

Abstract
Seasonal and pandemic influenza outbreaks remain a major human health problem. Inhibition of the endonuclease activity of influenza RNA-dependent RNA polymerase is attractive for the development of new agents for the treatment of influenza infection. Our earlier studies identified a series of 5- and 6-phenyl substituted 3-hydroxypyridin-2(1H)-ones that were effective inhibitors of influenza endonuclease. These agents identified as bimetal chelating ligands binding to the active site of the enzyme. In the present study, several aza analogues of these phenyl substituted 3-hydroxypyridin-2(1H)-one compounds were synthesized and evaluated for their ability to inhibit the endonuclease activity. In contrast to the 4-aza analogue of 6-(4-fluorophenyl)-3-hydroxypyridin-2(1H)-one, the 5-aza analogue (5-hydroxy-2-(4-fluorophenyl)pyrimidin-4(3H)-one) did exhibit significant activity as an endonuclease inhibitor. The 6-aza analogue of 5-(4-fluorophenyl)-3-hydroxypyridin-2(1H)-one (6-(4-fluorophenyl)-4-hydroxypyridazin-3(2H)-one) also retained modest activity as an inhibitor. Several varied 6-phenyl-4-hydroxypyridazin-3(2H)-ones and 2-phenyl-5-hydroxypyrimidin-4(3H)-ones were synthesized and evaluated as endonuclease inhibitors. The SAR observed for these aza analogues are consistent with those previously observed with various phenyl substituted 3-hydroxypyridin-2(1H)-ones.
Introduction
Two classes of agents, those targeting the M2 ion-channel and those targeting neuramindase, are available to treat or prevent influenza infection. The emergence of resistance to the M2 ion-channel inhibiting drugs, amantadine and rimantadine, has limited their clinical utility.1,2 Resistance to neuraminidase inhibitors, including oseltamivir, has also been observed in several seasonal influenza A strains.3,4 Drug resistance mutations can emerge in almost all influenza A subtypes/strains, including the pandemic 2009 H1N1 virus,5 limiting the clinical efficacy of the currently approved drugs.
Influenza A contains eight negative-stranded RNA genomic segments. The three largest genomic RNA segments encode the viral RNA-dependent RNA polymerase (RdRP) consisting of the polymerase acidic protein (PA) and polymerase basic proteins 1 (PB1) and 2 (PB2). The PA subunit: (i) has endonuclease activity, (ii) is involved in viral RNA (vRNA)/complementary RNA (cRNA) promoter binding, and (iii) interacts with the PB1 subunit.6 PA has two domains, PAN (a ∼25 kDa N-terminal domain; residues 1–197) and PAC (∼55 kDa C-terminal domain; residues 239–716). Crystal structures of PAC have been determined in complexes with N-terminal fragments of PB1.7 The structure of PAN has been solved in several crystal forms both unliganded and with various ligands.8−13
Influenza RdRP is responsible for the replication and transcription of the segmented viral RNA genes. Viral mRNA transcription involves a cap-snatching mechanism in which the polymerase binds to the host cellular mRNA via the 5′-cap and cleaves the mRNA 12–13 nucleotides downstream. This cleaved RNA fragment, which contains the 5′ cap, acts as a primer for viral mRNA synthesis.14 Cap-snatching is an important event in the life cycle of all members of the Orthomyxoviridae family of viruses, including influenza A, B, and C viruses. Because the host cell has no analogous activity, inhibitors of cap-snatching may be selective against all influenza subtypes and strains, including oseltamivir-resistant influenza A viruses, without interfering with the host cell.
Several small molecules have been identified as influenza endonuclease inhibitors. These include 2,4-dioxobutanoic acid derivatives,10,11,15,16 5-hydroxy-1,6-dihydropyrimidine-4-carboxylic acid derivatives,11 and flutimide and its derivatives,10,11,15−18 2-hydroxyphenyl amide derivatives,19 as well as tetramic acid derivatives.20 Recently, in silico screening with in vitro and ex vivo follow up studies identified hexanetetrones and trihydroxyphenyls as endonuclease inhibitors with anti-influenza activity.21 As correctly hypothesized, the endonuclease activity of influenza polymerase belongs to the two metal ion group of phosphate-processing enzymes.20 From an X-ray crystallographic screening campaign of a fragment library targeting the influenza A endonuclease enzyme, we identified the 5-chloro-3-hydroxypyridin-2(1H)-one as a bimetal chelating ligand at the active site of the enzyme.13 Using this information, we have reported the structure–activity relationships for two new series of compounds, 3-hydroxypyridin-2(1H)-ones and 3-hydroxyquinolin-2(1H)-ones, as endonuclease inhibitors (Figure 1).22,23
Figure 1.

Structures and of 5-(p-fluorophenyl-3-hydroxypyridin-2(1H)-one (5-FPhP), 6-(p-fluorophenyl)-3-hydroxypyridin-2(1H)-one (6-FPhP), 6-(p-fluorophenyl)-3-hydroxyquinolin-2(1H)-one (6-FPhQ), and 7-(p-fluorophenyl)-3-hydroxyquinolin-2(1H)-one (7-FPhQ).
Phenyl substituted derivatives of 3-hydroxypyridin-2(1H)-ones are among the more potent inhibitors of influenza A endonuclease. While 4-(4-fluorophenyl)-3-hydroxypyridin-2(1H)one did not exhibit significant activity, both 5-(4-fluorophenyl)-3-hydroxypyridin-2(1H)one and 6-(4-fluorophenyl)-3-hydroxypyridin-2(1H)one had IC50 values <1.0 μM. The present study was undertaken to determine the relative effects of aza substitution at various positions of phenyl substituted 3-hydroxypyridin-2(1H)-ones on their potential to inhibit influenza A endonuclease.
Chemistry
The synthesis of 5-(4-fluorophenyl)-1,4-dihydropyrazine-2,3-dione, 1a, is outlined in Scheme 1. This dione can tautomerize to either 5- or 6-(4-fluorophenyl)-3-hydroxypyrazin-2(1H)-one (1b, 1c). The 2,3-dichloropyrazine was treated with excess sodium methoxide to form 2,3-dimethoxypyrazine as previously described.24 Bromination of 2,3-dimethoxypyrazine with N-bromosuccimide in DMF provided the 5-bromo derivative,24 which under Suzuki coupling conditions with 4-fluorophenylboronic acid gave 5-(4-fluorophenyl)-2,3-dimethoxypyrazine. Treatment of this dimethoxypyrazine with a (1:1) mixture of dioxane and 2 N HCl provided 1.25
Scheme 1.

The method used to prepare 6-(4-fluorophenyl)-5-hydroxypyrimidin-4(3H)-one 2 is outlined in Scheme 2. 4,5-Dimethoxy-6-chloropyrimidine was prepared from 4,6-dichloro-5-methoxypyrimidine by treatment with sodium methoxide.26 Suzuki coupling of this intermediate with 4-fluorophenylboronic acid provided 6-(p-fluorophenyl)-4,5-dimethoxypyrimidine, which in a 1:1 mixture of dioxane and 2 N HCl produced 5-methoxy-6-(4-fluorophenyl)pyrimidin-4(3H)-one. Treatment of this 5-methoxypyrimidin-4(3H)-one with BBr3 in CH2Cl2 provided 2.
Scheme 2.

The synthetic approach employed for the preparation of various substituted 2-phenyl 5-hydroxypyrimidin-4(3H)-ones is outlined in Scheme 3. 2-Chloro-4,5-dimethoxypyrimidine, prepared from 2,4-dichloro-5-methoxypyrimidine,27 was a common intermediate for these 2-phenyl 5-hydroxypyrimidin-4(3H)-ones. Suzuki coupling with the appropriate phenylboronic acid provided the desired 2-phenyl 4,5-dimethoxypyrimidine derivative, which was initially hydrolyzed with 1:1 dioxane and 2 N HCl to provide the requisite 2-phenyl 5-methoxypyrimidin-4(3H)-one. Further treatment of these 5-methoxypyrimidin-4(3H)-ones with BBr3 in CH2Cl2 provided the 2-(fluorophenyl) derivatives 3–5 as well as the various 2-biphenyl derivatives 6–8. In a similar manner, the 2-(4-cyanophenyl) and 2-(3-cyanophenyl) 5-methoxypyrimidin-4(3H)-ones were prepared and treated with and BBr3 in CH2Cl2 to form the 5-hydroxypyrimidin-4(3H)-ones 9 and 10. Treatment of 9 or 10 with sodium azide in DMF in the presence of a catalytic amount of acetic acid gave the 5-tetrazoyl derivatives 11 and 12, respectively.
Scheme 3.

The preparation of 5-(4-fluorophenyl)-4-hydroxypyridazin-3(2H)-one is summarized in Scheme 4. Formation of the 2-methoxymethyl derivative of 4,5-dichloropyridazin-3(2H)-one, followed by selective replacement of the 4-chloro substituent with a methoxyl group, provided 2-methoxymethyl-5-chloro-4-methoxypyridazin-3(2H)-one.28 This MOM-protected chloropyridazin-3(2H)-one was reacted with 4-fluorophenylboronic acid under Suzuki coupling conditions to provide 2-methoxymethyl-5-(4-fluorophenyl)-4-methoxypyridazin-3(2H)-one. Treatment of this 5-(4-fluorophenyl)pyridazin-3(2H)-one with BBr3 in CH2Cl2 gave 13 in good yield.
Scheme 4.

The synthesis of various 6-phenyl substituted pyridazin-3(2H)-ones was accomplished as outlined in Scheme 5. Commercially available 3,4,6-trichloropyridazine was converted to 6-chloro-3,4-dimethoxypyridazine,29 which was subsequently used with either 4-fluorophenylboronic acid or 4-cyanophenylboronic acid under Suzuki coupling conditions to form the 6-phenyl-3,4-dimethoxypyridazine intermediates. Treatment of these dimethoxypyridazines with a 1:1 mixture of dioxane and 2 N HCl provided the 4-methoxypyridazine-3(2H)-ones, which were subsequently treated with BBr3 in CH2Cl2 to provide 14 and 15. Treatment of 15 with sodium azide in DMF in the presence of a catalytic amount of acetic acid provided the 6-(4-(tetrazol-5-yl)phenyl) derivative, 16 (Table 1).
Scheme 5.

Table 1. Inhibition Assay of Influenza A Endonuclease by Phenyl Substituted 3-Hydroxypyrazin-2(1H)-ones, 5-Hydroxypyrimidin-4(3H)-ones, and 4-Hydroxypyridazin-3(2H)-ones.
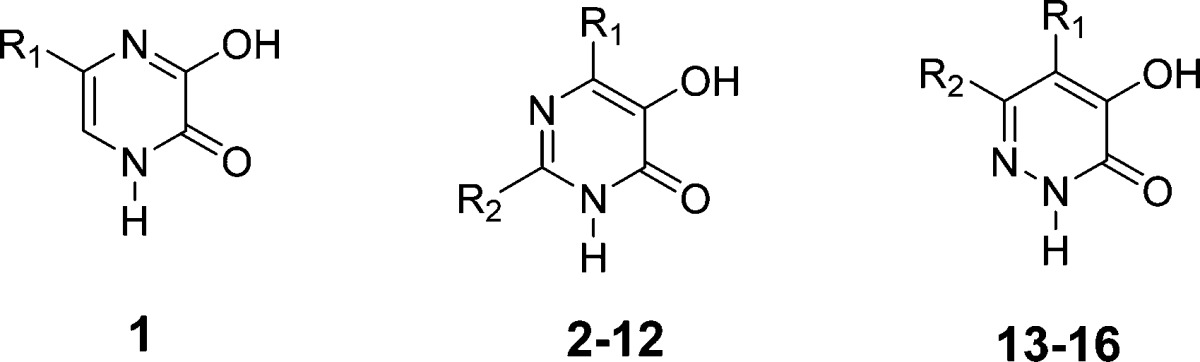
| compd | IC50 (μM) | R1 | R2 |
|---|---|---|---|
| 1 | 59.0 | 4-FC6H5 | |
| 2 | >193 | 4-FC6H5 | H |
| 3 | 0.58 | H | 4-FC6H4 |
| 4 | 1.56 | H | 3-FC6H4 |
| 5 | 1.67 | H | 2-FC6H4 |
| 6 | 2.24 | H | 4-[1,1′]-biphenyl |
| 7 | 0.40 | H | 3-[1,1′]-biphenyl |
| 8 | 1.10 | H | 2-[1,1′]-biphenyl |
| 9 | 0.52 | H | 4-CNC6H4 |
| 10 | 0.25 | H | 3-CNC6H4 |
| 11 | 0.15 | H | 4-(CN4H)phenyl |
| 12 | 0.48 | H | 3-(CN4H)phenyl |
| 13 | 177 | 4-FC6H4 | H |
| 14 | 6.0 | H | 4-FC6H4 |
| 15 | 9.3 | H | 4-CNC6H4 |
| 16 | 3.0 | H | 4-(CN4H)phenyl |
| 5-FPhP | 0.73 | ||
| 6-FPhP | 0.43 |
Results and Discussion
The tautomeric forms of 5-(4-fluorophenyl)-1,4-dihydropyrazine-2,3-dione, 1b and 1c (5- and 6-(4-fluorophenyl)-3-hydroxypyrazin-2(1H)-one), represent the 4-aza derivatives of both 5- and 6-(4-fluorophenyl)-3-hydroxypyridin-2(1H)-one, respectively. Upon the basis of its NMR spectra, however, the most dominant tautomeric form of 1 is the 1,4-dihydropyrazine-2,3-dione, 1a, with only minor amounts of tautomers 1b and 1c. This may explain the comparatively weak activity of 1 as an endonuclease inhibitor. Alternatively, should 1b or 1c form to an appreciable extent under the enzyme assay conditions, the presence of a 4-aza substituent adjacent to the 3-hydroxyl group of either tautomer 1b or 1c may be associated with reduced intrinsic activity as an inhibitor.
Compound 2 is the 5-aza analogue of 4-(4-fluorophenyl)-3-hydroxypyridin-2(1H)-one. The lack of activity observed for 2 is consistent with previous structure–activity relationships observed with various 4-substituted 3-hydroxypyridin-2-ones. The presence of a phenyl group or other relatively large substituents at the 4-position of various 3-hydroxypyridin-2-ones is associated with a dramatic loss in potency as an inhibitor. It is noteworthy that when the 4-fluorophenyl substituent is at the 2-postion of 5-hydroxypyrimidin-4(3H)-ones, which is comparable to the 6-position of 3-hydroxypyridin-2-ones, significant activity in terms of endonuclease inhibition is observed (IC50 = 0.58 μM).13,23 This 4-fluorophenyl derivative is more than twice as potent as either of the 3- or 2-fluorophenyl isomers, 4 and 5. A series of 2-biphenyl 5-hydroxypyrimidin-4(3H)-ones 6–8 was also evaluated. Steric factors may be responsible for the decreased activity observed for the 4- and 2-biphenyl derivatives 6 and 8, relative to 7.
The structure–activity studies performed with 3-hydroxypyridin-2-ones indicated 6-[(4-tetrazoyl)phenyl]-3-hydroxypyridin-2-one had pronounced activity as an endonuclease inhibitor (IC50 = 0.085 μM).23 On the basis of this observation, 2-[(tetrazoyl)phenyl] derivatives of 5-hydroxypyrimidin-4(3H)-one and their 2-(cyanophenyl) precursors were targeted for synthesis and evaluation as endonuclease inhibitors. The 3-cyanophenyl derivative 10 is approximately twice as potent compared to the 4-cyanophenyl isomer 9. However, the 4-(tetrazoly)phenyl derivative 11 is the most potent of 5-hydroxypyrimidin-4(3H)-ones evaluated (IC50 = 0.15 μM) with 3-fold greater potency than the 3-(tetrazoly)phenyl derivative 12.
Previously formed crystals of apo pandemic 2009 influenza endonuclease were soaked in a solution containing 11 as described previously (Supporting Information, Table 1S).13 The crystal structure revealed that 11 chelates to the two active site metal ions in a mode similar to the 3-hydroxypyridin-2(1H)-one containing compounds (Figure 2). The N1 atom of the pyrimidine ring forms hydrogen bond interactions with a network of water molecules present near the active site. N3 interacts with Tyr130 through two bridging waters. Similar to the 5-[4-(tetrazoly)phenyl] containing pyridinone compounds, the 2-[4-(tetrazoly)phenyl] makes bidentate hydrogen bond interactions with Arg124.23 The 2-phenyl ring is rotated ∼90° relative to the 5-phenyl seen previously and is coplanar with the pyrimidine ring due to the lack of steric restraint from additional ring substitutions as seen previously.13
Figure 2.
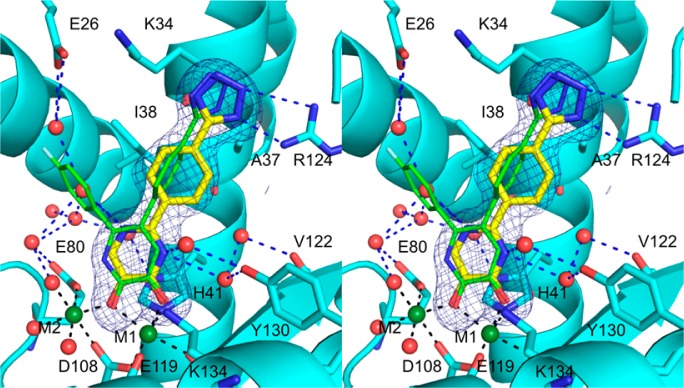
Stereoview image of a crystal structure of 11 (yellow) bound to PAN(cyan) superposed with a previously published structure (PDB ID: 4M5U) of 5-(4-(1H-tetrazol-5-yl)phenyl)-6-(4-fluorophenyl)-3-hydroxypyridin-2(1H)-one (green). Metal-coordinating bonds are depicted as black dashed lines whereas hydrogen or electrostatic bonds are depicted in blue. Electron density calculated from an omit map is contoured at 4.0σ (blue mesh).
5-(4-Fluorophenyl)-4-hydroxypyridazin-3(2H)-one 13 is the 6-aza analogue of 4-(4-fluorophenyl)-3-hydroxypyridin-2(1H)-one. Consistent with the structure–activity relationships observed with 4-phenyl substituted 3-hydroxy-pyridin-2(1H)-ones, 13 did not exhibit significant activity as an endonuclease inhibitor. The various 6-(phenyl)-4-hydroxypyridazin-3(2H)-ones evaluated in this study are the 6-aza analogues of 5-(phenyl)-3-hydroxypyridin-2(1H)-ones. These 6-phenylpyridazin-3(2H)-ones 14–16 are more active than the 4-fluorophenylpyrazine derivative 1 but are at least an order of magnitude less active than analogous 2-phenyl-5-hydroxypyrimidin-4(3H)-ones. The most active derivative was the 6-(4-tetrazoyl)phenyl-4-hydroxypyridazin-3(2H)-one 16 (IC50 = 3.0 μM). Interestingly, 5-substituted 3-hydroxypyridin-2(1H)-ones and 6-substituted 4-hydroxypyridazin-3(2H)ones as well as variously substituted 3-hydroxyquinolin-2(1H)ones have been previously established as d-amino acid oxidase (DAAO) inhibitors.30,31 However, none of these similarly structured aza-analogues of 3-hydroxypyridin-2(1H)-ones that were synthesized in the present study were evaluated for their relative activity as DAAO inhibitors.
Data on the relative activity of these aza 3-hydroxypyridin-2(1H)-one derivatives indicate that 2-phenyl-5-hydroxypyrimidin-4(3H)-ones, which are the 5-aza analogues of 6-phenyl-3-hydroxypyridin-2(1H)-ones, can exhibit comparable activity as inhibitors of endonuclease. However, 6-phenyl-4-hydroxypyridazin-3(2H)-ones, which are the 6-aza analogues of 5-phenyl-3-hydroxypyridin-2(1H)-ones, are 2.3–8.2-fold less active. Although the tautomers of 5-(4-fluorophenyl)-1,4-dihydropyrazine-2,3-dione can be viewed as 4-aza-analogues either 5- or 6-(4-fluorophenyl)-3-hydroxypyridin-2(1H)-one, they were more than 80 times less potent than either of these 3-hydroxypyridin-2(1H)-ones. The relative ranking of comparable phenyl substituted pyridin-2(1H)-ones and their aza derivatives in terms of endonuclease inhibition is 3-hydroxypyridin-2(1H)-one ≈ 5-hydroxypyrimidin-4(3H)-ones >4-hydroxypyridazin-3(2H)-ones ≫1,4-dihydropyrazine-2,3-dione.
Experimental Section
Chemistry: General Methods
All reactions, unless otherwise stated, were done under nitrogen atmosphere. Reaction monitoring and follow-up were done using aluminum backed Silica G TLC plates with UV254 (Sorbent Technologies), visualizing with ultraviolet light. Flash column chromatography was done on a Combi Flash Rf Teledyne ISCO using hexane, ethyl acetate, dichloromethane, and methanol. The 1H (400 MHz) and 13C (100 MHz) NMR spectra were done in CDCl3, methanol-d4, and DMSO-d6 and recorded on a Bruker Avance III (400 MHz) multinuclear NMR spectrometer. Data is expressed in parts per million relative to the residual nondeuterated solvent signals, spin multiplicities are given as s (singlet), d (doublet), dd (doublet of doublets), t (triplet), dt (doublet of triplets), q (quartet), m (multiplet), and bs (broad singlet), and coupling constants (J) are reported in hertz. Melting points were determined using Mel-temp II apparatus and are uncorrected. Analytical HPLC was performed on a Shimadzu LC-20AT Prominence liquid chromatograph using a 150 mm × 4.6 mm Princeton SPHER-100 RP C18 1000A 5 μ column using 0% water for 2 min and a 0–100% water/methanol gradient over a 5 min period and 5 min at 100% methanol at a 2.0 mL/min flow rate monitoring UV absorbance at 254 and 296 nm. Using this method of analysis, the purity of all compounds used in bioassays was determined to be ≥95%. HRMS experiments were conducted by Washington University Resource for Biomedical and Bioorganic Mass Spectrometry Department of Chemistry.
5-(4-Fluorophenyl)pyrazine-2,3(1H,4H)-dione (1)
5-(4-Fluorophenyl)-2,3-dimethoxypyrazine (100 mg, 0.43 mmol) was dissolved in a mixture of dioxane (5 mL) and 2 N HCl (5 mL). The reaction mixture was then refluxed for 21 h. After the reaction was completed, it was cooled to room temperature. Then solvent was removed under reduced pressure. The resulting white solid was dissolved in satd NaHCO3. The white suspension was filtered and solid was collected and dried to give 5-(4-fluorophenyl)pyrazine-2,3(1H,4H)-dione as a white solid (75 mg, 85%); mp 285–287 °C. 1H NMR (400 MHz, DMSO-d6) δ 11.55 (s, 1H), 11.46 (s, 1H), 7.57, (dd, J = 9 Hz, J = 5 Hz, 2H), 7.23–7.19 (m, 2H), 6.59 (d, J = 3 Hz, 1H). 13C NMR (100 MHz, DMSO-d6) δ 161.8 (JC,FJC,F = 243 Hz), 156.9, 155.7, 128.1, 127.6 (JC,F = 9 Hz), 121.0, 115.5 (JC,F = 21 Hz), 107.1. 19F NMR (376 MHz, DMSO-d6) δ −114.0. HRMS (ESI) calculated for C10H7FN2O2Na (M + Na)+ 229.0384, found 229.0382.
5-(4-Fluorophenyl)-2,3-dimethoxypyrazine
5-Bromo-2,3-dimethoxypyrazine (150 mg, 0.69 mmol), (4-fluorophenyl)boronic acid (144 mg, 1.03 mmol), Pd(PPh3)4 (80 mg, 0.069 mmol), and Na2CO3 (218 mg, 2.06 mmol) were dissolved in a mixture of dioxane (6 mL) and water (2 mL). The air was evacuated and replaced with N2. Then, the reaction mixture was refluxed for 5 h. Because the reaction was not completed, additional (4-fluorophenyl)boronic acid (48 mg, 0.34 mmol) was added. It was then again refluxed for 16 h. It was cooled to room temperature, and it was diluted with EtOAc and washed with satd NH4Cl, followed by brine. The organic layer was dried over Na2SO4 and concentrated under reduced pressure, and the resulting residue was purified by flash chromatography on silica gel, eluting with 0–10% EtOAc/hexane. This afforded 5-(4-fluorophenyl)-2,3-dimethoxypyrazine as a white solid (123 mg, 77%); mp 108–110 °C. 1H NMR (400 MHz, CDCl3) δ 8.03 (s, 1H), 7.90 (dd, J = 9 Hz, J = 5 Hz, 2H), 7.15–7.10 (m, 2H). 13C NMR (100 MHz, CDCl3) δ 163.06 (JC,F = 247 Hz), 149.43, 149.41, 140.29, 132.66 (JC,F = 3 Hz), 127.77, 127.73, 127.69 (JC,F = 8 Hz), 115. 68 (JC,F = 21 Hz), 54.09, 53.81. 19F NMR (376 MHz, CDCl3) δ −113.7. HRMS (ESI) calculated for C12H12FN2O2 (M + H)+ 235.0877, found 235.0880.
5-Bromo-2,3-dimethoxypyrazine
2,3-Dimethoxypyrazine (565 mg, 4.03 mmol) and NBS (754 mg, 4.23 mmol) were dissolved in DMF (5 mL). The mixture was stirred for 29 h at room temperature. After the reaction was completed, DMF was removed by Kugelrohr distillation. Then, the resulting residue was diluted with DCM, and the organic layer was washed with satd NaHCO3, followed by brine. The organic layer was dried over Na2SO4 and concentrated under reduced pressure to provide 5-bromo-2,3-dimethoxypyrazine as a white solid (384 mg, 43%); mp 54–56 °C. 1H NMR (400 MHz, CDCl3) δ 7.86 (s, 1H), 4.03 (s, 3H), 3.88 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 161.2, 150.0, 141.7, 138.1, 56.6, 55.1.
2,3-Dimethoxypyrazine
2,3-Dichloropyrazine (1.04 mL, 10 mmol) was added to MeOH (10 mL). Then the reaction mixture was cooled to 0 °C. It was treated with NaOMe (5.4 g, 100 mmol) and allowed to warm to room temperature. The reaction mixture was then stirred for 22 h at room temperature. After the reaction was completed, it was diluted with DCM. The resulting white suspension was filtered, and the filtrate was concentrated under reduced pressure. The residue was diluted with DCM and washed with water, followed by brine. The organic layer was dried over Na2SO4 and concentrated under reduced pressure to reveal a colorless liquid. 2,3-Dimethoxypyrazine was crystallized as a white solid (1.13 g, 81%); mp 21–23 °C. 1H NMR (400 MHz, CDCl3) δ 7.47 (s, 2H), 3.88 (s, 6H). 13C NMR (100 MHz, CDCl3) δ 150.3, 131.7, 53.4.
6-(4-Fluorophenyl)-5-hydroxypyrimidin-4(3H)-one (2)
6-(4-Fluorophenyl)-5-methoxypyrimidin-4(3H)-one (107 mg, 0.49 mmol) was dissolved in anhydrous DCM (5 mL). The reaction mixture was cooled to 0 °C, and the 1 M in DCM BBr3 (5 mL, 5 mmol) was added. It was then allowed to warm to room temperature and stirred for 24 h. Then the solvent was removed under reduced pressure. The resulting residue was diluted with EtOAc, which was washed with satd NaHCO3, followed by brine. The organic layer was dried over Na2SO4, followed by concentration under the vacuum. The resulting residue was purified by flash chromatography on silica gel, eluting with 0–10% MeOH/DCM to provide 6-(4-fluorophenyl)-5-hydroxypyrimidin-4(3H)-one as a white solid (50 mg, 50%); mp 285–287 °C. 1H NMR (400 MHz, DMSO-d6) δ 12.67 (bs, 1H), 9.83 (bs, 1H), 8.20 (dd, J = 9 Hz, J = 6 Hz, 2H), 7.87 (s, 1H), 7.29–7.25 (m, 2H). 13C NMR (100 MHz, DMSO-d6) δ 161.9 (JC,F = 245 Hz), 158.7, 141.1, 138.5, 136.0, 132.3, 130.7 (JC,F = 8 Hz), 114.8 (JC,F = 21 Hz). 19F NMR (376 MHz, DMSO-d6) δ −113.0. HRMS (ESI) calculated for C10H8FN2O2 (M + H)+ 207.0564, found 207.0568.
6-(4-Fluorophenyl)-5-methoxypyrimidin-4(3H)-one
4-(4-Fluorophenyl)-5,6-dimethoxypyrimidine (134 mg 0.57 mmol) was dissolved in a mixture of 2 N HCl (5 mL) and dioxane (5 mL). The reaction mixture was then refluxed for 12 h. It was then cooled to room temperature. The reaction mixture was put under the vacuum to remove the solvent, which gave white residue. This residue was diluted with water and filtered. The solid was collected and gave 6-(4-fluorophenyl)-5-methoxypyrimidin-4(3H)-one as a white solid (109 mg, 87%); mp 204–206 °C. 1H NMR (400 MHz, DMSO-d6) δ 12.76 (s, 1H), 8.07 (s, 1H), 8.04 (dd, J = 9 Hz, J = 6 Hz, 2H), 7.33–7.28 (m, 2H), 3.34 (s, 3H). 13C NMR (100 MHz, DMSO-d6) δ 162.5 (JC,F = 246 Hz), 158.8, 147.4, 143.7, 142.9, 131.7, 131.2 (JC,F = 8 Hz), 115.0 (JC,F = 21 Hz), 58.8. 19F NMR (376 MHz, DMSO-d6) δ −111.8. HRMS (ESI) calculated for C11H10FN2O2 (M + H)+ 221.0721, found 221.0721.
4-(4-Fluorophenyl)-5,6-dimethoxypyrimidine
4-Chloro-4,5-dimethoxypyrimidine (165 mg, 0.95 mmol), (4-fluorophenyl)boronic acid (99 mg, 1.42 mmol), Pd(PPh3)4 (110 mg, 0.095 mmol), and Na2CO3 (300 mg, 2.84 mmol) were dissolved in a mixture of dioxane (9 mL) and water (3 mL). The air was evacuated and replaced with N2. Then the reaction mixture was refluxed for 19 h. After the reaction was completed, it was cooled to room temperature and it was diluted with EtOAc and washed with satd NH4Cl, followed by brine. The organic layer was dried over Na2SO4 and concentrated under reduced pressure, and the resulting residue was purified by flash chromatography on silica gel, eluting with 0–10% EtOAc/hexane. This afforded 4-(4-fluorophenyl)-5,6-dimethoxypyrimidine as a white solid (181 mg, 82%); mp 62–64 °C. 1H NMR (400 MHz, CDCl3) δ 8.53 (s, 1H), 8.07 (dd, J = 9 Hz, J = 6 Hz, 2H), 7.17–7.11 (m, 2H), 4.07 (s, 3H), 3.73 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 163.7 (JC,F = 249 Hz), 164.0, 154.3, 152.0,139.4, 131.4 (JC,F = 8 Hz), 131.3, 115.3 (JC,F = 21 Hz), 60.2, 54.2. 19F NMR (376 MHz, CDCl3) δ −111.0. HRMS (ESI) calculated for C12H12FN2O2 (M + H)+ 235.0877, found 235.0882.
4-Chloro-5,6-dimethoxypyrimidine
4,6-Dichloro-5-methoxypyrimidine (300 mg, 1.68 mmol) was added to MeOH (10 mL). Then the reaction mixture was cooled to 0 °C. It was treated with NaOMe (99 mg, 1.85 mmol) and allowed to warm to room temperature. The reaction mixture was then stirred for 19 h at room temperature. After the reaction was completed, it was put under the vacuum to remove MeOH. The resulting residue was diluted with EtOAc, which was then washed with satd NH4Cl, followed by brine. The organic layer was dried over Na2SO4 and then concentrated. The residue was purified by flash chromatography on silica gel, eluting with 0–10% EtOAc/hexane to provide 4-chloro-5,6-dimethoxypyrimidine as a white solid (168 mg, 57%); mp 53–55 °C. 1H NMR (400 MHz, CDCl3) δ 8.27 (s, 1H), 4.03 (s, 3H), 3.88 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 163.6, 151.6, 151.3, 138.2, 60.7, 54.8.
2-(4-Fluorophenyl)-5-hydroxypyrimidin-4(3H)-one (3)
2-(4-Fluorophenyl)-5-methoxypyrimidin-4(3H)-one (58 mg, 0.26 mmol) was dissolved in anhydrous DCM (5 mL). The reaction mixture was cooled to 0 °C, and the 1 M in DCM BBr3 (3 mL, 3 mmol) was added. It was then allowed to warm to room temperature and stirred for 24 h. Then the solvent was removed under reduced pressure. The resulting residue was suspended in water. It was filtered, and the solid was collected and dried under vacuum to provide 2-(4-fluorophenyl)-5-hydroxypyrimidin-4(3H)-one as a white solid (23 mg, 42%); mp 252–254 °C. 1H NMR (400 MHz, DMSO-d6) δ 12.88 (bs, 1H), 9.64 (bs, 1H), 8.05 (dd, J = 9 Hz, J = 5 Hz, 2H), 7.54 (s, 1H), 7.33–7.29 (m, 2H). 13C NMR (100 MHz, DMSO-d6) δ 163.4 (JC,F = 246 Hz), 159.0, 146.9, 143.4, 131.7, 129.4 (JC,F = 9 Hz), 129.1, 115.5 (JC,F = 22 Hz). 19F NMR (376 MHz, DMSO-d6) δ −110.5. HRMS (ESI) calculated for C10H8FN2O2 (M + H)+ 207.0564, found 207.0566.
2-(4-Fluorophenyl)-5-methoxypyrimidin-4(3H)-one
2-(4-Fluorophenyl)-4,5-dimethoxypyrimidine (187 mg, 0.799 mmol) was dissolved in a mixture of 2 N HCl (5 mL) and dioxane (5 mL). The reaction mixture was then refluxed for 12 h. It was then cooled to room temperature. The reaction mixture was diluted with EtOAc and washed with satd NaHCO3, followed by brine. The organic layer was dried over Na2SO4 and concentrated. The resulting residue was purified by flash chromatography on silica gel, eluting with 50–100% EtOAc/hexane to give 2-(4-fluorophenyl)-5-methoxypyrimidin-4(3H)-one as a white solid (41 mg, 23%); mp 229–231 °C. 1H NMR (400 MHz, DMSO-d6) δ 12.79 (s, 1H), 7.68 (s, 1H), 8.08 (dd, J = 9 Hz, J = 5 Hz, 2H), 7.35–7.30 (m, 2H), 3.79 (s, 3H). 13C NMR (100 MHz, DMSO-d6) δ 163.6 (JC,F = 247 Hz), 158.1, 148.4, 145.4, 130.4, 129.6 (JC,F = 9 Hz), 129.1, 115.5 (JC,F = 22 Hz), 56.0. 19F NMR (376 MHz, DMSO-d6) δ −110.2. HRMS (ESI) calculated for C11H9FN2O2Na (M + Na)+ 243.0540, found 243.0546.
2-(4-Fluorophenyl)-4,5-dimethoxypyrimidine
2-Chloro-4,5-dimethoxypyrimidine (500 mg, 2.86 mmol), (4-fluorophenyl)boronic acid (601 mg, 4.30 mmol), Pd(PPh3)4 (330 mg, 0.29 mmol), and Na2CO3 (910 mg, 8.59 mmol) were dissolved in a mixture of dioxane (12 mL) and water (4 mL). The air was evacuated and replaced with N2. Then, the reaction mixture was refluxed for 5 h. After the reaction was completed, it was cooled to room temperature and it was diluted with EtOAc and washed with satd NH4Cl, followed by brine. The organic layer was dried over Na2SO4 and concentrated under reduced pressure, and the resulting residue was purified by flash chromatography on silica gel, eluting with 0–10% EtOAc/hexane. This afforded 2-(4-fluorophenyl)-4,5-dimethoxypyrimidine as a white solid (123 mg, 77%); mp 114–116 °C. 1H NMR (400 MHz, CDCl3) δ 8.37 (dd, J = 9 Hz, J = 6 Hz, 2H), 8.12 (s, 1H), 7.16–7.12 (m, 2H), 4.17 (s, 3H), 3.98 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 163.1 (JC,F = 248 Hz), 159.7, 155.3, 141.0, 137.2, 133.6 (JC,F = 3 Hz), 129.6 (JC,F = 8 Hz), 115.3 (JC,F = 22 Hz), 56.4, 54.0. 19F NMR (376 MHz, CDCl3) δ −111.9. HRMS (ESI) calculated for C12H12FN2O2 (M + H)+ 235.0877, found 235.0878.
2-Chloro-4,5-dimethoxypyrimidine
2,4-Dichloro-5-methoxypyrimidine (2.37 g, 13.2 mmol) and K2CO3 (1.8 g, 13.2 mmol) were dissolved in MeOH (50 mL) and stirred for 19 h at room temperature. The solvent was removed under reduced pressure. The resulting residue was dissolved in EtOAc and washed with distilled water, followed by brine. The organic layer was dried over Na2SO4 and concentrated. The resulting residue was purified by flash chromatography on silica gel, eluting with 0–20% EtOAc/hexane to give 2-chloro-4,5-dimethoxypyrimidine as a white solid (1.70 g, 73%); mp 65–67 °C. 1H NMR (400 MHz, CDCl3) δ 7.84 (s, 1H), 4.03 (s, 3H), 3.88 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 161.2, 150.0, 141.7, 138.2, 56.6, 55.0.
2-(3-Fluorophenyl)-5-hydroxypyrimidin-4(3H)-one (4)
2-(3-Fluorophenyl)-5-methoxypyrimidin-4(3H)-one (100 mg, 0.45 mmol) was dissolved in anhydrous DCM (5 mL). The reaction mixture was cooled to 0 °C, and then 1 M in DCM BBr3 (4.54 mL, 4.54 mmol) was added. It was then allowed to warm to room temperature and stirred for 18 h. Then the solvent was removed under reduced pressure. The resulting residue was diluted with EtOAc, which was washed with 2 N HCl, followed by brine. The organic layer was dried over Na2SO4, followed by concentration under the vacuum. The resulting residue was purified by flash chromatography on silica gel, eluting with 0–10% MeOH/DCM to give 2-(3-fluorophenyl)-5-hydroxypyrimidin-4(3H)-one as a white solid (87 mg, 93%); mp 210–212 °C. 1H NMR (400 MHz, DMSO-d6) δ 7.89 (d, J = 8 Hz, 1H), 7.84 (dd, J = 10 Hz, J = 2 Hz, 1H), 7.61 (s, 1H), 7.57–7.52 (m, 1H), 7.36 (td, J = 8 Hz, J = 2 Hz, 1H). 13C NMR (100 MHz, DMSO-d6) δ 162.1 (JC,F = 243 Hz), 158.9, 146.5, 143.8, 134.7, 131.5, 130.6 (JC,F = 8 Hz), 123.0, 117.2 (JC,F = 20 Hz), 113.6 (JC,F = 24 Hz). 19F NMR (376 MHz, DMSO-d6) δ −112.5. HRMS (ESI) calculated for C10H8FN2O2 (M + H)+ 207.0564, found 207.0565.
2-(3-Fluorophenyl)-5-methoxypyrimidin-4(3H)-one
2-(3-Fluorophenyl)-4,5-dimethoxypyrimidine (390 mg, 1.67 mmol) was dissolved in a mixture of 2 N HCl (10 mL) and dioxane (10 mL). The reaction mixture was then refluxed for 18 h. It was then cooled to room temperature. The reaction mixture was diluted with EtOAc and washed with water, followed by brine. The organic layer was dried over Na2SO4 and concentrated. The resulting residue was purified by flash chromatography on silica gel, eluting with 0–100% EtOAc/hexane to give 2-(3-fluorophenyl)-5-methoxypyrimidin-4(3H)-one as a white solid (237 mg, 63%); mp 122–124 °C. 1H NMR (400 MHz, DMSO-d6) δ 12.86 (bs, 1H), 7.91 (d, J = 8 Hz, 1H), 7.87–7.83 (m, 1H), 7.71 (s, 1H), 7.58–7.53 (m, 1H), 7.37 (td, J = 8 Hz, J = 2 Hz, 1H), 3.81 (s, 3H). 13C NMR (100 MHz, DMSO-d6) δ 162.1 (JC,F = 242 Hz), 157.9, 147.8, 145.5, 134.7, 130.7 (JC,F = 9 Hz), 130.1, 123.2 (JC,F = 9 Hz), 117.5 (JC,F = 22 Hz), 113.8 (JC,F = 24 Hz), 56.1. 19F NMR (376 MHz, DMSO-d6) δ −112.5. HRMS (ESI) calculated for C11H10FN2O2 (M + H)+ 221.0721, found 227.0722.
2-(3-Fluorophenyl)-4,5-dimethoxypyrimidine
2-Chloro-4,5-dimethoxypyrimidine (300 mg, 1.72 mmol), (3-fluorophenyl)boronic acid (343 mg, 2.58 mmol), Pd(PPh3)4 (199 mg, 0.17 mmol), and Na2CO3 (546 mg, 5.15 mmol) were dissolved in a mixture of dioxane (12 mL) and water (4 mL). The air was evacuated and replaced with N2. Then the reaction mixture was refluxed for 8 h. After the reaction was completed, it was cooled to room temperature and it was diluted with EtOAc and washed with satd NH4Cl, followed by brine. The organic layer was dried over Na2SO4 and concentrated under reduced pressure, and the resulting residue was purified by flash chromatography on silica gel, eluting with 0–10% EtOAc/hexane. This afforded 2-(3-fluorophenyl)-4,5-dimethoxypyrimidine as a white solid (393 mg, 98%); mp 83–85 °C. 1H NMR (400 MHz, CDCl3) δ 8.09 (dt, J = 8 Hz, J = 1 Hz, 1H), 8.03 (s, 1H), 8.01–7.98 (m, 1H), 7.38–7.33 (m, 1H), 7.07 (tdd, J = 8 Hz, J = 3 Hz, J = 1 Hz, 1H), 4.09 (s, 3H), 3.90 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 163.1 (JC,F = 242 Hz), 159.5, 154.6 (JC,F =3 Hz), 141.3, 139.8 (JC,F = 8 Hz), 136.9, 129.7 (JC,F = 8 Hz), 123.1 (JC,F = 3 Hz), 116.4 (JC,F = 22 Hz), 114.3 (JC,F = 23 Hz), 56.2, 53.9. 19F NMR (376 MHz, CDCl3) δ −113.5. HRMS (ESI) calculated for C12H12FN2O2 (M + H)+ 235.0877, found 235.0879.
2-(2-Fluorophenyl)-5-hydroxypyrimidin-4(3H)-one (5)
2-(2-Fluorophenyl)-5-methoxypyrimidin-4(3H)-one (100 mg, 0.45 mmol) was dissolved in anhydrous DCM (5 mL). The reaction mixture was cooled to 0 °C, and then 1 M in DCM BBr3 (4.54 mL, 4.54 mmol) was added. It was then allowed to warm to room temperature and stirred for 18 h. Then the solvent was removed under reduced pressure. The resulting residue was diluted with EtOAc, which was washed with 2 N HCl, followed by brine. The organic layer was dried over Na2SO4, followed by concentration under the vacuum. The resulting residue was purified by flash chromatography on silica gel, eluting with 0–10% MeOH/DCM to give 2-(2-fluorophenyl)-5-hydroxypyrimidin-4(3H)-one as a white solid (89 mg, 96%); mp 187–189 °C. 1H NMR (400 MHz, DMSO-d6) δ 12.80 (bs, 1H), 9.73 (bs, 1H), 7.65 (t, J = 7 Hz, 1H), 7.59 (s, 1H), 7.55 (t, J = 7 Hz, 1H), 7.36–7.30 (m, 2H). 13C NMR (100 MHz, DMSO-d6) δ 159.3 (JC,F = 248 Hz), 158.2, 144.6, 143.9, 132.2 (JC,F = 8 Hz), 132.1, 130.8, 124.5 (JC,F = 3 Hz), 121.8 (JC,F = 12 Hz), 116.1 (JC,F = 21 Hz). 19F NMR (376 MHz, DMSO-d6) δ −115.2. HRMS (ESI) calculated for C10H8FN2O2 (M + H)+ 207.0564, found 207.0565.
2-(2-Fluorophenyl)-5-methoxypyrimidin-4(3H)-one
2-(2-Fluorophenyl)-4,5-dimethoxypyrimidine (400 mg, 1.71 mmol) was dissolved in a mixture of 2 N HCl (10 mL) and dioxane (10 mL). The reaction mixture was then refluxed for 18 h. It was then cooled to room temperature. The reaction mixture was diluted with EtOAc and washed with water, followed by brine. The organic layer was dried over Na2SO4 and concentrated. The resulting residue was purified by flash chromatography on silica gel, eluting with 0–100% EtOAc/hexane, followed by 0–10% MeOH/DCM to give 2-(2-fluorophenyl)-5-methoxypyrimidin-4(3H)-one as a white solid (300 mg, 80%); mp 159–161 °C. 1H NMR (400 MHz, DMSO-d6) δ 12.81 (bs, 1H), 7.70 (s, 1H), 7.66 (t, J = 8 Hz, 1H), 7.60–7.55 (m, 1H), 7.37–7.31 (m, 1H), 3.80 (s, 3H). 13C NMR (100 MHz, DMSO-d6) δ 159.3 (JC,F = 248 Hz), 157.3, 145.9, 132.4 (JC,F = 9 Hz), 130.8 (JC,F = 8 Hz), 130.2, 124.5 (JC,F = 3 Hz), 121.8, 121.6, 116.1 (JC,F = 22 Hz), 56.0. 19F NMR (376 MHz, DMSO-d6) δ −115.2. HRMS (ESI) calculated for C11H10FN2O2 (M + H)+ 221.0721, found 221.0721.
2-(2-Fluorophenyl)-4,5-dimethoxypyrimidine
2-Chloro-4,5-dimethoxypyrimidine (300 mg, 1.72 mmol), (2-fluorophenyl)boronic acid (343 mg, 2.58 mmol), Pd(PPh3)4 (199 mg, 0.17 mmol), and Na2CO3 (546 mg, 5.15 mmol) were dissolved in a mixture of dioxane (12 mL) and water (4 mL). The air was evacuated and replaced with N2. Then, the reaction mixture was refluxed for 18 h. After the reaction was completed, it was cooled to room temperature and it was diluted with EtOAc and washed with satd NH4Cl, followed by brine. The organic layer was dried over Na2SO4 and concentrated under reduced pressure, and the resulting residue was purified by flash chromatography on silica gel, eluting with 0–30% EtOAc/hexane. This afforded 2-(2-fluorophenyl)-4,5-dimethoxypyrimidine as a white solid (402 mg, 100%); mp 72–74 °C. 1H NMR (400 MHz, CDCl3) δ 8.11 (s, 1H), 7.97 (td, J = 8 Hz, J = 2 Hz, 1H), 7.34–7.29 (m, 1H), 7.15 (td, J = 8 Hz, J = 1 Hz, 1H), 7.11–7.06 (m, 1H), 4.06 (s, 3H), 3.90 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 161.0 (JC,F = 253 Hz), 159.5, 154.2 (JC,F = 4 Hz), 140.9, 137.0, 131.4 (JC,F = 2 Hz), 130.9 (JC,F = 8 Hz), 126. 0 (JC,F = 9 Hz), 123.8 (JC,F = 4 Hz), 116.7 (JC,F = 22 Hz), 56.2, 54.1. 19F NMR (376 MHz, CDCl3) δ −114.6. HRMS (ESI) calculated for C12H12FN2O2 (M + H)+ 235.0877, found 235.0877.
2-(4-Biphenyl)-5-hydroxypyrimidin-4(3H)-one (6)
2-(4-Biphenyl)-5-methoxypyrimidin-4(3H)-one (50 mg, 0.18 mmol) was dissolved in anhydrous DCM (5 mL). The reaction mixture was cooled to 0 °C, and then 1 M in DCM BBr3 (1.80 mL, 1.80 mmol) was added. It was then allowed to warm to room temperature and stirred for 18 h. Then the solvent was removed under reduced pressure. The resulting residue was diluted with EtOAc, which was washed with 2 N HCl, followed by brine. The organic layer was dried over Na2SO4, followed by concentration under the vacuum. The resulting residue was purified by flash chromatography on silica gel, eluting with 0–10% MeOH/DCM to give 2-(4-biphenyl)-5-hydroxypyrimidin-4(3H)-one as a white solid (46 mg, 96%); dec 259–261 °C. 1H NMR (400 MHz, DMSO-d6) δ 12.85 (bs, 1H), 9.68 (bs, 1H), 8.13 (d, J = 8 Hz, 2H), 7.80 (d, J = 8 Hz, 2H), 7.75 (d, J = 7 Hz, 2H), 7.62 (s, 1H), 7.50 (t, J = 8 Hz, 2H), 7.41 (t, J = 7 Hz, 1H). 13C NMR (100 MHz, DMSO-d6) δ 159.1, 147.5, 143.5, 141.9, 139.0, 131.7, 131.3, 129.0, 128.0, 128.5, 126.7, 126.7. HRMS (ESI) calculated for C16H13N2O2 (M + H)+ 265.0972, found 265.0973.
2-(4-Biphenyl)-5-methoxypyrimidin-4(3H)-one
2-(4-Biphenyl)-4,5-dimethoxypyrimidine (343 mg, 1.73 mmol) was dissolved in a mixture of 2 N HCl (15 mL) and dioxane (15 mL). The reaction mixture was then refluxed for 18 h and became a white suspension. It was then cooled to room temperature. The suspension was filtered and solid was collected to give 2-(4-biphenyl)-5-methoxypyrimidin-4(3H)-one as white solid (256 mg, 78%); mp 292–294 °C. 1H NMR (400 MHz, DMSO-d6) δ 12.83 (bs, 1H), 8.14 (d, J = 8 Hz, 2H), 7.81 (d, J = 8 Hz, 2H), 7.77–7.72 (m, 3H), 7.50 (t, J = 8 Hz, 2H), 7.42 (t, J = 7 Hz, 1H), 3.81 (s, 3H). 13C NMR (100 MHz, DMSO-d6) δ 158.0, 148.8, 145.6, 142.1, 139.0, 131.2, 130.4, 129.0, 128.0, 127.7, 126.74, 126.70, 56.1. HRMS (ESI) calculated for C17H15N2O2 (M + H)+ 279.1128, found 279.1128.
2-(4-Biphenyl)-4,5-dimethoxypyrimidine
2-Chloro-4,5-dimethoxypyrimidine (400 mg, 2.29 mmol), 4-biphenylboronic acid (681 mg, 3.44 mmol), Pd(PPh3)4 (264 mg, 0.23 mmol), and Na2CO3 (728 mg, 6.87 mmol) were dissolved in a mixture of dioxane (12 mL) and water (4 mL). The air was evacuated and replaced with N2. Then the reaction mixture was refluxed for 8 h. After the reaction was completed, it was cooled to room temperature and it was diluted with EtOAc and washed with satd NH4Cl, followed by brine. The organic layer was dried over Na2SO4 and concentrated under reduced pressure, and the resulting residue was purified by flash chromatography on silica gel, eluting with 0–30% EtOAc/hexane. This afforded 2-(4-biphenyl)-4,5-dimethoxypyrimidine as a white solid (670 mg, 100%); mp 115–117 °C. 1H NMR (400 MHz, CDCl3) δ 8.43 (d, J = 8 Hz, 2H), 8.15 (s, 1H), 7.70 (d, J = 8 Hz, 2H), 7.67 (J = 7 Hz, 2H), 7.47 (t, J = 8 Hz, 2H), 7.37 (t, J = 7 Hz, 1H), 4.19 (s, 3H), 3.98 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 159.7, 156.0. 142.5, 141.1, 140.7, 137.3, 136.4, 128.8, 128.1, 127.5, 126.1, 56.4, 54.0. HRMS (ESI) calculated for C18H17N2O2 (M + H)+ 293.1285, found 293.1286.
2-(3-Biphenyl)-5-hydroxypyrimidin-4(3H)-one (7)
2-(3-Biphenyl)-5-methoxypyrimidin-4(3H)-one (100 mg, 0.36 mmol) was dissolved in anhydrous DCM (5 mL). The reaction mixture was cooled to 0 °C, and the 1 M in DCM BBr3 (3.60 mL, 3.60 mmol) was added. It was then allowed to warm to room temperature and stirred for 6 h. Then the solvent was removed under reduced pressure. The resulting residue was diluted with EtOAc, which was washed with 2 N HCl, followed by brine. The organic layer was dried over Na2SO4, followed by concentration under the vacuum. The resulting residue was purified by flash chromatography on silica gel, eluting with 0–10% MeOH/DCM to give 2-(3-biphenyl)-5-hydroxypyrimidin-4(3H)-one as a white solid (34 mg, 36%); mp 221–223 °C. 1H NMR (400 MHz, DMSO-d6) δ 8.30 (s, 1H), 7.99 (d, J = 7 Hz, 1H), 7.88 (d, J = 8 Hz, 1H), 7.81 (d, J = 8 Hz, 2H), 7.66 (s, 1H), 7.63 (t, J = 8 Hz, 1H), 7.52 (t, J = 8 Hz, 2H), 7.43 (t, J = 7 Hz, 1H). 13C NMR (100 MHz, DMSO-d6) δ 158.9, 147.5, 143.7, 140.4, 139.5, 133.1, 132.3, 129.2, 128.9, 128.5, 127.7, 126.8, 126.0, 125.0. HRMS (ESI) calculated for C16H13N2O2 (M + H)+ 265.0972, found 265.0973.
2-(3-Biphenyl)-5-methoxypyrimidin-4(3H)-one
2-(3-Biphenyl)-4,5-dimethoxypyrimidine (670 mg, 2.29 mmol) was dissolved in a mixture of 2 N HCl (15 mL) and dioxane (15 mL). The reaction mixture was then refluxed for 18 h. It was then cooled to room temperature. The reaction mixture was diluted with EtOAc and washed with water, followed by brine. The organic layer was dried over Na2SO4 and concentrated. The resulting residue was suspended in hot chloroform and was filtered. The solid was collected to give 2-(3-biphenyl)-5-methoxypyrimidin-4(3H)-one as a white solid (493 mg, 77%); mp 209–211 °C. 1H NMR (400 MHz, DMSO-d6) δ 12.97 (br s, 1H), 8.34 (s, 1H), 8.04 (d, J = 8 Hz, 1H), 7.83–7.80 (m, 3H), 7.73 (s, 1H), 7.59 (t, J = 8 Hz, 1H), 7.51 (t, J = 8 Hz, 2H), 7.41 (t, J = 7 Hz, 1H), 3.82 (s, 3H). 13C NMR (100 MHz, DMSO-d6) δ 157.9, 148.9, 145.8, 140.4, 139.4, 132.9, 130.5, 129.3, 128.9, 128.8, 127.8, 126.9, 126.2, 125.2, 56.1. HRMS (ESI) calculated for C17H15N2O2 (M + H)+ 279.1128, found 279.1129.
2-(3-Biphenyl)-4,5-dimethoxypyrimidine
2-Chloro-4,5-dimethoxypyrimidine (400 mg, 2.29 mmol), 3-biphenylboronic acid (681 mg, 3.44 mmol), Pd(PPh3)4 (264 mg, 0.23 mmol), and Na2CO3 (728 mg, 6.87 mmol) were dissolved in a mixture of dioxane (12 mL) and water (4 mL). The air was evacuated and replaced with N2. Then the reaction mixture was refluxed for 8 h. After the reaction was completed, it was cooled to room temperature and it was diluted with EtOAc and washed with satd NH4Cl, followed by brine. The organic layer was dried over Na2SO4 and concentrated under reduced pressure, and the resulting residue was purified by flash chromatography on silica gel, eluting with 0–30% EtOAc/hexane. This afforded 2-(3-biphenyl)-4,5-dimethoxypyrimidine as a white solid (670 mg, 100%); mp 72–74 °C. 1H NMR (400 MHz, CDCl3) δ 8.62 (s, 1H), 8.36 (d, J = 8 Hz, 1H), 8.15 (s, 1H), 7.71 (d, J = 7 Hz, 2H), 7.67 (d, J = 8 Hz, 1H), 7.54 (t, J = 8 Hz, 2H), 7.49–7.45 (m, 3H), 7.37 (t, J = 7 Hz, 1H), 4.18 (s, 3H), 3.97 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 159.7, 156.1, 141.4, 141.17, 141.15, 137.9, 137.2, 128.9, 128.8, 128.6, 127.4, 127.3, 126.6, 126.4, 56.4, 54.0. HRMS (ESI) calculated for C18H17N2O2 (M + H)+ 293.1285, found 293.1286.
2-(2-Biphenyl)-5-hydroxypyrimidin-4(3H)-one (8)
2-(2-Biphenyl)-5-methoxypyrimidin-4(3H)-one (50 mg, 0.18 mmol) was dissolved in anhydrous DCM (5 mL). The reaction mixture was cooled to 0 °C, and the 1 M in DCM BBr3 (1.80 mL, 1.80 mmol) was added. It was then allowed to warm to room temperature and stirred for 18 h. Then the solvent was removed under reduced pressure. The resulting residue was diluted with EtOAc, which was washed with 2 N HCl, followed by brine. The organic layer was dried over Na2SO4, followed by concentration under the vacuum. The resulting residue was purified by flash chromatography on silica gel, eluting with 0–10% MeOH/DCM to give 2-(2-biphenyl)-5-hydroxypyrimidin-4(3H)-one as a white solid (31 mg, 64%); mp 271–273 °C. 1H NMR (400 MHz, DMSO-d6) δ 7.70–7.65 (m, 2H), 7.56 (t, J = 8 Hz, 2H), 7.48 (s, 1H), 7.39 (t, J = 7 Hz, 2H), 7.33 (t, J = 7 Hz, 1H), 7.23 (d, J = 7 Hz, 2H). 13C NMR (100 MHz, DMSO-d6) δ 158.6, 156.9, 148.3, 140.8, 140.5, 139.4, 131.9, 130.4, 130.1, 129.9, 128.6, 128.2, 127.2. HRMS (ESI) calculated for C16H13N2O2 (M + H)+ 265.0972, found 265.0974.
2-(2-Biphenyl)-5-methoxypyrimidin-4(3H)-one
2-(2-Biphenyl)-4,5-dimethoxypyrimidine (309 mg, 1.06 mmol) was dissolved in a mixture of 2 N HCl (15 mL) and dioxane (15 mL). The reaction mixture was then refluxed for 18 h. It was then cooled to room temperature. The reaction mixture was diluted with EtOAc and washed with water, followed by brine. The organic layer was dried over Na2SO4 and concentrated. The resulting residue was purified by flash chromatography on silica gel, eluting with 0–10% MeOH/DCM to give 2-(2-biphenyl)-5-methoxypyrimidin-4(3H)-one as colorless oil (158 mg, 54%). 1H NMR (400 MHz, DMSO-d6) δ 12.51 (bs, 1H), 7.59 (t, J = 8 Hz, 1H), 7.55–7.47 (m, 4H), 7.36 (t, J = 7 Hz, 2H), 7.30 (t, J = 7 Hz, 1H), 7.23 (d, J = 7 Hz, 2H), 3.72 (s, 3H). 13C NMR (100 MHz, DMSO-d6) δ 157.1, 151.0, 145.4, 140.3, 139.8, 133.0, 131.5, 131.4, 130.03, 130.01, 129.8, 128.6, 128.2, 127.2, 55.8. HRMS (ESI) calculated for C17H15N2O2 (M + H)+ 279.1128, found 279.1129.
2-(2-Biphenyl)-4,5-dimethoxypyrimidine
2-Chloro-4,5-dimethoxypyrimidine (500 mg, 2.86 mmol), 2-biphenylboronic acid (851 mg, 4.30 mmol), Pd(PPh3)4 (266 mg, 0.29 mmol), and Na2CO3 (909 mg, 8.58 mmol) were dissolved in a mixture of dioxane (21 mL) and water (7 mL). The air was evacuated and replaced with N2. Then the reaction mixture was refluxed for 8 h. After the reaction was completed, it was cooled to room temperature and it was diluted with EtOAc and washed with satd NH4Cl, followed by brine. The organic layer was dried over Na2SO4 and concentrated under reduced pressure, and the resulting residue was purified by flash chromatography on silica gel, eluting with 0–30% EtOAc/hexane. This afforded 2-(2-biphenyl)-4,5-dimethoxypyrimidine as colorless oil (377 mg, 45%). 1H NMR (400 MHz, DMSO-d6) δ 8.26 (s, 1H), 7.84 (dd, J = 7 Hz, J = 1 Hz, 1H), 7.54–7.45 (m, 2H), 7.40 (dd, J = 7 Hz, J = 1 Hz, 1H), 7.30–7.24 (m, 3H), 7.08 (d, J = 7 Hz, 2H), 3.85 (s, 3H), 3.30 (s, 3H). 13C NMR (100 MHz, DMSO-d6) 158.1, 157.2, 142.2, 141.1, 140.0, 137.7, 137.3, 130.4, 130.2, 128.9, 128.6, 127.8, 127.2, 126.2, 56.0, 52.9. HRMS (ESI) calculated for C18H17N2O2 (M + H)+ 293.1285, found 293.1290.
4-(5-Hydroxy-6-oxo-1,6-dihydropyrimidin-2-yl)benzonitrile (9)
4-(5-Methoxy-6-oxo-1,6-dihydropyrimidin-2-yl)benzonitrile (50 mg, 0.22 mmol) was dissolved in anhydrous DCM (5 mL). The reaction mixture was cooled to 0 °C, and then 1 M in DCM BBr3 (2.2 mL, 2.2 mmol) was added. It was then allowed to warm to room temperature and stirred for 18 h. Then the solvent was removed under reduced pressure. The resulting residue was suspended in water. It was filtered, and the solid was collected and dried under vacuum to provide 4-(5-hydroxy-6-oxo-1,6-dihydropyrimidin-2-yl)benzonitrile as a white solid (15 mg, 32%); mp 324–326 °C. 1H NMR (400 MHz, DMSO-d6) δ 13.05 (bs, 1H), 9.91 (bs, 1H), 8.18 (d, J = 8 Hz, 2H), 7.95 (d, J = 8 Hz, 2H), 7.64 (s, 1H). 13C NMR (100 MHz, DMSO-d6) δ 158.8, 132.5, 129.4, 127.6, 127.2, 127.1, 126.7, 118.4, 112.5. HRMS (ESI) calculated for C11H8N3O2 (M + H)+ 214.0611, found 214.0618.
4-(5-Methoxy-6-oxo-1,6-dihydropyrimidin-2-yl)benzonitrile
4-(4,5-Dimethoxypyrimidin-2-yl)benzonitrile (53 mg, 0.22 mmol) was dissolved in a mixture of 2 N HCl (5 mL) and dioxane (5 mL). The reaction mixture was then refluxed for 5 h. It was then cooled to room temperature. The reaction mixture was diluted with EtOAc and washed with satd NaHCO3, followed by brine. The organic layer was dried over Na2SO4 and concentrated. The resulting residue was purified by flash chromatography on silica gel, eluting with 0–5% MeOH/DCM to give 4-(5-methoxy-6-oxo-1,6-dihydropyrimidin-2-yl)benzonitrile as a white solid (50 mg, 100%); mp 297–299 °C. 1H NMR (400 MHz, DMSO-d6) δ 12.99 (bs, 1H), 8.19 (d, J = 8 Hz, 2H), 7.96 (d, J = 8 Hz, 2H), 7.75 (s, 1H), 3.81 (s, 3H). 13C NMR (100 MHz, DMSO-d6) 157.8, 147.4, 146.2, 136.5, 132.5, 130.0, 127.8, 118.4, 112.8, 56.1. HRMS (ESI) calculated for C12H10N3O2 (M + H)+ 228.0768, found 228.0770.
4-(4,5-Dimethoxypyrimidin-2-yl)benzonitrile
2-Chloro-4,5-dimethoxypyrimidine (100 mg, 0.57 mmol), (4-cyanophenyl)boronic acid (126 mg, 0.86 mmol), Pd(PPh3)4 (66 mg, 0.06 mmol), and Na2CO3(182 mg, 1.72 mmol) were dissolved in a mixture of dioxane (9 mL) and water (3 mL). The air was evacuated and replaced with N2. Then the reaction mixture was refluxed for 4 h. After the reaction was completed, it was cooled to room temperature and it was diluted with EtOAc and washed with satd NH4Cl, followed by brine. The organic layer was dried over Na2SO4 and concentrated in vacuo, and the resulting residue was purified by flash chromatography on silica gel, eluting with 0–20% EtOAc/hexane. This afforded 4-(4,5-dimethoxypyrimidin-2-yl)benzonitrile as a white solid (69 mg, 74%); mp 170–172 °C. 1H NMR (400 MHz, CDCl3) δ 8.49 (d, J = 9 Hz, 2H), 8.18 (s, 1H), 7.76 (d, J = 9 Hz, 2H), 4.20 (s, 3H), 4.02 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 159.8, 154.0, 141.8, 141.5, 137.0, 132.3, 128.0, 119.0, 113.0, 56.4, 54.2. HRMS (ESI) calculated for C13H12N3O2 (M + H)+ 242.0924, found 242.0929.
3-(5-Hydroxy-6-oxo-1,6-dihydropyrimidin-2-yl)benzonitrile (10)
3-(5-Methoxy-6-oxo-1,6-dihydropyrimidin-2-yl)benzonitrile (50 mg, 0.22 mmol) was dissolved in anhydrous DCM (5 mL). The reaction mixture was cooled to 0 °C, and then 1 M in DCM BBr3 (2.2 mL, 2.2 mmol) was added. It was then allowed to warm to room temperature and stirred for 18 h. Then the solvent was removed under reduced pressure. The resulting residue was suspended in water. It was filtered, and the solid was collected and dried under vacuum to provide 3-(5-hydroxy-6-oxo-1,6-dihydropyrimidin-2-yl)benzonitrile as a white solid (27 mg, 58%); mp 294–296 °C. 1H NMR (400 MHz, DMSO-d6) δ 8.44 (s, 1H), 8.35 (d, J = 8 Hz, 1H), 7.93 (d, J = 8 Hz, 1H), 7.68 (t, J = 8 Hz, 1H), 7.61 (s, 1H). 13C NMR (100 MHz, DMSO-d6) δ 159.3, 146.4, 143.9, 134.3, 133.4, 131.9, 131.5, 130.4, 129.8, 118.4, 111.7. HRMS (ESI) calculated for C11H8N3O2 (M + H)+ 214.0611, found 214.0613.
3-(5-Methoxy-6-oxo-1,6-dihydropyrimidin-2-yl)benzonitrile
3-(4,5-Dimethoxypyrimidin-2-yl)benzonitrile (138 mg, 0.57 mmol) was dissolved in a mixture of 2 N HCl (5 mL) and dioxane (5 mL). The reaction mixture was then refluxed for 6 h. It was then cooled to room temperature. The reaction mixture was diluted with EtOAc and washed with satd NaHCO3, followed by brine. The organic layer was dried over Na2SO4 and concentrated. The resulting residue was purified by flash chromatography on silica gel, eluting with 0–10% MeOH/DCM to give 3-(5-methoxy-6-oxo-1,6-dihydropyrimidin-2-yl)benzonitrile as a white solid (56 mg, 43%); mp 255–257 °C. 1H NMR (400 MHz, DMSO-d6) δ 12.93 (bs, 1H), 8.44 (s, 1H), 8.34 (d, J = 8 Hz, 1H), 7.99 (d, J = 8 Hz, 1H), 7.74–7.70 (m, 2H), 3.82 (s, 3H). 13C NMR (100 MHz, DMSO-d6) δ 157.9, 147.5, 145.9, 133.9, 133.7, 131.7, 130.7, 130.4, 129.9, 118.3, 111.7, 56.1. HRMS (ESI) calculated for C12H10N3O2 (M + H)+ 228.0768, found 228.0767.
3-(4,5-Dimethoxypyrimidin-2-yl)benzonitrile
2-Chloro-4,5-dimethoxypyrimidine (100 mg, 0.57 mmol), (3-cyanophenyl)boronic acid (126 mg, 0.86 mmol), Pd(PPh3)4 (66 mg, 0.06 mmol), and Na2CO3(182 mg, 1.72 mmol) were dissolved in a mixture of dioxane (9 mL) and water (3 mL). The air was evacuated and replaced with N2. Then the reaction mixture was refluxed for 5 h. After the reaction was completed, it was cooled to room temperature and it was diluted with EtOAc and washed with satd NH4Cl, followed by brine. The organic layer was dried over Na2SO4 and concentrated in vacuo, and the resulting residue was purified by flash chromatography on silica gel, eluting with 0–20% EtOAc/hexane. This afforded 3-(4,5-dimethoxypyrimidin-2-yl)benzonitrile as a white solid (138 mg, 100%); mp 134–136 °C. 1H NMR (400 MHz, CDCl3) δ 8.61 (s, 1H), 8.54 (d, J = 8 Hz, 1H), 8.09 (s, 1H), 7.65 (d, J = 8 Hz, 1H), 7.51 (t, J = 8 Hz, 1H), 4.13 (s, 3H), 3.95 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 159.8, 153.6, 141.7, 138.5, 137.0, 132.8, 131.6, 131.3, 129.2, 118.9, 112.6, 56.4, 54.2. HRMS (ESI) calculated for C13H12N3O2 (M + H)+ 242.0924, found 242.0928.
2-(4-(1H-Tetrazol-5-yl)phenyl)-5-hydroxypyrimidin-4(3H)-one (11)
4-(5-Hydroxy-6-oxo-1,6-dihydropyrimidin-2-yl)benzonitrile (68 mg, 0.32 mmol) and NaN3 (79 mg, 1.21 mmol) were dissolved in anhydrous DMF (1 mL). The reaction mixture was treated with 2 drops of acetic acid. It was sealed, and then it was heated at 130 °C for 17 h. The reaction was cooled to room temperature and gave a brownish suspension. DMF was removed by Kugelrohr distillation. The resulting residue was suspended in water and filtered to give 2-(4-(1H-tetrazol-5-yl)phenyl)-5-hydroxypyrimidin-4(3H)-one as a dark-brown solid (42 mg, 51%); dec 290–292 °C. 1H NMR (400 MHz, DMSO-d6) δ 8.08 (d, J = 9 Hz, 2H), 8.03 (d, J = 8 Hz, 2H), 7.55 (s, 1H). 13C NMR (100 MHz, DMSO-d6) δ 161.0, 160.2, 148.9, 143.3, 133.8, 132.4, 131.6, 126.9, 125.5. HRMS (ESI) calculated for C11H9N6O2 (M + H)+ 257.0781, found 257.0791.
2-(3-(1H-Tetrazol-5-yl)phenyl)-5-hydroxypyrimidin-4(3H)-one (12)
3-(5-Hydroxy-6-oxo-1,6-dihydropyrimidin-2-yl)benzonitrile (108 mg, 0.50 mmol) and NaN3 (131 mg, 2.02 mmol) were dissolved in anhydrous DMF (1 mL). The reaction mixture was treated with 2 drops of acetic acid. It was sealed, and then it was heated at 130 °C for 18 h. The reaction was cooled to room temperature and gave a brownish suspension. It was filtered, and a greenish solid was obtained. The greenish solid was suspended in 2 N HCl, followed by second filtration to provide 2-(3-(1H-tetrazol-5-yl)phenyl)-5-hydroxypyrimidin-4(3H)-one as a beige solid (32 mg, 25%); dec 295–297 °C. 1H NMR (400 MHz, DMSO-d6) δ 8.67 (s, 1H), 8.11 (d, J = 8 Hz, 1H), 7.97 (d, J = 8 Hz, 1H), 7.63 (s, 1H), 7.56 (t, J = 8 Hz, 1H). 13C NMR (100 MHz, DMSO-d6) δ162.3, 159.1, 158.7, 148.0, 143.4, 133.1, 130.4, 128.9, 127.9, 126.5, 125.1. HRMS (ESI) calculated for C11H9N6O2 (M + H)+ 257.0781, found 257.0785.
5-(4-Fluorophenyl)-4-hydroxypyridazin-3(2H)-one (13)
5-(4-Fluorophenyl)-4-methoxy-2-(methoxymethyl)pyridazin-3(2H)-one (55 mg, 0.21 mmol) was dissolved in anhydrous DCM (10 mL). The reaction mixture was cooled to 0 °C, and then 1 M in DCM BBr3 (2.1 mL, 2.1 mmol) was added. It was then allowed to warm to room temperature and stirred for 24 h. Then the solvent was removed under reduced pressure. This gave 5-(4-fluorophenyl)-4-methoxypyridazin-3(2H)-one, which was again recharged with 1 M in DCM BBr3 (2.1 mL, 2.1 mmol). The reaction mixture was stirred for 24 h at room temperature. Then the solvent was again removed under reduced pressure. The resulting residue was suspended with water and filtered. The filtered solid was purified by flash chromatography on silica gel, eluting with 0–10% MeOH/DCM. This gave 5-(4-fluorophenyl)-4-hydroxypyridazin-3(2H)-one as a white solid (6.2 mg, 14%); mp 274–276 °C. 1H NMR (400 MHz, DMSO-d6) δ 12.70 (s, 1H), 7.76 (s, 1H), 7.58 (J = 8 Hz, J = 5 Hz, 2H), 7.21–7.17 (m, 2H). 13C NMR (100 MHz, DMSO-d6) δ 161.2 (JC,F = 243 Hz), 161.9, 154.6, 132.7, 132.4 (JC,F = 8 Hz), 127.2 (JC,F = 4 Hz), 115.8, 114.2 (JC,F = 21 Hz). 19F NMR (376 MHz, DMSO-d6) δ −114.1. HRMS (ESI) calculated for C10H8FN2O2 (M + H)+ 207.0564, found 207.0575.
5-(4-Fluorophenyl)-4-methoxy-2-(methoxymethyl)pyridazin-3(2H)-one
5-Chloro-4-methoxy-2-(methoxymethyl)pyridazin-3(2H)-one (58 mg, 0.28 mmol), (4-fluorophenyl)boronic acid (140 mg, 0.43 mmol), Pd(PPh3)4 (32 mg, 0.028 mmol), and Na2CO3 (90 mg, 0.85 mmol) were dissolved in a mixture of dioxane (9 mL) and water (3 mL). The air was evacuated and replaced with N2. Then the reaction mixture was refluxed for 14 h. After the reaction was completed, it was cooled to room temperature, and it was diluted with EtOAc and washed with satd NH4Cl, followed by brine. The organic layer was dried over Na2SO4 and concentrated in vacuo, and the resulting residue was purified by flash chromatography on silica gel, eluting with 0–30% EtOAc/hexane. This afforded 5-(4-fluorophenyl)-4-methoxy-2-(methoxymethyl)pyridazin-3(2H)-one as a white solid (56 mg, 74%); mp 123–125 °C. 1H NMR (400 MHz, CDCl3) δ 7.95 (s, 1H), 7.53 (dd, J = 9 Hz, J = 6 Hz, 2H), 7.11–7. 07 (m, 2H), 5.48 (s, 2H), 3.93 (s, 3H), 3.49 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 162.6 (JC,F = 247 Hz), 161.6, 154.8, 132.3 (JC,F = 8 Hz), 128.5, 125.7, 120.7, 112.9 (JC,F = 22 Hz), 81.6, 57.9, 57.3. 19F NMR (376 MHz, CDCl3) δ −112.6. HRMS (ESI) calculated for C13H14FN2O3 (M + H)+ 265.0983, found 265.0992.
5-Chloro-4-methoxy-2-(methoxymethyl)pyridazin-3(2H)-one
4,5-Dichloro-2-(methoxymethyl)pyridazin-3(2H)-one (84 mg, 0.40 mmol) was added to MeOH (10 mL). Then the reaction mixture was cooled to 0 °C. It was treated with NaOMe (24 mg, 0.44 mmol) and allowed to warm to room temperature. The reaction mixture was then stirred for 18 h at room temperature. After the reaction was completed, it was put under the vacuum to remove MeOH. The resulting residue was diluted with EtOAc, which was then washed with satd NH4Cl, followed by brine. The organic layer was dried over Na2SO4 and then concentrated. The residue was purified by flash chromatography on silica gel, eluting with 0–30% EtOAc/hexane to provide 5-chloro-4-methoxy-2-(methoxymethyl)pyridazin-3(2H)-one as a white solid (59 mg, 72%); mp 101–103 °C. 1H NMR (400 MHz, CDCl3) δ 7.85 (s, 1H), 5.45 (s, 2H), 4.08 (s, 3H), 3.44 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 159.1, 155.1, 127.1, 116.9, 81.9, 57.9, 57.8. HRMS (ESI) calculated for C7H10ClN2O3 (M + H)+ 205.0374, found 205.0384.
4,5-Dichloro-2-(methoxymethyl)pyridazin-3(2H)-one
4,5-Dichloropyridazin-3(2H)-one (200 mg, 1.21 mmol) and 4-dimethylaminopyridine (15 mg, 0.12 mmol) were dissolved in anhydrous DCM (20 mL). Then the reaction mixture was cooled to 0 °C and treated with NEt3 (0.29 mL, 1.70 mmol), followed by MOMCl (0.110 mL, 1.454 mmol). The reaction mixture was allowed to warm to room temperature and stirred for 17 h. It was then poured into DCM, and it was washed with satd NH4Cl, followed by brine. The organic layer was dried over Na2SO4, which was concentrated. The resulting residue was purified by flash chromatography on silica gel, eluting with 0–30% EtOAc/hexane to provide 4,5-dichloro-2-(methoxymethyl)pyridazin-3(2H)-one as a white solid (84 mg, 33%); mp 65–67 °C. 1H NMR (400 MHz, CDCl3) δ 7.78 (s, 1H), 5.41 (s, 2H), 3.43 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 156.9, 137.0, 136.0, 134.9, 82.4, 58.1.
6-(4-Fluorophenyl)-4-hydroxypyridazin-3(2H)-one (14)
6-(4-Fluorophenyl)-4-methoxypyridazin-3(2H)-one (16 mg, 0.074 mmol) was dissolved in anhydrous DCM (5 mL). The reaction mixture was cooled to 0 °C, and then 1 M in DCM BBr3 (0.74 mL, 0.74 mmol) was added. It was then allowed to warm to room temperature and stirred for 36 h. Then the solvent was removed under reduced pressure. The resulting residue was suspended in water. It was filtered, and the solid was collected and dried under vacuum to provide 6-(4-fluorophenyl)-4-hydroxypyridazin-3(2H)-one as a white solid (5 mg, 35%); mp 281–283 °C. 1H NMR (400 MHz, DMSO-d6) δ 13.10 (bs, 1H), 11.02 (bs, 1H), 7.87 (dd, J = 9 Hz, J = 5 Hz, 2H), 7.30–7.26 (m,2H), 7.19 (s, 1H). 13C NMR (100 MHz, DMSO-d6) δ 162.6 (JC,F = 245 Hz), 157.6, 155.4, 145.2, 132.0 (JC,F = 3 Hz), 128.0 (JC,F = 9 Hz), 115.6 (JC,F = 22 Hz), 106.0. 19F NMR (376 MHz, DMSO-d6) δ −113.0. HRMS (ESI) calculated for C10H8FN2O2 (M + H)+ 207.0564, found 207.0567.
6-(4-Fluorophenyl)-4-methoxypyridazin-3(2H)-one
6-(4-Fluorophenyl)-3,4-dimethoxypyridazine (122 mg 0.52 mmol) was dissolved in a mixture of 2 N HCl (5 mL) and dioxane (5 mL). The reaction mixture was then refluxed for 11 h. It was then cooled to room temperature. The reaction mixture was diluted with EtOAc and washed with satd NaHCO3, followed by brine. The organic layer was dried over Na2SO4 and concentrated. The resulting residue was purified by flash chromatography on silica gel, eluting with 0–10% MeOH/DCM to give 6-(4-fluorophenyl)-4-methoxypyridazin-3(2H)-one as a white solid (41 mg, 36%); mp 211–213 °C. 1H NMR (400 MHz, DMSO-d6) δ 13.05 (br s, 1H), 7.96 (dd, J = 9 Hz, J = 6 Hz, 2H), 7.34–7.29 (m, 2H), 7.28 (s, 1H), 3.92 (s, 3H). 13C NMR (100 MHz, DMSO-d6) δ 162.7 (JC,F =245 Hz), 156.2, 155.7, 144.3, 132.0 (JC,F = 3 Hz), 128.2 (JC,F = 8 Hz), 115.6 (JC,F = 22 Hz), 104.4, 56.2. 19F NMR (376 MHz, DMSO-d6) δ −112.9. HRMS (ESI) calculated for C11H10FN2O2 (M + H)+ 221.0721, found 221.0727.
6-(4-Fluorophenyl)-3,4-dimethoxypyridazine
6-Chloro-3,4-dimethoxypyridazine (101 mg, 0.58 mmol), (4-fluorophenyl)boronic acid (122 mg, 0.87 mmol), Pd(PPh3)4 (67 mg, 0.06 mmol), and Na2CO3(184 mg, 1.74 mmol) were dissolved in a mixture of dioxane (15 mL) and water (5 mL). The air was evacuated and replaced with N2. Then the reaction mixture was refluxed for 3 h. After the reaction was completed, it was cooled to room temperature and it was diluted with EtOAc and washed with satd NH4Cl, followed by brine. The organic layer was dried over Na2SO4 and concentrated under reduced pressure, and the resulting residue was purified by flash chromatography on silica gel, eluting with 0–20% EtOAc/hexane. This afforded 6-(4-fluorophenyl)-3,4-dimethoxypyridazine as a white solid (109 mg, 80%); mp 103–105 °C. 1H NMR (400 MHz, CDCl3) δ 7.88 (dd, J = 9 Hz, J = 5 Hz, 2H), 7.10–7. 06 (m, 2H), 7.00 (s, 1H), 4.14 (s, 3H), 3.93 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 163.7 (JC,F = 247 Hz), 156.5, 155.7, 148.9, 132.9 (JC,F = 3 Hz), 128.5 (JC,F = 9 Hz), 115.8 (JC,F = 21 Hz), 104.7, 55.7, 55.2. 19F NMR (376 MHz, CDCl3) δ −112.2. HRMS (ESI) calculated for C12H12FN2O2 (M + H)+ 235.0877, found 235.0885.
6-Chloro-3,4-dimethoxypyridazine
3,4,6-Trichloropyridazine (200 mg, 1.09 mmol) was added to MeOH (15 mL). Then the reaction mixture was cooled to 0 °C. It was treated with NaOMe (117 mg, 2.17 mmol) and allowed to warm to room temperature. The reaction mixture was then stirred for 10 h at room temperature. After the reaction was completed, it was put under the vacuum to remove MeOH. The resulting residue was diluted with EtOAc, which was then washed with satd NH4Cl, followed by brine. The organic layer was dried over Na2SO4 and then concentrated. The residue was purified by flash chromatography on silica gel, eluting with 0–30% EtOAc/hexane to provide 4-chloro-5,6-dimethoxypyrimidine as a white solid (101 mg, 53%); mp 117–119 °C. 1H NMR (400 MHz, CDCl3) δ 6.71 (s, 1H), 4.08 (s, 3H), 3.88 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 156.9, 151.1, 149.8, 108.4, 56.2, 55.3.
4-(5-Hydroxy-6-oxo-1,6-dihydropyridazin-3-yl)benzonitrile (15)
4-(5-Methoxy-6-oxo-1,6-dihydropyridazin-3-yl)benzonitrile (109 mg, 0.48 mmol) was dissolved in anhydrous DCM (5 mL). The reaction mixture was cooled to 0 °C, and then 1 M in DCM BBr3 (4.8 mL, 4.8 mmol) was added. It was then allowed to warm to room temperature and stirred for 20 h. Then the solvent was removed under reduced pressure. The resulting residue was suspended in water. It was filtered, and the solid was collected. The solid was dry loaded on silica gel and was purified by flash chromatography on silica gel, eluting with 0–10% MeOH/DCM. This afforded 4-(5-hydroxy-6-oxo-1,6-dihydropyridazin-3-yl)benzonitrile as a white solid (75 mg, 73%); mp 305–307 °C. 1H NMR (400 MHz, DMSO-d6) δ 13.29 (s, 1H), 11.15 (bs, 1H), 8.03 (d, J = 9 Hz, 2H), 7.92 (d, J = 9 Hz, 2H), 7.29 (s, 1H). 13C NMR (100 MHz, DMSO-d6) δ 157.7, 154.6, 144.4, 139.8, 132.7, 126.5, 118.6, 111.4, 106.0. HRMS (ESI) calculated for C11H8N3O2 (M + H)+ 214.0611, found 214.0615.
4-(5-Methoxy-6-oxo-1,6-dihydropyridazin-3-yl)benzonitrile
4-(5,6-Dimethoxypyridazin-3-yl)benzonitrile (125 mg, 0.52 mmol) was dissolved in a mixture of 2 N HCl (5 mL) and dioxane (5 mL). The reaction mixture was then refluxed for 4 h. It was then cooled to room temperature and then put under reduced pressure. The resulting residue was suspended in water and filtered. The product 4-(5-methoxy-6-oxo-1,6-dihydropyridazin-3-yl)benzonitrile was collected as a white solid (82 mg, 70%); mp 271–273 °C. 1H NMR (400 MHz, DMSO-d6) δ 13.97 (bs, 1H), 8.02 (d, J = 8 Hz, 2H), 7.94 (d, J = 8 Hz, 2H), 6.62 (s, 1H), 3.96 (s, 3H). 13C NMR (100 MHz, DMSO-d6) δ 156.3, 155.9, 143.5, 139.7, 132.7, 126.7, 118.6, 111.4, 104.3, 56.3. HRMS (ESI) calculated for C12H10N3O2 (M + H)+ 228.0768, found 228.0774.
4-(5,6-Dimethoxypyridazin-3-yl)benzonitrile
6-Chloro-3,4-dimethoxypyridazine (298 mg, 1.71 mmol), (4-cyanophenyl)boronic acid (376 mg, 2.56 mmol), Pd(PPh3)4 (198 mg, 0.17 mmol), and Na2CO3(543 mg, 5.12 mmol) were dissolved in a mixture of dioxane (15 mL) and water (5 mL). The air was evacuated and replaced with N2. Then the reaction mixture was refluxed for 15 h. After the reaction was completed, it was cooled to room temperature and it was diluted with EtOAc and washed with satd NH4Cl, followed by brine. The organic layer was dried over Na2SO4 and concentrated under reduced pressure, and the resulting residue was purified by flash chromatography on silica gel, eluting with 0–30% EtOAc/hexane. This afforded 4-(5,6-dimethoxypyridazin-3-yl)benzonitrile as a white solid (130 mg, 31%); mp 187–189 °C. 1H NMR (400 MHz, CDCl3) δ 8.10 (d, J = 8 Hz, 2H), 7.77 (d, J = 8 Hz, 2H), 7.14 (s, 1H), 4.23 (s, 3H), 4.03 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 157.1, 154.7, 149.1, 140.9, 132.6, 127.3, 118.5, 113.0, 104.9, 55.9, 55.3. HRMS (ESI) calculated for C13H12N3O2 (M + H)+ 242.0924, found 242.0931.
6-(4-(1H-Tetrazol-5-yl)phenyl)-4-hydroxypyridazin-3(2H)-one (16)
4-(5-Methoxy-6-oxo-1,6-dihydropyridazin-3-yl)benzonitrile (56 mg, 0.26 mmol) and NaN3 (69 mg, 1.06 mmol) were dissolved in anhydrous DMF (1 mL). The reaction mixture was treated with 2 drops of acetic acid. It was sealed, and then it was heated at 130 °C for 21 h. The reaction was cooled to room temperature and gave a brownish suspension. The suspension was filtered and gave a white solid, which was treated with 2 N HCl and filtered again. This afforded 6-(4-(1H-tetrazol-5-yl)phenyl)-4-hydroxypyridazin-3(2H)-one as a white solid (33 mg, 48%); dec 273–275 °C. 1H NMR (400 MHz, DMSO-d6) δ 11.13 (s, 1H), 8.09 (d, J = 8 Hz, 2H), 7.95 (d, J = 7 Hz, 2H), 7.25 (s, 1H). 13C NMR (100 MHz, DMSO-d6) δ 158.0, 157.7, 154.9, 145.7, 135.8, 128.9, 126.6, 126.2, 105.9. HRMS (ESI) calculated for C11H9N6O2 (M + H)+ 257.0781, found 257.0791.
Expression, Purification, and Crystallization
Pandemic H1N1 endonuclease (residues 1–204) was expressed and purified as described previously.13 The BL21 (RIL) cells (Stratagene) were grown to an OD600 of 0.8 and induced with 0.15 mM IPTG at 17 °C for 17 h. Cells were harvested by centrifugation and purified on Ni-NTA (Qiagen) according to manufacturers recommendations, except the final elution was completed with 500 mM imidazole containing buffer. The dual hexahis tag was then removed by HRV14 3C protease cleavage. The endonuclease was further purified by size exclusion chromatography using HiLoad 26/60 Superdex 75 (GE Healthcare). The buffer used for size exclusion and the final buffer for storage of the protein was 100 mM NaCl and 20 mM Tris pH 8.0. The protein was concentrated to 5 mg/mL using an Ultrafree 10K (Millipore), aliquoted, and stored at −80 °C.
Crystals are formed by mixing in a 1:1 ratio endonuclease (5 mg/mL) with crystallization buffer containing 200 mM MES pH 6.7, 27% PEG8000, 200 mM ammonium sulfate, 1 mM manganese chloride, 10 mM magnesium acetate, 10 mM taurine, and 50 mM sodium fluoride. Crystals form within a few hours and grow to maximum size in 1–2 weeks at 20 °C.
Compound Soaking, Data Collection, and Processing
Crystal structures of compounds 11 were determined in complex with endonuclease as previously described.1111 was soaked into preformed crystals by stepwise gradient shifting the surrounding crystallization solution to 1 mM manganese sulfate, 200 mM HEPES pH 7.7, 25% (w/v) PEG 8000, 50 mM ammonium sulfate, 5 mM magnesium acetate, 80 mM l-arginine, 10% DMSO containing 100 mM 11, and 10% (v/v) ethylene glycol. Crystals were then soaked with the ligand for 2 h at 20 °C before being placed into liquid nitrogen for storage. X-ray diffraction data collection was performed at the Cornell High Energy Synchrotron Source (CHESS) F1 beamline. The diffraction data were indexed, processed, scaled, and merged using HKL2000.32 The structure was solved and refined using the software PHENIX.33
Endonuclease Assay
A high throughput endonuclease assay was performed as described previously.11 The endonuclease assay used a single-stranded DNA probe with the sequence (6-FAM)TGGCAATATCAGCTCCACA(MGBNFQ) (Applied Biosystems). Reaction buffer contained 50 mM Tris pH 7.5, 50 mM sodium chloride, 1 mM DTT, 5 mM magnesium chloride, 0.5 mM manganese sulfate, and 1 mM CHAPS. DNA probe (50 nM), endonuclease (25 nM), and compound were mixed on ice and then placed in a Varioskan fluorimeter (Thermo Scientific) preincubated at 37 °C to detect fluorescence with an excitation of 488 nm and emission of 518 nm. Fluorescence is measured at various time points, and activity/inhibition is calculated with GraphPad Prism 6.0b.
Acknowledgments
The Bruker Avance III 400 MHz NMR spectrometer used for this study was purchased with funds from NCRR Grant no. 1S10RR23698-1A1. Mass spectrometry was provided by the Washington University Mass Spectrometry Resource with support from the NIH National Center for Research Resources grant no. P41RR0954. We thank the laboratories of Ann Stock and Gaetano Montelione for access to equipment used in this study. E.A.’s laboratory is grateful for support from NIH grants R37 AI027690 (MERIT AWARD) and P50 GM103368. X-ray data collection was conducted at the Cornell High Energy Synchrotron Source (CHESS). CHESS is supported by the NSF and NIH/NIGMS via NSF award DMR-0225180, and the MacCHESS resource is supported by NIH/NCRR award RR-01646.
Supporting Information Available
X-ray data and refinement statistics for the analysis performed with 11. The atomic coordinates and structure factors are deposited in the Protein Data Bank (PDB) with ID 4W9S. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare the following competing financial interest(s): Prof. LaVoie is a co-founder, together with Dr. Bauman, Dr. Das and Prof. Arnold, of Prodaptics Pharmaceuticals, Inc..
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Hayden F. G.; Hay A. J. Emergence and transmission of influenza A viruses resistant to amantadine and rimantadine. Curr. Top. Microbiol. Immunol. 1992, 176, 119–130. [DOI] [PubMed] [Google Scholar]
- Bright R. A.; Medina M.; Xu X.; Peresz-Oronoz G.; Wallis T. R.; Davis X. M.; Provinelli L.; Cox N. J.; Klimov A. I. Incidence of admantane resistance among influenza A (H3N2) viruses isolated worldwide from 1994 to 2005: a cause for concern. Lancet 2005, 366, 1175–1181. [DOI] [PubMed] [Google Scholar]
- Moscona A. Oseltamivir Resistance—Disabling our Influenza Defenses. N. Engl. J. Med. 2005, 353, 2633–2636. [DOI] [PubMed] [Google Scholar]
- Bloom J. D.; Gong L. I.; Baltimore D. Permissive secondary mutations enable the evolution of influenza oseltamivir resistance. Science 2010, 328, 1272–1275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Memoli M. J.; Davis A. S.; Proudfoot K.; Chertow D. S.; Hrabal R. J.; Bristol T.; Taubenberger J. K. Multidrug-resistant 2009 pandemic influenza A(H1N1) viruses maintain fitness and transmissibility in ferrets. J. Infect Dis. 2011, 203, 348–357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das K.; Aramini J. M.; Ma L.-C.; Krug R. M.; Arnold E. Structures of influenza A proteins and insights into antiviral drug targets. Nature Struct Mol. Biol. 2010, 17, 530–538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He X.; Zhou J.; Bartlam M.; Zhang R.; Ma J.; Lou Z.; Li X.; Li J.; Joachimiak A.; Zeng Z.; Ge R.; Rao Z.; Liu Y. Crystal structure of the polymerase PAC–PB1N complex from an avian influenza H5N1 virus. Nature 2008, 454, 1123–1126. [DOI] [PubMed] [Google Scholar]
- Yan P.; Bartlam M.; Lou Z.; Chen S.; Zhou J.; He X.; Lv Z.; Ge R.; Li X.; Deng T.; Fodor E.; Rao Z.; Liu Y. Crystal structure of an avian influenza polymerase PAN reveals an endonuclease active site. Nature 2009, 458, 909–913. [DOI] [PubMed] [Google Scholar]
- Dias A.; Bouvier D.; Crépin T.; McCarthy A. A.; Hart D. J.; Baudin F.; Cusack S.; Ruigrok R. W. H. The cap-snatching endonuclease of influenza virus polymerase resides in the PA subunit. Nature 2009, 458, 914–918. [DOI] [PubMed] [Google Scholar]
- DuBois R. M.; Slavish P. J.; Baughman B. M.; Yun M.-K.; Bao J.; Webby R. J.; Webb T. R.; White S. W. Structural and biochemical basis for development of influenza virus inhibitors targeting the PA endonuclease. PLoS Pathog. 2012, 8, e1002830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kowalinski E.; Zubieta C.; Wolkerstorfer A.; Szolar O. H. J.; Ruigrok R. W. H.; Cusack S. Structural Analysis of Specific Metal Chelating Inhibitor Binding to the Endonuclease Domain of Influenza pH1N1 (2009) Polymerase. PLoS Pathogens 2012, 88e1002831. 10.1371/journal.ppat.1002831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tefsen B.; Lu G.; Zhu Y.; Haywood J.; Zhao L.; Deng T.; Qi J.; Gao G. F. The N-terminal domain of PA from bat-derived influenza-like virus H17N10 has endonuclease activity. J. Virol. 2013, 88, 1935–1941 10.1128/JVI.03270-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauman J. D.; Patel D.; Baker S.; Vijayan R. S. K.; Xiang A.; Parhi A.; Martinez-Sobrido L.; LaVoie E. J.; Das K.; Arnold E. Crystallographic fragment screening and structure-based optimization yields a new class of influenza endonuclease inhibitors. ACS Chem. Biol. 2013, 8, 2501–2508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plotch S. J.; Bouloy M.; Ulmanen I.; Krug R. M. A unique Cap(m7CppXm)-dependent influenza virion endonuclease cleaves capped RNAs to generate the primers that initiate viral RNA transcription. Cell 1981, 23, 847–858. [DOI] [PubMed] [Google Scholar]
- Tomassini J.; Selnick H.; Davies M. E.; Armstrong M. E.; Bladwin J.; Bourgeois M.; Hastings J.; Hazuda D.; Lewis J.; McClements W.; Ponticello G.; Radzilowski E.; Smith G.; Tebben A.; Wolfe A. Inhibition of Cap (m7GpppXm)-dependent endonuclease of influenza virus by 4-substituted 2,4-dioxobutanoic acid compounds. Antimicrob. Agents Chemother. 1994, 38, 2827–2837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hastings J. C.; Selnick H.; Wolanski B.; Tomassini J. E. Anti-influenza virus activities of 4-substituted 2,4-dioxobutanoic acid inhibitors. Antimicrob. Agents Chemother. 1996, 40, 1304–1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomassini J.; Davies M. E.; Hastings J.; Lingham R.; Mojena M.; Raghoobar S. L.; Singh S. B.; Tkacz J. S.; Goetz M. A. A novel antiviral agent which inhibits the endonuclease of influenza viruses. Antimicrob. Agents Chemother. 1996, 40, 1189–1193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh S. B. Total synthesis of flutimide, a novel endonuclease inhibitor of influenza virus. Tetrhedron Lett. 1995, 36, 2009–2012. [Google Scholar]
- Carcelli M.; Rogolino D.; Bacchi A.; Rispoli G.; Fisicaro E.; Compari C.; Sechi C.; Stevaert A.; Naesens L. Metal-Chelating 2-Hydroxyphenyl Amide Pharmacophore for Inhibition of Influenza Virus Endonuclease. Mol. Pharmaceutics 2013, 11, 304–316 10.1021/mp400482a. [DOI] [PubMed] [Google Scholar]
- Parkes K. E. B.; Ermert P.; Fässler J.; Ives J.; Martin J. A.; Merrett J. H.; Obrecht D.; Williams G.; Klumpp K. Use of a pharmacophore model to discover a new class of influenza endonuclease inhibitors. J. Med. Chem. 2002, 46, 1153–1164. [DOI] [PubMed] [Google Scholar]
- Chen E.; Swift R. V.; Alderson N.; Feher V. A.; Feng G.-S.; Amaro R. E. Computation-guided discovery of influenza endonuclease inhibitors. ACS Med. Chem. Lett. 2014, 5, 61–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sagong H. Y.; Parhi A.; Bauman J. D.; Patel D.; Das K.; Vijayan R. S. K.; Arnold E.; LaVoie E. J. 3-Hydroxyquinolin(1H)-2-ones: potential inhibitors of influenza A endonuclease. ACS Med. Chem. Lett. 2013, 4, 547–550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parhi A.; Xiang A.; Bauman J. D.; Patel D.; Das K.; Vijayan R. S. K.; Arnold E.; LaVoie E. J. Phenyl substituted 3-hydroxypyridin-2(1H)-ones: potential inhibitors of influenza A endonuclease. Bioorg. Med. Chem. 2013, 21, 6435–6446. [DOI] [PubMed] [Google Scholar]
- Aronov A.; Bandarage U. K.; Cottrell K.; Davies R.; Krueger E.; Ledeboer M.; Ledford B.; Le Tiran A.; Liao Y.; Messersmith D.; Wang T.; Xu J.. Tetrahydrothiazolopyridine Inhibitors of Phosphatidylinositol 3-kinase. (Vertex Pharmaceuticals, Inc.) Patent WO/2010/096389, 2010.
- Fray M. J.; Bull D. J.; Carr C. L.; Gautier E. C. L.; Mowbray C. E.; Stobie A. Structure–Activity Relationships of 1,4-dihydro-(1H,4H)-quinoxalin-2,3-diones as N-methyl-d-aspartate (glycine site) receptor antagonists. 1. Heterocyclic substituted 5-alkyl derivatives. J. Med. Chem. 2001, 44, 1951–1962. [DOI] [PubMed] [Google Scholar]
- Darout E.; Robinson R. P.; McClure K. F.; Corbett M.; Li B.; Shavnya A.; Andrews M. P.; Jones C. S.; Li Q.; Minich M. L.; Mascitti V.; Guimarães C. R. W.; Munchhof M. J.; Bahnck K. B.; Cai C.; Price D. A.; Liras S.; Bonin P. D.; Cornelius P.; Wang R.; Bagdasarian V.; Sobota C. P.; Hornby S.; Masterson V. M.; Joseph R. M.; Kalgutkar A. S.; Chen Y. Design and synthesis of diazatricyclodecane agonists of the G-protein-coupled receptor 119. J. Med. Chem. 2013, 56, 301–319. [DOI] [PubMed] [Google Scholar]
- Wolkenberg S.; Harrison S. T.; Barrow J. C.; Zhao Z.; Kett N.; Zartman A.. Inhibitors of Catechol O-methyl Transferase and their Use in the Treatment of Psychotic disorders. (Merck Sharp & Dohme Corp.) Patent WO/2011/109267, 2011.
- Guerrero M.; Urbano M.; Schaeffer M.-T.; Brown S.; Rosen H.; Roberts E. SAR analysis of novel non-peptide NPBWR1 (GPR7) antagonists. Bioorg. Med. Chem. Lett. 2013, 23, 614–619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagashima H.; Ukai K.; Oda H.; Masaki Y.; Kaji K. Studies on syntheses and reactions of methoxypyridazines. II. Methoxylation of 3,4,6-trichloropyridazine. Chem. Pharm. Bull. 1987, 35, 350–356. [Google Scholar]
- Hondo T.; Warizaya M.; Niimi T.; Namatame I.; Yamaguchi T.; Nakanishi K.; Hamajima T.; Harada K.; Sakashita H.; Matsumoto Y.; Orita M.; Takeuchi M. 4-Hydroxypyridazin-3(2H)-one derivatives as novel d-amino acid oxidase inhibitors. J. Med. Chem. 2013, 56, 3582–3592. [DOI] [PubMed] [Google Scholar]
- Duplantier A. J.; Becker S. L.; Bohanon M. J.; Bozilleri K. A.; Chrunyk B. A.; Downs J. T.; Hu L.-Y.; El-Kattan A.; James L. C.; Liou S.; Lu J.; Maklad N.; Mansour M. N.; Mente S.; Piotrowski M. A.; Sakaya S. M.; Sheehan S.; Steyn; Strick S. J.; Williams V. A.; Zhang Z. Discovery, SAR, and pharmacokinetics of a novel 3-hydroxyquinolin-2(1H)-one series of potent d-amino acid oxidase (DAAO) inhibitors. J. Med. Chem. 2009, 52, 3576–3585. [DOI] [PubMed] [Google Scholar]
- Otwinowski Z.; Minor W.. Processing of X-ray Diffraction Data Collected in Oscillation Mode. In Methods in Enzymology, 276; Carter C. W. J., Sweet R. M., Eds.; Academic Press: New York, 1997; Vol. 276, pp 307–326. [DOI] [PubMed] [Google Scholar]
- Adams P. D.; Afonine P. V.; Bunkoczi G.; Chen V. B.; Davis I. W.; Echols N.; Headd J. J.; Hung L. W.; Kapral G. J.; Grosse-Kunstleve R. W.; McCoy A. J.; Moriarty N. W.; Oeffner R.; Read R. J.; Richardson D. C.; Richardson J. S.; Terwilliger T. C.; Zwart P. H. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2010, 66, 213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.