing zeocin and normocin. Stable expression of secreted alkaline phosphatase (sAP) under control of NF-κB/AP-1 promoters is inducible by appropriate TLR agonists, and extracellular sAP in the supernatant is proportional to NF-κB induction. HEK-Blue cells were incubated at a density of ∼105 cells/mL in a volume of 80 μL/well, in 384-well, flat-bottomed, cell culture-treated microtiter plates until confluency was achieved and subsequently stimulated with graded concentrations of stimuli. sAP was assayed spectrophotometrically using an alkaline phosphatase-specific chromogen (present in HEK-detection medium as supplied by the vendor) at 620 nm.
Immunoassays for Interferon (IFN) α and Cytokines
Fresh human peripheral blood mononuclear cells (hPBMCs) were isolated from human blood obtained by venipuncture with informed consent and as per institutional guidelines on Ficoll–Hypaque gradients as described elsewhere.36 Aliquots of PBMCs (105 cells in 100 μL/well) were stimulated for 12 h with graded concentrations of test compounds. Supernatants were isolated by centrifugation and were assayed in triplicate using either high-sensitivity multisubtype IFN-α ELISA kits (PBL Interferon Source, Piscataway, NJ, and R&D Systems, Inc., Minneapolis, MN) or analyte-specific multiplexed cytokine/chemokine bead array assays as reported by us previously.11 PBMC supernatants were also analyzed for 41 chemokines and cytokines (EGF, eotaxin, FGF-2, Flt-3 ligand, fractalkine, G-CSF, GM-CSF, GRO, IFN-α2, IFN-γ, IL-10, IL-12 (p40), IL-12 (p70), IL-13, IL-15, IL-17, IL-1ra, IL-1α, IL-1β, IL-2, IL-3, IL-4, IL-5, IL-6, IL-7, IL-8, IL-9, IP-10, MCP-1, MCP-3, MDC (CCL22), MIP-1α, MIP-1β, PDGF-AA, PDGF-AB/BB, RANTES, TGFα, TNF-α, TNF-β, VEGF, and sCD40L) using a magnetic bead-based multiplexed assay kit (Milliplex MAP human cytokine/chemokine kit). Data were acquired and processed on a MAGPIX instrument (EMD Millipore, Billerica, MA) with intra-assay coefficients of variation ranging from 4% to 8% for the 41 analytes.
Flow Cytometric Immunostimulation Experiments
CD69 up-regulation was determined by flow cytometry using protocols published by us previously12 and modified for rapid throughput. Briefly, heparin-anticoagulated whole blood samples were obtained by venipuncture from healthy human volunteers with informed consent and as per guidelines approved by the University of Kansas Human Subjects Experimentation Committee. Serial dilutions of selected compounds were performed using a Bio-Tek Precision 2000 XS liquid handler in sterile 96-well polypropylene plates, to which were added 100 μL aliquots of anticoagulated whole human blood. The plates were incubated at 37 °C for 16.5 h. Negative (endotoxin free water) controls were included in each experiment. Following incubation, fluorochrome-conjugated antibodies (CD3-PE, CD19-FITC, CD56-APC, CD69-PE-Cy7, 10 μL of each antibody, Becton-Dickinson Biosciences, San Jose, CA) were added to each well with a liquid handler and incubated at 37 °C in the dark for 30 min. Following staining, erythrocytes were lysed and leukocytes fixed by mixing 200 L of the samples in 2 mL of prewarmed whole blood lyse/fix buffer (Becton-Dickinson Biosciences, San Jose, CA) in 96-deep-well plates. After washing the cells twice at 200 g for 8 min in saline, the cells were transferred to a 96-well plate. Flow cytometry was performed using a BD FACSArray instrument in the tricolor mode (tricolor flow experiment) and two-color mode (two-color flow experiment) for acquisition on 100 000 gated events. Compensation for spillover was computed for each experiment on singly stained samples. CD69 activation in the major lymphocytic populations, viz., natural killer lymphocytes (NK cells, CD3–CD56+), cytokine-induced killer phenotype (CIK cells, CD3+CD56+), nominal B lymphocytes (CD19+CD56–), and nominal T lymphocytes (CD3+CD56–) were quantified using FlowJo, version 7.0, software (Treestar, Ashland, OR).
Protein Expression, Purification, and Crystallization
The extracellular domain of human TLR8 (hTLR8, residues 27–827) was prepared as described previously27 and was concentrated to 16 mg/mL in 10 mM MES (pH 5.5), 50 mM NaCl. The protein solutions for the crystallization of hTLR8/compound complexes contained hTLR8 (8.5 mg/mL) and compound (protein/compound molar ratio of 1:10) in a crystallization buffer containing 7 mM MES (pH 5.5), 35 mM NaCl. Crystallization experiments were performed with sitting-drop vapor-diffusion methods at 293 K. Crystals of hTLR8/compound were obtained with reservoir solutions containing 9–12% (w/v) PEG3350, 0.3 M potassium formate, and 0.1 M sodium citrate (pH 4.8–5.2).
Data Collection and Structure Determination
Diffraction data sets were collected on beamlines PF-AR NE3A (Ibaraki, Japan) and SPring-8 BL41XU under cryogenic conditions at 100 K. Crystals of hTLR8/compound were soaked into a cryoprotectant solution containing 15% (w/v) PEG3350, 0.23 M potassium formate, 75 mM sodium citrate, pH 4.8–5.2, 7.5 mM MES, pH 5.5, 38 mM NaCl, and 25% glycerol. Data sets were processed using the HKL2000 package37 or imosflm.38 HTLR8/compound structures were determined by the molecular replacement method using the Molrep program39 with the hTLR8/CL097 structure (PDB code 3W3J) as a search model. The model was further refined with stepwise cycles of manual model building using the COOT program40 and restrained refinement using REFMAC41 until the R factor was converged. Compound molecule, N-glycans, and water molecules were modeled into the electron density maps at the latter cycles of the refinement. The quality of the final structure was evaluated with PROCHECK.42 The statistics of the data collection and refinement are also summarized in Table S1. The figures representing structures were prepared with PyMOL.43 Coordinates have been deposited in the Protein Data Bank of the Research Collaboratory for Structural Bioinformatics; PDB codes for compounds 9 and 53 are, respectively, 4QBZ and 4QC0.
Quantum Chemical Computations and Linear Discriminant Analyses
Calculations were performed using the NWChem44 quantum chemical software for the electronic structure, electrostatic charge, and property calculations. All the compounds were fully optimized at the density functional theory (DFT) level of theory using the M06-2X45 functional and correlation consistent cc-pVDZ basis set. The optimized structures were verified as minima by calculating the second-order Hessian matrices. The molecular electrostatic potentials were calculated on the DFT-optimized geometries and superimposed onto a constant electron density (0.002 e/Å3) to provide a measure of the electrostatic potential at roughly the van der Waals surface of the molecules using the GaussView03 software.46 The color-coded surface provides a location of the positive (blue, positive) and negative (red, negative) electrostatic potentials. The regions of positive charge indicate relative electron deficiency, and regions of negative potential indicate areas of excess negative charge. Fisher’s linear discriminant analyses were performed using SPSS v22 (IBM, Armonk, NY); classification function coefficients, classification results, and casewise statistics are given in Table S4.
Acknowledgments
This work was supported by the U.S. National Institutes of Health (NIH)/U.S. National Institute of Allergy and Infectious Diseases (NIAID) Contract HSN272200900033C (S.A.D.), a Grant-in-Aid from the Japanese Ministry of Education, Culture, Sports, Science, and Technology (U.O., H.T., and T.S.), the Takeda Science Foundation (U.O. and T.S.), the Mochida Memorial Foundation for Medical and Pharmaceutical Research (U.O.), and Core Research for Evolutional Science and Technology (CREST), Japan Science & Technology Agency (JST) (T.S.). Support for the NMR instrumentation was provided by NIH Shared Instrumentation Grant S10RR024664 and NSF Major Research Instrumentation Grant 0320648. The computational work was supported by NIFA/USDA Grant 2011-10006-30362. We gratefully acknowledge valuable suggestions from an anonymous reviewer.
Glossary
Abbreviations Used
- APC
antigen-presenting cell
- AIBN
azobisisobutyronitrile
- cDC
conventional dendritic cell
- DFT
density functional theory
- DIPEA
diisopropylethylamine
- DMF
N,N-dimethylformamide
- EC50
half-maximal effective concentration
- EDC
N-(3-dimethylaminopropyl)-N′-ethylcarbodiimide
- ESI-TOF
electrospray ionization-time-of-flight
- HEK
human embryonic kidney
- IFN-α
interferon α
- IFN-γ
interferon γ
- IL
interleukin
- m-CPBA
meta-chloroperoxybenzoic acid
- MESP
molecular electrostatic potential
- MHC
major histocompatibility complex
- NBS
N-bromosuccinimide
- NF-κB
nuclear factor κB
- NK
natural killer
- NMP
N-methyl-2-pyrrolidone
- PAMP
pathogen associated molecular pattern
- PBMC
peripheral blood mononuclear cell
- pDC
plasmacytoid dendritic cell
- PRR
pattern recognition receptor
- QSAR
quantitative structure–activity relationship
- SAP
secreted alkaline phosphatase
- SAR
structure–activity relationship
- ssRNA
single-stranded RNA
- TLR
Toll-like receptor
- TNF-α
tumor necrosis factor α
Supporting Information Available
Summary crystallographic data of compounds 9 and 53, Mulliken charges and electron density maps, statistical summary of linear discriminant analyses, characterization data (1H, 13C, mass spectra), and LC–MS analysis results of final compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
Accession Codes
PDB codes for compounds 9 and 53 are, respectively, 4QBZ and 4QC0.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Iwasaki A.; Medzhitov R. Toll-like receptor control of the adaptive immune responses. Nat. Immunol. 2004, 5, 987–995. [DOI] [PubMed] [Google Scholar]
- Kawai T.; Akira S. The role of pattern-recognition receptors in innate immunity: update on Toll-like receptors. Nat. Immunol. 2010, 11, 373–384. [DOI] [PubMed] [Google Scholar]
- Janeway C. A. Jr.; Medzhitov R. Innate immune recognition. Annu. Rev. Immunol. 2002, 20, 197–216. [DOI] [PubMed] [Google Scholar]
- Akira S.; Takeda K.; Kaisho T. Toll-like receptors: critical proteins linking innate and acquired immunity. Nat. Immunol. 2001, 2, 675–680. [DOI] [PubMed] [Google Scholar]
- Kumagai Y.; Takeuchi O.; Akira S. Pathogen recognition by innate receptors. J. Infect. Chemother. 2008, 14, 86–92. [DOI] [PubMed] [Google Scholar]
- Takeda K.; Akira S.. Toll-like receptors. Curr. Protoc. Immunol. 2007, DOI: 10.1002/0471142735.im1412s77. [DOI] [PubMed] [Google Scholar]
- Yu L.; Wang L.; Chen S. Endogenous Toll-like receptor ligands and their biological significance. J. Cell. Mol. Med. 2010, 14, 2592–2603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spyvee M.; Hawkins L. D.; Ishizaka S. T. Modulators of Toll- like receptor (TLR) signaling. Annu. Rep. Med. Chem. 2010, 45, 191–207. [Google Scholar]
- O’Neill L. A.; Golenbock D.; Bowie A. G. The history of Toll-like receptors—redefining innate immunity. Nat. Rev. Immunol. 2013, 13, 453–460. [DOI] [PubMed] [Google Scholar]
- Meyer T.; Stockfleth E. Clinical investigations of Toll-like receptor agonists. Expert Opin. Invest. Drugs 2008, 17, 1051–1065. [DOI] [PubMed] [Google Scholar]
- Kimbrell M. R.; Warshakoon H.; Cromer J. R.; Malladi S.; Hood J. D.; Balakrishna R.; Scholdberg T. A.; David S. A. Comparison of the immunostimulatory and proinflammatory activities of candidate Gram-positive endotoxins, lipoteichoic acid, peptidoglycan, and lipopeptides, in murine and human cells. Immunol. Lett. 2008, 118, 132–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warshakoon H. J.; Hood J. D.; Kimbrell M. R.; Malladi S.; Wu W. Y.; Shukla N. M.; Agnihotri G.; Sil D.; David S. A. Potential adjuvantic properties of innate immune stimuli. Hum. Vaccines 2009, 5, 381–394. [DOI] [PubMed] [Google Scholar]
- Agnihotri G.; Ukani R.; Malladi S. S.; Warshakoon H. J.; Balakrishna R.; Wang X.; David S. A. Structure–activity relationships in nucleotide oligomerization domain 1 (Nod1) agonistic gammaglutamyldiaminopimelic acid derivatives. J. Med. Chem. 2011, 54, 1490–1510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu W.; Li R.; Malladi S. S.; Warshakoon H. J.; Kimbrell M. R.; Amolins M. W.; Ukani R.; Datta A.; David S. A. Structure–activity relationships in Toll-like receptor-2 agonistic diacylthioglycerol lipopeptides. J. Med. Chem. 2010, 53, 3198–3213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agnihotri G.; Crall B. M.; Lewis T. C.; Day T. P.; Balakrishna R.; Warshakoon H. J.; Malladi S. S.; David S. A. Structure–activity relationships in Toll-like receptor 2-agonists leading to simplified monoacyl lipopeptides. J. Med. Chem. 2011, 54, 8148–8160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salunke D. B.; Shukla N. M.; Yoo E.; Crall B. M.; Balakrishna R.; Malladi S. S.; David S. A. Structure–activity relationships in human Toll-like receptor 2-specific monoacyl lipopeptides. J. Med. Chem. 2012, 55, 3353–3363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salunke D. B.; Connelly S. W.; Shukla N. M.; Hermanson A. R.; Fox L. M.; David S. A. Design and development of stable, water-soluble, human Toll-like receptor 2 specific monoacyl lipopeptides as candidate vaccine adjuvants. J. Med. Chem. 2013, 56, 5885–5900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ukani R.; Lewis T. C.; Day T. P.; Wu W.; Malladi S. S.; Warshakoon H. J.; David S. A. Potent adjuvantic activity of a CCR1-agonistic bis-quinoline. Bioorg. Med. Chem. Lett. 2012, 22, 293–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vasilakos J. P.; Tomai M. A. The use of Toll-like receptor 7/8 agonists as vaccine adjuvants. Expert Rev. Vaccines 2013, 12, 809–819. [DOI] [PubMed] [Google Scholar]
- Akira S.; Takeda K. Toll-like receptor signaling. Nat. Rev. Immunol. 2004, 4, 499–511. [DOI] [PubMed] [Google Scholar]
- Shukla N. M.; Kimbrell M. R.; Malladi S. S.; David S. A. Regioisomerism-dependent TLR7 agonism and antagonism in an imidazoquinoline. Bioorg. Med. Chem. Lett. 2009, 19, 2211–2214. [DOI] [PubMed] [Google Scholar]
- Shukla N. M.; Malladi S. S.; Mutz C. A.; Balakrishna R.; David S. A. Structure–activity relationships in human Toll-like receptor 7-active imidazoquinoline analogues. J. Med. Chem. 2010, 53, 4450–4465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoo E.; Crall B. M.; Balakrishna R.; Malladi S. S.; Fox L. M.; Hermanson A. R.; David S. A. Structure–activity relationships in Toll- like receptor 7 agonistic 1H-imidazo[4,5-c]pyridines. Org. Biomol. Chem. 2013, 11, 6526–6545. [DOI] [PubMed] [Google Scholar]
- Kokatla H. P.; Yoo E.; Salunke D. B.; Sil D.; Ng C. F.; Balakrishna R.; Malladi S. S.; Fox L. M.; David S. A. Toll-like receptor-8 agonistic activities in C2, C4, and C8 modified thiazolo[4,5-c]quinolines. Org. Biomol. Chem. 2013, 11, 1179–1198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salunke D. B.; Yoo E.; Shukla N. M.; Balakrishna R.; Malladi S. S.; Serafin K. J.; Day V. W.; Wang X.; David S. A. Structure–activity relationships in human Toll-like receptor 8-active 2,3-diamino-furo[2,3-c]pyridines. J. Med. Chem. 2012, 55, 8137–8151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kokatla H. P.; Sil D.; Malladi S. S.; Balakrishna R.; Hermanson A. R.; Fox L. M.; Wang X.; Dixit A.; David S. A. Exquisite selectivity for human Toll-like receptor 8 in substituted furo[2,3-c]quinolines. J. Med. Chem. 2013, 56, 6871–6885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanji H.; Ohto U.; Shibata T.; Miyake K.; Shimizu T. Structural reorganization of the Toll-like receptor 8 dimer induced by agonistic ligands. Science 2013, 339, 1426–1429. [DOI] [PubMed] [Google Scholar]
- Kokatla H. P.; Sil D.; Tanji H.; Ohto U.; Malladi S. S.; Fox L. M.; Shimizu T.; David S. A. Structure-based design of novel human Toll-like receptor 8 agonists. ChemMedChem 2014, 9, 719–723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boehm H. J.; Flohr A.; Stahl M. Scaffold hopping. Drug Discovery Today: Technol. 2004, 1, 217–224. [DOI] [PubMed] [Google Scholar]
- Lloyd D. G.; Buenemann C. L.; Todorov N. P.; Manallack D. T.; Dean P. M. Scaffold hopping in de novo design. Ligand generation in the absence of receptor information. J. Med. Chem. 2004, 47, 493–496. [DOI] [PubMed] [Google Scholar]
- Sun H.; Tawa G.; Wallqvist A. Classification of scaffold hopping approaches. Drug Discovery Today 2012, 17, 310–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Murray J. S.; Politzer P.. The use of molecular electrostatic potential in medicinal chemistry. Quantum Medicinal Chemistry; Wiley-VCH: New York, 2002; Vol. 17, pp 233–254. [Google Scholar]; b Höltje H.-D.; Höltje M.. Applications of quantum chemical methods in drug design. Quantum Medicinal Chemistry; Wiley-VCH: New York, 2002; Vol. 17, pp 255–274. [Google Scholar]
- Lai J. J.; Salunke D. B.; Sun C. M. Multistep microwave-assisted divergent synthesis of indolo-fused pyrazino-/diazepinoquinoxalinones on PEG support. Org. Lett. 2010, 12, 2174–2177. [DOI] [PubMed] [Google Scholar]
- Borchardt A. J.; Beauregard D.; Cook T.; Davis R. L.; Gamache D. A.; Yanni J. M.. Heterocyclic inhibitors of histamine receptors for the treatment of disease. World Patent WO 2010/030785 A2, 2006.
- Salio M.; Speak A. O.; Shepherd D.; Polzella P.; Illarionov P. A.; Veerapen N.; Besra G. S.; Platt F. M.; Cerundolo V. Modulation of human natural killer T cell ligands on TLR-mediated antigen-presenting cell activation. Proc. Natl. Acad. Sci. U.S.A. 2007, 104, 20490–20495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- David S. A.; Smith M. S.; Lopez G.; Mukherjee S.; Buch S.; Narayan O. Selective transmission of R5-tropic HIV type 1 from dendritic cells to resting CD4+ T cells. AIDS Res. Hum. Retroviruses 2001, 17, 59–63. [DOI] [PubMed] [Google Scholar]
- Otwinowski Z.; Minor W. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997, 276, 344–358. [DOI] [PubMed] [Google Scholar]
- Vagin A.; Teplyakov A. Molecular replacement with MOLREP. Acta Crystallogr. 2010, D66, 22–25. [DOI] [PubMed] [Google Scholar]
- Battye T. G.; Kontogiannis L.; Johnson O.; Powell H. R.; Leslie A. G. iMOSFLM: a new graphical interface for diffraction-image processing with MOSFLM. Acta Crystallogr. 2011, D67, 271–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P.; Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr. 2004, D60, 2126–2132. [DOI] [PubMed] [Google Scholar]
- Murshudov G. N.; Vagin A. A.; Dodson E. J. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. 1997, D53, 240–255. [DOI] [PubMed] [Google Scholar]
- Laskowski R. A.; Macarthur M. W.; Moss D. S.; Thornton J. M. Procheck—a program to check the stereochemical quality of protein structures. J. Appl. Crystallogr. 1993, 26, 283–291. [Google Scholar]
- DeLano W. L.The PyMOL Molecular Graphics System; DeLano Scientific LLC: Palo Alto, CA, U.S., 2008; http://www.pymol.org.
- Valiev M.; Bylaska E. J.; Govind N.; Kowalski K.; Straatsma K. P.; van Dam H. J. J.; Wang D.; Nieplocha J.; Apra E.; Windus T. L.; de Jong W. A. NWChem: a comprehensive and scalable open-source solution for large scale molecular simulations. Comput. Phys. Commun. 2010, 181, 1477–1489. [Google Scholar]
- Zhao Y.; Truhlar D. G. Density functionals with broad applicability in chemistry. Acc. Chem. Res. 2008, 41, 157–167. [DOI] [PubMed] [Google Scholar]
- GaussView03; Gaussian Inc.: Pittsburgh, PA, 2003.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.